
Abstract
One of the most often utilized methods for drug discovery is molecular docking. With docking, one may discover new therapeutically relevant molecules by targeting the molecule and predicting the target–ligand interactions as well as different conformation of ligand at various positions. The prediction signifies the effectiveness of the molecule or the developed molecule having different affinity with target. Drug discovery plays an important role in the development of a new drug molecule of different moiety attached to it, which leads us in the management of several diseases. In silico approach led us to identification of numerous diseases caused by virus, fungi, bacteria, protozoa, and other microorganisms that affect human health. By means of computational approach, we can categorize disease symptoms and use the drugs available for such types of warning signs. After the docking process, molecular dynamics computational technique helps in the simulation of the physical movement of atoms and molecules for a fixed period of time, giving a view of the dynamic evaluation of the system. This review is an attempt to illustrate the role of molecular docking in drug development.

INTRODUCTION
The computational process of finding the ligand or a moiety that is fit geometrically and energetically to the receptor active site is known as molecular docking. The activity of the drug depends on the molecular affinity of the drug to the receptor (protein) active site. Molecular docking plays a key role in recognition of drug (ligand)–receptor interaction. 1 The free energy modification unassociated and associated states of the drug with the target designates its molecular recognition. Hydrogen bond, electrostatic, Van der Waal, pi–pi, and hydrophobic interaction are significant in molecular recognition. The in silico study helps in understanding the nature of these ligand–receptor interactions, which may provide a potential drug molecule.
More and new therapeutic targets are being studied using in silico methods such as chemoinformatics, molecular modeling, pharmacophore modeling, homology modeling, and artificial intelligence (AI), which can be explained by recent computer technology advancement and restoration of biological, chemical, and structural data. 2 It is now possible to conduct virtual screenings of millions of compounds in a definite period, hence diminishing the expenses of hit identification and increasing the probability of finding a desirable drug molecule. In drug development, there are a variety of molecular modeling tools that may be divided into structure-based and ligand-based approaches (Fig. 1). 3
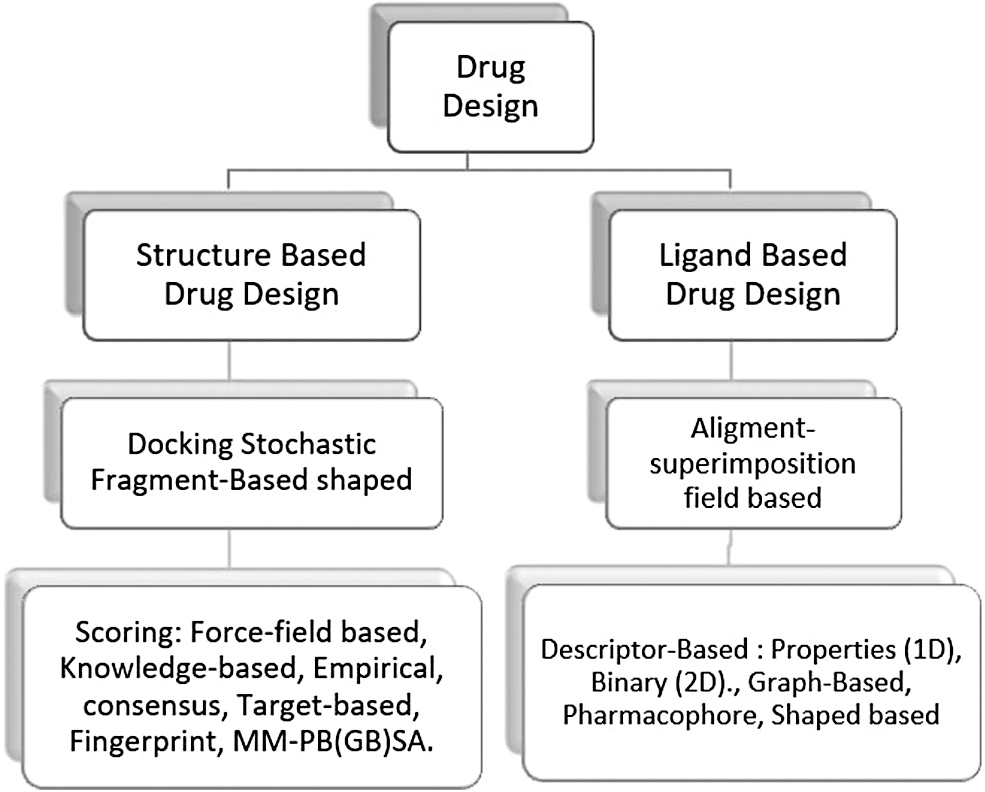
Schematic representation of Drug Design method. MM-PB(GB)SA, Molecular Mechanic-Poisson Boltzmann (Generalized Born) Surface Area.
Types of Docking
Rigid docking
In this docking process, the target protein and the selected ligand conformations are meant to be rigid. Flexibility in the molecular bond angle, bond length, and torsion angle is not permitted or they are constant. This substantial change at molecular level is not permitted in this type of docking.
Flexible ligand docking
This docking simulation describe about the target protein or receptor that are constant at the place but the ligand has the degree of freedom to bind with protein at different conformation, translational and rationally.
Flexible docking
This molecular docking provides all the manifestation of freedom to the target and the ligand under process. They account for complete conformational changes at the molecular level. 4
MOLECULAR DOCKING IN DISEASE MANAGEMENT
Understanding the role of target in the diseases mode of action is very important. Remarkable advancement in molecular biology (genomic and proteomics) offers new and well-characterized drug target as in Alzheimer's disease (AD) in which the most common symptom is dementia, which is frequently characterized by decreased episodic memory and cognitive functions. 5 Caspase 8 (Protein Data Bank [PDB]-3KJN) (Fig. 2) is a significant therapeutic target in AD. In this study, molecular docking was employed to evaluate the natural compounds that have the affinity to bind with caspase 8.

Caspase 8 protein.
Recently rutaecarpine (PubChem CID: 65752) (Fig. 3) showed maximum binding energy (−5.806 kcal/mol) and was verified with molecular dynamics (MD) simulation studies. Caspase 8 interacts with rutaecarpine with eight amino acid residue TRP420, ARG413, ARG143, ARG413, GLY318, CYS360, GLN358, SER411 with receptor (Fig. 4). The data show that rutaecarpine may be a lead molecule with anti-Alzheimer's activity by inhibiting caspase 8. 6 The treatment of obesity with low toxicity and adverse effect by the natural compound Taraxacum officinale shows antioxidant strength by inhibiting pancreatic lipase. 7
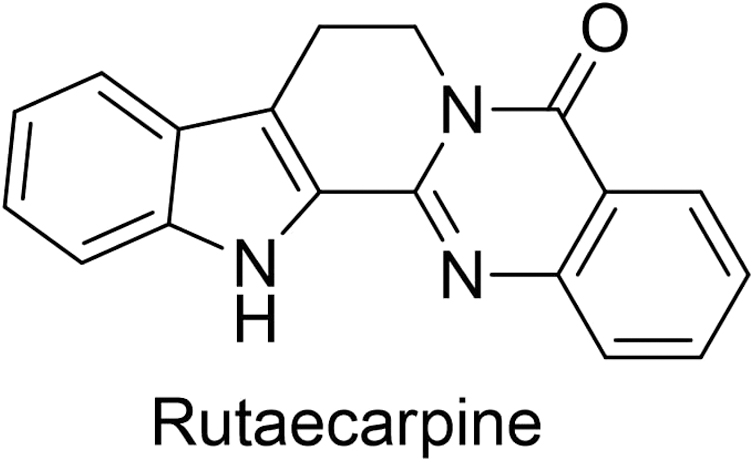
Structure of anti-Alzheimer's drug.

Two- and three-dimensional structure of molecular interaction of rutaecarpine with 3KJN protein.
In December 2019, the onset of COVID-19 caused by SARS-CoV-2 showed illness in human with the same symptoms as in the existing diseases such as pneumonia, developed by the use of computational study. The drugs used in the treatment of pneumonia are azithromycin and doxycycline that prohibit the spread of pneumonia in the body. These antibacterial drugs inhibit the spread but upto a certain extent. Further study shows that it is a virus that binds to the Angiotensin Converting Enzyme 2 receptor (PDB-6M18) (Fig. 5).
ACE 2 receptor.
This virus is located in the kidney and then it spreads to the lung through blood stream and causes illness. In silico studies are done and old antiviral drugs such as remdisivir (PubChem CID: 121304016), ivermectin (PubChem CID: 6321424), and boceparavir are used for the treatment of COVID-19 (Fig. 6). Our structure-based chemical screening study led us to propose a number of structurally unique medications that could display antiviral efficacy against SARS-CoV-2. 8

Structures of SARS-CoV-2 inhibitors.
Drug development for cancer is more complicated especially when it comes to molecular pharmacology, which is not yet understood. The discovery and development of a new drug molecule are considered very expensive and time consuming. The computational method is constructive for performing various tasks that include protein interaction with molecule, predicting binding site, and virtual screening (Table 1).
Virtual Screening Databases
ChEBI, chemical entities of biological interest; DTP, development therapeutic program; DUD-E, directory of useful decoy; KEGG, Kyoto Encyclopedia of Genes and Genomes; NRDBSM, nonredundant database of small molecule; TTD, therapeutic target database.
In cancer therapy, development of drug by the computational approach helped in developing crizotinib (PubChem CID: 11626560), axitinib (PubChem CID: 6450551), selinexor (PubChem CID: 71481097), glasdegib maleate (PubChem CID: 122640033), apalutamide (PubChem CID: 24872560), and alpelisib (PubChem CID: 56649450) (Fig. 7). So the computational approach plays a vital role in the diagnosis of various life-threatening diseases. 9
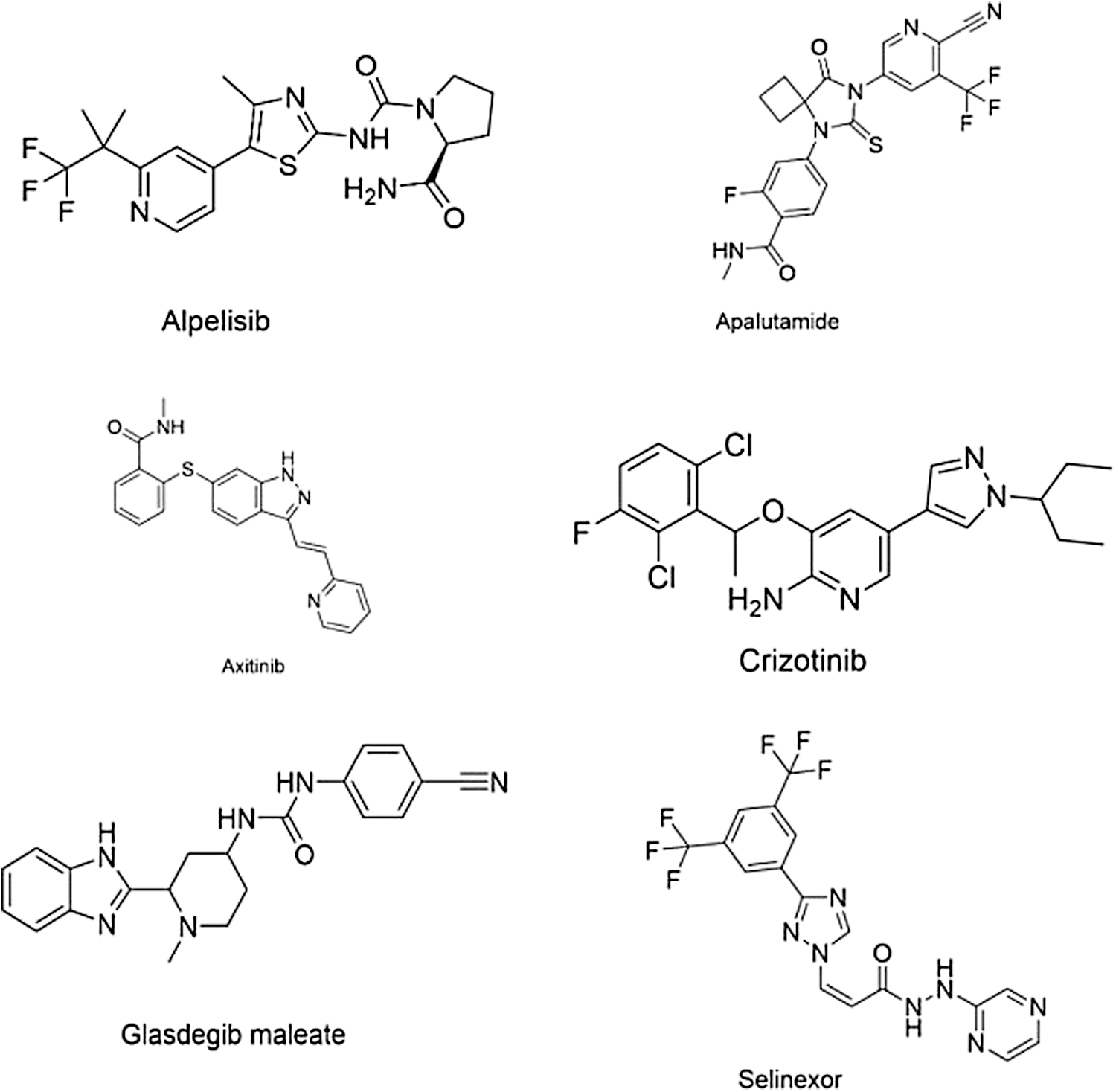
Structure of anticancer drugs.
Psoriasis is an autoimmune disorder that makes the people physically, socially, and psychologically ill. It is an immune-mediated disease with multiple signaling targets. Recently tofacitinib (PubChem CID: 9926791) (Fig. 8) is used to treat the disease by inhibiting the JAK-signaling pathway (PDB id-7F7W) (Fig. 9). Tofacitib acts as a pharmacophore for the development of new derivatives that are under clinical trial and may be useful in the treatment of psoriasis. Tofacitinib shows the interaction with Janus kinase protein with three amino acid residues ASP912, CYS909, and LEU905 (Fig. 10). 10
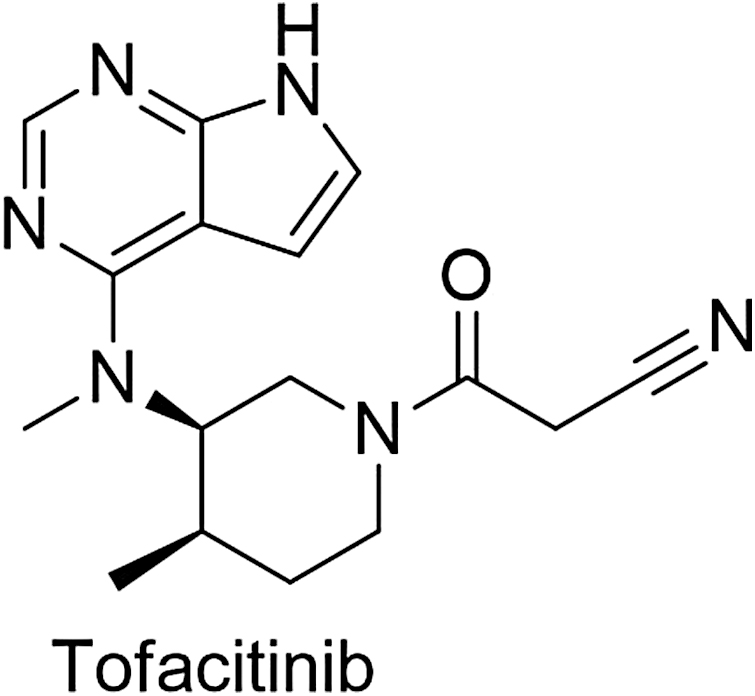
Structure of antipsoriatic drug.
JAK protein.
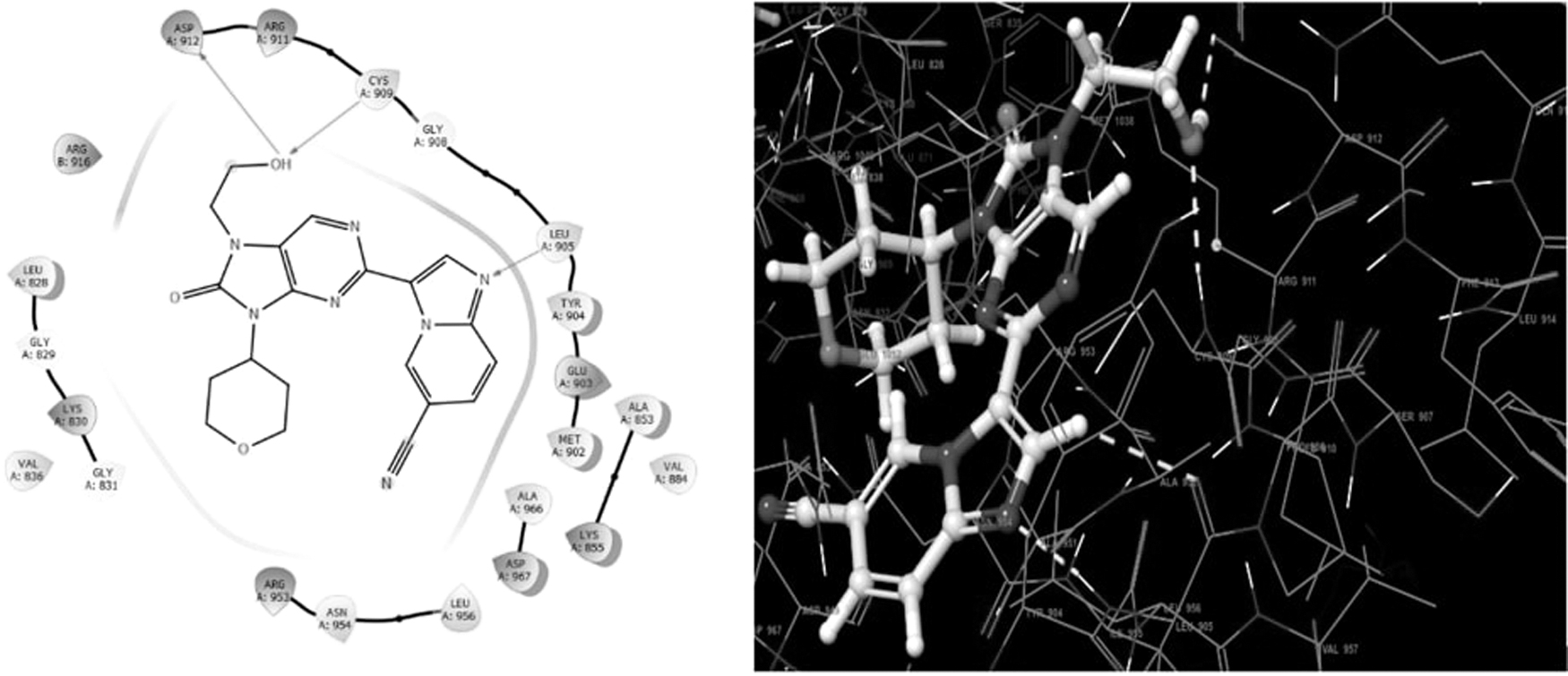
Two- and three-dimensional structure of molecular interaction of tofacitinib with 7F7W protein.
DATABASE SEARCH
Molecules with appropriate pharmacokinetic and pharmacodynamics properties are known as leads. The leads can be identified from the chemical database. Structure–activity relationship and binding affinity analysis are useful in the identification of leads. Most chemical databases store in two-dimensional and three-dimensional structural models in different domains and can be searched separately. 11
SOFTWARE USED IN MOLECULAR DOCKING
AutoDock
For the computational study, AutoDock provides the most modeling and docking solution for protein and computational chemistry. It is a software that is publically available for the docking purpose. Blind docking as well as flexible ligand docking can be done in AutoDock, but flexible docking in which both protein and ligand have the degree of freedom to bind at different conformation, translational and rationally, with each other cannot be done. AutoDock has been used in X-Ray crystallography structure-based drug design, virtual screening, chemical mechanism studies, and lead optimization (Table 2). 12
List of Molecular Docking Software
ASP, Application Service Provider; BFGS, Broyden Fletcher Goldfarb Shanno; GNU, General Public License; ICM, Internal Coordinate Mechanics; XP, extra precision.
AutoDock Vina
It is the most advanced version of AutoDock, which is faster and reliable for the docking study. The results of the AutoDock Vina are more accurate and can also be used for the structure–activity relationship. AutoDock Vina does not require choosing atom types and precalculation grid maps. It calculates the grades internally for the atom types that are needed. 13
Chimera
Chimera is a program for interactive visualization and analysis of molecular structure and related data, including density map, trajectories, and sequence alignment. It is used for energy minimization of the selected ligand so that it can bind freely with the protein and at different positions. It is available free of charge for noncommercial use. 14
Discovery Studio
Discovery Studio provides modeling and simulation solution for protein modeling and computation chemistry. Discovery Studio provides the most advanced software solution for the life science researcher today. From project conception to lead optimization, Discovery Studio includes a diverse collection of sophisticated software applications to take researchers to the next level, all conveniently packaged into a single, easy-to-use, Linux and Window-based environment because it is built on pipeline pilot and scientific operating platform, and any software that you need can be integrated into the research environment. 15
Genetic Optimization for Ligand Docking
Genetic optimization for ligand docking (GOLD) is based on the genetic algorithm for docking flexible ligand into the protein binding site. It is the most recent and highly expensive software for the computational approach. Common feature of this software is pose prediction, virtual screening protein ligand docking in the kmine interface to easily buildup into pipeline of work and understand irreversible binding with co-valent docking to exlire cancer, immunology and infectious diseases target. 16
Schrodinger
This is extensively used in combinatorial chemistry and high throughput screening in the pharmaceutical and biotechnology industries that means that huge amount of compound can now be routinely used for biological activity. Thus, screening of a large chemical library remains an expensive and time-consuming process with significant rates of both positive and negative falls.
Glide offers high range of speed and accuracy from the high-throughput virtual screening mode for efficiently evaluating million compound libraries to the standard precision mode for reliable docking tens of hundreds of thousands of ligands with high accuracy to accomplish an extensive sampling and advanced scoring result. Glide consistently finds the correct binding modes for a large set of target molecules. It performs the docking program in lower route mean square deviation from native cocrystalized structure. iGEMDOCK performs molecular docking with less than 2.0 Angstrom route mean square deviation value. 17
iGEMDOCK
It is a graphical representation for recognizing pharmacological interaction and virtual screening. For virtual screening, the pharmacological interaction derived by iGEMDOCK software often involved the biological function in which the hitrate on the three public sets (antagonist, agonist and kinase-PK). iGEMDOCK understand the ligand binding mechanism and discover lead compound. 18
Smina
It is fork of AutoDock Vina that is customized to scoring development and high-performance energy minimization. It works on the Linux operating system. It should be placed in a path of command line with no dependency and libraries. Smina does not used the scoring function and the caches for the default scoring function with easy to script the possibility to calculate the box from the existing ligand. 19
Pymol
It is a open source molecular visualization software for structural biology, macromolecule analysis, protein–ligand docking, molecular docking simulation, and pharmacophore modeling. It is primarily used to view, rotate, translate, and change the orientation of the object. Pymol program is based on the Python programming language. 20
Open Babel
It is a chemical toolbox designed to convert, analyze, and store data from molecular modeling chemistry, solid state material, biochemistry, and/or bioinformatics areas. It facilitates interconversion of chemical data from one format to another including file format of various types such as Structure Data Format, Simplified input line entry system, and International Chemical Identifier. 21
The highest scoring predictions were from AutoDock Vina, GOLD, and Schrodinger. The proper ligand binding postures were found using GOLD and Discovery Studio. There is a 90% correlation between the postures predicted by Glide (extra precision) and GOLD. Recent research has shown that, on average, these docking systems can predict experimental postures with Root Mean Square Deviation (RMSDs) of just 1.5–2.0 Angstrom. However, the flexibility of receptors, particularly their backbones, remains a significant obstacle for current docking approaches. 7
To obtain protein conformations for docking screening, more sophisticated sampling techniques may also be used, such as replica exchange MD, metadynamics, and umbrella sampling. It has previously been used to examine protein flexibility and function, as well as to uncover new binding sites that might be utilized in the creation of novel inhibitors. There have been multiple docking scoring systems established based on various techniques and ideas, including knowledge-based, experimental, and force field-based techniques. 38 Improved docking score methods have also made use of machine learning (ML) technologies.
The earliest ML-based scoring systems, called RF-Score, uses Random Forest to improve predictions of protein–ligand binding affinity. Based on this, an RF-based scoring method was created using the ligand–protein complexes from the PDB bind database and previously released activity data. In addition to this, docking results were improved utilizing “RF-Score” compared with other scoring methods used in currently available docking tools in both virtual screening and lead optimization activities. 39
MOLECULAR DYNAMICS
It describes about the electronic clouds in the molecule that is inherent to drug receptor binding processes called MD. It enables in analyzing multiples energy minima. It has a capacity to separate the molecular motion into inter- and intramolecular interactions. MD stimulates molecular motion at high temperature (heating dynamics). This thermal energy overcomes the conformational barrier. Slow heating of the system followed by the energy equilibration among the constituent atom permits this process. MD provides insight about the molecular level interaction between ligand (drug) and receptor (protein).
It utilizes the Newton's law of motion (second law) in MD. MD generates a conformation through the energy minimization process. It calculates the force on each atom throughout the specific period (∼10–12 seconds). MD requires lesser time (∼1–4 femtoseconds). MD calculation validates the efficiency of molecular docking predictions. 40
PREDICTION OF DRUG-INDUCED SIDE EFFECTS
For predicting off-target actions of previously published medicines, docking and statistical techniques have been combined. Docking and ML models have lately been used together. According to further experiment, >1,200 compounds from Drug Bank were tested against 600 human proteins to predict or rationalize drug side effects. Before building ML-based models on adverse reaction, data retrieve from the side effect resource database. They next compared the measured ligand–protein scores with the training ML models to predict side effects. Absorption, distribution, metabolism, excretion, and toxicity can be predicted by using the different computational software available for the prediction of their hepatotoxicity, cardiotoxicity, urinary toxicity, and its skin permeability parameters such as pkCSM (Table 3). 41
Software Tools for Absorption, Distribution, Metabolism and Excretion Analysis
ADME, Absorption, distribution, metabolism and excretion; ADMET, Absorption, distribution, metabolism, excretion, and toxicity; HIAP, human intestinal absorption percentage; HOA, human oral absorption; PSA, polar surface area; QSAR, quantitative structure activity relationship; SASA, solvent accessible surface area.
POLYPHARMACOLOGY
The pharmaceutical industry has concentrated on developing extremely selective medications to minimize any negative side effects. Polypharmacology, which refers to the discovery of ligands that hit a variety of selected therapeutically significant sites, has become more common in modern drug design as a result of high attrition rates in late clinical trials.
Molecular docking may be a beneficial tool in this situation since it permits the development of chemical scaffolds that effectively and concurrently attach to a variety of different target molecules. Several research studies on the utilization of docking for the production of new multitarget ligands have previously been conducted. It is challenging to rationally create multitarget ligands with the protein conformations so that the in vitro study of the multiple targeted ligand can be done followed by the in vivo study. The process of drug development will be faster and available for the welfare of humans.
For compounds with many targets, docking is increasingly used in conjunction with other ligand design techniques. The discovery of the first Hsp90/B-Raf dual inhibitors, which demonstrated the efficiency of combining substructure prefiltering with pharmacophore-guided docking to produce polypharmacology ligands that bind to structurally dissimilar targets, is one example. 41 The findings suggested that docking may be utilized to drive the development of selective potentially safer multitarget drugs capable of circumventing the adverse effects frequently associated with medications.
A further contribution to our understanding of disease-related targets is the discovery of novel selective ligands by combining diverse in silico techniques. 11 CANDO, or the Computational Analysis of Novel Drug Opportunities, platform is used to investigate polypharmacology and identify several targets for ligand effects; these two web servers, CANDO and Drug Positioning and Drug Repositioning via Chemical Protein Interactome, may anticipate the possible multitarget interactions of compounds, and help to find potential new targets for drugs, as well as aid in medication repositioning.
DRUGS REPROCESSING
Molecular docking is the most often utilized computational techniques for repurposing drugs for new therapeutic targets. Docking may be utilized in reverse screening approaches to uncover novel molecular targets for known ligands based on structural complementarity. Through docking, pharmaceutical, natural product, and synthetic chemical databases may be quickly and affordably screened against one or more relevant biological targets. The possibility of repurposing the Parkinson's disease-treating drugs entacapone and tolcapone for use against tuberculosis is being further investigated using in-depth structure-based research on nine different Mycobacterium tuberculosis structures. 42
Despite having a significant potential for drug development, naturally occurring chemicals are rarely exploited for drug repositioning. Recently, novel therapeutic uses for natural compounds have been found using repositioning algorithms that incorporate chemocentric target identification and docking analysis. For instance, to find novel therapeutic targets for the two nonpsychoactive cannabinoids cannabigerol and cannabichromene, we recently combined shape-based similarity screening with rigid and flexible docking simulations. This study was the first to do a computational shape-based similarity screening using the Drug Bank database.
This conclusion was verified by further in vitro testing. There are several advantages using this strategy, which may be used to repurpose ligands from both natural and synthetic sources, to make more accurate predictions about medication repositioning. 43 Using the MTiOpenScreen web service and the “Drugs-lib” library they built, they ran docking screens to see whether the strategy they had followed had been successful in identifying five authorized ligands with prior reports of drug repositioning against cancer-related targets.
CONCLUSION AND FUTURE PROSPECTS
Molecular biology research identifies >10,000 potential drug targets in the human system. The newer potential target pool shows promising prospects for molecular docking-assisted drug development. The functional and positional information of the target derived from bioinformatics supports the target identification and validation process. Number of informatics tools enabled the in silico cloning of the target molecule.
The computational approach is used in a variety of fields such as bioinformatics, pharmaceutical sciences, biotechnology and transcriptomics, which reduce the direct in vivo study of the drug, time reducing in lead discovery of molecule and more potent molecule are designed that are efficacious, safer, and least toxic to the human system. In cancer, various chemical scaffolds are developed by the use of AI. The orphan diseases such as psoriasis, adenosine deaminase deficiency is occurring in human being that makes them feel low self-esteem, socially and psychologically ill. In silico approach helps in the development of new molecules that have the potential to treat them.
Footnotes
AUTHORs' CONTRIBUTIONS
D.S. contributed to methodology and writing–original draft. L.S.R. was involved in docking approach and software-based data validation. S.D.D. took charge of conceptual work. K.S. carried out data curation. N.S.C. was in charge of validation, writing–reviewing and editing, and formal analysis. M.R.S. was in charge of conceptualization, resources, supervision, visualization, investigation, writing–reviewing and editing, and funding acquisition. D.S. took charge of supervision, validation, writing–reviewing and editing, and visualization.
DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
No funding was received for this article.