
Abstract
The rodent neuroblastoma cell line, ND7-23, is used to express voltage-dependent sodium (Nav) and other neuronal ion channels resistant to heterologous expression in Chinese hamster ovary (CHO) or human embryonic kidney (HEK) cells. Their advantage is that they provide endogenous factors and signaling pathways to promote ion channel peptide folding, expression, and function at the cell surface and are also amenable to automated patch clamping. However, ND7-23 cells exhibit endogenous tetrodotoxin (TTX)-sensitive Nav currents, and molecular profiling has revealed the presence of Nav1.2, Nav1.3, Nav1.6, and Nav1.7 transcripts, but no study has determined which subtypes contribute to functional channels at the cell surface. We profiled the repertoire of functional Nav channels endogenously expressed in ND7-23 cells using the QPatch automated patch clamp platform and selective toxins and small molecules. The potency and subtype selectivity of the ligands (Icagen compound 68 from patent US-20060025415-A1-20060202, 4,9 anhydro TTX, and Protoxin-II) were established in human Nav1.3, Nav1.6, and Nav1.7 channel cell lines before application of selective concentrations to ND7-23 cells. Our data confirm previous studies that >97% of macroscopic Nav current in ND7-23 cells is carried by TTX-sensitive channels (300 nM TTX) and that Nav1.7 is the predominant channel contributing to this response (65% of peak inward current), followed by Nav1.6 (∼20%) and negligible Nav1.3 currents (∼2%). In addition, our data are the first to assess the Nav1.6 potency (50% inhibitory concentration [IC50] of 33 nM) and selectivity (50-fold over Nav1.7) of 4,9 anhydro TTX in human Nav channels expressed in mammalian cells, confirming previous studies of rodent Nav channels expressed in oocytes and HEK cells.
Introduction
The sodium channel (Nav) gene family is classified into tetrodotoxin-sensitive (TTX-S; Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7) and TTX-resistant (TTX-R) channels (Nav1.5, Nav1.8, and Nav1.9), each of which is associated with specific therapeutic indications based on their expression pattern, function, and genetic mutations (reviewed in Refs. 1 –3 ). Neuronal voltage-gated sodium channels are important drug discovery targets for pain (Nav1.3, Nav1.7, Nav1.8, Nav1.9), epilepsy (Nav1.1, Nav1.2), and multiple sclerosis (Nav1.6). 4,5 High-throughput screening, hit validation, lead optimization, and gene family selectivity now all largely rely on heterologous expression of specific Nav ion channel subunits in a limited set of mammalian cell backgrounds amenable to cell-based assay and automated patch clamp (APC) electrophysiology platforms. For example, most TTX-sensitive Nav channels express well in human embryonic kidney (HEK) cells, 6,7 but it is noteworthy that HEK cells also exhibit significant levels (100–500 pA) of endogenous TTX-S and TTX-R Nav currents and express Nav1.2, Nav1.3, Nav1.7, and Nav1.5 subunits. 8,9 In contrast, mutant Nav1.6 channels associated with ataxia and epilepsy 10,11 and TTX-resistant Nav1.8 and Nav1.9 channels implicated in neuropathic, inflammatory, and visceral pain have proven resistant to heterologous expression in fibroblast-like Chinese hamster ovary (CHO) or HEK cells. 7,12 –15 Several groups have therefore turned to immortalized neuroblastoma cell lines that contain a more diverse and appropriate set of accessory proteins, 16 successfully expressing mutant Nav1.6 channels in rodent ND7-23 neuroblastoma cells 11 and Nav1.8 channels in human SH-SY5Y 17 and rodent ND7-23 cell lines 7,18 –22 and more recently the recalcitrant hNav1.9 subunit in ND7-23 cells. 23 –25
Although the heterologous expression of Nav1.6 mutant and TTX-resistant Nav channels is higher in neuroblastoma cell lines compared with HEK cells, both of these cell types exhibit a background of endogenous Nav channel activity. This can reduce the signal window as well as compromise the fidelity of drug discovery assays designed to detect subtype selective Nav ligands with improved therapeutic and side effect profiles. 3,4 It is therefore important that both the level of background expression and mix of Nav ion channel subtypes are determined in the various cell lines being used as hosts for heterologous expression of human Nav channels to ensure reliable ion channel drug screening. There are a variety of subtype-selective Nav antagonists available that originated from such drug discovery efforts, which can be used to define Nav1.x expression profiles in native systems. In this study, we used an Nav1.3-selective small molecule patented by Icagen, 26 and the Nav1.7-selective tarantula spider toxin Protoxin-II that was used by Merck & Co., Inc., in their pain drug discovery program, 27 which derives its selectivity through binding to divergent voltage sensor domains on Nav1.x channels. 28 Finally, we used the naturally occurring TTX metabolite 4,9 anhydro TTX 29 that was first shown to be selective for rodent Nav1.6 channels expressed in Xenopus oocytes 30 to define the proportion of Nav1.x channels in ND7-23 neuroblastoma cells.
The ND7-23 immortalized rat dorsal root ganglion (DRG)-mouse N18Tg2 neuroblastoma hybrid cell line 31 has shown great utility for transient and stable expression of exogenous ion channels and receptors in a native milieu, yielding useful fluorescence and electrophysiology assays for academic and drug discovery applications. Given the frequent use of ND7-23 neuroblastoma cells to express TTX-resistant Nav channels and their suitability for APC electrophysiology screening assays, there is a lack of consensus in the literature on the endogenous Nav subunits expressed in this cell line. Several groups have used molecular techniques to identify the presence of mRNA or protein for β1 and β3 accessory subunits 7 and Nav1.2, Nav1.3, Nav1.6, and Nav1.7 pore-forming α subunits in ND7-23 cells. 32,33 However, none of these studies confirmed the contribution of these particular Nav channel subtypes to functional macroscopic currents. Similarly, patch clamp studies of TTX-resistant Nav channels heterologously expressed in ND7-23 cells did not profile the underlying endogenous Nav currents other than to establish that they were completely blocked by TTX. 7,18,20,21,23,32 We therefore undertook to define the repertoire of endogenous Nav1.x channels functionally expressed on ND7-23 neuroblastoma cells using subtype-selective pharmacological ligands.
Materials and Methods
Compounds
The Icagen Nav1.3 compound 68 was synthesized according to Gonzalez et al. 26 and details are given below (Fig. 1). 4,9 Anhydro TTX was synthesized commercially and kindly shared by Mr. Ari Alexandrou (Pfizer-Neusentis). Synthetic Protoxin-II was purchased from Peptanova GmbH. All remaining physiological salts and reagents were obtained from commercial suppliers (Sigma-Aldrich and Calbiochem).
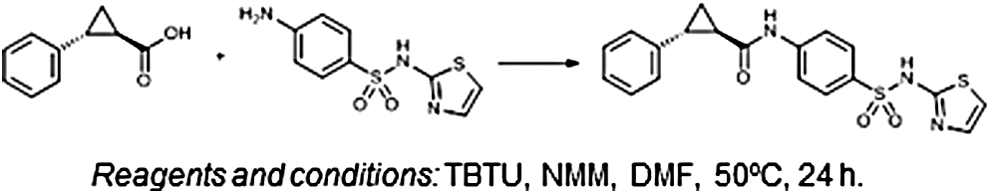
Synthetic route for Icagen compound 68. Schematic showing starting materials and experimental conditions leading to synthesis of Icagen patent compound 68 (1R,2R)-2-phenyl-N-(4-(N-(thiazol-2-yl)sulfamoyl)phenyl)cyclopropane-1-carboxamide. DMF, dimethyl formamide; NMM, N-methylmorpholine, TBTU, N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate.
Synthesis of Icagen Nav1.3 Compound 68
Chemicals were obtained from Acros Organics and Sigma-Aldrich and used without further purification. Analytical thin-layer chromatography was performed on silica gel Merck 60 F254 plates (0.25 mm), using visualization with ultraviolet light and spray reagents. High-performance liquid chromatography (HPLC) analyses were performed on an Agilent Technologies 1100 instrument (Agilent Technologies) with a G1365B UV-Vis detector, a G1316A thermostat, and a G1313A autosampler using a Phenomenex Luna 5 μm C18 column (4.6 × 150 or 4.6 × 250 mm; Phenomenex) and a flow rate of 1.0 mL/min. The eluent consisted of trifluoroacetic acid (TFA; 0.1% in water) as solvent A and acetonitrile as solvent B. Melting points were determined on a Reichert hot-stage microscope and are uncorrected. 1 H and 13 C NMR (nuclear magnetic resonance) spectra were recorded at 400 and 100 MHz, respectively, on a Bruker AVANCE III 400 spectrometer (Bruker Corporation) in dimethyl sulfoxide (DMSO)-d6, with TMS as the internal standard. Infrared (IR) spectra were recorded on a PerkinElmer Spectrum BX FT-IR spectrometer (PerkinElmer, Inc.) or Thermo Nicolet Nexus 470 ESP FT-IR spectrometer (Thermo Fisher Scientific). Mass spectra were obtained using an Advion Expression CMS mass spectrometer (Advion).
The synthetic procedure for (1R,2R)-2-phenyl-N-(4-(N-(thiazol-2-yl)sulfamoyl)phenyl)cyclopropane-1-carboxamide (Fig. 1) is as follows. To the solution of (1R,2R)-2-phenylcyclopropane-1-carboxylic acid (100 mg, 0.617 mmol) and TBTU (238 mg, 0.740 mmol) in DMF (3 mL), N-methylmorpholine (203 μL, 1.85 mmol) was added, and the mixture was stirred at room temperature for 0.5 h. After that 4-amino-N-(thiazol-2-yl)benzenesulfonamide (157 mg, 0.617 mmol) was added and the mixture was stirred at 50°C overnight. To the reaction mixture, ethyl acetate (30 mL) was added and the organic phase was washed with water (2 × 10 mL), 10% citric acid (2 × 10 mL), 1 M HCl (3 × 10 mL), and brine (2 × 10 mL), dried over Na2SO4, and evaporated under reduced pressure. To the residue, ether (20 mL) was added, the obtained suspension was sonicated, the undissolved solid was filtered off, washed with ether (10 mL), and dried to obtain (1R,2R)-2-phenyl-N-(4-(N-(thiazol-2-yl)sulfamoyl)phenyl)cyclopropane-1-carboxamide as a pale yellow solid (142 mg); yield was 58% and melting point was 191°C–195°C. Spectra are as follows: IR (ATR) ν = 3245, 3103, 3057, 2899, 1652, 1593, 1566, 1519, 1406, 1313, 1282, 1254, 1193, 1138, 1085, 931, 932 cm−1; 1 H NMR (400 MHz, DMSO-d6) δ 1.39–1.44 (m, 1H, CH), 1.49–1.54 (m, 1H, CH), 1.92–2.00 (m, 1H, CH), 2.07–2.12 (m, 1H, CH), 6.82 (d, 1H, J = 4.8 Hz, thiazole-H), 7.18–7.32 (m, 6H, Ar-H), 7.73 (s, 4H, Ar-H), 10.60 (s, 1H, CONH), 12.69 (s, 1H, SO2NH); 13 C NMR (100 MHz, DMSO-d6) δ 15.85, 25.32, 26.80, 108.05, 118.37, 124.35, 125.89, 126.18, 126.97, 128.36, 136.12, 140.55, 142.27, 168.67, 170.39. MS (ESI) m/z (%) = 397.87 ([M-H]-). HPLC was carried out using a Phenomenex Luna 5 μm C18 column (4.6 × 150 mm); mobile phase: 30%–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL/min; injection volume: 10 μL; retention time: 8.475 min (98.7% at 280 nm).
Cell Culture
CHO and HEK heterologous cell lines stably expressing hNav1.3 (CYL3003), hNav1.6 (CYL3010), and hNav1.7 channels (CYL3011) were grown according to the manufacturer's instructions (Cytomyx/Millipore). All of the Nav1.x cell lines express the α pore-forming subunit under a single selection agent, with the Nav1.6 and Nav1.7 cell lines presumably interacting with endogenous β1 subunits in HEK cells. 6,7 Routine passaging every 2–3 days using agents such as divalent-free phosphate-buffered saline and Accutase® (ThermoFisher) and growth in suitable tissue culture-treated flasks (Corning®) ensure that cells are maintained in log-phase growth and do not form islands or exceed 75% confluency, critical parameters for automated patch clamping. ND7-23 cells were obtained from European Collection of Authenticated Cell Cultures and grown according to guidelines to maintain uniform confluency and morphology; cells were not differentiated. ND7-23 cells were passaged in a similar manner to heterologous Nav1.x cells lines to ensure optimum patchability (gigaseal and whole-cell formation, and seal quality) on the QPatch APC platform.
APC Electrophysiology
All of the Nav channel electrophysiology experiments in this study were carried out on the QPatch APC system (Sophion Bioscience A/S), which we have used to make high-fidelity Nav1.x recordings at Xention since 2006. The QPatch is a silicon chip-based planar patch clamp device allowing for up to 48 parallel recordings during one experimental run. Cells are added to each well on the QPlate chip and drawn by suction onto a small aperture to obtain a gigaohm seal (>100 MΩ) between the cell membrane and treated silicon surface, after which a whole-cell recording is achieved by applying a combination of suction and voltage pulses to break through the cell membrane and allow perfusion of the cell interior. Most of our experiments employed single-hole QPlates (2.5 MΩ resistance), but 10-hole X-Plates were employed for heterologous hNav1.3 recordings to give improved current amplitude and more consistent stability. The QPatch platform and silicon chip QPlates enable high-fidelity recordings of large and rapid Nav currents due to the combination of gigaohm seal quality recordings and amplifier circuits offering continuous voltage clamp with capacitive and series resistance compensation (≥65%). All Nav1.x cell lines used in this study have been validated in-house on the QPatch by comparing their voltage dependence, biophysics, and pharmacology to manual and APC data in the literature and information available from APC and cell line vendors and other contract screening companies.
Cells were prepared for QPatch experiments by manual dissociation from T175 cell culture flasks using either 0.05% trypsin-EDTA (ThermoFisher) or Accutase followed by centrifugation to remove enzyme and debris and resuspension in serum-free media (CHO-III from ThermoFisher or Excel-302 from Sigma-Aldrich). Cells were kept in suspension by constant stirring in the cell hotel on the QPatch before they were washed and resuspended in extracellular recording solution using the Q-fuge unit on-board the QPatch immediately before application to recording wells on the QPlate chip.
Extracellular solution contained (in millimolars) 140 NaCl, 5 KCl, 1 CaCl2, 1.2 MgCl2, and 5 HEPES (pH 7.4), unless inward Nav currents were too large, in which case, we reduced the external Na+-permeant cation by making an appropriate admixture from a similar solution in which 140 NaCl was substituted with 140 mM choline chloride. Standard intracellular solution contained (in millimolars) 15 NaCl, 120 CsF, 10 HEPES, 10 EGTA, and 10 HEPES (pH 7.2). The combination of these solutions was also sufficient to isolate inward Nav currents in ND7-23 cells as they effectively inhibited contaminating potassium currents (also present in HEK cells).
The QPatch platform uses static perfusion, where a small volume of recording solution or drug is added to a reservoir on the chip and passes through quartz-lined microfluidic channels to perfuse each cell recording site. This volume is removed by capillary action when the next sample application is made, and excess cells are also removed from the recording chamber by vehicle washes before any compound is added. After achieving the whole-cell configuration, the appropriate vehicle (0.1% DMSO v/v or 0.05% bovine serum albumin [BSA] w/v) was applied to each cell to acquire a control recording of Nav amplitude (usually 4-min period); achieving a stable baseline during the vehicle period is a critical quality control parameter. The potency (IC50, the 50% inhibitory concentration) of compounds against sodium channel subtypes was determined by cumulatively applying three or four escalating concentrations (typically in half log-unit increments) to each cell to construct individual concentration–response relationships. We employed either a single bolus application (10 μL) of each test concentration or double addition (at t = 0 and t = 2 min) to expose each cell to a 4-min incubation at each test concentration. Compounds were prepared by dilution (from a 1,000× stock) in extracellular solution (in appropriate vehicle) to yield a top concentration that was serially diluted into glass-lined wells of a microtiter plate compound plate containing extracellular solution and vehicle to avoid any exposure to excess plastic and loss of compound or changes in osmolality. All stock solutions were frozen as aliquots from the master and thawed and used fresh on each day, ensuring only a single freeze–thaw cycle per use.
Currents were elicited from Nav1.3, Nav1.6, and Nav1.7 cell lines and ND7-23 cells using a standard two-pulse voltage protocol. From a holding potential of −100 mV, a 20-ms activating step to the peak of the IV for each Nav1.x channel (0, −10, −20, and −10 mV for Nav1.3, Nav1.6, Nav1.7, and TTX-S, respectively) was applied to assess the effect of compounds on channels opened from the resting (closed) state (Peak 1). The second activating pulse was applied following a 5-s prepulse to a membrane potential that induces ∼50% inactivation of available channels to assess block on the inactivated state of the channel. The half-inactivating potential varied depending on Nav1.x type and tissue culture conditions and was adjusted each day for each target (typically ranging between −70 and −50 mV). The two-step voltage protocol was applied at a sweep interval of 0.067 Hz throughout the duration of the experiment. Leak subtraction was applied between the voltage sweeps, but the length of the voltage protocol only allowed two leak sweeps per epoch, limiting their utility and our use of this method. Rather, sweep subtraction was employed in the QPatch software that enables a single or averaged number of sweeps to be subtracted from all other sweeps in the experiment; applying a saturating dose of 300 nM TTX to each cell at the end of every experiment enabled effective sweep subtraction and removal of capacitive artifacts and contaminating (e.g., outward) currents.
For analysis of hNav1.3, hNav1.6, and hNav1.7 currents in heterologous cells and endogenous TTX-S channels in ND7-23 cells, the peak inward current amplitude was measured using QPatch assay software (v5.0) by placing cursors over the two test pulses evoking resting- (Peak 1) and half-inactivated state (Peak 2) currents during each voltage protocol sweep. The % inhibition of peak current was calculated from the mean peak current value for the last three sweeps in each concentration period in the application protocol divided by the average of the last three sweeps recorded at the end of the initial vehicle period. Sigmoidal concentration–response curves (four parameter logistic curve) were fitted to the % inhibition data using Prism (v6; GraphPad) to determine the mean IC50 value. Fits were constrained to include 0% and 100% inhibition at very low and high compound concentrations, respectively, and the hillslope was a free parameter (QC filter was 0.5 < nH >2.0). Data are presented as mean ± standard deviation (SD) or standard error of the mean (SEM; as indicated) for a minimum of three independent observations. All compounds were tested in at least two independent experiments conducted with separate cell preparations on different days and separate QPlates.
Results
Confirming Nav1.x Selectivity of Pharmacological Antagonists
The consensus from previous molecular characterizations of endogenous Nav channels expressed in ND7-23 cells indicates the presence of Nav1.3, Nav1.6, Nav1.7, and possibly Nav1.2 α subunit channel transcripts. 32,33 To confirm the presence of functional Nav currents carried by these pore-forming subunits and determine their relative contribution to total inward Nav current, we would need to make single and additive applications of selective antagonists to individual ND7-23 cells on the QPatch APC platform. As most of these ligands had been characterized previously in Xenopus oocytes or by manual patch clamp, we needed to establish their efficacy and Nav1.x selectivity on the QPatch screening platform before using them on ND7-23 cells. Selective ligands were available for Nav1.3, Nav1.6, and Nav1.7 channels, but selective antagonists for Nav1.2 channels were not available at the time of these experiments (e.g., conotoxin TIIIA, tarantula toxins, PaurTx3 or CcoTx 34,35 ). To assess the state-dependent inhibition of Nav1.x channels by the chosen ligands, we employed a two-pulse voltage protocol, whereby the first test pulse (Peak 1) was evoked from a negative holding potential (typically −100 mV) to assay all channels available from their resting state, and the second test pulse (Peak 2) was delivered 5 s after a prepulse to a voltage chosen to induce ∼50% inactivation of available Nav1.x channels. There was no return to holding potential after the inactivating prepulse, so this protocol measures ligand potency against both fast and slow inactivated states of each Nav1.x channel. There is a sufficient interval upon return to the negative holding potential after the second pulse and the next application of the two-pulse voltage protocol to allow complete recovery from fast and slow inactivation (data not shown).
Icagen compound 68 (Nav1.3)
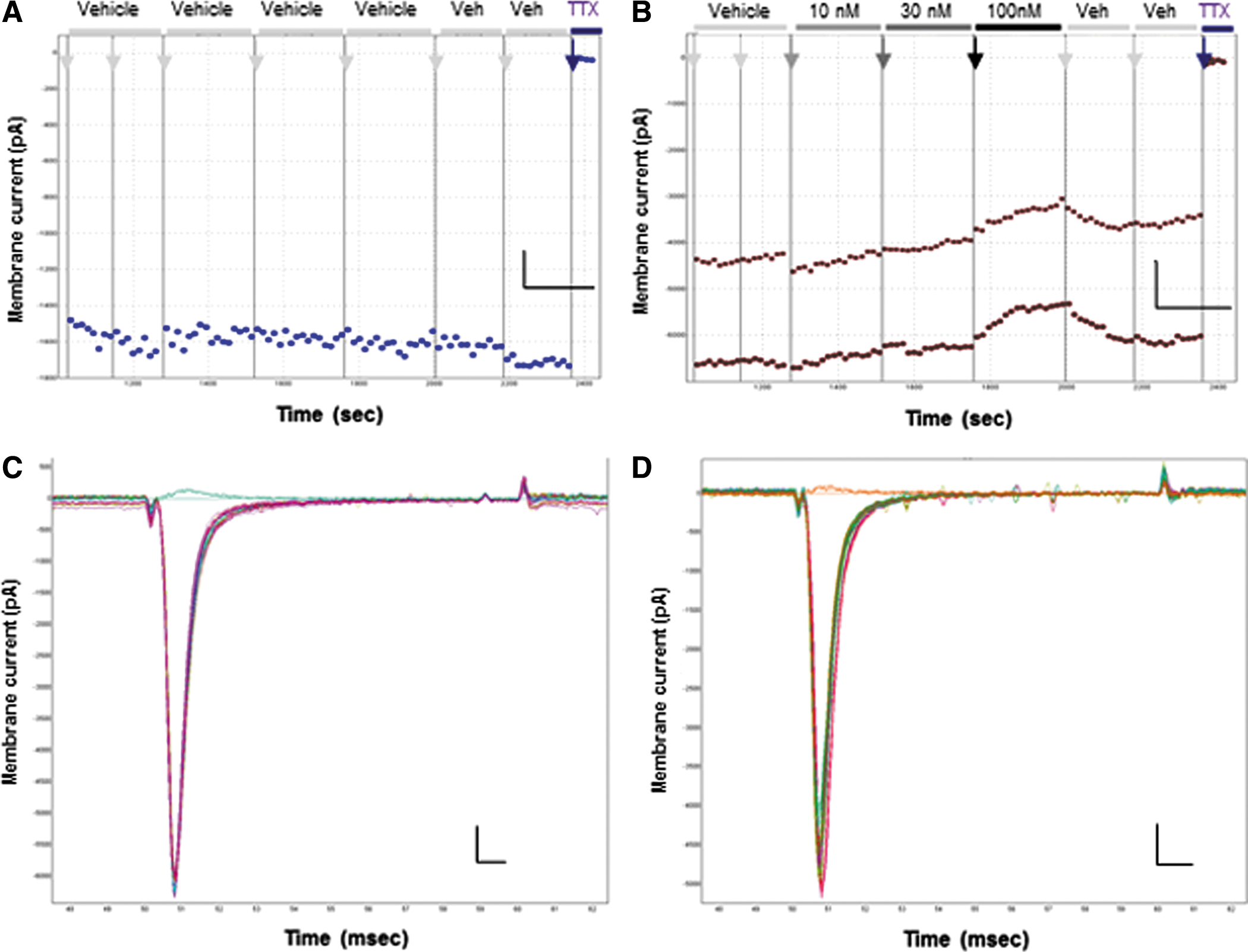
Icagen, Inc., has published a number of patents and articles describing potent and selective antagonists of Nav1.3 channels. 26,36 We synthesized several examples and analogs of their arylsulfonamide series as part of our work to discover and design marine-derived inhibitors of ion channels within the EU Framework Program 7 grant-funded MAREX consortium. 37 Icagen patent compound 6826 was chosen to be the selective Nav1.3 inhibitor in this study as it is a potent antagonist of Nav1.3 channels with no effects on Nav1.4, Nav1.5, Nav1.6, or Nav1.7 channels. 38,39 The efficacy of Icagen compound 68 against hNav1.3 channels expressed in CHO cells was determined using the QPatch (Fig. 2 and Table 1). As expected, 36 this compound demonstrates strong state-dependent inhibition of Nav1.3 (Fig. 2B), weakly reducing the amplitude of resting-state (Peak 1) currents with an estimated IC50 of 18 μM (∼20% inhibition at 10 μM), while dose-dependently inhibiting inactivated (Peak 2) Nav1.3 currents (Fig. 2C) with an IC50 of 56 nM (Fig. 2D). Both resting- and inactivated-state Nav1.3 currents are stable in 0.1% DMSO vehicle during the course of the QPatch IC50 testing protocol (Fig. 2A), with only a small amount of rundown (∼15%) seen in inactivated Peak2 currents during the 20-min time course of the experiment, which is typical for whole-cell Nav1.x recordings on QPatch. The average amplitude of inactivated Peak 2 currents at the end of the initial vehicle period in both vehicle (DMSO) and compound 68 datasets (∼45%–50% of resting-state Peak 1 amplitude, respectively) indicates that the prepulse voltage chosen for these Nav1.3 recordings achieved close to the desired level of 50% steady-state inactivation.

Characterization of Icagen compound 68 as an Nav1.3 channel inhibitor. The Icagen patent compound
Summary of Nav1.x-Selective Pharmacology and ND7-23 Cell TTX-S Current Components
Percent of inhibition produced by selective concentrations of Icagen compound 68 (300 nM), 4,9 anhydro TTX (100 nM), or Protoxin-II (10 nM) applied to different cells.
Determined against mouse Nav1.6 channels. 39
From Ref. 39
IC50, 50% inhibitory concentration; ND, not determined; NE, no effect; TTX-S, tetrodotoxin-sensitive.
The Nav1.3 selectivity of Icagen compound 68 was previously determined on QPatch (hNav1.5) and in voltage clamp recordings from mammalian Nav1.x channels expressed in Xenopus oocytes (rNav1.4, mNav1.6, hNav1.7) by ourselves and our MAREX collaborators, with no significant effects on resting or inactivated current amplitudes in any of the Nav1.x isoforms tested. 39
4,9 Anhydro TTX (Nav1.6 vs. Nav1.7)
Given the interest in subtype-selective Nav1.x channel ligands for various therapeutic drug discovery efforts and the role of Nav1.6 channels in axonal excitability 40 and diseases such as multiple sclerosis and epilepsy, 11,41 it was surprising that an initial finding that the natural TTX metabolite 4,9 anhydro TTX is a selective inhibitor of Nav1.6 channels 30 had not been confirmed in human Nav1.6 channels. As previous studies had predominantly used rodent Nav1.x channels expressed in Xenopus oocytes or HEK cells, 42 we first confirmed the potency and selectivity of 4,9 anhydro TTX against human Nav1.x channels expressed in mammalian cells before using it to dissect out endogenous TTX-S channels in ND7-23 cells.
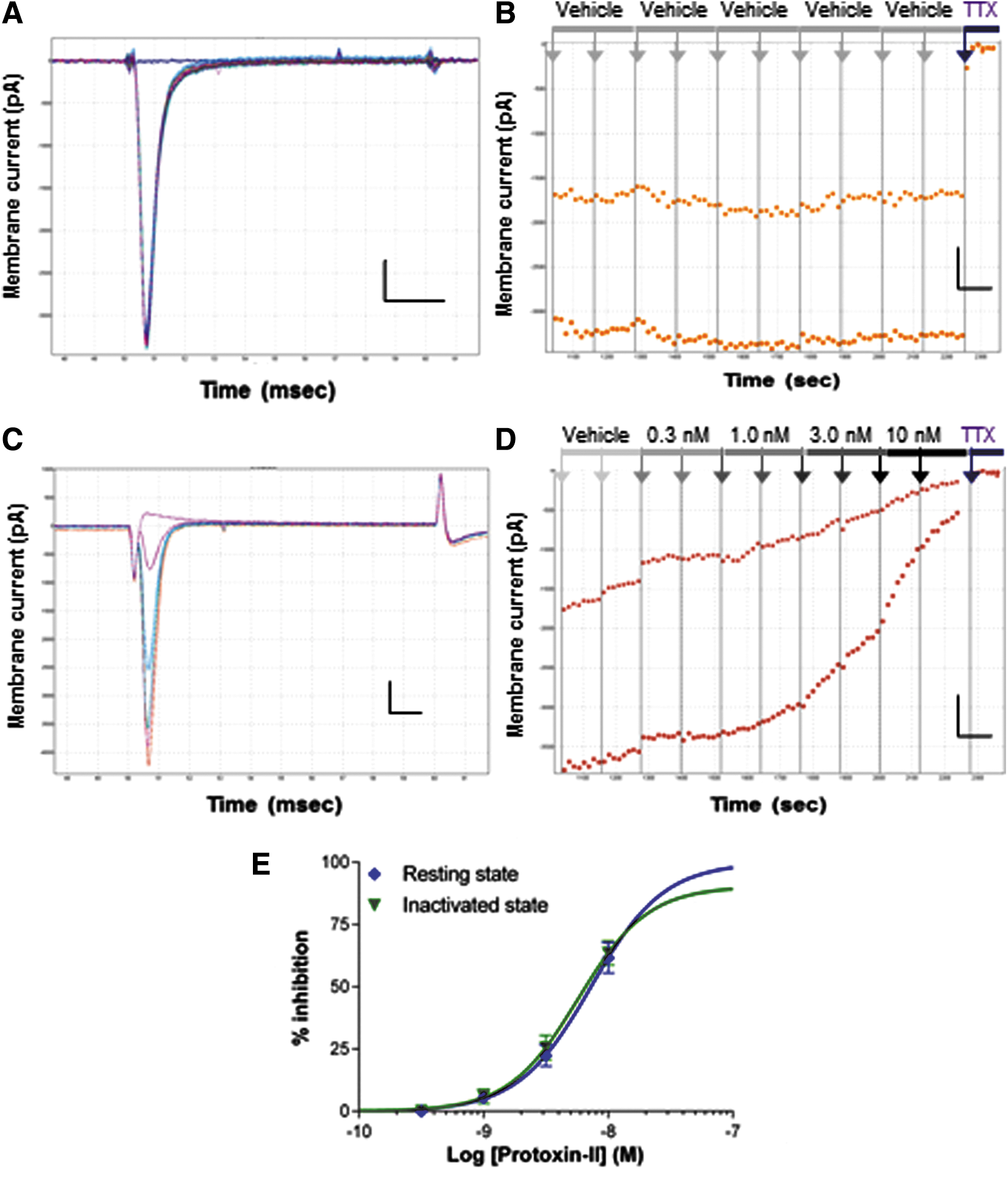
Whole-cell QPatch recordings from HEK cells stably expressing the human Nav1.6 channel revealed inward currents (1–5 nA amplitude) with a rapidly activating and fully inactivating profile evoked by positive test pulses from a negative holding potential of −100 mV. Nav1.6 currents were stable in vehicle control throughout the whole-cell recording (Fig. 3A, B), while those recorded in cells on the same QPlate, but exposed to 4,9 anhydro TTX, were inhibited in a dose-dependent manner (Fig. 3C). An overlay of macroscopic currents recorded in vehicle and in the presence of increasing concentrations of 4,9 anhydro TTX show no discernible differences in activation or inactivation kinetics (Fig. 3D). The onset of block by 4,9 anhydro TTX (Fig. 3C) was only slightly slower than that seen with TTX, taking several sweeps to reach saturating effect at each concentration. The mean IC50 of 32.9 nM for inhibition of resting hNav1.6 channels (Fig. 3E and Table 1) was slightly less potent than originally obtained for mouse Nav1.6 channels in oocytes (7.8 nM), 30 but fourfold more potent than found by manual patch of rat Nav1.6 channels expressed in HEK cells. 42 Both the level of maximal block (65% at 100 nM) and hillslope (0.6) were slightly lower than in the original oocyte study, but similar to the hillslope for rat Nav1.6 channels in HEK cells. 42 In determining the IC50, we assumed that maximal block could be achieved at concentrations higher than the 100 nM limit we imposed due to material availability, and the adequate curve fit to the mean data in Figure 3E (r 2 = 0.94, data not shown) indicates that this is a reasonable assumption when using a somewhat shallow hillslope.
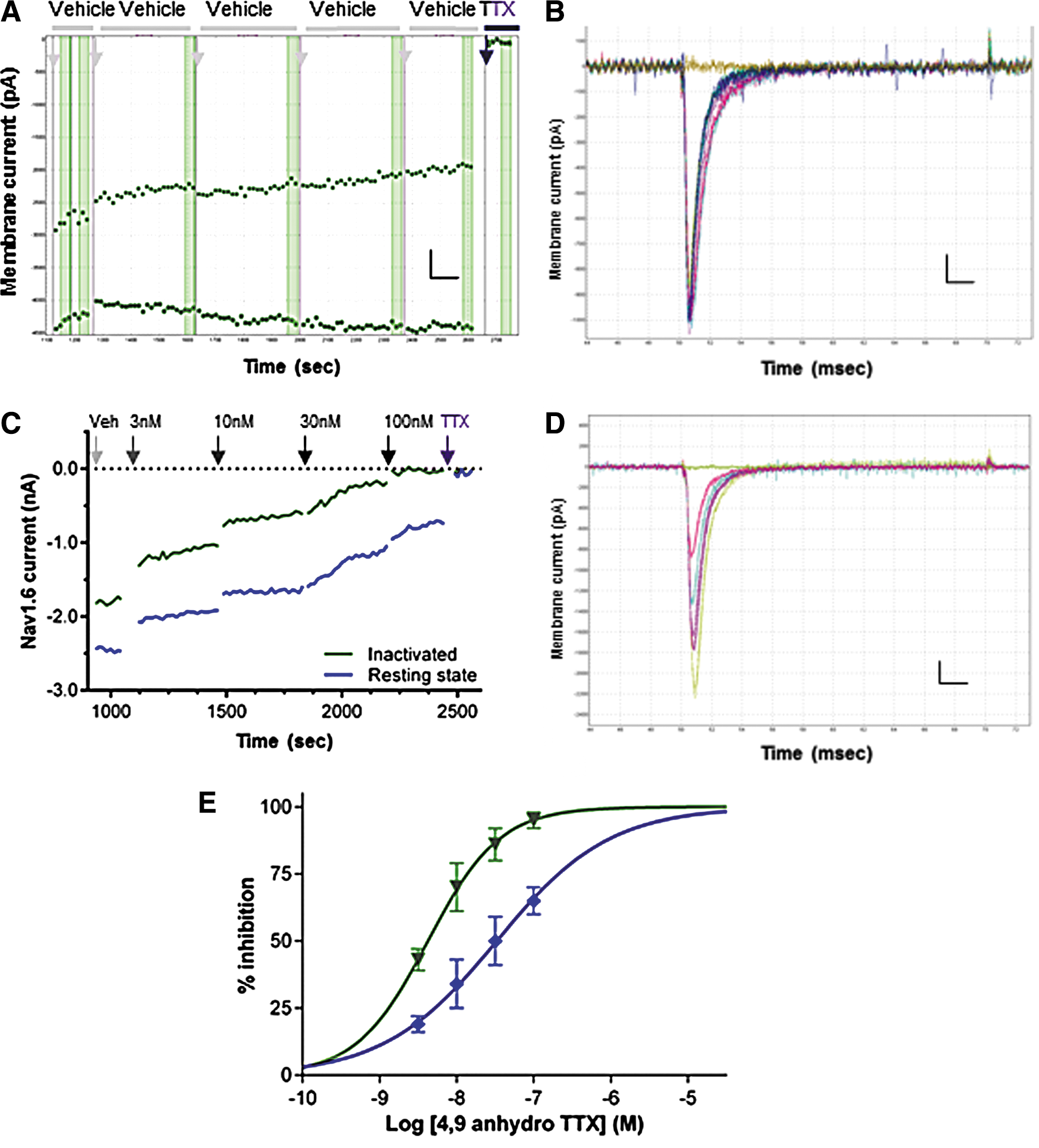
Potency of 4,9 anhydro TTX against human Nav1.6 channels.
We observed that the potency of 4,9 anhydro TTX was significantly higher for Nav1.6 currents evoked after an inactivating prepulse (Peak 2 IC50 of 4.3 nM, Fig. 3E) than that for block of the open state evoked from a negative resting potential. This is indicative of a state-dependent mechanism of 4,9 anhydro TTX to bind with higher affinity to a conformational state associated with the inactivated state, similar to observations with TTX. 43,44 Thus, to simplify the use of 4,9 anhydro TTX as a ligand to discriminate between Nav1.x channel subtypes in ND7-23 cells, we concentrated on its effects on the resting state of Nav1.x channels.
As molecular profiling indicates that Nav1.7 is the only other major TTX-S Nav subtype expressed in ND7-23 cells, we limited our selectivity profiling of 4,9 anhydro TTX to human Nav1.6 and Nav1.7 channels. Inward currents evoked in HEK cells stably expressing hNav1.7 channels using the same voltage protocol described above exhibit rapid activation and inactivation kinetics (Fig. 4C, E). Both hNav1.7 current amplitude (Fig. 4A) and kinetics are stable in vehicle, indicating that the rapid inward currents of ≤5 nA amplitude are well clamped in single-hole gigaseal recordings on the QPatch platform. All inward currents in hNav1.7-expressing cells were completely inhibited by addition of 300 nM TTX to each cell at the end of the experiment (Fig. 4A–E). Repeating the experiment of Figure 3C on cells expressing hNav1.7 channels shows no inhibition of resting-state Nav1.7 current amplitude by 4,9 anhydro TTX up to a test concentration of 100 nM (Fig. 4B, C), whereas the same concentration reduced hNav1.6 currents by >50%. Increasing the 4,9 anhydro dose range by 30-fold reveals dose-dependent inhibition of hNav1.7 currents (Fig. 4D, E) to yield an IC50 for resting-state Peak 1 currents of 1.62 μM (Fig. 4F). As with hNav1.6 channels, the kinetics of hNav1.7 channel block by 4,9 anhydro TTX are slightly slower than seen with TTX (300 nM) added at the end of each experiment.
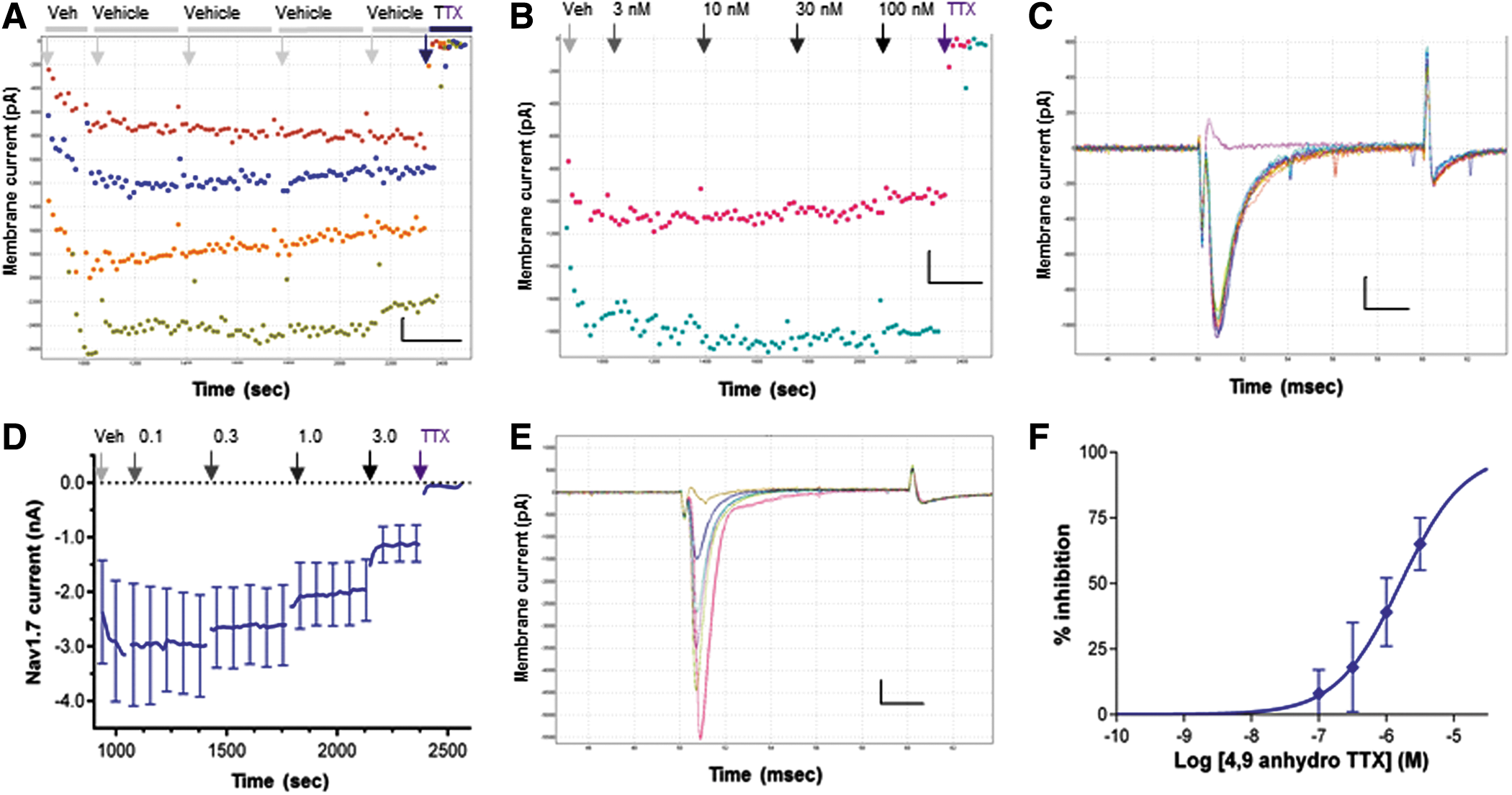
Potency and selectivity of 4,9 anhydro TTX against human Nav1.7 channels.
These data reveal that 4,9 anhydro TTX is a potent inhibitor of human Nav1.6 channels with 50-fold selectivity over hNav1.7 channels (Table 1), confirming the selectivity shown previously in rodent Nav1.x channels expressed in oocytes. 30 We conclude that we can use 4,9 anhydro TTX at a moderate concentration of 100 nM to distinguish between the activity of Nav1.6 and Nav1.7 channels in ND7-23 cells.
Protoxin-II (Nav1.7)
The Nav1.7 selectivity of Protoxin-II has been established by several groups using manual patch clamp of human Nav1.x channels expressed in mammalian cells, 27 so we first confirmed its efficacy against human Nav1.7 channels in HEK cells on the QPatch before using it as a tool to probe Nav currents in ND7-23 cells. Protoxin-II is not as water soluble as TTX, but to prevent any nonspecific loss of peptide activity or absorption onto glass surfaces, we added 0.05% BSA to all solutions used in the QPatch experiments with this ligand. Accordingly, we first determined that 0.05% BSA vehicle would not affect Nav current stability or create artifacts (e.g., bubbles) with the small volumes and laminar flow channels employed on the QPlates. Figure 5A and B illustrates that hNav1.7 currents in HEK cells (and TTX-S currents in ND7-23 cells, Fig. 8A, B) are stable in the presence of 0.05% BSA with no change in inward current amplitude or kinetics during the course of a ∼20-min QPatch experiment, thereby enabling reliable screening and pharmacological profiling of agents dissolved in this vehicle. Rapidly activating and inactivating resting-state hNav1.7 currents (1.5–6 nA peak amplitude) were well clamped by the QPatch (Fig. 5A, C). Nav1.7 currents exhibited dose-dependent inhibition by nanomolar concentrations of Protoxin-II (Fig. 5C, D), yielding an IC50 of 3.7 nM for Peak 1 currents (Fig. 5E). Inactivated-state Peak 2 Nav1.7 currents were inhibited by Protoxin-II (Fig. 5D) with little state dependence, yielding an average IC50 of 2.3 nM (Fig. 5E, Table 1).
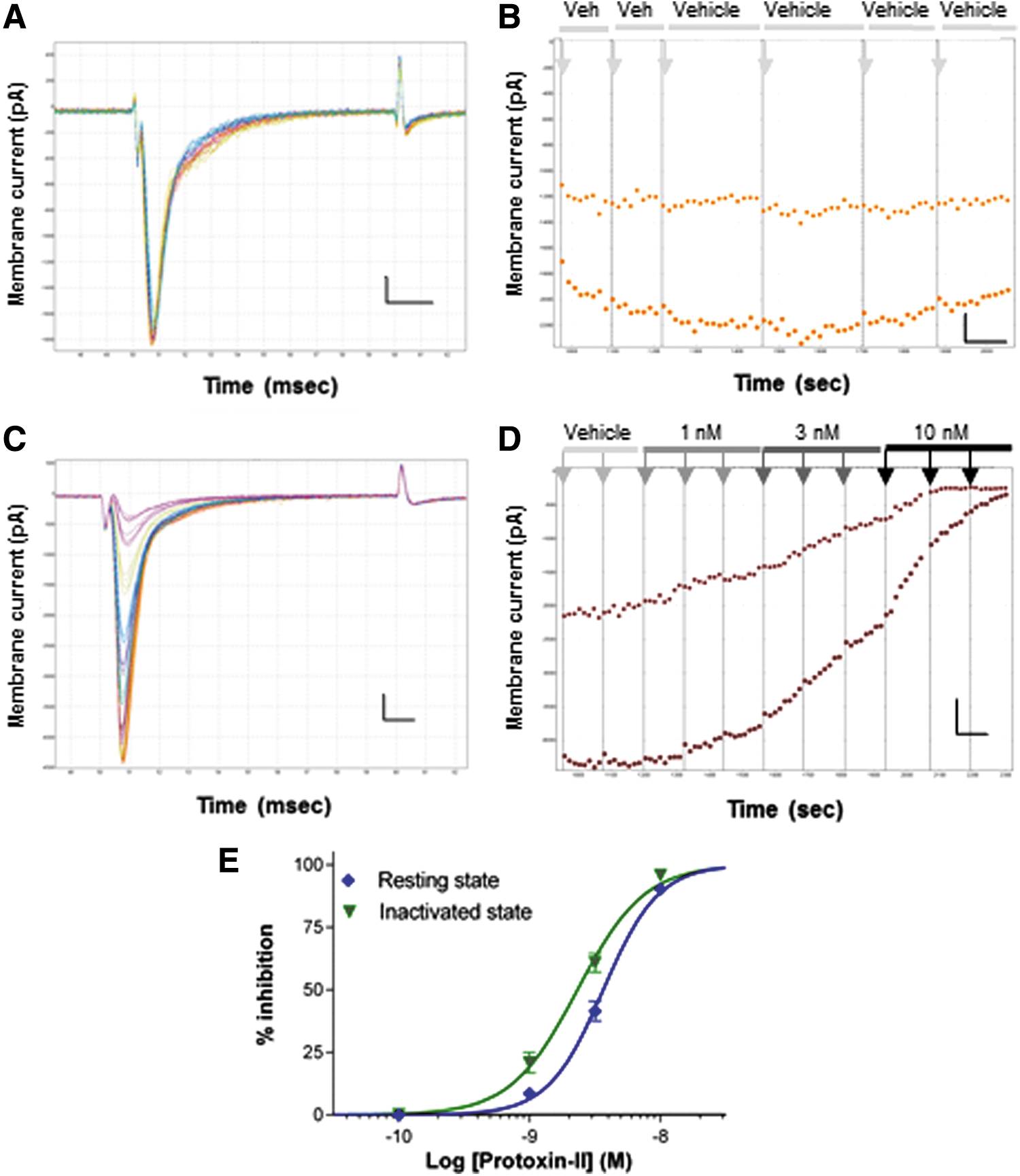
Confirming potency and state dependency of Protoxin-II for hNav1.7 channels on QPatch.
To maximize current block by this slow-acting toxin, we employed triple additions of each test concentration. The rate of block of Nav1.7 currents by Protoxin-II increased with higher peptide concentrations and saturated at >90% inhibition at the top concentration tested of 10 nM. Using higher concentrations of Protoxin-II to achieve full block of Nav1.7 was avoided for two reasons. First, higher doses (≥100 nM) can induce sustained currents through Nav1.7 channels. 45 Second, although the 70- to 100-fold selectivity of Protoxin-II for Nav1.7 over other TTX-S channels is exceptional, 46 Nav1.6 is the second most sensitive Nav1.x channel to Protoxin-II with an IC50 of 26 nM. 27 This profile creates limited selectivity of Protoxin-II for Nav1.7 over other TTX-S channels known to be expressed in ND7-23 cells and puts an upper limit of 10 nM on the concentration of Protoxin-II that can be used to maintain specificity for Nav1.7 channels.
The fact that our QPatch validation studies were conducted on human Nav1.7 channels, but the profiling of endogenous Nav1.x channels was to be carried out in a rodent cell line, raises the potential for species differences in Nav1.x selectivity. However, previous data 27,45 indicate that there is no significant difference in the potency or Nav1.x selectivity of Protoxin-II between human and rat TTX-S sodium channels. In addition, the potency for 4,9 anhydro TTX inhibition of hNav1.6 channels was similar in this study to that obtained for mouse Nav1.6 channels 30 and although the potency against rat Nav1.6 channels expressed in HEK cells was lower (122 nM), those authors selected 100 nM 4,9 anhydro TTX as the specific concentration. 42 Taking these observations together, a concentration of 100 nM 4,9 anhydro TTX and 10 nM Protoxin-II was considered to be selective for Nav1.6 and Nav1.7 channels, respectively, during subsequent experiments to determine the profile of functional Nav1.x channels in ND7-23 cells.
Determining the Functional Repertoire of Nav1.x Channels in ND7-23 Cells
Undifferentiated ND7-23 cells can be dissociated into single-cell suspensions that resemble HEK cell preparations and perform extremely well on APC platforms, yielding a high proportion of gigaohm seals that successfully convert into stable whole-cell recordings with good access and low series resistance suitable for high-fidelity recordings of endogenous ion channel currents. In contrast to QPatch recordings from CHO and HEK cells expressing heterologous Nav channels that can show runup or rundown of resting and inactivated current amplitude (e.g., Figs. 2A , 4A , and 5C), endogenous Nav currents in dissociated ND7-23 cells are remarkably stable throughout the 20-min duration of a typical IC50 experiment (Figs. 6A , 7A , and 8C). A more negative resting membrane potential in neuronal ND7-23 cells could account for this stability as it allows a greater proportion of endogenous Nav channels to reside in a fully available resting state with little inactivation. In contrast, the depolarized resting potential in CHO and HEK cells leads to accumulation of heterologous Nav channels in a long-lived inactivated state from which they slowly recover at the beginning of a whole-cell recording (runup) and quickly return to during the experiment (rundown).
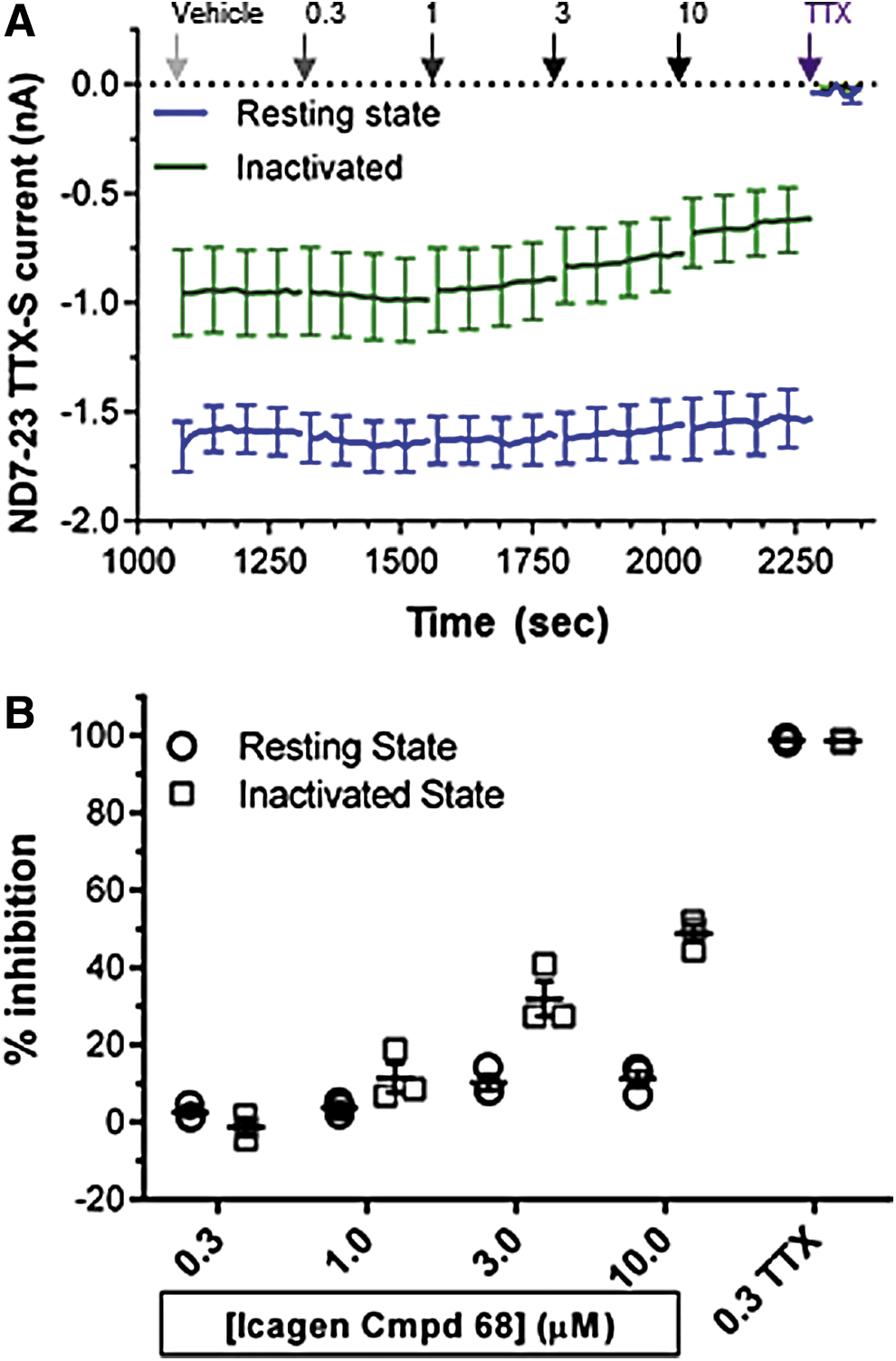
Effect of an Nav1.3 inhibitor on TTX-S currents in ND7-23 cells.
Inhibition of TTX-S currents in ND7-23 cells by selective Nav1.6 antagonist 4,9 anhydro TTX.
Inhibition of TTX-S currents in ND7-23 cells by selective Nav1.7 antagonist Protoxin-II.
Consistent with previous molecular, biophysical, and pharmacological studies, our results show that all of the endogenous Nav currents in ND7-23 cells are carried by TTX-S channels. Application of 300 nM TTX at the end of each experiment in every ND7-23 cell inhibited 98.4% ± 2.0% (mean ± SD from N = 33 cells) of peak inward resting-state Peak 1 currents and 300 nM TTX inhibited inactivated-state Peak 2 currents to a similar extent (Figs. 9 and 10B).
Individual components of TTX-S current in ND7-23 cell inhibited by selective concentrations of Nav1.3, Nav1.6, and Nav1.7 antagonists. Individual cell and mean % inhibition (±SEM) of resting-state (filled circles) and inactivated-state TTX-S currents (open squares) in the presence of the indicated μM concentrations of the Nav1.3 inhibitor ICA (N = 5) and nanomolar concentrations of the Nav1.6 antagonist, 4,9 anhydro TTX (N = 4), and Nav1.7 blocker, Protoxin-II (N = 6), are shown from independent samples of ND7-23 cells. Note the moderate inactivated state preference of the Icagen compound and 4,9 anhydro TTX compared with equipotent inhibition of resting- and inactivated-state TTX-S currents by Protoxin-II. A concentration of 300 nM TTX added to each cell at the end of every experiment completely reduced the remaining resting- and inactivated-state currents (resting state 98.5% ± 0.64% of vehicle control amplitude (N = 26). ICA, Icagen compound 68.
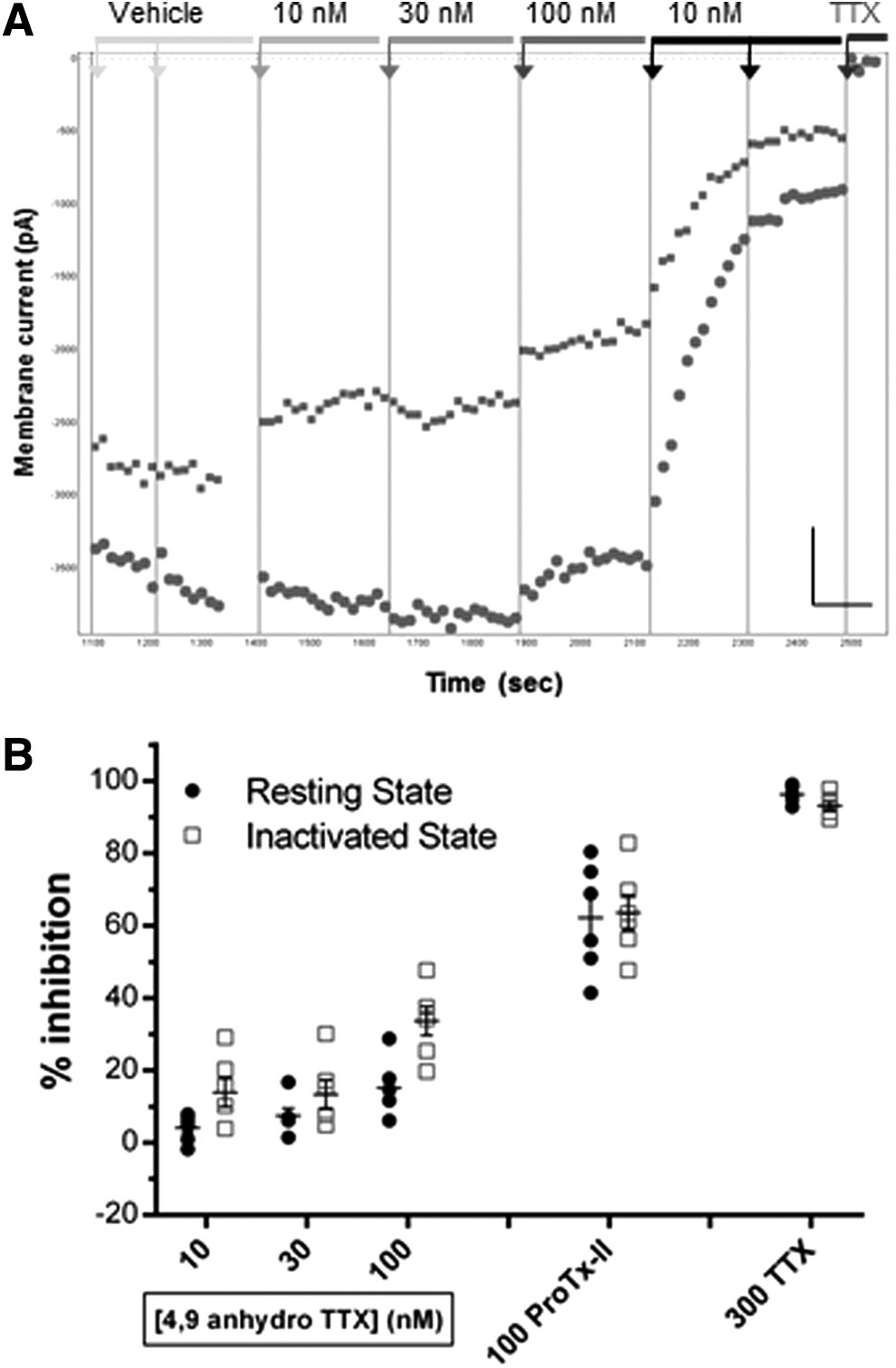
Additive effects of selective Nav1.6 and Nav1.7 antagonists on TTX-S currents in ND7-23 cells.
Application of a high, but moderately selective, concentration (300 nM) of the Icagen Nav1.3 compound 68 produced little discernible effect on either resting-state or inactivated-state TTX-S currents in ND7-23 cells (Fig. 6), even though this was 5× higher than the inactivated-state IC50 determined with a similar voltage protocol using heterologous Nav1.3 channels (Fig. 2). The full four-point cumulative IC50 application protocol exposed ND7-23 cells to half log-unit increases in concentration of the Icagen compound, and as these reached levels that were no longer selective for Nav1.3 channels, there was an incremental increase in inward current inhibition, with the expected state-dependent profile favoring inhibition of inactivated-state (Peak 2) currents compared with resting-state (Peak 1) currents (Fig. 6B). As the maximum % inhibition values were <50% and the higher concentrations were no longer selective for Nav1.3 channels (especially inactivated Peak 2 data), we did not attempt to measure potency or calculate an IC50 from this dataset.
We then determined the fraction of endogenous TTX-S currents carried by Nav1.6 channels by applying selective concentrations of 4,9 anhydro TTX (10–100 nM). As noted above, inward currents evoked in ND7-23 cells from resting (Fig. 7A, C) and half-inactivated membrane potentials (Fig. 7B) were remarkably stable during control experiments in the presence of vehicle (0.1% DMSO) and during the initial control period of the IC50 application protocol before application of 4,9 anhydro TTX. There were minor effects of 10 and 30 nM 4,9 anhydro TTX on resting-state Peak 1 and inactivated-state Peak 2 currents (Fig. 7B), while the top concentration tested of 100 nM 4,9 anhydro TTX (which is ∼3× the Nav1.6 IC50, but remains selective for Nav1.6 over Nav1.7 channels) produced significant inhibition of both resting-state and half-inactivated state currents. Although there is clear evidence for dose-dependent inhibition of ND7-23 cell TTX-S currents by selective concentrations of 4,9 anhydro TTX, the low % inhibition values precluded us from determining accurate potency data or calculating an IC50 value. However, the moderate effects of 4,9 anhydro TTX were reasonably reversible after application of small volumes of vehicle to the QPlate recording chamber (Fig. 7B), suggesting they were not due to rundown or other artifacts.
Protoxin-II is less water soluble than TTX and to preserve its low nanomolar potency in the small volume static flow chambers on the QPlate, we used 0.05% BSA as the vehicle in all external solutions; all activity of Protoxin-II was lost in the absence of BSA as a protein carrier. It was therefore necessary to first establish that the BSA vehicle had no effect on endogenous TTX-S currents in ND7-23 cells on the QPatch platform. Figure 8A and C shows that both resting-state Peak 1 and half-inactivated Peak 2 currents remain stable throughout the 20-min experimental recording period in the presence of 0.05% BSA, as seen previously for Nav current recordings from ND7-23 cells made in external saline and 0.1% DMSO vehicle (Fig. 7). A full four-point IC50 application protocol (0.3–10 nM in half log-unit increments) was employed to establish the potency of Protoxin-II in ND7-23 cells and determine the proportion of endogenous Nav current carried by Nav1.7 channels. There was no significant effect of 0.3 or 1 nM Protoxin-II on inward currents, but higher concentrations of Protoxin-II that began to exceed the IC50 determined against hNav1.7 channels (∼3.5 nM) produced significant inhibition of endogenous TTX-S currents in ND7-23 cells (Fig. 8C, D). Inhibition by 3 and 10 nM Protoxin-II was relatively slow (Fig. 8D), and maximal block by the saturating, but selective, concentration of 10 nM Protoxin-II ranged between 40% and 80% in individual cells. In contrast to the effects of 4,9 anhydro TTX, both resting-state Peak 1 (Fig. 8C) and half-inactivated-state Peak 2 currents (Fig. 8D) were equally sensitive to Protoxin-II, yielding IC50 values of 7.2 and 5.7 nM against the resting-state Peak 1 and half-inactivated-state Peak 2 currents, respectively (Fig. 8E).
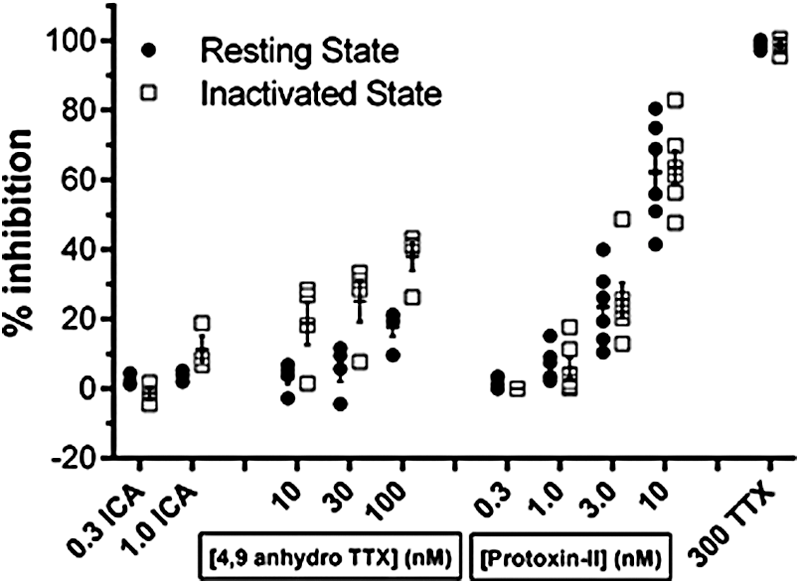
The average effects on TTX-S currents of selective Nav1.3, Nav1.6, or Nav1.7 antagonists added alone to ND7-23 cells are summarized in Figure 9 and Table 1. An Nav1.3-selective concentration of 300 nM of the Icagen compound 68 produced no effect on inward currents, while 1 μM reduced resting-state Peak 1 current by 0.7% ± 3.0% and inactivated Peak 2 current by 8.5% ± 1.7% (mean ± SEM, N = 5). However, as this concentration was nearly 20× higher than the heterologous Nav1.3 Peak 2 IC50, these values should be considered an upper estimate on the contribution of native Nav1.3 channels to TTX-S currents in ND7-23 cells. The Nav1.6 inhibitor 4,9 anhydro TTX produced moderate, but dose-dependent, effects on TTX-S currents, reducing resting-state Peak 1 current by 3.3% ± 2.1% at 10 nM, 5.6% ± 3.6% at 30 nM and by 17.8% ± 2.8% at 100 nM (mean ± SEM, N = 4). As noted above and by Rosker et al., 30 this antagonist had greater potency against the inactivated state of Nav1.6 channels, with Peak 2 currents in ND7-23 cells inhibited by 18.7% ± 6.2%, 25.1% ± 5.9%, and 37.9% ± 3.9% in the presence of 10, 30, and 100 nM 4,9 anhydro TTX, respectively. In contrast, Protoxin-II reduced the amplitude of resting- and inactivated-state TTX-S currents equally well, with minor effects at 1 nM (average of 5.6% ± 3.6% inhibition of Peak 1 and 6.0% ± 7.0% inhibition of Peak 2, respectively; mean ± SEM from N = 6 cells) increasing to 22.4% ± 10.5% and 25.5% ± 12.1% at 3 nM, and reaching a maximum inhibition of 62.2% ± 14.9% for Peak 1 resting-state currents and 63.6% ± 12.0% of Peak 2 half-inactivated currents at 10 nM (Fig. 9 and Table 1). Summing the effects of maximal, but selective, concentrations of Nav1.3, Nav1.6, and Nav1.7 inhibitors added individually to single ND7-23 cells reveals an overall reduction of resting TTX-S currents of 80.2% and complete (100.9%) inhibition of inactivated-state currents.
Finally, we additively applied selective concentrations of 4,9 anhydro TTX and Protoxin-II to the same ND7-23 cells to assess the variation in contribution of Nav1.6 and Nav1.7 channels to endogenous TTX-S currents in single cells. Figure 10A illustrates the result for an exemplar cell, showing the minor, but discernibly, dose-dependent inhibition of both resting-state and half-inactivated state currents with ascending doses of 4,9 anhydro TTX (10–100 nM), followed by slow, but profound, inhibition of the majority of the remaining endogenous Nav current by 10 nM Protoxin-II. Addition of a saturating dose of 300 nM TTX at the end of the experiment reduced peak inward currents to <5% of initial values, consistent with the fact that essentially all of the endogenous currents in ND7-23 cells are carried by TTX-S channels. The average data from these experiments are shown in Figure 10B, showing that a minor component (∼20%) of total inward current is inhibited by a concentration of 100 nM 4,9 anhydro TTX that is selective for Nav1.6 channels. The majority of the remaining TTX-S currents in these cells are blocked by a concentration of Protoxin-II (10 nM) selective for Nav1.7 channels, reducing total peak inward current by 73.7% ± 3.5% compared with the vehicle period (mean ± SEM from N = 6 cells). As noted above (Fig. 9), the inhibition of inactivated-state Peak 2 currents by 100 nM 4,9 anhydro TTX is somewhat greater than its inhibition of resting-state Peak 1 currents, leading to a small increase in the overall proportion of endogenous inactivated-state TTX-S current that is blocked after the additive exposure of single ND7-23 cells to 4,9 anhydro TTX and Protoxin-II (82.1% ± 2.7%, mean ± SEM from N = 6 cells). Thus, we conclude that ∼75%–80% of the total inward TTX-S currents in ND7-23 cells are carried by a combination of Nav1.7 > Nav1.6 channels.
Discussion
Our first significant finding was to confirm the potency and selectivity of 4,9 anhydro TTX for human Nav1.6 channels that were originally demonstrated using rodent channels 30,42 and to demonstrate its utility as a subtype-selective ligand for studying the function of rodent and human Nav1.6 channels implicated in pain signaling, epilepsy, and multiple sclerosis. Our second major finding was that functional patch clamp electrophysiology largely confirms previous molecular profiling to reveal that 75%–80% of the endogenous inward sodium currents in ND7-23 cells are carried by Nav1.7 > Nav1.6 >> Nav1.3 channels. We also confirmed previous findings that >95% of the total Nav currents in these cells were due to activity of TTX-S channels. The remaining small fraction (∼20%) of TTX-S currents could not be accurately assigned to a particular Nav1.x subtype owing to limits in the selectivity of the pharmacological ligands available. Finally, we demonstrate that native ND7-23 neuronal cells display very stable endogenous TTX-S Nav currents in whole-cell recordings and are amenable for automated patch clamping on robot platforms normally employed for ion channel screening. This combination of physiological and biophysical features makes ND7-23 cells a useful native neuronal system suitable for the Nav1.x gene family and species selectivity testing in ion channel drug discovery screening cascades as well as translational phenotypic drug discovery assays.
Selectivity for Human Nav1.6 Channels and Mechanism of Action of 4,9 Anhydro TTX
When designing these experiments, we were surprised that the initial finding of the Nav1.6 potency and selectivity of 4,9 anhydro TTX 30 had not been extended to human Nav1.x channels expressed in a mammalian cell background, although moderate potency and markedly reduced selectivity had been shown in rat Nav1.6 and human Nav1.2 channels expressed in HEK cells. 42 Nav1.6 is an important target for the regulation of axonal excitability 40 and pain transmission 47 and is implicated in diseases such as multiple sclerosis and epilepsy, 11,41 so a ligand with a reported 150-fold selectivity for Nav1.6 should be attractive for drug discovery purposes. We therefore set out to confirm data from the previous studies (which predominantly used rodent Nav1.x channels) using human Nav1.x channels expressed in mammalian cells before using 4,9 anhydro TTX as a selective ligand to reveal the repertoire of endogenous TTX-S channels in ND7-23 cells.
We determined the potency and selectivity of 4,9 anhydro TTX for heterologous hNav1.6 channels using the same QPatch APC platform and voltage protocols we would employ for Nav1.x profiling in ND7-23 cells. We confirmed many features of inhibition of Nav1.6 channels by 4,9 anhydro TTX originally described, 30 including the slightly slower block compared with TTX (Fig. 3C), low nanomolar potency (Fig. 3E), and additional effects on the inactivated state (Fig. 3C, E; also see Figs. 9 and 10). We also confirmed the significant selectivity of 4,9 anhydro TTX for Nav1.6 over Nav1.7 channels (Fig. 4) using human channels expressed in a mammalian cell background, although the 50-fold selectivity ratio we calculated is lower than the ∼150-fold ratio reported in oocytes, 30 but higher than the 5-fold selectivity for Nav1.x channels expressed in HEK cells. 42 The QPatch IC50 for inhibition of resting-state hNav1.6 Peak 1 currents (32.9 nM) was fourfold less potent than that reported for mouse channels expressed in Xenopus oocytes, but fourfold lower than that against rat Nav1.6 channels in HEK cells, 42 which can be attributed to a number of experimental differences (e.g., solutions, whole-cell perfusion vs. intracellular impalement, compound flow rate, and volume) as well as biological, species, and expression system variables. However, the hNav1.7 potency of 4,9 anhydro TTX (1.62 μM) obtained in mammalian cells on the QPatch is very close to that reported for hNav1.7 channels expressed in oocytes (1.27 μM), suggesting that Nav1.6 species differences may be the most important issue for this ligand.
Species differences in the efficacy of 4,9 anhydro TTX may be hard to explain if one assumes it acts like its parent TTX as a simple pore blocker binding to a specific tyrosine residue in the S5–S6 pore loop of domain I. However, we have observed and others have published that TTX exhibits moderate use- and state-dependent inhibition of Nav channels, and the underlying mechanism of this effect is still being determined. 43,44 We note that 4,9 anhydro TTX had greater potency against the inactivated state compared with the resting/open state of hNav1.6 channels in heterologous HEK cells (Fig. 3), as well as native TTX-S currents in ND7-23 cells (Figs. 9 and 10), indicative of some additional state-dependent mechanism of action. Similarly, although Rosker et al. 30 noted that most of the effects of TTX and 4,9 anhydro TTX on Nav1.x channels were due to open-state tonic block, supplemental data showed that TTX and 4,9 anhydro TTX (10 nM) produced a statistically significant leftward shift in the inactivation curve of mouse Nav1.6 channels. This would indicate that 4,9 anhydro TTX (and TTX) exhibits a more complex mechanism of action, which also involves a state-dependent preference for the inactivated conformation of Nav1.6 channels. This could involve binding to a more divergent set of amino acid residues of the Nav1.6 channel protein in or adjacent to the outer vestibule, enhancing the possibility of Nav1.6 subtype specificity, as well as species differences. Therefore, the lower potency and efficacy (i.e., partial block, Fig. 3) of 4,9 anhydro TTX against human Nav1.6 channels in this study compared with previous work on mouse channels may be explained by species differences in inactivated-state residues and channel conformations, leading to abrogation of an additional binding mechanism.
Repertoire of Functional Nav1.x Currents Endogenously Expressed in ND7-23 Cells
Nav1.3
There is a consistent pattern for very low expression of Nav1.3 channels in ND7-23 cells as evidenced by molecular, biophysical, and pharmacological methods. Polymerase chain reaction (PCR) transcripts are low, 32,33 and use-dependent block of TTX-S currents by ambroxol in ND7-23 cells is more similar to that seen with heterologous Nav1.7 than Nav1.3 channels. 32 Our QPatch results are consistent with these observations as we found negligible effects of saturating concentrations of a selective Nav1.3 small-molecule antagonist on whole-cell TTX-S currents in ND7-23 cells (Figs. 6 and 9). As the Nav1.3-selective Icagen compound 68 is strongly state dependent (Fig. 2), we based our analysis on its effects on inactivated Peak 2 current amplitude. Thus, a saturating, but selective, concentration of 300 nM of the Icagen compound 68 (∼5-fold over the heterologous Nav1.3 Peak 2 IC50 concentration of 56 nM) had no significant effect on inactivated-state Peak 2 TTX-S current amplitude in ND7-23 cells (−0.5%), while a potentially less selective concentration of 1 μM reduced ND7-23 inactivated-state current by only 8.5%. We conclude that Nav1.3 channels contribute at most ∼5% of the macroscopic TTX-S current in ND7-23 cells, but in all likelihood, they represent an even smaller fraction.
Nav1.6
In two separate sets of experiments, we determined that Nav1.6 channels carry a minimum of 15%–20% of TTX-S current evoked in ND7-23 cells from a resting state (Figs. 7B , 9 , and 10), and based on the fact that inactivated-state Peak 2 Nav currents exhibit a higher affinity for block by 4,9 anhydro TTX, we estimate that a maximum of ∼35% of the endogenous TTX-S current in ND7-23 cells flows through Nav1.6 channels. We used a maximal concentration of 100 nM 4,9 anhydro TTX to selectively block Nav1.6 channels without affecting Nav1.7 currents, similar to a previous study in HEK cells expressing rat and human Nav1.x channels. 42 Although 4,9 anhydro TTX also blocks Nav1.3 channels with a potency of 341 nM in oocytes, 30 we think it is unlikely that any of the effects of 4,9 anhydro TTX attributed to inhibition of Nav1.6 in this study are due to inhibition of Nav1.3 for two reasons. First, the selective Icagen Nav1.3 antagonist had no significant effect on macroscopic currents in ND7-23 cells at 300 nM, five times its IC50 for heterologous Nav1.3 channels. Second, both Leffler et al. 32 and Kennedy et al. 33 reported seeing low or barely detectable levels of Nav1.3 transcript expression in ND7-23 cells. Thus, we conclude that the previous molecular and our pharmacological data suggest that there is negligible expression of functional Nav1.3 channels in ND7-23 cells and that the effects of moderate concentrations of 4,9 anhydro TTX (up to 100 nM) can be attributed to selective inhibition of Nav1.6 channels.
The intermediate level of Nav1.6 channel function that we detected is consistent with the PCR study of Kennedy et al., 33 but contradicts the negative result obtained by Leffler et al. 32 Unpublished western blots indicate the presence of Nav1.6 protein in ND7-23 cells, but the relative levels compared with other Nav1.x channels were not described. 48 Nav1.6 channels can generate characteristic resurgent currents during repolarization of the membrane potential, 42 but this diagnostic feature appears to depend on the presence of β4 accessory subunits, 3,49 which are lacking in ND7-23 cells, 7 and there is no evidence for such a biophysical phenomenon in our QPatch recordings (data not shown). Nevertheless, the presence of functional Nav1.6 channels in ND7-23 cells, alongside Nav1.7 channels, makes them a useful model of sensory neuron action potential generation and axonal conduction for translational studies of pain signal transmission and drug discovery.
Nav1.7
The effects of Protoxin-II on TTX-S currents in ND7-23 were quite consistent, with application of cumulative concentrations in the IC50 experiments producing dose-dependent reductions in resting- and inactivated-state current amplitude that reached a maximum inhibition of 62% (Figs. 8 and 9), while the net inhibition by a saturating dose of 10 nM Protoxin-II (applied after 100 nM 4,9 anhydro TTX) was also ∼60% (75% total −15% after 4,9 anhydro TTX, Fig. 10). As the QPatch IC50 for Protoxin-II inhibition of heterologous hNav1.7 channels (3.6 nM) is very close to that for ND7-23 TTX-S currents (7.2 nM), and the top concentration of 10 nM inhibits >90% of macroscopic Nav1.7 currents without affecting other human Nav1.x channels, 27 we are confident in our estimate that 60% of the TTX-S current in ND7-23 cells is carried by Nav1.7 channels. Our pharmacological data that Nav1.7 is the predominant functional TTX-S species in ND7-23 cells are also consistent with previous molecular studies, which detected Nav1.7 as the most abundant PCR transcript. 32,33
Unlike the pore blocker TTX, Protoxin-II is a gating modifier that binds to voltage sensor paddles in several domains of the Nav1.7 channel to retard voltage-dependent activation. 28,50 Although the binding mechanism of Protoxin-II is state dependent, it does not necessarily elicit a preference for inhibiting resting-state or inactivated-state Nav1.7 currents, as reflected in the similar maximal inhibition values and IC50 potencies for hNav1.7 and ND7-23 TTX-S currents ( Figs. 5, 8, 9, and 10). Protoxin-II has to access more distant and cryptic binding pockets within the channel than TTX and so the kinetics of block are relatively slow, as seen in our QPatch experiments (Figs. 5, 8, and 10). We failed to achieve complete block with the highest (selective) concentration of 10 nM Protoxin-II tested, and our Nav1.7 IC50 of 3 nM was 10-fold less potent than that reported using longer applications of larger volumes of peptide under manual patch clamp conditions, 27 although it is identical to that recently reported by Amgen scientists using similar protocols on the PatchXpress APC platform. 50 Subsaturating block of Nav1.7 currents by Protoxin-II has been observed by others 51 and our QPatch data are very similar to the 3 nM IC50 and 80% maximal block seen with 10 nM Protoxin-II in manual patch experiments reported by Smith et al. 46 We expected slow kinetics of block from previous studies in oocytes and heterologous cells and, for this reason, designed the QPatch application protocol to use multiple additions during each 2-min application of each concentration to try and saturate the system, but although the perfusion capabilities of the QPatch system are well suited to studies of small volumes of expensive reagents such as toxins, it will always be less efficacious than the open-ended perfusion properties of a manual patch clamp system. Nevertheless, our data prove that it is possible to work with potent and sticky peptide toxins on an APC system and obtain results that are comparable with the gold standard of manual patch clamp and preserve the characteristic binding properties and blocking mechanisms of such important ion channel ligands.
In summary, a significant amount, but not all, of the endogenous TTX-S currents in ND7-23 cells can be accounted for by the fractions inhibited in different cells by application of selective concentrations of each of the Nav1.x antagonists (Fig. 9). The Icagen compound (300 nM, Nav1.3) inhibited ∼0.5% of inward TTX-S currents, while 4,9 anhydro TTX (100 nM, Nav1.6) reduced resting-state current amplitude by 18% and Protoxin-II (10 nM, Nav1.7) led to a 62% reduction in Peak 1 current magnitude. Summing these independent Nav1.x subtype inhibitions yields an overall fraction of peak TTX-S currents of 80%, while addition of the fractions of Peak 2 inactivated currents affected by the same three compounds yields an overall inhibition of exactly 100% (the major difference stemming from the greater effect of 4,9 anhydro TTX on the inactivated state of heterologous and presumably also endogenous Nav1.6 TTX-S channels). Similarly, additive application of selective concentrations of 100 nM 4,9 anhydro TTX (Nav1.6), followed by 10 nM Protoxin-II (Nav1.7) in the same ND7-23 cells (Fig. 10), inhibits 73.7% ± 3.5% of total inward resting-state current and 82.1% ± 2.7% of half-inactivated Peak 2 current (mean ± SEM, N = 6). Therefore, we can account for 75%–100% of total TTX-S current in ND7-23 cells through a combination of Nav1.7 > Nav1.6 >> Nav1.3 channels.
Significantly, the repertoire and rank order of functional Nav1.x channels we identified in ND7-23 cells (Nav1.7 > Nav1.6) are similar to the major composition of TTX-S currents in small-diameter DRG neurons and the pharmacological sensitivity of action potentials in unmyelinated C-fibers of the rodent sciatic nerve. 40,52 It should be noted that the third and minor component of adult rodent DRG TTX-S responses was attributed to Nav1.1 channels, not Nav1.3, as found by molecular and pharmacological studies in ND7-23 cells. The presence of functional Nav1.1 channels in ND7-23 cells remains to be determined with appropriately selective antagonists, but the possibility remains as the molecular data are equivocal with PCR experiments failing to detect Nav1.1 transcripts, 32 but unpublished western blots indicating the presence of Nav1.1 protein in ND7-23 cells. 48 It may be that ND7-23 cells represent a less mature phenotype and express low levels of the embryonic Nav1.3 channel rather than adult Nav1.1; this may also explain the lack of endogenous TTX-R Nav1.8 and Nav1.9 channels in ND7-23 cells. This DRG-like profile of TTX-S current expression suggests that the ND7-23 cell line is a useful reagent in the design of in vitro ion channel assays for pain drug discovery, including medium- and high-throughput high content electrophysiology screens on APC platforms. Having a native cell reagent that not only works so well on these platforms but also captures the physiology and biophysics of native rodent DRG neurons could enable the translational benefits of a phenotypic assay to be realized higher up a traditional ion channel screening cascade and therefore allow the testing of more compounds earlier in the drug discovery process. This could in turn highlight suitable scaffolds with efficacy in preclinical animal models of pain physiology, reducing animal use (NC3R) both through reduced need for testing in native neuronal cells and higher in vivo success rates.
Remaining TTX-S Current
It is difficult to unequivocally define the small fraction (∼15%–25%) of macroscopic TTX-S current that remains in ND7-23 cells in the presence of selective concentration of Nav1.x subtype-specific ligands. The first issue to consider is the efficacy and selectivity of the pharmacological tools we employed. For example, the remaining TTX-S current may be carried by unblocked Nav1.6 channels as 100 nM 4,9 anhydro TTX does not completely inhibit these channels (Fig. 3). However, higher concentrations of 4,9 anhydro TTX could not be used as they begin to inhibit Nav1.7 channels (Fig. 4). Similarly, the top concentration of 10 nM Protoxin-II chosen for this study did not completely block all Nav1.7 channels expressed in HEK cells (Fig. 5), and the rate of inhibition was relatively slow in the ND7-23 experiments (Figs. 8D and 10A), so we may have underestimated the full fraction of TTX-S current carried by Nav1.7 channels. Higher concentrations of Protoxin-II would presumably have faster onset and more complete block of TTX-S currents, but would have lost selectivity against Nav1.6 channels and may also activate sustained Nav1.7 currents. 45
We also assume that the concentration–response curves for the selective antagonists we obtained against heterologous human Nav1.x channels in CHO and HEK cells can be translated to the same subtypes of rodent Nav1.x channels in ND7-23 cells. Although this is likely to be true for Protoxin-II, 27,45 and our IC50 value for 4,9 anhydro TTX was fairly close to that obtained against mouse Nav1.6 channels, 30 it was fourfold lower than that reported for rat Nav1.6 channels expressed in HEK cells, 42 although those authors also used 100 nM 4,9 anhydro TTX as the top concentration that maintained specificity for Nav1.6. We did not confirm the species selectivity of Icagen compound 68, but studies with the close analog ICA-121431 suggest that its potency and selectivity are maintained between human and mouse, but not rat, Nav1.x channels. 36 In this case, the otherwise Nav1.3-selective ICA-121431 had low nanomolar potency against rat Nav1.7 channels, but if the same were true for Icagen compound 68, we would expect application of 300 nM to inhibit >50% of TTX-S current as the Nav1.7-selective antagonist Protoxin-II affects rodent and human Nav1.7 channels equally, but this was not seen (Fig. 6). Thus, the first explanation for the residual TTX-S current in ND7-23 cells in the current experiments is that it is carried by Nav channels of the most common endogenous subtypes (Nav1.7 > Nav1.6) that remain unblocked in the presence of specific, but subsaturating, concentrations of their selective ligands, Protoxin-II (10 nM) and 4,9 anhydro TTX (100 nM). Alternatively, the residual TTX-S current may be carried by rat rather than mouse Nav1.3 channels resistant to the effects of Icagen compound 68, 4,9 anhydro TTX, or Protoxin-II, but we regard this as unlikely given the extremely low levels of Nav1.3 expression detected in ND7-23 cells by molecular methods. 32,33
Another explanation for the residual TTX-S current is that it is carried by Nav1.x channels other than Nav1.3, Nav1.6, or Nav1.7. We doubt it is Nav1.1 as PCR experiments do not detect any such transcripts, 32 although another study suggests the presence of Nav1.1 protein in unpublished western blots from ND7-23 cells. 48 Similarly, the Icagen compound ICA-121431 is a close analog of Nav1.3 patent compound 68 and is an almost equipotent (∼20 nM IC50) antagonist of Nav1.3 and Nav1.1, 36 but application of a saturating concentration of 300 nM of Icagen compound 68 had negligible effects on resting- or inactivated-state TTX-S currents in ND7-23 cells (Fig. 6). In contrast, it is possible that Nav1.2 channels may underlie the remaining 20% of macroscopic Nav activity in ND7-23 cells as moderate molecular signals for Nav1.2 channels were seen in ND7-23 cells using PCR 32 and western blotting. 48 However, the lack of any commercially available selective Nav1.2 ligands at the time of these experiments made their identification difficult from a functional perspective; it has also been claimed that the pattern of opioid modulation of endogenous TTX-S currents in ND7-23 cells suggests that no functional Nav1.2 channels are present. 32
Endogenous TTX-S Channels in Other Neuroblastoma Cell Lines
ND7-23 neuroblastoma cells are one of several immortalized neuroectodermal cell lines to express endogenous Nav1.7 channels, but they have proven to be the most amenable to automated patch clamping. In contrast, the mouse NIE-115 mouse neuroblastoma cell line predominantly expresses Nav1.3 channels, 53 although previous biophysical articles claimed it was Nav1.2. 46 Rat GH3 pituitary neuroendocrine cells express Nav1.1, Nav1.2, Nav1.3, and Nav1.6 channel transcripts, 54,55 but their relative contribution to functional macroscopic currents in these cells remains to be determined. The F11 neuronal cell line (similar to ND7-23 cells being a hybrid between mouse N18TG-2 neuroblastoma and embryonic rat DRG neurons) expresses Nav1.2, Nav1.3, and Nav1.6 transcripts, but not Nav1.7 channels, based on the effects of scorpion toxins and unpublished PCR data. 56 The NG108-15 cell line is another mouse N18TG-2 neuroblastoma hybrid (with rat glioma cells), which expresses endogenous calcium and sodium currents; although the former are well characterized, there is uncertainty about the underlying Nav channel subtypes. PCR failed to detect Nav1.7 transcripts in undifferentiated NG108-15 cells, 34 but differentiation produces a marked increase in Nav current density associated with expression of Nav1.7 as detected by immunohistochemistry, real-time PCR, and western blotting. 57,58 Finally, the SH-SY5Y neuroblastoma cell line has been used extensively in pain drug discovery owing to its human origin. Ionix relied on it for expression of Nav1.8 for their pain program, 17 and several groups at the University of Queensland used various molecular and pharmacological techniques to establish that human SH-SY5Y cells express hNav1.7 > hNav1.2 > hNav1.3 channels. 34,59 They designed several Nav subtype-specific fluorescence screening assays, including one to detect novel Nav1.7-selective spider toxins as part of their pain drug discovery program. 60,61
The lack of Nav1.7 channel expression at the molecular or functional level in various neuroectodermal cell lines is surprising given the original cloning of the hNE Nav1.7 gene from a neuroendocrine carcinoma 62 and the fact that Nav1.7 is in the same duplicated TTX-S gene cluster as Nav1.1, Nav1.2, and Nav1.3, 1 all of which are expressed in the various cell lines described above. A recent publication provides a possible explanation as it was reported that Nav1.7 gene products are regulated by a natural antisense transcript; this factor is found in the brain, spinal cord, and several subtypes of DRG neurons that do not express Nav1.7 channels, but is absent in SH-SY5Y cells that do express hNav1.7 channels. 63 Thus, it may be that Nav1.7 gene expression and transcript levels are selectively downregulated in neuroectodermal cell lines that express this antisense factor or that the process of hybridization between different rodent species and neuroblastoma cell backgrounds has introduced or activated the Nav1.7 silencing factor that was previously absent or quiescent in rat DRG progenitor cells.
Lack of Endogenous TTX-R Channels in ND7-23 Cells
The complete lack of expression of endogenous Nav1.8 or Nav1.9 TTX-resistant channel transcripts or currents in ND7-23 cells was unexpected given their hybridization from rat DRG neurons. For example, Zhou et al. 18 found that 98.8% of endogenous Nav currents were blocked by 300 nM TTX and our results are consistent with the application of 300 nM TTX (a concentration selective for TTX-S channels 7 ), which at the end of every QPatch experiment inhibited >97% of macroscopic inward Nav current in all cells analyzed. ND7-23 cells can, however, heterologously express human (and rat) Nav1.8 channels 7,18 –22 and also the more difficult human Nav1.9 channels. 23 –25 Icagen recently showed that hNav1.9 channels can be expressed in HEK cells through the coexpression of two undefined (presumably human) β subunits; 64 as ND7-23 cells express both β1 and β3 accessory subunits, 7 this could suggest that although the latter rodent β subunits can support the insertion and function of human TTX-R channels, it is β2 and/or β4 subunits that are required for expression of recalcitrant rodent TTX-R channels. Similarly, other accessory proteins and modulatory pathways necessary for surface expression of functional rodent TTX-R channels may have been lost during the hybridization of rat DRG neurons with mouse neuroblastoma cells. For example, Annexin p11 is absent from ND7-23 cells, but present in SH-SY5Y cells, which can express rodent Nav1.8 channels, 17 and coexpression of p11 enables the transient expression of rodent Nav1.8 in ND7-23 cells. 7,15,22 The recent success in achieving low levels of transient 24 and stable expression 23 of hNav1.9 in ND7-23 cells without transfection of additional subunits suggests that expression of human, but not rodent, Nav1.9 is less dependent on Annexin p11 than hNav1.8 expression. Finally, the lack of any endogenous TTX-R Nav1.8 or Nav1.9 currents or PCR transcripts in ND7-23 cells may indicate fundamental inactivation of the endogenous rodent Nav1.8 and Nav1.9 genes in this cell line, perhaps occurring through effects on the TTX-R gene cluster 1 or reflecting an embryonic differentiation state (hence the low-level expression of Nav1.3 transcripts and channels). Application of new generation gene-editing techniques such as CRISPR-Cas9 to ND7-23 cells may be able to resurrect the expression of endogenous TTX-R channels and create a more complete in vitro model of rodent DRG neurons suitable for APC ion channel screening and pain drug development that will meet NC3R guidelines to reduce animal use.
Footnotes
Acknowledgments
The authors gratefully acknowledge Louise Webdale and Toni Cooper for their cell culture expertise, and Richard Davies and Neil Bruce for assistance with setting up and optimizing the QPatch Nav1.x and ND7-23 automated patch clamp assays at Xention. Dr. Tony Rush provided insightful advice and helpful feedback on the manuscript. The authors would also like to acknowledge permissions and materials obtained from their colleagues, Dr. Takashi Sasamura and Mrs. Yoko Sekioka (nee Ono), and Mr. Ari Alexandrou and Dr. Eddy Stevens. This work was partially supported by the Slovenian Research Agency (Grant No. Z1-5458).
Disclosure Statement
M.R. and R.W.K. were employees at Xention Ltd., when this work was carried out and are now employees and shareholders in Metrion Biosciences Ltd. The authors declare that they do not have any conflicting interests concerning the present study. For clarity, they wish to highlight the fact that this work follows on from an EU-funded grant (MAREX) that all of the authors were participants in, but the experiments described here were not part of, or funded by, this collaboration.