
Abstract
Contamination of pharmaceutical products and medical devices with pyrogens such as endotoxins is the most common cause of systemic inflammation and, in worst cases, of septic shock. Thus, quantification of pyrogens is crucial. The limulus amebocyte lysate (LAL)-based assays are the reference tests for in vitro endotoxin detection, in association with the in vivo rabbit pyrogen test (RPT), according to European Pharmacopoeia (EP 2.6.14), and U.S. Pharmacopoeia (USP <85>). However, several substances interfere with LAL assay, while RPT is not accurate, not quantitative, and raises ethical limits. Biological assays, as monocyte activation tests, have been developed and included in European Pharmacopoeia (EP 7.0; 04/2010:20630) guidelines as an alternative to RPT and proved relevant to the febrile reaction in vivo. Because this reaction is carried out by endogenous mediators under the transcriptional control of nuclear factor-kappaB (NF-kappaB), we sought to determine whether a NF-kappaB reporter-gene assay, based on MonoMac-6 (MM6) cells, could reconcile the basic mechanism of innate immune response with the relevance of monocytoid cell lines to the organism reaction to endotoxins. This article describes both optimization and characterization of the reporter cells-based assay, which overall proved the linearity, accuracy, and precision of the test, and demonstrated the sensitivity of the assay to 0.24 EU/mL endotoxin, close to the pyrogenic threshold in humans. Moreover, the assay was experimentally compared to the LAL test in the evaluation of selected interfering samples. The good performance of the MM6 reporter test demonstrates the suitability of this assay to evaluate interfering or false-positive samples.
Introduction
A heterogeneous class of compounds can cause fever and stimulate the inflammatory response in the host. 1,2 Pyrogens can derive from various microorganisms: peptidoglycan and other carbohydrates from bacterial cell wall, lipoteichoic acid, enterotoxins, exotoxins, and other components of viruses, bacteria, fungi, protozoa, and helminths. 3 Pyrogen contamination of preparations destined to parenteral administration can lead to severe reactions such as organ failure and septic shock, depending on type and amount of the contaminant, and on the sensitivity of the recipient.
Bacterial cell wall lipopolysaccharide (LPS) is the most potent immune stimulator 4 and the major concern to the pharmaceutical industry due to the large use of gram-negative strains in the production of recombinant drugs. Thus, pharmacopoeias indicate that all products and devices intended to overcome the natural barriers of the body have to be tested before release (European Pharmacopoeia, 7th edition 2010 chapters 2.6.8 and 2.6.14; British Pharmacopoeia, volume V 2012, appendix XIV C and D; U.S. Pharmacopoeia, 37th edition. 2012, chapters 85 and 151). To this aim, the rabbit pyrogen test (RPT), introduced at the beginning of the 20th century, 5,6 detects various types of contaminants.
Despite the wide use, the RPT is not accurate and not quantitative, is poorly reproducible, depending on age, weight, and health of rabbits, and raises ethical concerns on the use of animals. 7 Another established test is the limulus amebocyte lysate (LAL) test, specific for endotoxin and β-glucans from yeast cells, 8 but not suited for the detection of other kinds of pyrogen. Moreover, there is phylogenetic distance between Limulus polyphemus and higher vertebrates, and the molecular mechanism at the basis of LAL does not reflect the physiologic reactions underlying fever in humans. 9
In addition, routine LAL test on biologicals (e.g., plasmid DNA, vector preparation, and recombinant proteins) may be sometimes cumbersome due to artifacts and false-positive results and needs to be confirmed by the RPT. The principal interference mechanisms include suboptimal pH conditions, aggregation or adsorption of endotoxins, unsuitable cation concentrations, enzyme or protein modification, β-glucan, and samples containing endotoxin. 10 As recently evidenced by Gnauck et al., the LAL assays may be unsuitable for the determination of endotoxin levels in blood samples or other human fluids. 11 Finally, LAL test—yet indirectly—is an animal-based test using rare animals. In light of the debate regarding the in vitro alternatives to animal tests, a cell-based assay, which allows release tests to be performed sparing animal pain and distress, would be welcome. 12
To overcome the lack of specificity and the ethical concerns, and to better represent the organism reaction to pyrogens, a number of tests based on human whole blood or isolated primary cells have been developed, commonly referred to as monocyte activation tests (MAT). The interaction between pathogen-associated molecular patterns and Toll-like receptors on the surface of monocytoid cells triggers intracellular signaling, resulting in nuclear factor-kappaB (NF-kappaB) activation, expression of genes encoding cytokines, chemokines and adhesion molecules, and release of inflammation mediators. 13 In particular, the febrile reaction is mediated by interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor α (TNFα), whose expression is dependent on NF-kappaB engagement.
Two international validation studies led to the development of four variants of MAT: the human whole blood test measuring IL-1β, its variant using cryopreserved whole blood, and the peripheral blood mononuclear cell test and the MonoMac-6 (MM6) cell line, measuring IL-6. 14 –18 Despite several limitations due to the cumbersome method, the MAT were approved by European Pharmacopoeia as an alternative or in addition to RPT (EP 7.0; 04/2010:20630). Recently, other tests have been proposed, based on monocytic/macrophage cell lines, overall demonstrating their suitability for representing the in vivo reaction to pyrogens. 9,19,20
We show here a test based on the luciferase reporter gene under the control of a NF-kappaB-responsive promoter. This mechanism mimics the physiological response to pathogens at an upstream step, respect to the production of cytokines, and makes the reporter system potentially responsive to a large array of pyrogens and immune stimulators. In addition, the test is based on a clone of MM6 cells, expressing large amounts of CD14 coreceptor of TLR4 (Toll-like receptor 4), 21 and therefore, particularly sensitive to endotoxins. As shown here, the optimized assay was characterized for several parameters according to official guidelines, verifying its reliability. This allowed the method to be applied to selected biologicals, particularly difficult to evaluate by LAL assays due to interference, demonstrating the suitability of the cell-based test and its potentiality as a support test.
Materials and Methods
Cell Culture
Human MM6 from acute monocytic leukemia (DMSZ, Braunschweig, Germany) were grown in suspension at 37°C in a 5% CO2 atmosphere, using RPMI1640 supplemented with 10% (v/v) fetal bovine serum, 2 mM L-glutamine, 1X nonessential aminoacids, 9 μg/mL insulin (Sigma-Aldrich, St. Louis, MO, USA), and 1 mM sodium pyruvate (Lonza, Basel, Switzerland). The cells were maintained at a density of 0.5–1 × 106 cells/mL and diluted with fresh medium 1:3 (v/v) at 3-day intervals. The master cell bank of MM6 clone #119 was prepared expanding the cells until passage no. 21; the working bank was created expanding until passage no. 23 the cells thawed from the master bank. All the subsequent analyses were performed on cells of the working bank.
Lentiviral Vector Construct and Cell Transduction
The reporter construct was obtained from a plasmid encoding a third generation self-inactivating vector, originally bearing the green fluorescent protein (GFP) reporter gene. The GFP coding sequence was excised with XhoI and SalI and replaced with the firefly luciferase coding sequence, driven by a minimal promoter responsive to NF-kappaB, and featuring a transcription blocker. The luciferase expression cassette has been inserted antisense, respect to the vector. Lentiviral vector particles (LVVINV#1) were obtained after transfection of four different constructs in 293FT cells (two core packaging, envelope, and transfer vector constructs). 22
Physical titer was measured as the amount of p24 produced (1.8E + 09 pp/mL) by the HIV-1 p24 ELISA (enzyme-linked immunosorbent assay) Kit (PerkinElmer, Waltham, MA, USA). Infectivity titer was quantified as the copy number of lenti-proviral DNA by real-time quantitative polymerase chain reaction (qPCR) after transduction of reference CEM A3.01 cell line (2.3E + 06 TU/mL). MM6 cells were then transduced by spinoculation with the obtained vector, and subsequently cloned, starting from 0.3 cells/well. The 64 clones obtained were evaluated by real-time qPCR for copy number of integrated vector, considering the cells as tetraploid, as described in manufacturer's instructions, and 25 clones out of 64 resulted to be positive.
Stimulation with Bacterial Endotoxin
Endotoxin treatments were performed, for the times and with the doses each time indicated, with reference standard endotoxin (RSE) from Escherichia coli serotype 0113:H10:K (Charles River, Wilmington, MA, USA). In addition to RSE, 1 μg/mL LPS from E. coli serotype 0111:B4 (Sigma-Aldrich) was used, where indicated.
Luciferase Assay Procedure
The assay protocol was optimized as shown in Table 1. Cells from the working bank are thawed on day 1 and seeded at a density of 106 cells/mL. On day 3, cells are diluted 1:2 (v/v) and expanded until day 7, when cells are seeded at 0.5 × 106 cells/mL. Assay is performed on day 8, utilizing 50 μL of 4 × 106 cells/mL suspension in complete RPMI1640 without phenol red (PR; Sigma-Aldrich) per well of a 96-well black plate (Costar, Corning, NY, USA). Fifty microliters of the same medium, containing either the desired concentrations of RSE for the standard curve or the appropriate dilution of the test samples, is added to the cells, and the plate is incubated at 37°C ± 1°C for 4 h ± 15 min. After incubation, the plate is maintained at 22°C ± 1°C for 30 ± 5 min, then 100 μL of Neolite Reporter Gene Assay reagent (PerkinElmer) is added to each test well, according to the manufacturer's instructions, and the plate is read for luminescence in a plate luminometer (BMG Labtech, Offenburg, Germany) using the software FLUOstar OPTIMA. As a positive control, 100 μL of the purified luciferase enzyme QuantiLum (Promega, Madison, WI, USA) is used, previously diluted 1:106 (v/v) in RPMI1640 without PR. Raw data from the standard curve, expressed as relative light units (RLU), are plotted as a five parameter curve, and sample data are analyzed accordingly. To determine sample interference, sample dilutions are analyzed in the presence or absence of 0.7 EU/mL of RSE. A percentage of recovery of the spiked RSE concentration, calculated as x = [sample +0.7 EU/mL]–[sample] × 100/0.7, ranging between 50% and 200% (0.35 × 1.40 EU/mL), is considered as evidence of no interference. Sample dilutions that satisfy this requirement are selected for the calculation of endotoxin content.
Assay Protocol
RSE, reporter standard endotoxin.
Immunofluorescence
MM6 cells and transduced MM6 reporter cells were washed in phosphate-buffered saline (PBS), fixed on coverslips with 4% (v/v) formaldehyde in PBS for 15–30 min at room temperature. The samples were subsequently blocked for 30 min with 50 μg/106 cells of human IgG in blocking buffer, that is, PBS with 3% (v/v) heat-inactivated fetal bovine serum and 10 mM Hepes, then incubated with 25 μg/mL of monoclonal anti-CD14 (R&D Systems, Inc., Minneapolis, MN, USA) in blocking buffer for 1 h. The primary antibody was then detected by incubating the samples for 1 h with the secondary fluoresceine isothiocyanate (FITC)-conjugated rabbit anti-mouse IgG (Sigma-Aldrich) diluted 1:100 (v/v) in a blocking buffer. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) at a dilution of 1:10,000 (v/v) for 30 s. The samples were observed in a fluorescence microscope Leica DMLB (Wetzlar, Germany) equipped with a CCD camera DC300F. The images were processed with the software NIH ImageJ (Bethesda, MD, USA).
Flow Cytometry
MM6 cells and transduced MM6 reporter cells were washed with PBS, resuspended at a concentration of 5 × 105 cells/mL, and blocked 30 min at room temperature in blocking buffer added with 50 μg/106 cells of human IgG. The cells were then incubated in blocking buffer for 30 min with monoclonal antibody anti-CD14, following the manufacturer's instructions, and subsequently for 30 min with the secondary FITC-conjugated rabbit antibody, diluted 1:100 (v/v) in a blocking buffer. After washes in PBS, the cells were resuspended in 300 μL PBS and read in a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA).
Inhibition of the CD14/TLR4 Receptor Complex
MM6 reporter cells (5 × 106) were preincubated for 1 h at 37 ± 1°C with 0, 0.6, 1.2, 2.5, 5, 10, 20, or 40 μg/mL of monoclonal antibodies anti-CD14 (R&D Systems) or anti-TLR4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The antibodies were serially diluted 1:2 (v/v) in RPMI1640 without PR. After incubation, the cells were resuspended in complete medium without PR at a concentration of 4 × 106 cells/mL and processed for the luciferase assay as described above, with an RSE standard curve in the range 5.88–0.05 EU/mL. Unstimulated cells and cells not pretreated with antibodies were considered as negative controls. Anti-CD14 and anti-TLR4 were also administered simultaneously, both at a concentration of 10 μg/mL and their combined effect assayed on the same standard curve.
LAL Assay
Endotoxin content of the compounds tested was determined by a kinetic LAL assay using an Endochrome-K assay kit (Charles River Laboratories International, Inc., Wilmington, MA, USA), according to the manufacturer's instructions.
Statistical Analysis
Data are presented as the mean ± standard deviation (SD) of at least three independent experiments. The significance was calculated either by the Friedman statistical test followed by nonparametric multiple comparison or by the Student t-test, as appropriate. P values <0.05 were considered to be significant. A five-parameter standard curve was used to calculate endotoxin content of unknown samples.
Results
MM6 Transduction and Clones Characterization
MM6 monocytic cell lines were chosen for their high response to bacterial endotoxin. 9 MM6 were transduced with LVVINV#1 vector (Fig. 1) with a transduction efficiency of 39% and a copy number ranging between 0.96 and 10.22 copies/cell. Each positive clone was expanded and selected for further characterization, testing the response to endotoxin and the luciferase activity after incubation with different dilutions of RSE, ranging between 10 and 0.16 EU/mL. Table 2 shows that among the clones tested, clones #2, #44, #95, #104, and #119 were more responsive, as they yielded better signal/background ratios. Since, among the most responsive clones, clone #119 was the only one bearing a single integrated copy of the reporter vector, it was definitively chosen for subsequent expansion and creation of a cell bank.
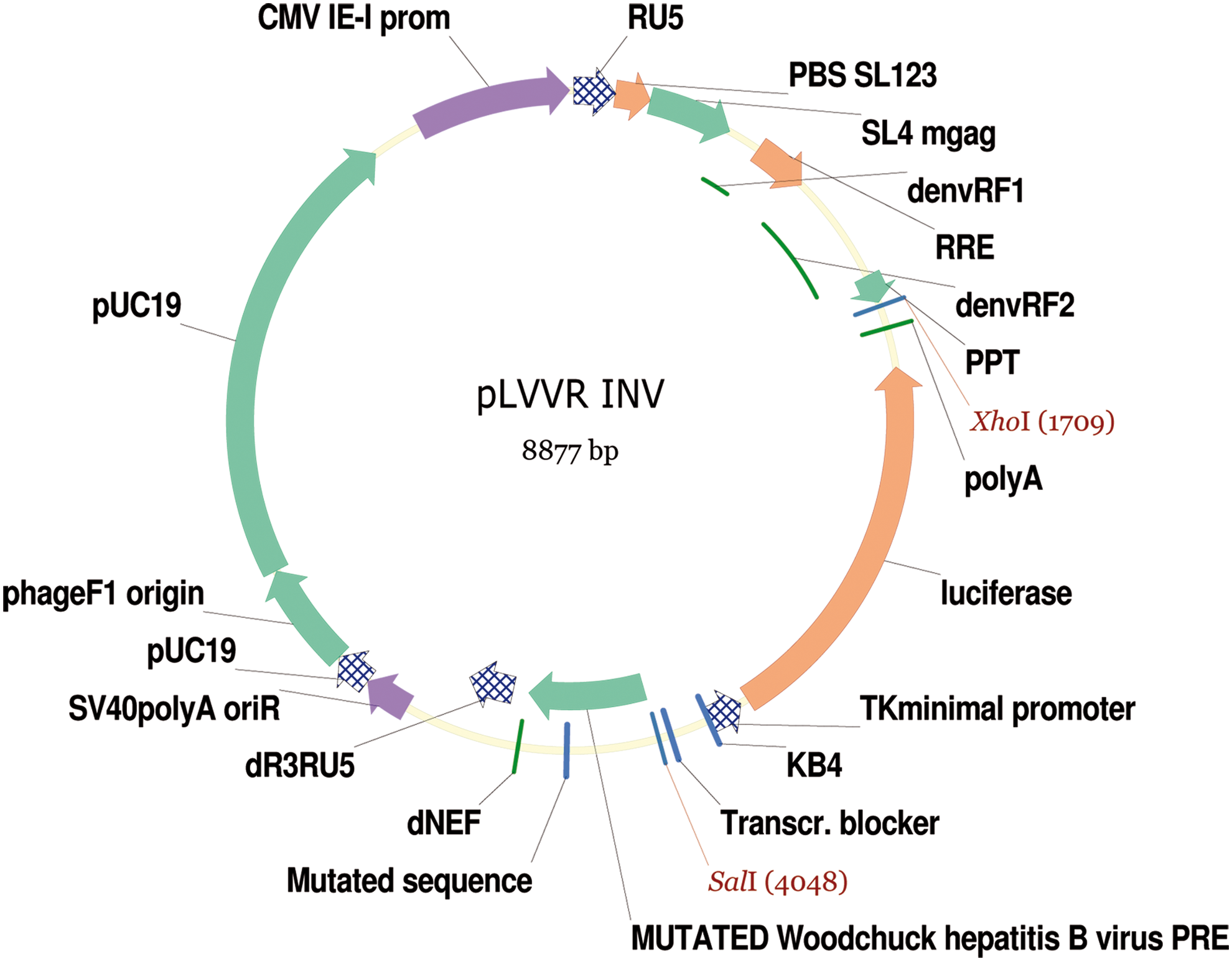
Map of the lentiviral vector-based construct LVVINV#1, containing the luciferase reporter gene driven by a minimal-kappaB promoter. LVV, lentiviral vector. Color images available online at
Responsiveness of the Reporter Cell Clones
RLU, relative light units.
Since PR in culture media negatively affects the light output, dose–response curves in presence or absence of PR were compared. The dose–response curves in Figure 2 confirmed the interference of PR, because it yielded 40%–50% (P < 0.001) lower signals from cells as well as from positive control, represented by recombinant luciferase (47,057 vs. 51,394 RLU).
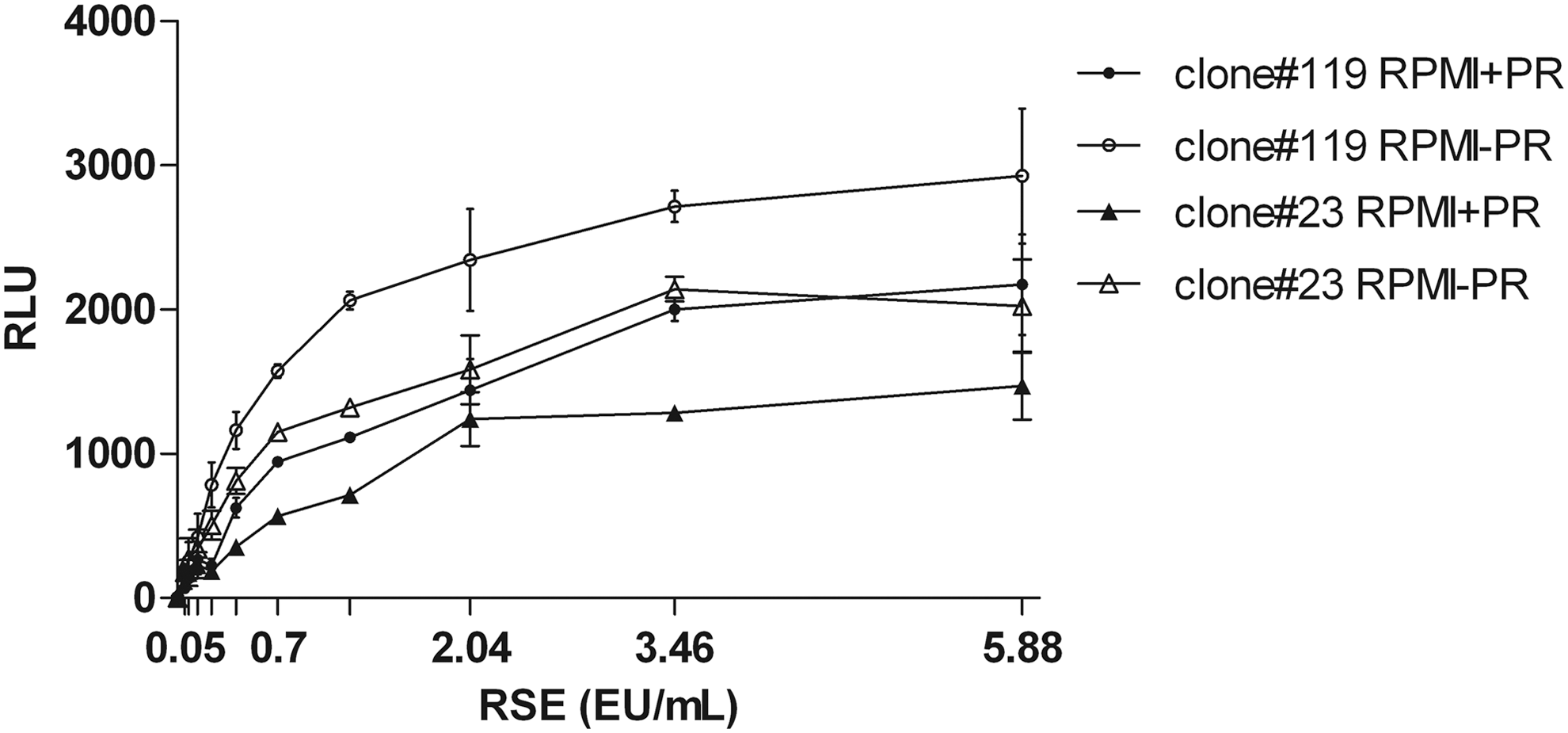
Line chart shows luminescent signal generated by RSE standard curve of the endotoxin reporter assay. The assay was performed on two MM6 reporter cell clones (#23 and #119) with culture medium either lacking or containing PR. Signal is expressed as mean RLU ± SD from three independent experiments. MM6, MonoMac-6; PR, phenol red; RLU, relative light units; RSE, reference standard endotoxin; SD, standard deviation.
CD14 Expression by Reporter Cells
The reporter cells, compared to not-transduced MM6, were characterized for membrane expression of CD14 by cytofluorimetric and immunofluorescence analyses. Thus, the effect of cell prestimulation with LPS (1 μg/mL, 1 h) was also tested in terms of CD14 expression, both on MM6 and on MM6 reporter clone #119. Figure 3a shows that MM6 reporter cells are CD14+ already in basal conditions, compared to MM6 cells. In addition, LPS stimulation remarkably increases the fluorescent signal of MM6 reporter cells. Although a slight increase occurs also in not-transduced MM6 upon prestimulation, these results indicate that our reporter system is particularly competent for endotoxin detection. These observations were confirmed by flow cytometry (Fig. 3b): basal MM6 are extremely heterogeneous in terms of CD14 expression, whereas MM6 reporter cells show significantly higher amounts of CD14 (21.5 ± 2.1% vs. 1.7 ± 0.3%, P < 0.05). In addition, LPS prestimulation raises to 36.5% ± 4.9% the percentage of CD14+ reporter cells.
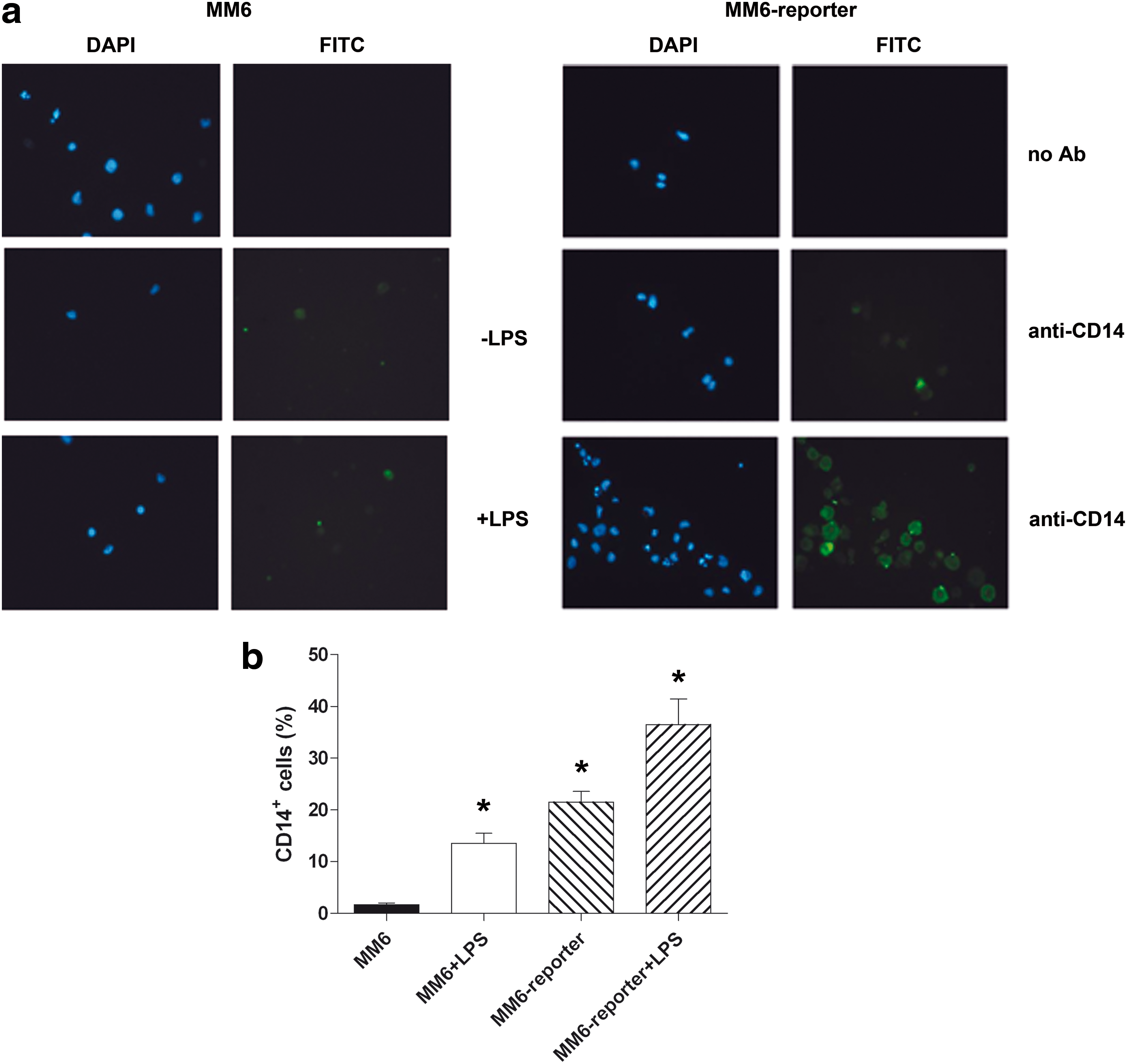
Optimization of the Reporter System
The reporter system was optimized for the number of seeded cells per well, the time of incubation, the 96-well plate plastic, and the reagent used to detect the light emission. Three cell concentrations were tested (5 × 104, 2 × 105, and 5 × 105 cells/well) by treatment with several dilutions of RSE. The concentration 2 × 105 cells/well was selected because it showed the highest signal/background ratio, as shown in Table 3, whereas the concentration 5 × 104 cells/well lacked sensitivity, and 5 × 105 cells/well—although the most sensitive—yielded too high a background.
Determination of Optimal Cell Concentration
The concentration 2 × 105 cells/well was therefore tested at different incubation times at 37 ± 1°C: 0, 4, 5 h, and overnight. Four hours of RSE treatment was established as incubation time for all the subsequent analyses, because it ensures a high signal, within a reasonable duration of the total assay, whereas overnight stimulation causes a considerable reduction of light emission, probably as a consequence of cell suffering (data not shown).
Although both black and white tissue culture plates are indicated for luminescence analyses, black 96-well plates were preferred over white plates. In fact, although they generally dampen the light emission, yielding lower background as well as lower specific signals, black plates ensure reduction of eventual cross-well interference, as indicated by the manufacturers (Table 4).
Comparison Between Cell Culture Plates
Four reporter assay reagents have been tested on cells treated with RSE dilutions for 4 h: Steady-Glo® Luciferase Assay System (Promega, Catalog #E2510), Neolite, Britelite, and Steadylite (PerkinElmer, Catalog #6016726, #6066766, and #6066756, respectively). Of the two suppliers, PerkinElmer was chosen because of easier execution, and a longer shelf-life; in fact, the PerkinElmer reagents are ready-to-use, while the Steady-Glo Luciferase Assay System needs to be reconstituted, and its shelf-life is 15 days. Neolite was chosen out of the three PerkinElmer reagents, because it generates a luminescent signal that is both bright and stable, whereas the Britelite yielded the highest signal, but with a very fast decay (Table 5).
Comparison Between Luciferase Assay Reagents
Accuracy
The accuracy of the test was assessed running nine independent RSE standard curves and calculating the percentage of difference between the obtained results and the expected values (Table 6). The percentage of difference resulted to be ≤28%, for each RSE concentration ≥0.24 EU/mL, of each curve, and percent coefficients of variation (% CV) calculated on at least three replicates were ≤25%. These results demonstrate the accuracy of the assay in the range of RSE standard curve between 5.88 and 0.24 EU/mL.
Accuracy of the Assay
CV, coefficient of variation.
Repeatability and Intermediate Precision of the Assay
Intra-assay precision and interassay precision were assessed as the ability of a sample to yield the same signal from replicates read, respectively, in the same plate or in independent runs. To this end, two different RSE concentrations, 3.46 and 0.41 EU/mL, were run as at least three replicates in the same plate, and the assay was repeated eight independent times. The independent runs were performed in different days, starting from different vials of frozen cells and different preparations of RSE and samples. For each sample, intra- and inter-assay percentage of CV is reported in Table 7. In addition, intra-assay precision was verified on four samples, represented by LVV (LVV-A, LVV-B, LVV-C, and LVV-D) that were run as quadruplicates on the same plate (Table 8). The inter-assay precision was also evaluated on a recombinant protein, Protein 1, tested as triplicates of a 1:100 dilution, in three independent runs (Table 9). Overall, these analyses demonstrated the repeatability of the assay, because the percentage of CV values was ≤29% in all the tests performed.
Intra- and Inter-Assay Precision of the Test on Reporter Standard Endotoxin Samples
Intra-Assay Precision of the Test on Lentiviral Vector Samples
LVV, lentiviral vector.
Inter-Assay Precision of the Test on Recombinant Protein
Limit of Detection, Linearity, and Range of the Assay
According to the United States and European Pharmacopoeias, the lower limit of detection (LOD) was determined considering the SD of the blank (Table 10). The mean background signal from eight independent runs was used to set the LOD at (mean +3x SD) 1,227 RLU, which corresponds to the signal of 0.14 EU/mL, as determined by eight independent runs of the standard curve. On the contrary, taking into account that the lower and upper limits of quantification are, respectively, the lowest and the highest amount of analyte in a sample that can be quantitatively determined with suitable precision and accuracy, the analyses on accuracy of the assay allowed us to set the range of the procedure between 0.24 and 5.88 EU/mL (Fig. 4). On these premises, we have also evaluated two alternative standard curves, extended to 10 or to 17 EU/mL RSE. The results obtained provided evidence for nonlinearity when concentrations of RSE higher than 5.88 EU/mL were added to the curve (0–17 EU/mL RSE: R2 = 0.8959; 0–5.88 EU/mL RSE: R2 = 0.9582).

Sensitivity of the endotoxin reporter system. Line chart shows luminescent signal generated by RSE standard curve of the endotoxin reporter assay. The assay was performed with culture medium without PR. Signal is expressed as average RLU ± SD from 15 independent experiments.
Determination of the Limit of Detection of the Assay
LOD, limit of detection; SD, standard deviation.
Interference of Media
Interference of the assay must be evaluated before testing a new sample, or a new buffer composition. To this aim, three common buffers were analyzed after addition of a known RSE concentration. We selected 0.7 EU/mL to spike our samples, as it is an intermediate dilution of the RSE curve. RPMI1640 medium, CellGRO GMP SCGM medium (CellGenix GmbH, Freiburg im Breisgau, Germany), and PBS were tested either undiluted or at different dilutions in RPMI1640 without PR (Sigma-Aldrich) (Table 11). The recovery of spiked RSE ranged within 50%–200% in undiluted RPMI1640 and CellGRO media, whereas undiluted PBS caused interference. From this evaluation, we concluded that, to be analyzed, samples containing RPMI1640 and CellGRO media do not need prior dilution, while samples suspended in PBS must be diluted at least 1:2 (v/v) in RMPI1640 without PR.
Interference of Buffer Composition
PBS, phosphate-buffered saline; PR, phenol red.
Specificity
We next assessed whether the reporter system was specifically activated upon endotoxin binding to the CD14/TLR4 complex. The CD14 receptor was blocked by different concentrations of a monoclonal anti-CD14 antibody (0, 0.6, 1.2, 2.5, 5, 10, 20, and 40 μg/mL), before assaying the endotoxin standard curve optimized as described above. The luminescent signal is maintained at background level by all the antibody concentrations tested (Fig. 5a), demonstrating the specificity of the endotoxin-mediated signal, because inhibition of receptor engagement prevents signal transduction and reporter gene expression. Similarly, specificity was demonstrated blocking the TLR4, also involved in endotoxin recognition, with anti-TLR4 antibody concentrations in the same range used for anti-CD14. The luminescent signal obtained in the presence of blocking antibody is 50% of that produced by cells in the absence of antibody upon stimulation with endotoxin concentrations in the upper range of the curve (5.88–2.04 EU/mL), and the signal is further dampened to background levels in cells stimulated with the lower range of endotoxin concentrations (Fig. 5b). Moreover, we evaluated the combined effect of 10 μg/mL of both anti-CD14 and anti-TLR4, demonstrating that the luminescent signal of double antibody-treated cells is comparable to background levels for all the endotoxin concentrations tested, thus providing additional evidence of the specificity of the reporter system (Fig. 5c).

Specificity of the endotoxin reporter assay. The indicated concentrations of
Determination of Endotoxin Contamination of Protein Samples
Different substances interfere with LAL assay and generate artifacts, such as IGVENA, which contains IgG proteins and requires dilution before analysis with conventional LAL test, because its protein content interferes with the enzymatic reaction. Similarly, LVV-based constructs, purified and suspended in phosphate buffer for direct in vivo infusion, may yield false-positive results in LAL tests. From this point of view, the use of a MM6 reporter assay may allow to overcome such interference issues. Therefore, four LVV, of which two produced in phosphate buffer (LVV1 and LVV4) and two produced in CellGRO medium (LVV2 and LVV3), and IGVENA were analyzed by both LAL assay and MM6 reporter assay to detect their endotoxin content and to establish eventual product interference. To this aim, several dilutions of the five samples were prepared in RPMI1640 without PR and tested—as such or spiked with 0.7 EU/mL of RSE—by MM6 reporter assay as shown in Table 12. The recovery was calculated as [spiked sample]–[sample], as described in the LAL guidelines by Food and Drug Administration (December 1987). For each sample, the spiked dilution(s) yielding a percentage of recovery in the range 50%–200% (0.35 × 1.4 EU/mL) were considered as not interfering and were then used to calculate endotoxin content of the sample. When assayed by LAL test, 1:100 (v/v) dilutions of LVV1, LVV2, and LVV3 were found to contain, respectively, 11, 210, and 6 EU/mL of endotoxin, whereas they showed a 0.3, 1.3, and 0.2 EU/mL content when assayed by the MM6 reporter test. These results, in light of the valid percentage of recovery resulting from the reporter assay, support the hypothesis that some vector purification procedures may lead to false-positive results by LAL assay. Different results were obtained with the LVV4 vector, for which analysis performed with MM6 reporter test (28 EU/mL) was consistent with the results of LAL test (52 EU/mL). On the contrary, IGVENA, which sometimes shows high interference with conventional tests, was analyzed by LAL assay upon 1:80 (v/v) dilution and by MM6 reporter assay upon 1:5 (v/v) dilution, demonstrating in both cases absence of endotoxin contamination and absence of interference.
Interfering Samples Tested by MonoMac-6-Reporter Assay, Compared to Limulus Amebocyte lysate Assay
MM6, MonoMac-6; LAL, limulus amebocyte lysate.
Discussion
Because pyrogens from microorganisms can occur as contaminations of parenterals, the RPT and the LAL tests are available to estimate their pyrogen content. As an alternative, monocytoid cells have been proposed as bioindicators for these contaminants. The sensitivity of rabbits toward pyrogens depends on the strain used and the experimental conditions. 7 However, the most sensitive rabbit strains develop a significant temperature increase upon exposure to 0.5 EU/mL of endotoxin, whereas the human fever threshold is approximately 0.3 EU/mL. Furthermore, the RPT is not suitable for many products such as radiopharmaceuticals, chemotherapics, analgesics, antipyretics, cytokines, dopamines, and immunosuppressive agents. In addition, RPT cannot be used for cellular preparations, blood components, and stem cells. Above all, the use of in vitro tests is strongly encouraged by animal welfare laws.
Indeed, sterile pharmaceuticals are commonly assayed for contaminations by the LAL test, which does not consume animals, is very sensitive, and allows for semiquantitative or quantitative measurement of endotoxins. The detection limit is usually around 0.03 EU/mL, and the most sensitive test variants detect down to 0.005 EU/mL. Nonetheless, the use of LAL test is limited by its restricted specificity to few types of pyrogens and by the high susceptibility of the clotting system to interference by different drugs. 23 Moreover, although LAL correlates well with the monocytoid cell test, it is less reliable when applied to protein-rich samples and to blood or serum. 9,11 The use of LAL as a pyrogen test is further limited by the fact that LAL activities displayed by the various LPS species are not consistent to their pyrogenicity in humans.
To this regard, the use of human fever reaction of blood-derived cells for in vitro tests was pioneered by Dinarello. 2 This led to the optimization of different qualitative and quantitative MAT, able to detect distinct classes of pyrogens, with a sensitivity limit of 0.5 EU/mL. So far, the MAT rely on a multistep protocol, in which the contaminant readout consists of cytokine detection by ELISA. The main limits of these tests are the heterogeneity of the response, due to individual susceptibility of donors, and the execution of the method, from cell isolation and culture to final readout. 9 On the contrary, several authors have focused on the release, from blood cells and monocytic cell lines, of prostaglandin E2 (PGE2) as a measure of the febrile reaction. It appears, for certain endotoxins, that their PGE2-inducing activity correlates with pyrogenicity better than their LAL-activating properties. 20,24 Altogether, the mediators of fever IL-6, IL-1β, TNFα, and the cyclooxigenase-2 that produce PGE2 are under the transcriptional control of NF-kappaB. 19
Of note, as results from ENCODE data sets, their promoters are constitutively acetylated, suggesting that these genes may be immediately available for transcription upon induction of NF-kappaB and recruitment of the transcriptional machinery. Indeed, tests based on cytokine release by monocytes demonstrate that the protein products of the above-mentioned genes are increased after as little as 4 h of incubation. Thus, we reasoned that the extent of a pyrogenic reaction could be measured also by means of its upstream step represented by NF-kappaB activation, and we aimed at verifying if a reporter system, able to directly measure the engagement of NF-kappaB, could be useful in the detection of endotoxin contamination of biological products.
In light of these premises, we optimized a test that reconciles the features of monocyte cellular assays with a targeted focus on the engagement of NF-kappaB. The test described in this article is based on monocytoid cells MM6 transduced with a single copy of a luciferase reporter construct, responsive to the proinflammatory factor NF-kappaB. MM6 are particularly competent for CD14/TLR4 signaling, thereby providing a sensitive tool for endotoxin tests. Indeed, mouse macrophage reporter cells have been demonstrated to be biologically relevant to the immune response in vivo. 19 Although professional antigen-presenting cells are equipped with a whole array of receptors for known and unknown impurities, we preferred to develop the assay on a cell line instead of blood or peripheral blood mononuclear cells (PBMC), for its easier handling. 19,25 Moreover, it was reported that MM6 are sensitive to Pam3CSK4, flagellin, FSL.1, zymosan, and muramyl dipeptide in a comparable way to PBMC, which makes these cells suitable candidates for the detection of different pyrogens and immune stimulators. 19
Compared to the MAT, MM6 reporter assay has the advantage of being less variable, as whole blood and PBMC-based MAT show variability due to the donor sensitivity. 26 The high reproducibility of the system is ensured by selection of a stably transduced cellular clone, cryoconserved in a homogeneous cell bank. In fact, we observed that the performance of frozen cells is unaltered for at least 2 years. In addition, while the MM6-based MAT bear intrinsic variability due to the heterogeneous nature of the MM6 cells, and to the fact that culture conditions may affect the expression of cell receptors, our MM6 reporter assay, being developed on a selected clone, may assure a higher level of homogeneity, and, above all, a high level of expression of the CD14 receptor, as demonstrated by cytofluorimetric analyses. To this regard, it has been demonstrated that membrane expression of CD14 increases upon endotoxin binding to the TLR4-CD14 complex in monocyte-like cells. 27 –29
Through cytofluorimetric and immunofluorescence analyses, we observed that not only do MM6 increase CD14 coreceptor expression following LPS stimulation, thereby ensuring that cells maintain their competence for CD14/TLR4 signaling upon transduction, but also that the selected clone expresses higher levels of CD14 compared to untransduced cells already at basal levels. Although we preferred not to introduce the LPS prestimulation step in the assay, the more prominent CD14 expression of transduced cells versus normal MM6 may be the consequence of the selection procedure that favored the expansion of a homogeneous CD14-expressing clone, despite the known heterogeneous nature of these cells. 30
Our reporter test was conceived to ensure a rapid and easy execution, providing quantitative results within 6 h in total, of which at least four of incubation. In fact, we observed that cells produce a dose-dependent signal after 4 h from endotoxin addition to culture media. RSE standard curve dilutions in a range comparable to the sensitivity of commercial pyrogen tests allowed to demonstrate a limit of quantification of the assay of 0.24 EU/mL. Although higher than the limit of LAL tests, this concentration is very close to the pyrogenicity threshold of endotoxin in humans, and it almost equals the sensitivity of MAT, 14 thereby providing further evidence of the relevance of the MM6 reporter test to the in vivo mechanisms of innate immunity. 20
The assay is characterized by accuracy, precision, and specificity, as selective inhibition of the CD14/TLR4 receptor complex completely blunts the response induced by endotoxin. Finally, the assay proved effective in endotoxin detection when applied to samples, such as IGVENA or LVV, difficult to analyze by traditional methods—although more sensitive—due to frequent interference with the assay reaction. Because media and buffers may also interfere with the assay, RSE spiked dilutions of buffers, media and samples must be tested each time. This approach allowed us to evaluate the percentage of recovery of the spiked RSE and to exclude aberrant results of the assay.
When LAL and MM6 reporter assays were combined to evaluate endotoxin content of unknown samples, the cell-based test highlighted interfering condition on LAL assay, demonstrating the usefulness of a rapid and reliable support test. Although further characterization and optimization steps are required to fully validate our assay, and to compare it with standard tests, the results obtained so far encourage to consider this method as an advantageous tool in the release test phase of pharmaceutical products intended for human use, when the routine test, although more sensitive, fails owing to interferences or artifacts. This cell-based approach has relevant consequences also in an animal-sparing perspective, since researchers are encouraged to consider designing, developing, utilizing, and exploiting biomimetic alternatives to in vivo animal models, and toxicity and release tests are considered more reliable, more accurate, more practical, and cost-effective when performed on tissues, cells, and cell components of human origin, rather than on whole animals.
Footnotes
Acknowledgments
Authors would like to thank Prof. Laura Chiarantini for cytofluorimetric analyses. Plasmids for vector production were kindly provided by Luigi Naldini. This work was supported by FanoAteneo.
Disclosure Statement
No competing financial interests exist.