
Abstract
Glycosyltransferase enzymes play diverse metabolic and regulatory roles by catalyzing the transfer of sugar molecules to protein, lipid, and carbohydrate acceptors, and they are increasingly of interest as therapeutic targets in a number of diseases, including metabolic disorders, cancer, and infectious diseases. The glycosyltransferases are a challenging target class from an assay development perspective because of the diversity of both donor and acceptor substrates and the lack of suitable glycan detection methods. However, many glycosyltransferases use uridine 5′-diphosphate (UDP) sugars as donor substrates, and detection of the free UDP reaction product provides a generic approach for measuring the activity of those enzymes. To exploit this approach for a broadly applicable high-throughput screening (HTS) assay for discovery of glycosyltransferase inhibitors, we developed a Transcreener® assay for immunodetection of UDP with a time-resolved Förster resonance energy transfer (TR-FRET) signal. We optimized the assay for detection of glycosyltransferase activity with nucleotide diphosphate (NDP) sugars at concentrations from 10 μM to 1 mM, achieving Z' values of 0.6 or higher. The assay was validated by orthogonal pooled screening with 8,000 compounds using polypeptide N-acetylgalactosaminyltransferase T3 as the target, and the hits were confirmed using an orthogonal readout. The reagents and signal were both stable for more than 8 h at room temperature, insuring robust performance in automated HTS environments. The TR-FRET-based UDP detection assay provides a broadly applicable approach for screening glycosyltransferases that use a UDP-sugar donor.
Introduction
Glycosyltransferases catalyze the covalent attachment of a sugar molecule to an acceptor substrate to form a glycosylated product. 1,2 Depending on their acceptor substrate specificity, glycosyltransferases play widely diverse metabolic and regulatory roles, including conjugation of small molecule drugs and other xenobiotics, glycosylation of cell surface and extracellular matrix proteins, and formation of sugar polymers such as glycogen and hyaluronic acid. 3 –5 An activated sugar is required to drive the reaction, and in humans, uridine 5′-diphosphate (UDP)-sugars are the most commonly used donor molecules; other donors for the human enzymes include guanosine diphosphate, cytidine monophosphate, and dolichol phosphate sugars. The 5′-hydroxyl group of the sugar forms a glycosidic linkage to a hydroxyl or amino group on the acceptor molecule, and the dinucleotide is released.
Glycosyltransferases have been linked with a number of diseases, and significant effort has gone toward developing specific inhibitors, mostly targeting enzymes that use UDP-activated donor molecules. 6 –10 Metabolic glycosyltransferases are considered promising targets for drug discovery in a number of monogenic diseases, where the absence or malfunction of a downstream enzyme causes accumulation of the glycosyltransferase product to toxic levels. 11 For example, glucosylceramide synthetase (GCS) is a UDP-glucosyltransferase in the sphingolipid biosynthetic pathway that is being targeted to prevent the buildup of toxic levels of glucosylceramide in lysosomal storage disorders, such as Gaucher and Fabry diseases. 9,12 –14 Glycogen synthase, also a UDP-glucosyltransferase, is under investigation as a potential target for reducing glycogen accumulation in Pompe disease. 15
Glycosylated proteins participate in key biological process that impact tumorigenesis, including cell adhesion and migration and modulation of immune responses, and aberrant glycosylation of cell surface proteins occurs in nearly all types of cancers. 16 In many cases, one or more of the enzymes in the polypeptide N-acetylgalactosaminyltransferase (ppGalNAc) family, which catalyze the initial glycosylation of proteins at Ser or Thr resides using UDP-N-acetyl-galactosamine (UDP-GalNAc) as a donor, are over expressed. 16 –21
For example, multiple lines of evidence indicate that ppGalNAc-T3 drives cell proliferation, migration and invasion in pancreatic cancer, renal cell carcinoma, high-grade serous epithelial ovarian tumors, and oral squamous cell carcinoma, and its overexpression is correlated with poor prognosis. 17,18,20,21 Another ppGalNAc family member, ppGalNAc-T6, has been proposed to be responsible for alterations in O-type glycosylation that contribute to tumor cell growth, survival, and invasion in breast cancers. 19
O-GlcNAc transferase (OGT) is a distinct type of UDP-sugar-dependent peptide glycosyltransferase that has been intensely investigated for a range of diseases, including cancer, diabetes, and cardiovascular disease, and very recently as an antiviral target. 6,16,22 OGT is an essential enzyme that, in contrast to the selectivity of the ppGalNAc enzymes, catalyzes glycosylation of more than 1,000 proteins at serine and threonine residues. 22 Increased O-GlcNAcylation and OGT levels have been reported in many cancers, including breast, lung, prostate, pancreatic, and colorectal cancers, and OGT knockdown has been shown to inhibit tumor growth in some cases. 16
Despite their links to diverse pathologies, very few glycosyltransferase inhibitors have been developed into drugs. Approved drugs include the two GCS inhibitors miglustat and eliglustat, and a few compounds that inhibit microbial or parasitic enzymes involved in cell wall biosynthesis; for example, ethambutol, an inhibitor of mycobacterial UDP-arabinosyltransferase that is used as an integral part of antituberculosis combination therapy. 13,14,23
Development of biochemical assays for glycosyltransferases that are amenable to high-throughput screening (HTS) has been difficult because of the diversity of the acceptor substrates, and much of the inhibitor discovery efforts have focused on the design of nucleotide diphosphate (NDP)-sugar mimetics, which tend to have low affinity and selectivity. 5,10 Traditional methods to measure UDP-glycosyltransferase enzyme activity utilize radiolabeled substrates (e.g., UDP-1-[ 3 H]-glucose) and subsequent extraction to recover and measure glycosylated product formation, but this assay format is not amenable to HTS because of the radioactive disposal issues and a cumbersome separation step. 24
Nonradioactive assays for glycosylated products have been developed, but they generally are specific for a single enzyme, and require the use of modified sugar–nucleotide donors or a separation step. 25 –28 An example of this detection strategy is the use of 7-hydroxy-4-trifluoromethylcoumarin to monitor the activity of hepatic UDP-glucuronosyltransferases, based on its increased fluorescence upon glucuronidation. 28 This substrate is used with varying efficiency by several of the hepatic xenobiotic-metabolizing enzymes because they have broad substrate specificity for small molecules, however it would not be useful for other types of glycosyltransferases. An innovative immunoassay method for OGT recently was developed using immobilized His-tagged substrates and a chemically reactive UDP-N-acetyl-glucosamine (UDP-GlcNAc) derivative that allows postreaction attachment of immunodetectable tag. 29 This assay can be used for multiple OGT substrates and most likely could be adapted to other peptide-modifying UDP-glycosyltransferases. However, it is a complicated format that also requires wash steps.
Monitoring either UDP formation or UDP-sugar depletion is a universal approach that can be applied to any UDP-sugar glycosyltransferase. Most of the assays that have been developed for this purpose rely on coupling enzymes that convert the UDP or UDP-sugar to a detectable product, such as phosphate, ATP, NADH, or pyruvate. 30 –32 However, coupled assays are inherently prone to false positives from compound interference with the coupling enzymes, and thus require additional wells for counterscreening.
A xanthene-based Zn(II) fluorescent ATP sensor has been adapted for detection of NDPs in glycosyltransferase reactions, which overcomes the coupled assay interference issue. 33,34 Although the assay was not validated for HTS, it may be useful with the caveats that the micromolar sensitivity could be limiting for enzymes with intrinsically low activity, and the fluorescence emission at 520 nM will be more susceptible to interference from fluorescent small molecules than emission from a far red dye, which is much more attractive for HTS applications.
A competitive ligand displacement assay with a fluorescent UDP-GlcNAc probe has also been developed and used to screen OGT and MurG, a bacterial UDP-GlcNAc transferase involved in cell wall biosynthesis. 26 This fluorescence polarization (FP)-based assay is well suited for HTS, it does not rely on coupling enzymes, and it was used to identify the first cell permeable OGT inhibitors. 35 However, the assay biases screening toward inhibitors that bind at the UDP-GlcNAc site; and therefore, compounds that inhibit activity by binding at the acceptor substrate site or at allosteric sites might be missed (i.e., false HTS negatives).
Transcreener ® is an extensively validated HTS assay platform developed at BellBrook Labs (Madison, WI), which has been used in large-scale industrial screening for kinases and other enzyme targets since 2006. 36 –38 The Transcreener UDP assay is a competitive immunoassay that relies on an antibody with more than 500-fold selectivity for UDP over UDP-sugars. The high selectivity allows detection of UDP in the presence of excess UDP-sugars, as is required for initial velocity measurements of glycosyltransferases. We initially developed the assay with a FP readout and demonstrated its utility for detection of glycosyltransferases. We now have developed a time-resolved Förster resonance energy transfer (TR-FRET)-based assay because the time-gated detection minimizes interference from fluorescent compounds, making it a preferred detection format for some HTS applications. 39
One limitation of the TR-FRET format is the need to optimize spacing between the donor and acceptor molecules in the “on” state of the assay, as FRET decreases with the sixth power of distance. This distance requirement is usually easily met in a competitive immunoassay format, which relies on a donor-labeled antibody and a small, acceptor-labeled tracer. An additional limitation is the instability of commonly used lanthanide donor complexes in the presence of heavy metals that may be required as enzyme cofactors; for example, Mn2+ for glycosyltransferases. However, HTS assays are typically run in endpoint mode, and addition of a chelator, such as ethylenediaminetetraacetic acid (EDTA) as a quencher, is usually sufficient to stabilize the lanthanide complex.
In this study, we describe the development and characterization of the reagents, the optimization of assay conditions for different UDP-sugars, acceptor substrates, and glycosyltransferases, and validation of the assay in a pilot screen of 8,000 compounds in an orthogonally pooled screening format using ppGalNAc-T3 as a target protein. 40
Materials and Methods
Materials
The components for the Transcreener UDP 2 TR-FRET assay were produced at BellBrook Labs. A monoclonal antibody for UDP (UDP 2 antibody), was produced using an NDP-protein conjugate and purified from ascites fluid. A LanthaScreen® amine-reactive terbium chelate (Thermo Fisher Scientific, Waltham, MA) was conjugated to the UDP 2 antibody and the resulting UDP 2 antibody-Tb was dialyzed and concentrated. A tracer was produced by attaching HiLyte647 N-hydroxysuccinimide ester from AnaSpec (Fremont, CA) to a reactive NDP derivative. The 10X Stop and Detect Buffer contains 500 mM HEPES (pH 7.5), 200 mM EDTA, and 0.2% Brij-35.
All nucleotides and UDP-sugars were purchased from Sigma (St. Louis, MO): uridine, uridine 5′-monophosphate (UMP), uridine 5′-diphosphate(UDP), uridine 5′- triphosphate (UTP), UDP-Glucose (UDP-Glc), UDP-Galactose (UDP-Gal), UDP-Glucuronic Acid (UDP-GA), UDP-N-acetyl-galactosamine (UDP-GalNAc), UDP-GlcNAc, and N-Acetyl-d-glucosamine (D-GlcNAc). Mucin 10 (153–165) EA2 peptide was purchased from Anaspec.
All enzymes were obtained from R&D Systems (Minneapolis, MN): human N-terminal 6-His tagged Polypeptide GalNAc Transferase2 (ppGalNAc-T2), human N-terminal 6-His tagged Polypeptide GalNAc Transferase3 (ppGalNAc-T3), human N-terminal 6-His β-1,4-Galactosyltransferase 1 (β4GalT1), and C-terminal 6-His tagged Clostridium difficile Toxin B.
The orthogonal pooled screening (OPS) library was obtained from the Lankenau Institute for Medical Research (LIMR) Chemical Genomics Center (Wynnewood, PA), and was prepared as the authors and others have described in detail. 40 –42 Basic buffer components were purchased from Sigma and Fisher (Hampton, NH).
Instrumentation and Data Analysis
TR-FRET assays were performed in white Corning 384-well, round bottom, low-volume polystyrene nonbinding surface microplates (Part # 3673), and plates were read using a PerkinElmer EnVision© at an excitation of 320 nm and emission at 615 nM (from Tb) and 665 nm (from HiLyte 647). The TR-FRET signal is expressed as a ratio of emission at 665/615 nm. All experiments were run in replicates as indicated in the Figure Legends, and error bars indicate standard deviation (SD) from the mean.
The primary OPS data were deconvoluted to impute orthogonally active hits with software obtained from CeuticalSoft (Hudson, NY). Data from concentration–response experiments were analyzed and plotted using GraphPad Prism (San Diego, CA). Δ Ratios are the difference between signal with or without enzymes or difference between 0% and X% conversion for standard curves. ECx was calculated by using the EC50 and Hill slope values, calculated from fitting the equilibrium binding data to a variable slope sigmoidal dose–response curve using the equation: ECx = [(x/(100-x))1/hillslope] × EC50. The Z' value was derived from the equation Z' = 1-[(3 × StdDev(High Signal Mixture) + 3 × StdDev(Low Signal Mixture))/(mean(High Signal Mixture)–mean(LowSignal Mixture))]. 43 Z' values were determined with 16 replicates and a benchmark of >0.5 was used for a robust assay. The lower limit of detection (LLD) is defined as the concentration of UDP that generates a Z' >0.
UDP 2 TR-FRET Assay Methods
A similar UDP detection protocol and buffer conditions were used for all experiments. Reactions containing UDP-sugars and other indicated reagents, with or without enzyme, were incubated at room temperature using a standard buffer (25 mM Tris pH 7.5, 2.5 mM MgCl2, 1.0% DMSO) or a buffer for a specific enzyme (see Enzyme Assays, below), in a 15 μL volume. Then 5 μL of UDP Detection Mix was added, consisting of 8 nM UDP 2 antibody-Tb (final concentration of 2 nM) and UDP-HiLyte 647 tracer at indicated concentrations in 1X Stop and Detect Buffer [50 mM HEPES (pH 7.5), 20 mM EDTA, and 0.2% Brij-35].
Competitive Binding Curves
Uridines and UDP-sugars were tested for their relative ability to displace tracer from antibody. First, twofold serial dilutions were carried out by transferring 10 μL of a nucleotide or UDP-sugar mix down a series of wells containing 10 μL of buffer alone. Then 10 μL UDP Detection mix was added with a final concentration of 2 nM UDP 2 antibody-Tb, 9 nM UDP-HiLyte 647 tracer, and 1X Stop and Detect Buffer. Duplicate curves were performed with TR-FRET measured after 1 h at room temperature. IC50 values were determined and selectivity ratios were calculated by dividing the IC50 from each sugar nucleotide by the IC50 for UDP.
UDP-HiLyte 647 Tracer Optimization
With the UDP 2 antibody-Tb held constant, the concentration of the UDP-HiLyte 647 tracer determines the dynamic range for the assay. The EC85 concentration from equilibrium binding curves generated in the presence of the appropriate UDP-sugar generally provides a good signal and dynamic response for detection of enzymes under initial velocity conditions. To determine EC85 values, twofold, 24-point serial dilutions of 10 μL of UDP-HiLyte 647 Tracer in the indicated buffer were prepared covering a concentration range of 0.01–750 nM. UDP-sugars were then added at the desired concentration in a 5 μL volume followed by 5 μL of UDP 2 -antibody-Tb for a final concentration of 2 nM in 1X Stop and Detect Buffer. TR-FRET was measured after incubation for 1 h at room temperature.
Standard Curves
Nucleotide mixtures mimicking enzymatic conversion of UDP-sugar to UDP in standard buffer, or in specific enzyme buffers as noted, were added to wells to a 15 μL final volume. Five microliters of UDP Detection mixture, containing UDP 2 antibody-Tb, UDP-HiLyte 647 tracer, and Stop and Detect Buffer, were then added in succession as in the standard assay procedure. UDP-sugar/UDP mixtures contained a constant total concentration of substrate and UDP product. For example, for a 100 μM UDP-Glucose standard curve representing 0%, 1.0%, 2.5%, 5.0%, 7.5%, 10.0%, 12.5%, 15.0%, 25%, 50%, 75%, and 100% conversion of substrate, the 0% reaction contains 100 μM UDP-Glc, the 1% reaction contains 1.0 μM UDP +99 μM UDP-Glc, the 10% reaction contains 10 μM UDP +90 μM UDP-Glc, and so forth. TR-FRET was measured after 1 h of incubation at ambient temperature. Standard curves were run with 16 replicates to allow determination of Z' values for each point in the curve. 43
Deck and Signal Stability
Deck stability represents the ability of the detection mixture (UDP 2 antibody-Tb and tracer in Stop and Detect Buffer C) to remain functional over time when placed on the reagent deck of an automated liquid dispenser. The detection mixture was formulated to working concentration (34 nM tracer, 8 nM antibody, and 1X Stop and Detect Buffer C), placed in translucent containers (subject to ambient light and temperature), and sampled over an 8-h period for generation of UDP-Glc/UDP standard curve data, with a 1-h equilibration period at room temperature. Five microliters of the detection mixture was added to 15 μL of 10 μM UDP-Glc/UDP standard curves in standard buffer contained in an assay plate. Twelve-point 10 μM UDP-Glc/UDP standard curves were performed, with four replicates, representing 0%, 1.0%, 2.5%, 5.0%, 7.5%, 10%, 12.5%, 15%, 25%, 50%, 75%, and 100% conversion of substrate.
Plate stability demonstrates the stability of signal after detection reagents have been added to nucleotides in an assay plate. The detection mixture (same as above) was added to standard curve mixtures in an assay plate and held sealed at room temperature. The TR-FRET signal was measured periodically over 24 h.
Enzyme Assays
Reaction conditions for glycosyltransferase enzyme reactions were obtained from the supplier's product literature. UDP-sugars and acceptor substrate concentrations used were at or near their Km values. The final enzyme reaction conditions were as follows: ppGalNAc-T2: 50 mM Tris (pH 7.5), 2.5 mM MnCl2, 100 μM UDP-GalNAc, and 10 μM EA2 peptide. ppGalNAc-T3: 25 mM Tris (pH 7.5), 5 mM MnCl2, 0.15 mM NaCl, 2.5 mM CaCl2, 100 μM UDP-GalNAc, and 10 μM EA2. β4GalT1: 25 mM Tris (pH 7.5), 0.15 mM NaCl, 10 mM MgCl2, 10 mM MnCl2, 250 μM UDP-Gal, and 1.2 mM D-GlcNAc. Toxin B: 25 mM Tris (pH 7.5), 10 mM MnCl2, 0.15 mM NaCl, 5 mM CaCl2, 75 mM K2SO4, and 400 μM UDP-Glc.
Enzyme reactions, in 15 μL were incubated at room temperature followed by the addition of 5 μL of the UDP 2 antibody-Tb and tracer mix in Stop and Detect Buffer. All of the glycosyltransferases used require Mn2+ as a cofactor; the EDTA present in the Stop and Detect Buffer chelates the Mn2+, thereby quenching glycosyltransferase enzyme activity. After plates were read, UDP product formation was determined from FRET values by interpolating from the appropriate standard curve.
Validation Studies
For the pilot screen with ppGalNAc-T3 (Table 1), we used 8,000 compounds in an orthogonally pooled format from the Lankenau Chemical Genomics Center (LCGC). The compounds were provided as 0.25 μL aliquots in columns 3–22 in 384-well plates, with 10 compounds per well, each compound appearing twice in a unique set of nine other compounds to allow data deconvolution. The OPS library subset used here has been used successfully in previously reported HTS campaigns by others. 44 –47
Protocol for Pilot Screen with Orthogonal Pooled Screening Plates
1. Thaw out 384-well assay ready plates received from LCGC containing 0.25 μL compounds or DMSO control. Spin plates for 2 min at ∼1,000 RCF in a swinging bucket centrifuge. Compounds are in columns 3–22 and DMSO control wells are in columns 1, 2, 23, 24.
2. Add 7.3 μL of enzyme at 2.1X concentration needed for 15 μL reaction to columns 2–22 and add 7.3 uL of no enzyme control to column 23 in 1X enzyme buffer, excluding UDP-sugar and acceptor substrate.
3. Gently mix enzyme and compound, cover the plate, and incubate at room temperature for 30 min.
4. Add 7.5 μL of 2X UDP-sugar and acceptor substrate in 1X enzyme buffer to columns 2–23 to start reaction. All wells should now have 15 μL.
5. Mix plate well and place at room temperature after this addition for 1 h. Cover the plate to avoid evaporation.
6. Add 5 μL of 4X detection mixture to plate with the 4X UDP-Tracer determined in Figure 3B.
7. Cover plate to avoid evaporation for 1 h at RT.
8. Read the plate in a plate reader equipped with TR-FRET filters at Ex320/Em615/Em665.
OPS, orthogonal pooled screening; TR-FRET, time-resolved Förster resonance energy transfer; UDP, uridine 5′-diphosphate.
Compounds were screened at a concentration of 8.3 μM in the enzyme reaction; DMSO was at 1.7%; the reaction volume was 15 μL. UDP-GalNAc and EA2 peptide were used at their Km concentrations of 150 and 50 μM, respectively and ppGalNAc-T3 was used at 1.5 ng/μL in the screen and in the follow-up dose–response experiments. The ppGalNAc-T3 enzymatic reactions were incubated for 1 h at room temperature, followed by the addition of 5 μL of UDP Detection Mix containing UDP 2 antibody-Tb and UDP-HiLyte 647 tracer in 1x Stop and Detect Buffer. After 1 h incubation at room temperature, FRET was measured. Positive control reactions lacked inhibitor (DMSO-solvent control wells) and the negative control wells lacked enzyme.
Raw data were deconvoluted to identify individual compounds that caused an increase in FRET (indicating inhibition) more than three SDs from mean of all compound-containing wells. Hits were cherry picked from the same stock solutions as was used to prepare the OPS library and used for dose–response measurements starting at either 500 or 100 μM; assays were run using the same conditions used for the screen. To assess interference with the detection reagents, a mock screen was performed with the same 8,000 OPS compounds in reactions lacking enzyme and containing 135 μM UDP-GalNAc and 15 μM UDP to represent 10% conversion of 150 μM UDP-GalNAc. The fivefold greater efficiency of the OPS method was particularly helpful in this regard.
Results and Discussion
Assay Development and Optimization
Reagents
The Transcreener UDP 2 TR-FRET assay relies on detection of the invariant product of a UDP-glycosyltransferase reaction using a competitive immunoassay with a TR-FRET readout (Fig. 1). Displacement of a UDP HiLyte 647 tracer from a terbium-linked monoclonal antibody by UDP causes a decrease in the TR-FRET signal. The terbium chelate used is excited from 300–350 nm and has four sharp emission peaks centered at 490, 545, 580, and 620 nm; the excitation spectrum of the UDP-HiLyte 647 tracer overlaps with the 620 nM terbium emission, and it emits at 650–700 nm. We typically excite at 320 nm and read emissions at 615 from the terbium, and 665 nm from the HiLyte 647 tracer. FRET occurs when the tracer is bound to antibody, resulting in quenching of the 615 nm terbium signal and enhancement of the 665 nm tracer signal. The TR-FRET signal is expressed as a ratio of emission at 665/615 nm.
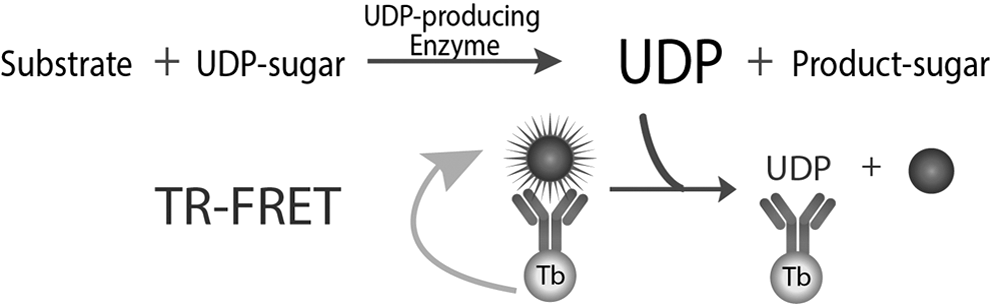
Transcreener UDP 2 TR-FRET assay principle. The assay relies on selective immunodetection of UDP in a homogenous format. Displacement of a UDP-fluorophore tracer from an antibody-Tb complex results in decreased FRET, which is read using time-gated detection to allow dissipation of background fluorescence from screening compounds. TR-FRET, time-resolved Förster resonance energy transfer; UDP, uridine 5′-diphosphate.
The long luminescence lifetime of the terbium chelate compared with fluorescence from organic dyes (hundreds of microseconds vs. several nanoseconds, respectively) allows time-gated detection, so that the TR-FRET signal is read well after prompt fluorescence from library compounds has dissipated.
The assay typically is formatted with an excess of HiLyte 647 tracer, which binds to the antibody with low nanomolar affinity, and the more expensive UDP 2 antibody-Tb is used at a constant concentration of 2 nM. The working range of the assay for detection of UDP is approximately 30-fold, as is typical for a competitive equilibrium binding assay, and the dynamic range can be tuned by varying the tracer concentration, allowing detection of UDP concentrations in the low nanomolar to high micromolar range.
Antibody specificity
High selectivity for UDP versus UDP-sugars is a critical requirement for use of the assay in measuring glycosyltransferase activity, especially under initial velocity conditions, where UDP-sugars will be present in at least 10-fold excess over UDP. To assess selectivity, competitive equilibrium binding assays were used to measure the relative affinity of the antibody for UDP, UDP-sugars, and other uridine nucleotides (Fig. 2). The IC50 value for UDP displacing the tracer was 0.29 ± 0.02 μM under these conditions, and the UDP-sugars tested had IC50 values at least 500-fold higher, indicating high selectivity for the target analyte. Note that we included many of the UDP-sugars that are used by glycosyltransferases in these experiments; therefore, the results suggest that the assay should be broadly applicable. The antibody exhibited 32-fold selectivity versus UTP, with an IC50 of 9.3 ± 0.4 μM, whereas uridine and UMP did not displace the tracer at the highest concentration tested.
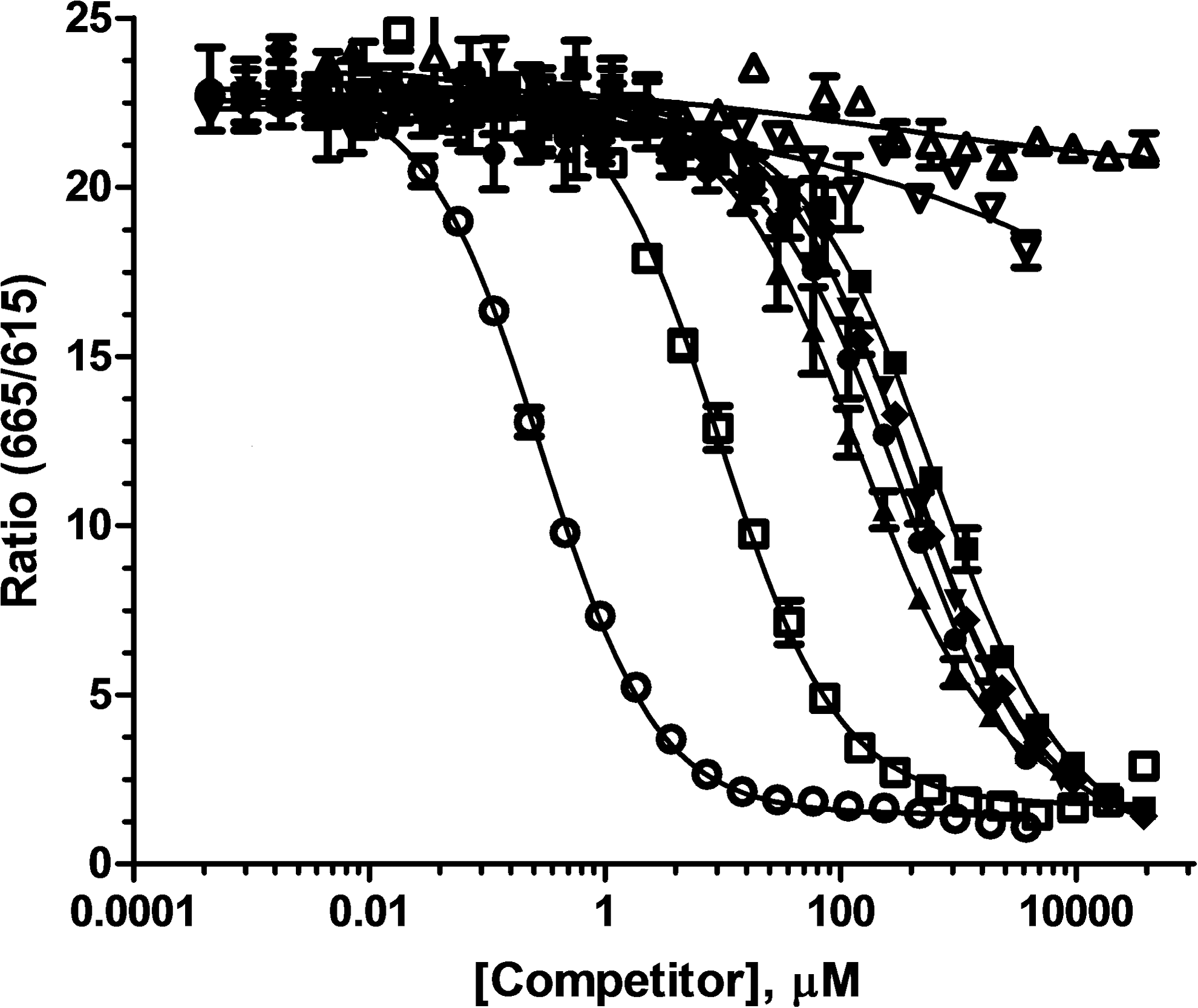
Competitive binding curves for uridine nucleotides and UDP-sugars. (◯) UDP, (□) UTP, (Δ) uridine, (▽) UMP, (●) UDP-GA, (■), UDP-Glc, (▲), UDP-Gal, and (◊) UDP-GalNAc were serially diluted in 50 mM Tris buffer and 5 mM MgCl2. Detection mix consisting of UDP-HiLyte 647 tracer and UDP 2 antibody-Tb was added and plates were read after 90 min of incubation at room temperature. The data presented are mean ± SD of duplicate wells (n = 2). SD, standard deviation; UDP-GA, UDP-glucuronic acid; UDP-Gal, UDP-Galactose; UDP-GalNAc, UDP-N-acetyl-galactosamine; UDP-Glc, UDP-glucose; UMP, uridine 5′-monophosphate; UTP, uridine 5′-triphosphate.
Optimizing tracer concentration
The assay is formatted with the UDP 2 antibody-Tb at a constant concentration of 2 nM, and the tracer concentration is varied to adjust the dynamic range. As the tracer concentration is increased with a proportional increase in the fractional occupancy of the antibody-Tb, more UDP is required to displace the tracer and the assay becomes less sensitive. In this way, the assay can be tuned for glycosyltransferases with different intrinsic Km and Vmax values. The tracer–antibody interaction can also be affected by buffer components such as metals and salts and by small amounts of UDP present in commercial UDP-sugar preparations, so it is important to determine empirically the optimal tracer concentration for the specific enzyme reaction conditions that will be used. For initial velocity enzyme detection (e.g., ≤10% substrate conversion), a concentration of tracer that produces 85% of the maximal FRET in an antibody–tracer binding curve (EC85), is a good balance between signal magnitude and sensitivity of detection.
Accordingly, we titrated the HiLyte 647-UDP tracer in the presence of UDP-Glc concentrations from 1 to 1,000 μM in a standard enzyme buffer (Fig. 3A). The binding curves were essentially superimposable at 1 and 10 μM UDP-Glc, reflecting tracer EC85 values of 7.3 ± 0.04 nM and 8.5 ± 0.05 nM, respectively; the EC85 was approximately twice as high at 100 μM UDP-Glc (15.7 ± 0.1 nM) and almost ten times higher at 1,000 μM UDP-Glc (67 ± 0.5 nM).

Determining optimal tracer concentration.
We then titrated the tracer in the specific reaction conditions for the four enzymes used in this study, ppGalNAc-T2, ppGalNAc-T3, β4GalT1, and Toxin B (Fig. 3B). The different conditions used reflect differences in the enzymes' requirements for UDP-sugar donors and acceptor substrates, as well as additives such as salts, metals, and detergents (see Enzyme Assays in Materials and Methods section). As expected, the curves for the two peptide glycosyltransferases, ppGalNAc-T2 and ppGalNAc-T3, are nearly identical; their buffers both contain 100 μM UDP-GalNAc with only minor differences in the salts and metals present. The curves for the β4GalT1 and Toxin B buffers, which contain 250 μM UDP-Gal and 400 μM UDP-Glc, respectively, are significantly shifted to the right, accordingly. Interpolation of EC85 values from these binding curves indicate optimal tracer concentrations of 16.5 ± 0.2, 20.9 ± 0.1, 173.8 ± 0.1, and 56.1 ± 0.1 nM for detection of ppGalNAc-T2, ppGalNAc-T3, β4GalT1, and Toxin B, respectively, in their specific reaction conditions.
These results demonstrate the ability to optimize the Transcreener UDP 2 TR-FRET for detection of glycosyltransferases with diverse donor, acceptor, and effector requirements simply by adjusting the tracer concentration. We note that most glycosyltransferases require Mn2+ as a cofactor, and metals such as Mn2+, Cr+, Co2+, Fe2+/3+, and Cu2+ can quench negatively charged lanthanide chelates by an ion-pair mechanism. This effect was mitigated by the addition of EDTA, which is present in the Stop and Detect buffer both to stop the enzyme reaction and to prevent quenching of the terbium complex.
Standard curves
Standard curves mimicking enzyme reactions were used to assess the assay response and robustness over a range of initial UDP-Glc concentrations. The standard curves were performed with 16 replicates to allow determination of Z' values at each point in the mock reaction progress curves. The LLD is at the point where Z' >0, and a Z' of 0.7 is considered to be the benchmark for HTS assay robustness. Standard curves mimicking the enzymatic conversion of UDP-Glc to UDP, at initial UDP-Glc concentrations from 1 to 1,000 μM, were constructed (Fig. 4). The dynamic range for each standard curve was set by using the tracer at the appropriate EC85 concentration determined, as described in Figure 3A.
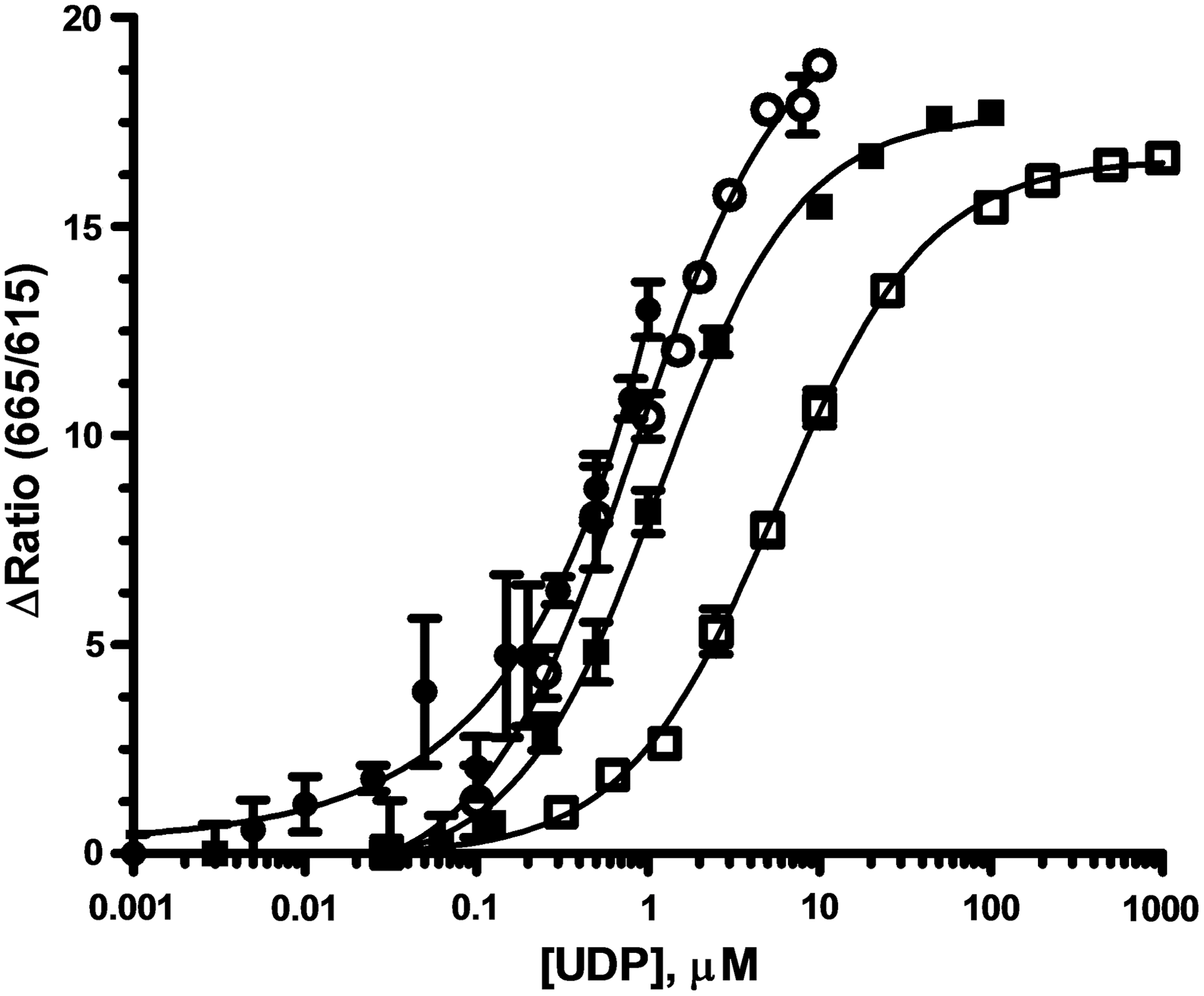
UDP-Glucose/UDP standard Curves and Robustness. Standard curves mimicking enzyme progress curves were constructed starting with 1 (●), 10 (◯), 100 (■), or 1,000 (□) μM UDP-Glc concentrations, with 16 replicates. UDP was added and UDP-Glc was decreased proportionally. For each curve, the EC85 concentration of UDP-HiLyte 647 Tracer was used (Fig. 3A) and UDP 2 antibody-Tb was present at 2 nM. The data presented are mean ± SD of sixteen wells (n = 16).
The same trend was observed for the standard curves starting at 10, 100, and 1,000 μM UDP-Glc: as UDP was added and UDP-Glc was decreased proportionally, the 665/615 emission ratio increased in a sigmoidal fashion to a maximum of 15–20. The total change in the FRET signal was very similar despite the 10-fold differences in the UDP concentration ranges because the dynamic range was adjusted by using different tracer concentrations. The Z' values determined from these standard curves (Table 2) indicate that the assay can be expediently tuned to provide a robust response for initial velocity enzyme activity measurements (i.e., ≤10% conversion) at these UDP-Glc concentrations. The sensitivity limits of the assay became apparent in the standard curve starting at 1 μM UDP-Glc, where 30% conversion was required to generate a sufficient signal for a Z' of 0.7 (Table 2), and the maximum FRET observed was <15 (Fig. 4). This result is consistent with the observed LLDs for UDP, which ranged from 150 to 500 nM, depending on how the dynamic range was adjusted.
Z' Values for Standard Curves
Each standard curve (Fig. 4) mimicked enzymatic conversion of UDP-glucose to UDP starting at the indicated initial concentration of UDP-glucose.
LLD, lower limit of detection.
Deck and signal stability
To assess the suitability of the detection reagents for an automated HTS environment, we tested their stability before dispensing into plates (deck stability) and after they were added to wells containing UDP, UDP-Glc and other components typically used in glycosyltransferase reactions (signal stability). To measure deck stability the UDP 2 antibody-Tb and tracer were mixed, stored at room temperature, and dispensed at intervals into wells containing standard curves mimicking conversion of 10 μM UDP-Glc to UDP. The standard curves are superimposed (Fig. 5A), demonstrating that the reagents are stable at room temperature for at least 8 h before dispensing. Signal stability was measured by intermittently reading a plate containing a similar UDP-Glc/UDP standard curve kept at room temperature. These standard curves also superimposed (Fig. 5B), demonstrating that the assay signal is stable for at least 24 h.

Stability of UDP
2
TR-FRET detection reagents and assay signal.
Day to day variability was also determined from standard curves. In standard curves for conversion of 10 μM UDP-Glc to UDP run on three separate days, we observed an average high signal of 19.2 (4.7% CV), an average low signal of 2.2 (11.2% CV), and an average Z' of 0.74 at 10% conversion or UDP-Glc to UDP. The tolerance of the assay to commonly used reagents was tested in dose–response mode in mock reactions representing 10% conversion of UDP-Glc to UDP. The maximal tolerated concentrations, defined as those which caused less than 10% change in the assay signal, were 3% for DMSO, 6.25% for ethanol, 5% for Triton™ X-100, 1% for Brij-35, 300 mM for NaCl, and 0.5 mg/mL for bovine serum albumin.
Validation for Detection of Enzyme Activity and Inhibitor Screening
Enzymes
We tested the UDP 2 TR-FRET assay for detection of glycosyltransferase activity using four enzymes that have diverse requirements for UDP-sugar donors, acceptor substrates, and buffers/additives. ppGalNAc-T2, ppGalNAc-T3, B4GalT1, and Toxin B were titrated (Fig. 6A) in their optimal reaction buffers (see Enzyme Assays), and the reactions were initiated by the addition of 100 μM UDP-GalNAc (ppGalNAc-T2 & T3), 250 μM UDP-Galactose (B4GalT1), or 400 μM UDP-Glc (Toxin B). Following a 60-min incubation, the enzyme reactions were stopped and detection reagents were added. The FRET ratio increased with the enzyme concentration for all four enzymes, with a robust response at a concentration in the 2–20 nM range (Fig. 6A). This indicates that the assay should be suitable for accurate measurement of inhibitor potencies in the low nanomolar range. Note that the acceptor substrates for these four enzymes include two peptides, a sugar, and water, reflecting the broad applicability of the UDP detection assay format for diverse types of glycosyltransferases.
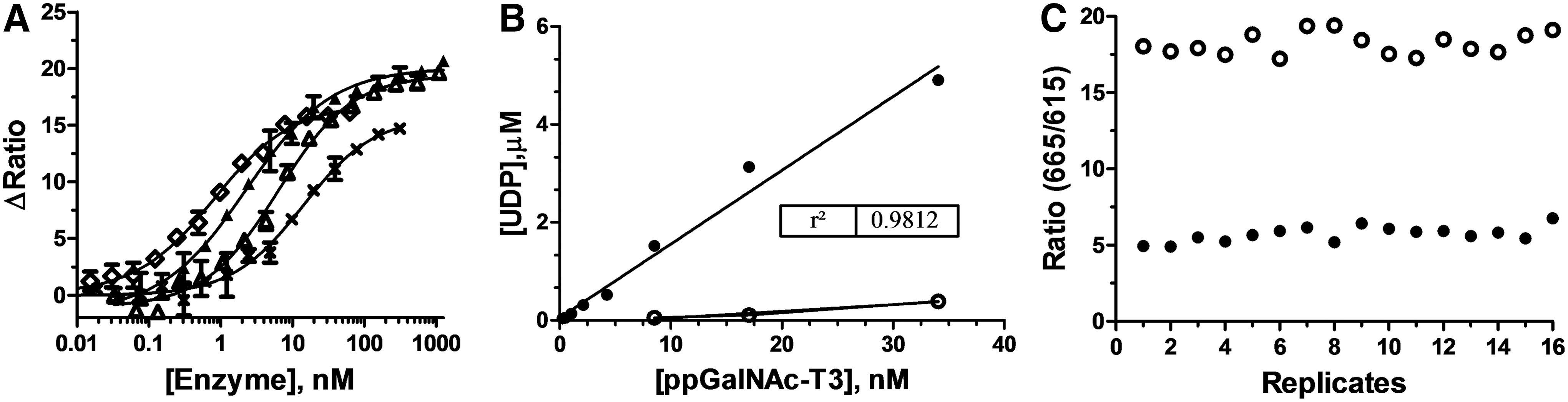
Detection of glycosyltransferases.
Demonstration of a linear response to enzyme concentration is an important requirement for a quantitative assay. Because the detection reaction in this assay is a saturable binding event, the raw TR-FRET signal is not expected to be linear. However, after converting the FRET data from the ppGalNAc-T3 titration into UDP formation, a good linear correlation between enzyme concentration and product formation was observed (Fig. 6B). Additionally, very little UDP was formed in the absence of the acceptor peptide (Fig. 6B). Taken together, these data show that the UDP 2 TR-FRET assay is measuring ppGalNAc-T3 enzymatic activity. Finally, the EC80 concentration of ppGalNAc-T3 from the enzyme titration, which occurred at ∼10% conversion, was used to determine the Z' value for enzyme detection. The observed Z' of 0.70 confirmed that the assay is suitable for screening with ppGalNAc-T3 (Fig. 6C).
Pilot screens
To test the robustness of the TR-FRET-based UDP detection assay, a pilot screen was performed using an 8,000 compound subset of an OPS library (Table 1). The OPS approach combines 10 compounds in each well, with each compound appearing twice among a unique combination of nine other compounds. Individual compound hits in screening data are imputed from their correlated activity at each location (X and Y), avoiding the need for retesting each component of active compound mixtures. 40 –42 Importantly, screening in this way requires only 1/5th of the resources for HTS compared with single-compound screening formats. We used a hit threshold of three SDs from the mean of the compound-containing wells, and in order for a compound to be selected as a hit, this score had to be achieved in both wells of the orthogonal arrays.
Before screening for ppGalNAc-T3 inhibitors, we screened the OPS subset in mock reactions representing 10% conversion of 150 μM UDP-GalNAc (135 μM UDP-GalNAc/15 μM UDP) to assess the potential for compound interference with the detection reagents. High and low controls for all the plates representing 0% and 10% conversion of UDP-GalNAc to UDP had average respective signals of 20.9 and 3.47 and SDs of 1.0 and 0.33, yielding a Z' value of 0.77. Only one compound out of 8,000 caused a change in the FRET ratio more than three SDs from the mean for the low controls, indicating a very low level of interference.
The 8,000 compound OPS subset was screened for inhibition of ppGalNAc-T3 enzyme activity under initial velocity conditions with UDP-GalNAc and the EA2 acceptor peptide at their Km concentrations. The statistics for the screen were indicative of a robust assay (Table 3), as reflected in Z and Z' values of 0.6. Seven compounds increased the FRET ratio for both the X and Y scores above the hit threshold of 6.6 for a raw hit rate of 0.09% (Table 4). The X and Y scores for the hits in the pilot screen were in good agreement, as shown by ratios close to unity, except for one outlier (compound #7). Such consistency in the orthogonally duplicated values for the compound hits is consistent with the OPS outcomes published by others, and is a hallmark of a well-developed HTS assay. 41,42 The “interfering” compound from the mock screen was not identified as a hit in the ppGalNAc-T3 screen, most likely because the CV for the positive controls in the live screen was considerably higher.
Pilot Screen Statistics
Pilot Screen Hit Scores
Inhibitors
The seven hits were cherry picked, dispensed as pure compounds from the same DMSO stock solutions used to prepare the pooled library, and used in dose–response experiments with ppGalNAc-T3 (Fig. 7A). The dose–response curves were repeated using a Transcreener UDP 2 assay with an FP readout (Fig. 7B), which uses the same antibody without the Tb conjugate and a UDP-Alexa 633 tracer rather than the HiLyte 647 tracer. Five of the seven hit compounds exhibited dose–response curves using either assay format for a confirmation rate of 71%. The IC50 values obtained with the two assay formats were within twofold in all cases except compound #5, which exhibited fivefold higher potency in the FP assay relative to the TR-FRET assay (Table 5). Despite this difference, the rank order of inhibitor potency for the five hits was the same in both assays.
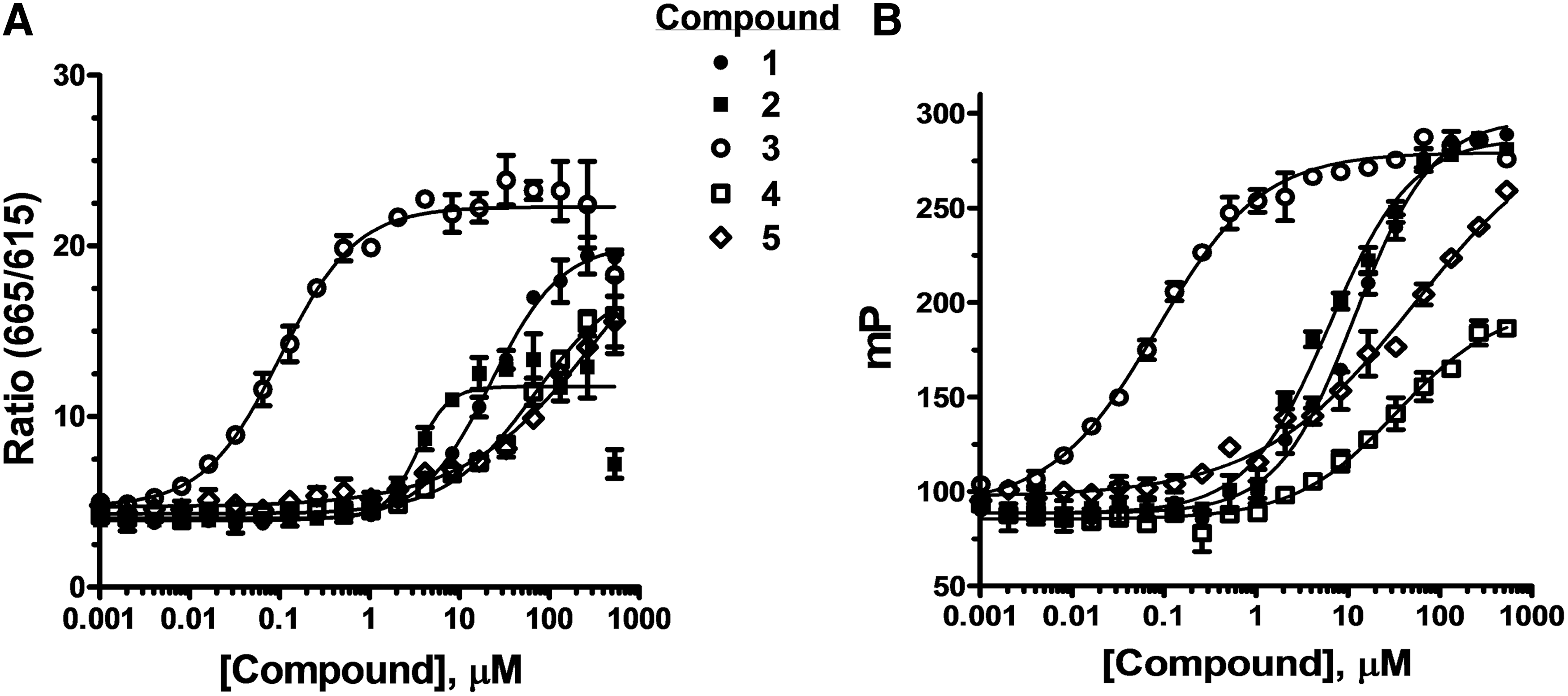
Confirmation of hits from ppGalNAc-T3 pilot screen. Dose–response experiments with hits from the pilot screen were performed using the
Potency of Confirmed Hits
FP, fluorescence polarization.
Summary
We developed a Transcreener assay for detection of UDP produced by glycosyltransferases with a far red TR-FRET readout. Because the assay relies on direct immunodetection of UDP, it eliminates the need to counter-screen for compounds that interfere with the coupling enzymes as is required for indirect UDP detection methods. Moreover, in contrast with ligand displacement assays, which bias screening toward inhibitors that bind to a specific site, the assay should be agnostic with respect to the inhibitor binding site or mechanism.
The antibody used in the assay was shown to be highly selective for UDP versus several UDP-sugars used as glycosyltransferase donors, enabling detection of UDP in the presence of a large excess of UDP-sugar. The LLD was 150 nM, and the dynamic range could be tuned for use of UDP-sugars from 1 μM to 1 mM by adjusting the tracer concentration. These are the key parameters for initial velocity glycosyltransferase enzyme assays, and the method was validated by detecting four different UDP-glycosyltransferases using UDP-GalNAc, UDP-Glc, and UDP-Gal near their Km concentrations.
In a pilot screen with ppGalNAc-T3, the assay showed robust performance (Z' ≥0.6), with a low-to-null level of compound interference, and five of the seven hits were confirmed in dose–response with an orthogonal assay.
These results provide strong validation for use of the Transcreener UDP 2 TR-FRET assay in HTS efforts targeting glycosyltransferases. In addition, both the reagents and the signal were stable at room temperature overnight, which will provide flexibility for automation protocols and minimize run-to-run variability. The UDP 2 TR-FRET assay should provide a general approach for screening glycosyltransferases that use a UDP-sugar donor.
Footnotes
Disclosure Statement
BellBrook Labs manufactures and markets the Transcreener UDP Assay Kits used in the research reported here. BellBrook also has an agreement with the Lankenau Institute for Medical Research to represent their Orthogonal Pooled Screening Library to potential fee-for-service clients. The authors, who collaborated in the research, declare no other potential conflicts of interest with respect to the research, authorship, and/or publication of this article.