
Abstract
The focal adhesion kinase–growth factor receptor 2 (FAK–Grb2) protein–protein interaction is implicated in pathogenesis of stress-induced cardiac hypertrophy. The focal adhesion targeting (FAT) domain of FAK unfolds to form a structural intermediate that interacts with a multibinding hot spot in the SH2 domain of Grb2. Disruption of the Grb2–FAT interaction is a therapeutic strategy for prevention of pathological cardiac hypertrophy. A pharmacophore was generated on the basis of structural and electrostatic properties of FAT bound to FAK using the Forge tool (Cresset). This pharmacophore was used as a query for Blaze server (Cresset) to screen a selectively enriched chemical library of 4,32,508 small molecules. The compounds selected were further filtered by hierarchical flexible docking approach using AutoDock v4. From the favorably docked compounds, five were selected on the basis of good adsorption, distribution, metabolism, excretion, and toxicity (ADMET) properties using SwissADME, MedChem Designer v.3, and MOLINSPIRATION. Stability of the binding mode of the inhibitors was further confirmed by molecular dynamic simulation study with AMBER v15 for a simulation time of 50 ns in aqueous environment. PM2307 was identified as the best inhibitor in terms of pharmacophoric features, dock score, and in silico ADMET analysis. The calculated binding affinity of PM2307 was better than that of the FAT–Grb2 complex as well as a previously reported small molecule inhibitor. PM2307 is also a quinolyl derivative sharing a similar scaffold with ofloxacin drugs, asserting its drug-like properties. Thus, it was proposed as a lead compound for development of drugs for pathological cardiac hypertrophy.
Introduction
Pathological cardiac hypertrophy (PAH) is caused by increased hemodynamic or metabolic stress leading to a maladaptive cellular process. PAH serves as a prominent marker of cardiac disease. Inhibition of several effective therapeutic targets involved in PAH has been shown to slow down the disease progression. Angiotensin II receptor, phospholipase C, mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase are some of the several established drug targets tested for pharmacological modulation of PAH. 1,2 Antihypertensive drugs such as enalapril, verapamil, nifedipine, indapamina, losartan, angiotensin-converting enzyme inhibitors, and atenolol have been tested against PAH-diagnosed mouse models, but were unsuccessful during the clinical trials. 3
Growth factor receptor (Grb) 2–Src homology (SH) 2 domain plays an essential role in pressure-overloaded cardiac muscle. The interaction between Grb2 and focal adhesion kinase (FAK) is pivotal for the development of PAH. 4 –6 Grb2 is an adaptor molecule that connects FAK to the MAPK signaling pathway by binding to Son-of-Sevenless, a guanine nucleotide exchange factor for Ras and Rac. In a pressure-overloaded heart, the adaptor molecule Grb2 is recruited to the C-terminal focal adhesion targeting (FAT) domain of FAK. Transverse aortic constriction data and coimmunoprecipitation assays on mice showed elevated expression and phosphorylation of FAK protein, as well as the presence of the FAT–Grb2 complex in stress-overloaded cardiomyocytes. Studies on mice models showed that Grb2 activation is directly responsible for the thickening of the myocardium and Grb2-deficient mice showed insufficient cardiac hypertrophy in response to pressure overload. 6,7 A malonyl-based phosphomimetic small molecule inhibitor with nanomolar affinity was proposed but failed to qualify for human trials. 8,9 Since then, not much progress has been made for targeting the Grb2–SH2 domain.
Structurally, the FAT domain has a four helical bundle conformation that shows metamorphic characteristics. 10 –16 Support for the existence of a folding intermediate came from observations that surface presentation and turn structure are required for Y925 phosphorylation by Src kinases and binding to Grb2–SH2 domain, respectively. 17,18 Both these features are not present in X-ray structure of FAT from human FAK. 12 The folding intermediate state likely allows the region around Y925 to attain the exposed turn-like structure required for interaction with Src, 12,13,19,20 optimal phosphorylation of FAT, and subsequent interaction with Grb2. 14
In a previous study, we reported targeted molecular dynamic (MD) simulations of the FAT domain to explore the conformational transition and to capture the structure of the intermediate (iFAT). The pathologically relevant iFAT–Grb2 complex was produced using docking refinement to get insights into their mode of binding and interaction. The binding mode of iFAT–Grb2 protein–protein interaction (PPI) complex was validated by superposition with 1TZE, a crystal structure of Grb2 in complex with high-affinity phosphotyrosyl peptide (BCR-Abl). The iFAT–Grb2 PPI interface is shown in Figure 1. The Grb2–SH2 binding site comprised two pockets: charged and hydrophobic. The charged pocket is responsible for binding, whereas the adjacent hydrophobic pocket mediates specificity. 20 Several phosphopeptide mimetic and small molecule inhibitors targeting the Grb2–SH2 domain have been reported, 21,22 but none could qualify for clinical testing due to poor adsorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. 23

oFAT–Grb2 docked complex generated using HADDOCK web server. 18 Grb2 and FAT 925pYENV928 peptide are shown in black and gray cartoon representation, respectively. The charged and hydrophobic residues of Grb2–SH2 domain are shown in light gray and black ball with stick representation, respectively, and labeled accordingly. The bound BCR-Abl peptide from 1TZE is shown in light gray cartoon representation and the side chain of the phosphotyrosyl is switched on for comparison. 32 FAT, focal adhesion targeting domain; Grb2, growth factor receptor 2; SH, Src homology.
In view of the lack of existing known drug-like compounds for disruption of the Grb2–FAT interaction, hot spot analysis was done to identify the important residues mediating the binding. The hot spot analysis and the previously modeled iFAT–Grb2 complex guided the pharmacophore generation process followed by virtual screening of a chemical library with 4,32,508 small molecules. A flexible docking was performed on filtered ligands followed by binding site analysis to identify potential inhibitors. Furthermore, the efficacy of the top compounds was analyzed using in silico ADMET studies to propose drug-like inhibitors of Grb2–SH2 domain.
Materials and Methods
Hot Spot Identification
Hot spot identification was done using PredHS, 24 a high-performance structure-based hot spot prediction server. In addition to conventional features such as physicochemical properties, evolutionary conservation, and solvent accessible area, the method combines novel structural and energetic features that represent the specific biological properties of the hot spot residues. The server extracts 108 site features from the PPI interface, 108 Euclidean, and 108 Voronoi neighborhood properties. During feature selection, a set of 38 optimal features were selected using a random forest and a sequential backward elimination method. It was found that the topographical score, side chain energy score, atom/residue contacts, four-body pseudopotential, and local structural entropy contribute most to the hot spot identification. Based on the features selected, both a support vector machine classifier and an ensemble model were constructed for prediction. The ensemble is generated from n submodels using a bootstrap resampling method to generate subsets. The online interface for the server 25 was used to upload the pdb file of the modeled iFAT–Grb2 complex as input.
Pharmacophore Generation
To design and identify small-molecule inhibitors against the iFAT–Grb2 PPI interface, a pharmacophore was generated on the basis of electrostatic field and surface properties (e.g., hydrogen bonding and hydrophobic properties). The iFAT–Grb2 complex obtained from HADDOCK web server in pdb format was imported into Forge (Cresset, United Kingdom). Forge is a molecular design and structure–activity relationship interpretation tool that generates three-dimensional (3D) pharmacophore using FieldTemplater tool. Within Forge-FieldTemplater, the pYENV peptide was selected as reference molecule, and the field point pattern was calculated. The field points can be considered as the areas on the ligand where there is some likelihood of interaction with the target. The field points (negative—blue, positive—maroon, shape—yellow, and hydrophobic—red) were derived from the molecular interaction potentials. 26 The size of the field point was proportional to the magnitude of the field at that point in space. The 3D pattern of distribution of field points in the 925pYENV928 was considered as a pharmacophore for iFAT–Grb2 interaction.
Virtual Screening
The pYENV peptide, with its field point pattern was saved in SDF format—to be used for virtual screening by rigid docking of a library of 4,32,508 molecules 27 using the Blaze server (Cresset). * The chemical library was taken from DUD data set 28 and ChEMBL v2.0. 29 To enrich the library, 379 potent inhibitors from BindingDB 30 with reported inhibition against Grb2–SH2 domain were incorporated. Duplicates were removed using the PAINS server 31 and high molecular weight compounds were removed by keeping the heavy atom count <35. The top scoring compounds were retrieved in the form of a 3D alignment showing query field points along with their similarity score. Hits were then quantified on the basis of their score in the distribution curve. 27
Batch Docking
The large number of filtered ligands from rigid virtual screening using Blaze was further subjected to flexible docking against the Grb2–SH2 binding pocket using AutoDock Vina v4. 32 Ligands obtained in SDF format from Blaze search were further processed using Open Babel v2.2.3 33 for file format conversion to PDBQT format. The ligands were kept flexible, and individual PDBQT files were generated using vina_split.exe from MGLTools 1.5.4. Protein preparation for Grb2, including addition of polar hydrogen and Gasteiger charge, was done using graphical user interface (GUI) of AutoDock v4. 34 Grid box and docking parameter files were also prepared using AutoDock4-GUI. The charged and hydrophobic pocket-forming residues in Grb2–SH2 domain as determined previously 35 were considered as binding site residues during docking runs. During the docking runs, the side chains of charged and hydrophobic residues in the Grb2–SH2 binding site were treated as flexible.
The configuration files that include the grid box information were made manually for the AutoDock Vina run. The cutoff taken for AutoDock binding energy was −8.0 kcal/mol. 36 The docked inhibitor complexes were validated by superposition with the crystal complex of Grb2–SH2 domain with phophotyrosyl peptidic inhibitor BCR-Abl (PDB ID: 1TZE). 35 The hydrogen bonding, lipophilic interactions, and nonbonded interactions of the shortlisted ligands were confirmed using the protein–ligand interaction profiler (PLIP) server. 37 Docking runs were made on the supercomputing facility provided by SCFBio at Indian Institute of Technology Delhi.
Evaluation of ADMET Properties of Top Compounds from Virtual Screening Studies
The drug likeness of the ligands shortlisted in the previous step was calculated using three tools, namely: SwissADME, 38 MedChem Designer v.3, and MOLINSPIRATION 39 tool. In both SwissADME and MedChem Designer, the input ligands were in SDF format, whereas the SMILES format was required for MOLINSPIRATION. The various molecular properties calculated using each software, their biological significance, and the recommended values are shown in Table 1.
The Molecular Properties Calculated Using SwissADME, MedChem Designer v3, and MOLINSPIRATION Tool to Predict the Drug Likeliness
BBB, blood brain barrier; GI, gastrointestinal; HBAs, hydrogen bond acceptors; HBDs, hydrogen bond donors.
Classical Molecular Dynamics Simulation of the Best Proposed Inhibitors
Classical molecular dynamics (CMD) simulations were carried out using the PMEMD module of AMBER v14 40,41 and ff99SB all atom force field parameters. 42 The Antechamber module was used to generate partial atomic charges and force field parameters using restrained electrostatic potential and general amber force field, respectively. Three Cl− ions were used to neutralize the complex in the CMD run. 20 Each of the systems was explicitly solvated using the TIP3P water potential inside an octahedral periodic box of water molecules with a minimum solute–wall distance of 10 Å.
The CMD simulations were carried out as follows: (1) The systems were energetically minimized to remove unfavorable contacts. (2) Four cycles of minimization with 10,000 steps each were performed. Harmonic restraints were relaxed in a stepwise manner by 10 kcal/mol per cycle. (3) The fifth cycle consisted of 20,000 steps of unrestrained minimization before the heating process. The cutoff distance used for the nonbonded interactions was 12 Å. The SHAKE algorithm was used to restrain the bonds containing hydrogen atoms, (4) Each energy-minimized structure was heated for 100 ps from 0 to 300 K, whereas the position of both the complexes was restrained by a small value (20 kcal/mol). Constant volume was maintained during the process. (5) Unrestrained equilibration of 500 ps with constant pressure and temperature conditions was carried out for each system. Langevin dynamics was used to control the temperature using a collision frequency of 2 ps. The temperature and pressure were allowed to fluctuate ∼300 K and 1 bar, respectively, with the corresponding coupling of 2 ps. For each simulation, an integration step of 2 fs was used. (6) Finally, CMD production run of 50 ns for both the systems was carried out to generate 1,000 frames each. Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were calculated using CPPTRAJ commands in AMBER tools.
Binding Site Analysis
To determine the interacting residues and the nature of the interaction, the Grb2 docked complex obtained from AutoDock Vina and 50 ns MD simulations were analyzed using PLIP and protein interfaces, surfaces, and assemblies servers. 43,44 To see the similarity of the proposed drug with the approved drugs, a search of all the approved drugs was done in Drug Bank v5.1 45 with Tanimoto similarity threshold of 0.8.
Binding Affinity Comparison of Top Ligands with the Crystal Complex of Grb2–SH2 Domain and Peptidic Inhibitor (PDB ID: 2HUW)
The two ligands selected on the basis of interaction with Grb2–SH2 domain and pharmacokinetics were further compared with the crystal structure of Grb2–SH2 domain in complex with a small molecule inhibitor (PDB ID: 2HUW 46 ) on the basis of AutoDock binding energy. 31 Hydrogen bonds, binding energy, and estimated inhibition values were analyzed using AutoDock Tools. 34
Results
Residues from Charged and Hydrophobic Pocket Together Formed the Hot Spot of the iFAT–Grb2 Interaction
Structure-based hot spot identified using PredHS returned two lists of residues on the basis of support vector machine classifier and ensemble score. The list is prepared in the order of the predicted score that contributes toward the affinity and specificity of the iFAT–Grb2 interaction. The results showed higher propensity of F108, H107, L120, R67, K109, and W121 as hot spot residues. R67 and H107 were found to be a part of the charged pocket, whereas F108 and K109 were a part of the hydrophobic pocket identified on by the previous analysis from our laboratory. 18 Significantly, a X-ray crystallographic study of Grb2–SH2 domain in complex with a peptidic inhibitor BCR-Abl shows that W121 was a part of the Grb2–SH2 domain binding site, but was not found to make direct interaction with the peptidic ligand. Instead, the bulky W121 group is responsible for mediating specificity by selectively allowing binding of ligands with β-turn conformation. 35
iFAT–Grb2 PPI-Based Pharmacophore Showed Charged and Hydrophobic Field Points
The iFAT–Grb2 complex was used to calculate the molecular features, yielding a pharmacophore as depicted in Figure 2. The field-based pharmacophore calculated using the Forge tool included electrostatic, steric, hydrophobic, and van der Waals fields. The pharmacophore mostly showed the presence of negatively charged field points (cyan balls), a few positive field points (red balls), and hydrophobic centroids (orange dots). The size of the field points indicates the potential strength of the interaction. This emphasized that the high-affinity inhibitors should possess these characteristic features. The pharmacophore was further validated by superposition with the crystal structure of Grb2–SH2 domain in complex with a phosphotyrosyl peptide BCR-Abl (PDB ID: 1TZE). The superposition of the two pharmacophores using FieldTemplater in Forge software (Cresset) showed a shape-based similarity score of 0.709 on a scale of 0–1. The positioning of the pharmacophore in the Grb2–SH2 domain binding pocket showed that all the field points in the pharmacophore matched with positioning of charged and hydrophobic residues in the Grb2–SH2 binding site (Figs. 1 and 2). This pharmacophore served as the query to search the enriched chemical library for matching spatiochemical features.

The field point pattern of oFAT–Grb2 complex used for virtual screening. Light and dark gray balls indicate negative and positive ionic field, respectively. White points (smaller in shape as compared to ionic field points) represent high accessible van der Waals surface area and small gray points represent hydrophobic centroids. Oxygen and nitrogen atoms are shown in light and dark gray sticks. The phosphotyrosyl peptide of oFAT is in dark gray and the binding site residues of Grb2 are atom typed in ball and stick representation, respectively.
Pharmacophore-Based Library Search, Docking Refinement, and Contact Map Analysis Produced ∼1,000 Ligands
A field-based pharmacophore derived from iFAT–Grb2 complex was used to query the compound library to identify novel scaffolds with similar structural and chemical properties. The chemical library screening was done using Blaze server 27 to search the ligand library for discovering new candidates with intrinsic drug-like properties. Out of 4,32,508 inhibitors from the Blaze ligand library, 3,866 molecules were selected as they matched with the properties of the query pharmacophore and heavy atom count. The duplicates with same CHEMBL ID were removed using PAINS server, and 3,861 ligands were finalized for the next step. The schema for the entire chemical library search and steps during small molecule filtering are shown in Figure 3.
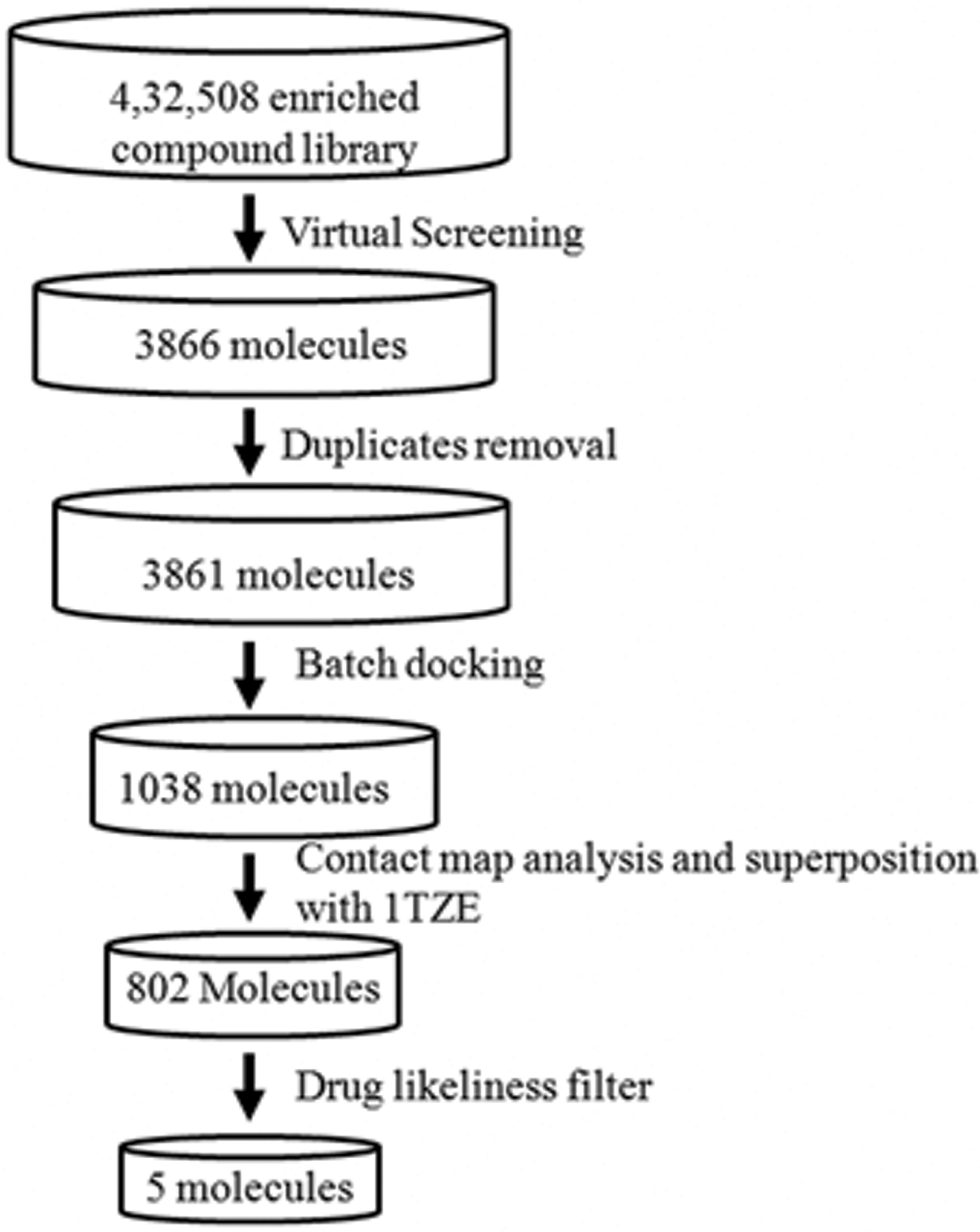
Steps involved in virtual screening for the identification of potent inhibitors against oFAT–Grb2 complex. The flow chart depicts a step-wise procedure employed for the virtual screening to propose six promising bioactive compounds against oFAT–Grb2 complex.
To refine the rigid docked positions obtained from virtual screening, flexible batch docking using AutoDock Vina was carried out. During the run, only 1,038 ligands showed binding energy values less than −8.0 kcal/mol and were selected for further analysis. On the basis of interaction of the ligand with Grb2–SH2 binding site in the 5 Å vicinity, 802 molecules were selected (Fig. 3).
In Silico ADMET Analysis Produced Only Five High-Affinity Ligands
The molecular properties, drug likeliness, and Lipinski rule of five calculated using SwissADME, MedChem Designer, and MOLINSPIRATION tool are listed in Table 2. The filtering generated only five ligands (hereby referred to as PM2305–PM2309). Moreover, the binding site was populated with charged and hydrophobic residues. The selected ligands also showed good superposition with the phosphotyrosyl peptide in 1TZE (Supplementary Fig. S1).
The Adsorption, Distribution, Metabolism, Excretion, and Toxicity Properties of Five Potent Ligands Identified from Blaze Virtual Screening
Partition coefficient (logP), TPSA, MWt, total number of atoms in the ligand molecule (Natoms), HBAs, HBDs, Moriguchi octanol–water partition coefficient (MlogP), logarithm of the distribution coefficient (logD), GI absorption, and blood brain barrier penetration are the criteria considered for ADMET profiling.
ADMET, adsorption, distribution, metabolism, excretion, and toxicity; MWt, molecular weight; TPSA, total polar surface area.
All the five compounds had low log P values (<5), indicating their hydrophilic nature and better permeability. The molecular weight of these compounds was within the desired range (150–500 g/mol). 47 The rotatable bonds were also <9 and the MlogP values were <4 along with lower logD values. Compounds with low logD values show acidic behavior. Previous reports suggest that acidic compounds exhibit high oral bioavailability and good solubility. 48 The total count of nitrogen and oxygen atoms was >7 in all the set of compounds. In addition, the total count of hydrogen bond acceptors (HBAs) and hydrogen bond donors (HBDs) was found to be >7 in the five sets of compounds. Except PM2309, the remaining four compounds show total polar surface area (TPSA) <120 Å2 as shown in Table 2. The HBAs, HBDs, total count of nitrogen and oxygen atoms, and TPSA values show that excluding PM2309, the remaining four compounds show a higher propensity for hydrogen bond formation.
Bioavailability of the drug molecule is primarily driven by gastrointestinal (GI) tract absorption. 49 GI absorption of a drug molecule happens in two stages: (1) dissolution and (2) membrane transfer. In the first stage, dissolution of drug molecule in the aqueous environment of GI track happens. In the second stage, the dissolved molecule penetrates through the intestinal membrane that acts as a barrier and reaches the blood circulation. The drug has to show better solubility and GI tract absorption such that it reaches the blood circulation. All five compounds showed high-predicted GI tract absorption and no blood brain barrier penetration, but only one compound, that is, PM2307, showed acceptable water solubility features.
Two Inhibitors Showed Maximum Binding Affinity for Grb2–SH2 Domain and Good Drug Likeness
Out of the five proposed ligands, PM2305 showed maximum binding with Grb2–SH2 domain. In addition, PM2307 showed chemical similarity (Tanimoto score: 0.85) with drugs such as DB01165 (ofloxacin), DB01137 (levofloxacin), and DB03034 (dextrofloxacine). All the three drugs are fluoroquinolone derivatives showing antibacterial activity. 50 PM2307 is also a quinoline analogue and the chemical similarity score suggests that PM2307 can be synthesized.
PM2305 and PM2307 Show Stable Binding and Have Higher Binding Affinity Than iFAT–Grb2 Complex as Well as Small Molecule Inhibitor
Considering that PM2305 and PM2307 show most promising results on the basis of superposition on 1TZE, contact map, and ADMET analysis, these two molecules were subjected to MD simulations. The docked poses obtained were further simulated for 50 ns to check for ligand stability in the Grb2 binding site. The RMSD plot showed that the trajectories tend to stabilize during the first 4 ns of the simulation (Fig. 4). Comparatively, PM2307 (Fig. 4, black line) is more stable showing least RMS fluctuations. In PM2305 (Fig. 4, gray line), the RMSD showed a leap toward the end of the simulation. The simulation was extended for 5 ns to conclude that the interaction with S96 was lost. The RMSF plot for both PM2305 and PM2307 showed a local drift in charged and hydrophobic residues of Grb2 (Fig. 5). The atomic fluctuations in these residues can be reasoned due to the interaction with PM2305 and PM2307. The binding energy values of PM2305 and PM2307 were compared with an already known small molecule inhibitor with available crystal coordinates and binding mode (PDB ID: 2HUW). The AutoDock binding energy for a cyclopropane-derived crystal ligand in complex with Grb2–SH2 domain 42 was found to be −6.01 kcal/mol. The binding energy of 50 ns MD-simulated PM2305 and PM2307 complexed with Grb2–SH2 domain was found to be −11.4 and −9.8 kcal/mol, respectively, with an estimated nanomolar inhibition, that is, 7.75 and 22.5 nM. In addition, the AutoDock binding energy for iFAT–Grb2 complex was found to be −9.2 kcal/mol. The results indicate that both PM2305 and PM2307 show strong binding with Grb2 comparatively to the existing small molecule inhibitor and the binding is stronger than iFAT–Grb2 interaction.
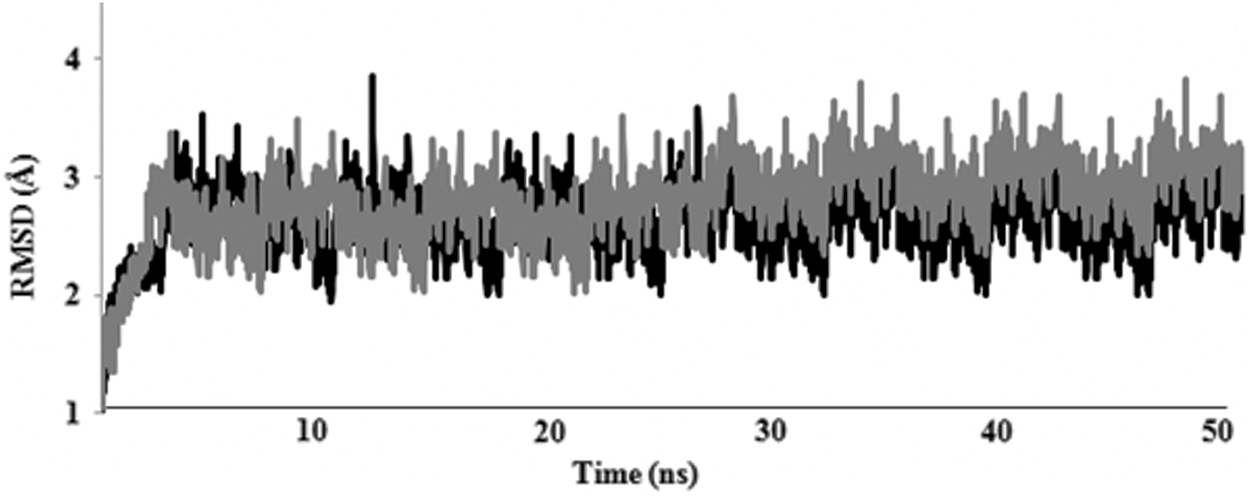
RMSD plot of 99Cα of Grb2 SH2 for 50 ns. Gray line depicts PM2305and black line depicts PM2307 docked complex. RMSD, root mean square deviation.
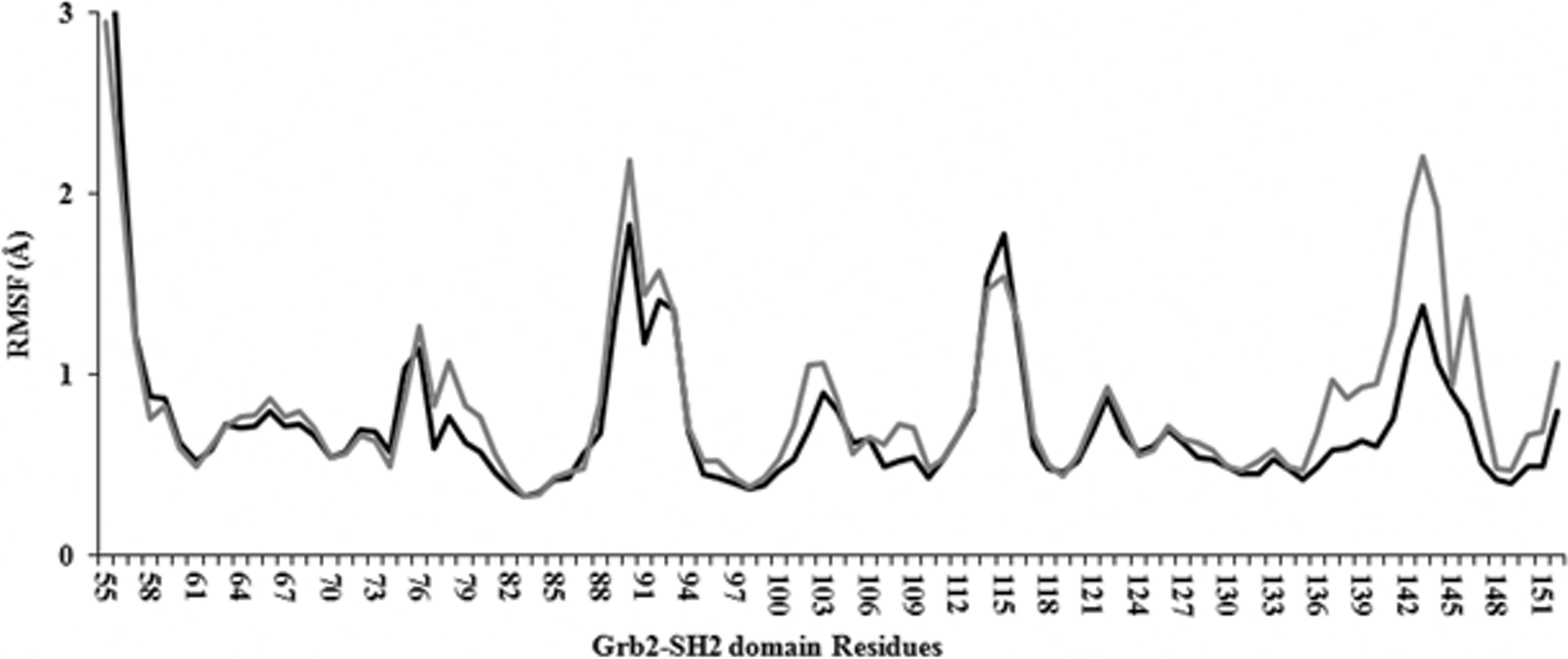
RMSF plot of 99Cα of Grb2 SH2 for 50 ns. RMS fluctuation of Grb2 complexed with PM2305 is shown in gray line whereas the black line depicts Grb2-PM2307 docked complex. RMSF, root mean square fluctuation.
Quinoline Derivative PM2307 Showed Stable Interaction with Grb2–SH2 Domain as Compared in Oxochromene PM2305 Derivative
PM2305 is an oxochromene derivative and the benzoic acid group of ligand formed two hydrogen bonds each with S96 and K109 of the charged and hydrophobic pocket of Grb2, respectively. The oxochromene group and the benzoic group of PM2305 together formed hydrophobic interactions with the side chain of R67, R86, F108, L111, and W121 of Grb2–SH2 domain (Fig. 6 and Table 3). While R67 and R86 were found to be conserved, F108 and W121 were averagely conserved, and L111 was a nonconserved residue within Grb2–SH2 domain. 20 Moreover, PM2305 also formed three salt bridges with the charged pocket residues, that is, R67, R86, and S96 (Table 3). Interaction of PM2305 with conserved and nonconserved residues confirms the strength of binding and specificity. 51
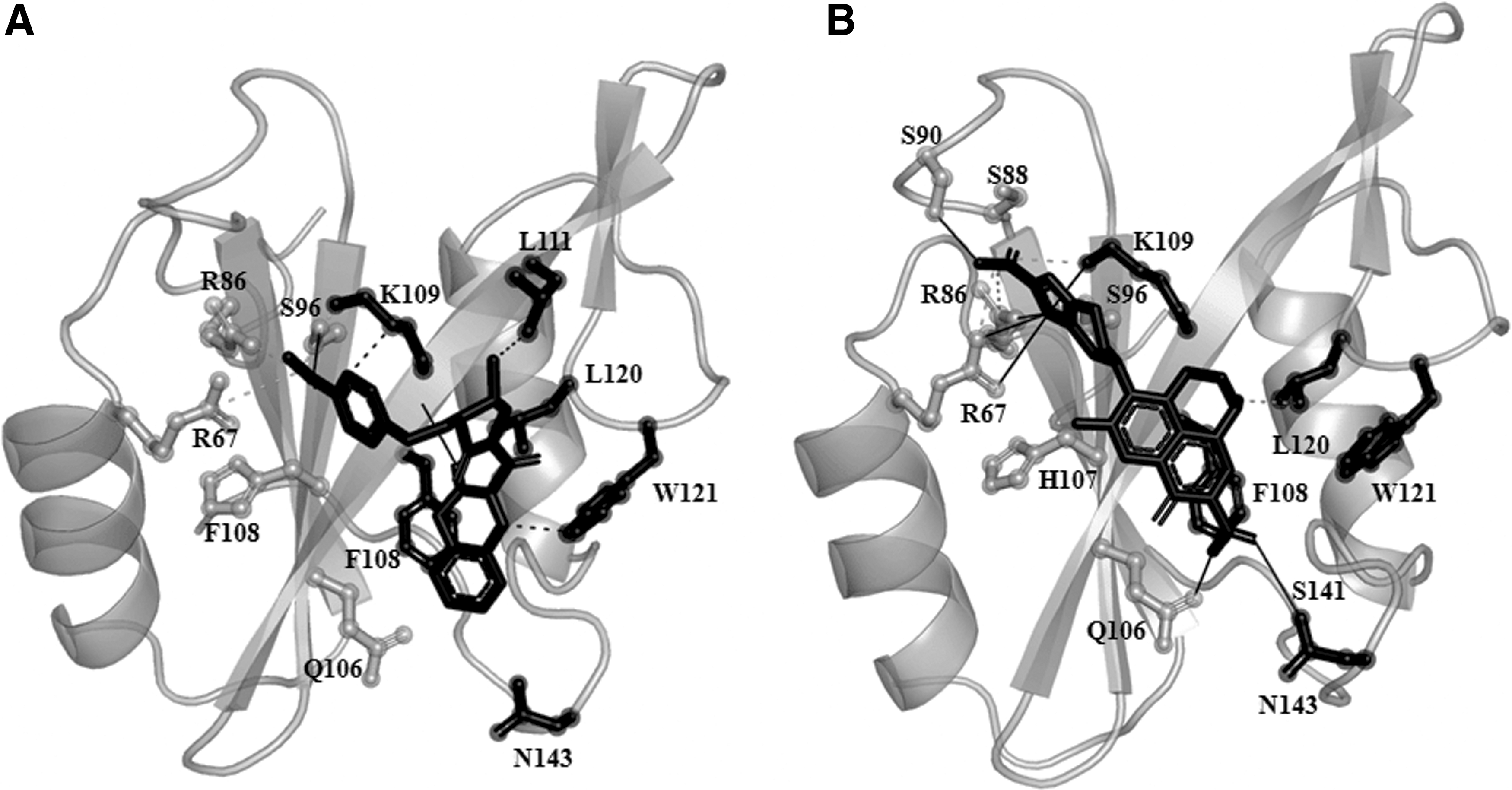
Binding site analysis.
Hydrogen Bond and Salt Bridging Interactions of Growth Factor Receptor 2-Src Homology 2 Domain with PM2305 and PM2307
The charged, hydrophobic, and specificity-determining residues are in bold, underlined, and italics, respectively.
Grb2, growth factor receptor2; SH, Src homology.
In contrast, PM2307 is a quinoline derivative with a binding energy of −10.1 kcal/mol. The imadazole ring of the quinoline derivative stably formed six hydrogen bonds with R67, R86, S88, S90 and K109, L120 from charged and hydrophobic pocket, respectively. Along with the active site residues, the carboxylate of quinoline group in PM2307 formed a hydrogen bond with the side chain of N143 (Table 3). PM2307 interactions also mediated binding and specificity. The specificity-determining residue was N143. Our results are in agreement with a recent crystallographic study on Grb2–SH2 domain complexed with phosphotyrosyl peptide with a consensus of YXNX (PDB ID: 3MXC). 52 The results showed the presence of Q106, W121, S141, R142, N143, and Q144 in mediating specificity along with the other active site residues (charged and hydrophobic). 52 PM2307 also formed three salt bridges and four hydrophobic interactions with R67, R86, K109 and L111, L120, W121, N143, respectively. Thus, interaction of PM2307 with R67, R86, S88, S90, K109, L111, L120, W121, and N143 supports its strength of binding and specificity of the interaction. Moreover, the spatial orientation and conformation of PM2307 mimic FAT interaction with Grb2–SH2 domain (Fig. 6b).
Conclusion
Grb2–SH2 domain interaction with an intermediate conformation of FAT domain is known to cause PAH. The PPI complex that was generated to model this is part of our previously published study. 20 In this study, a pharmacophore model was generated using this PPI complex as an input in FieldTemplater of Forge tool. Furthermore, BLAZE screening was done against a chemical library comprising 4,32,508 ligands. The inhibitor database was further populated by adding a ligand library from BindingDB that showed specific inhibition against Grb2. Results were further refined using AutoDock Vina flexible docking. Selected ligands showing binding values more than −8.0 kcal/mol were inspected on the basis of interaction with the charged, hydrophobic, and hot spot residues to propose five ligands having acceptable binding affinity and pharmacokinetics. Out of the five potent inhibitors, compound PM2307 showed high affinity and selective interaction with the Grb2–SH2 domain. Moreover, PM2307 is a quinolyl derivative sharing a similar scaffold with ofloxacin drugs asserting its drug-like properties. Thus, PM2307 mimics the binding of the FAT peptide at the Grb2 binding pocket. It is likely to have the desirable qualities of a drug-like compound, namely (1) higher binding affinity than the FAT peptide substrate, (2) good ADME properties as it passed the various filters applied, and (3) is easy to synthesize due to its chemical similarity to other drugs.
Future studies for biological validation of PM2307 as a lead compound to disrupt the FAT–Grb2 interaction can be carried out using ELISA as previously reported by us in case of inhibitors of MoxXT interaction. 53,54 Furthermore, the in vitro binding affinity of PM2307 and its other substituents will be measured using a combination of surface plasmon resonance, isothermal titration calorimetry, and nuclear magnetic resonance spectroscopy.
Footnotes
Acknowledgments
The authors thank Professor B. Jayaram and Indian Institute of Technology, Delhi, for use of the supercomputing facility. Pallavi Mohanty acknowledges financial support from Indian Council of Medical Research–Senior Research Fellowship (IRIS-ID 2013_17590).
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Abbreviations Used
*