
Abstract
The progression of type II diabetes (T2D) is characterized by a complex and highly variable loss of beta-cell mass, resulting in impaired insulin secretion. Many T2D drug discovery efforts aimed at discovering molecules that can protect or restore beta-cell mass and function have been developed using limited beta-cell lines and primary rodent/human pancreatic islets. Various high-throughput screening methods have been used in the context of drug discovery, including luciferase-based reporter assays, glucose-stimulated insulin secretion, and high-content screening. In this context, a cornerstone of small molecule discovery has been the use of immortalized rodent beta-cell lines. Although insightful, this usage has led to a more comprehensive understanding of rodent beta-cell proliferation pathways rather than their human counterparts. Advantages gained in enhanced physiological relevance are offered by three-dimensional (3D) primary islets and pseudoislets in contrast to monolayer cultures, but these approaches have been limited to use in low-throughput experiments. Emerging methods, such as high-throughput 3D islet imaging coupled with machine learning, aim to increase the feasibility of integrating 3D microtissue structures into high-throughput screening. This review explores the current methods used in high-throughput screening for small molecule modulators of beta-cell mass and function, a potentially pivotal strategy for diabetes drug discovery.
INTRODUCTION
Diabetes is a global health crisis affecting over 537 million people worldwide. It is quickly becoming one of the most pressing health concerns of our time. By 2030, it is predicted that more than 643 million people will be diagnosed with prediabetes or insulin-dependent type II diabetes mellitus (T2D). 1,2 The cost of dealing with diabetes-related health problems is currently approaching a staggering $1 trillion globally. In the United States, on average, each person impacted by diabetes spent about $11,779 on related health expenses in 2021. 2 When it comes to managing T2D, current therapeutic strategies combine a mix of lifestyle changes and medications, including both noninsulin drugs and insulin treatments. These approaches aim to control blood glucose homeostasis, but they do not address the loss of beta-cell mass, the root cause of the disease.
The beta cell performs a distinct physiological role as the sole cell type within the body to secrete insulin. In concert with the alpha cells, the endocrine cell system within the pancreas governs glucose homeostasis. The progression of T2D is correlated with an average of 30% loss in beta-cell mass (24%–60%), which ultimately leads to the disruption of glycemic regulation. 3 The precise mechanistic underpinnings leading to this cellular attrition have only been partially elucidated. The prevailing hypothesis suggested that beta-cell loss emanates from apoptosis, triggered by the stress of heightened insulin production to overcome the resistance posed by insulin-resistant tissues and glucolipotoxicity (GLT). 4 However, recent advancements have unveiled an alternative perspective, suggesting that a subset of the diminished beta-cell population may be undergoing a regressive transition toward a fetal state, marked by an inability to produce and secrete insulin. 5,6 Significant drug discovery efforts have been made to discover and develop molecules with the ability to protect or restore beta-cell mass and function.
The implementation of high-throughput screening (HTS) has been pivotal in the search for these agents. Within HTS, expansive libraries of chemical compounds are subjected to rapid evaluation through molecular-targeted or phenotypic bioassays. These chemical libraries, derived via parallel or combinatorial synthesis, can encompass millions of compounds. Execution of these assays occurs within standardized well plates containing 96, 384, or 1,536 wells. Although the adaptation to a reduced scale yields a lower cost per data point, it requires a higher level of precision as volumes decrease to the order of a few microliters per well. In tandem with the miniaturization of bioassays to accommodate the plate format, the integration of automation emerges as an indispensable tool, affording enhanced reproducibility and expediting logistics management. 7 Although limited in number, several insulin-expressing and glucose-responsive immortalized beta-cell lines have been developed and facilitated diabetes screening in general. 8,9 This review aims to summarize the current methods used in cell-based HTS for small molecule modulators of beta-cell mass and function.
LUCIFERASE-BASED REPORTER AND VIABILITY ASSAYS
Luciferase assays, a heavily used assay technology, are regularly leveraged to explore gene expression in reporter cell systems, assess cell viability, and explore other relevant end points. Highlighting its widespread use, nearly 22% of the 4,000 assays recorded in the PubChem database utilize the luminescence detection method. 10 The key to this assay is the luminescence-producing oxidation of luciferin by the luciferase enzyme—a reaction necessitating adenosine triphosphate (ATP), thereby serving as an effective measure of cell viability. The simplicity of luciferase assays is what makes it an efficient choice for HTS.
Luciferase assays have been used to identify compounds that alter the viability of beta cells in terms of stimulated growth or protection from cytotoxicity. A workflow for high-throughput luciferase assays is shown in Figure 1. In one example from 2009, a modified mouse beta-cell line, R7T1, was used to screen approximately 850,000 compounds. 11 The cells were immortalized using the SV40 T antigen oncoprotein allowing cell production for HTS, and cell cycle arrest was then triggered upon tetracycline withdrawal. This system enabled the identification of compounds that stimulate the entry of beta cells into a proliferative state, determined via a CellTiter-Glo readout. Results from this screen led to the subsequent discovery that a combination of DYRK1A and GSK-3B inhibition can stimulate beta-cell proliferation. 12,13
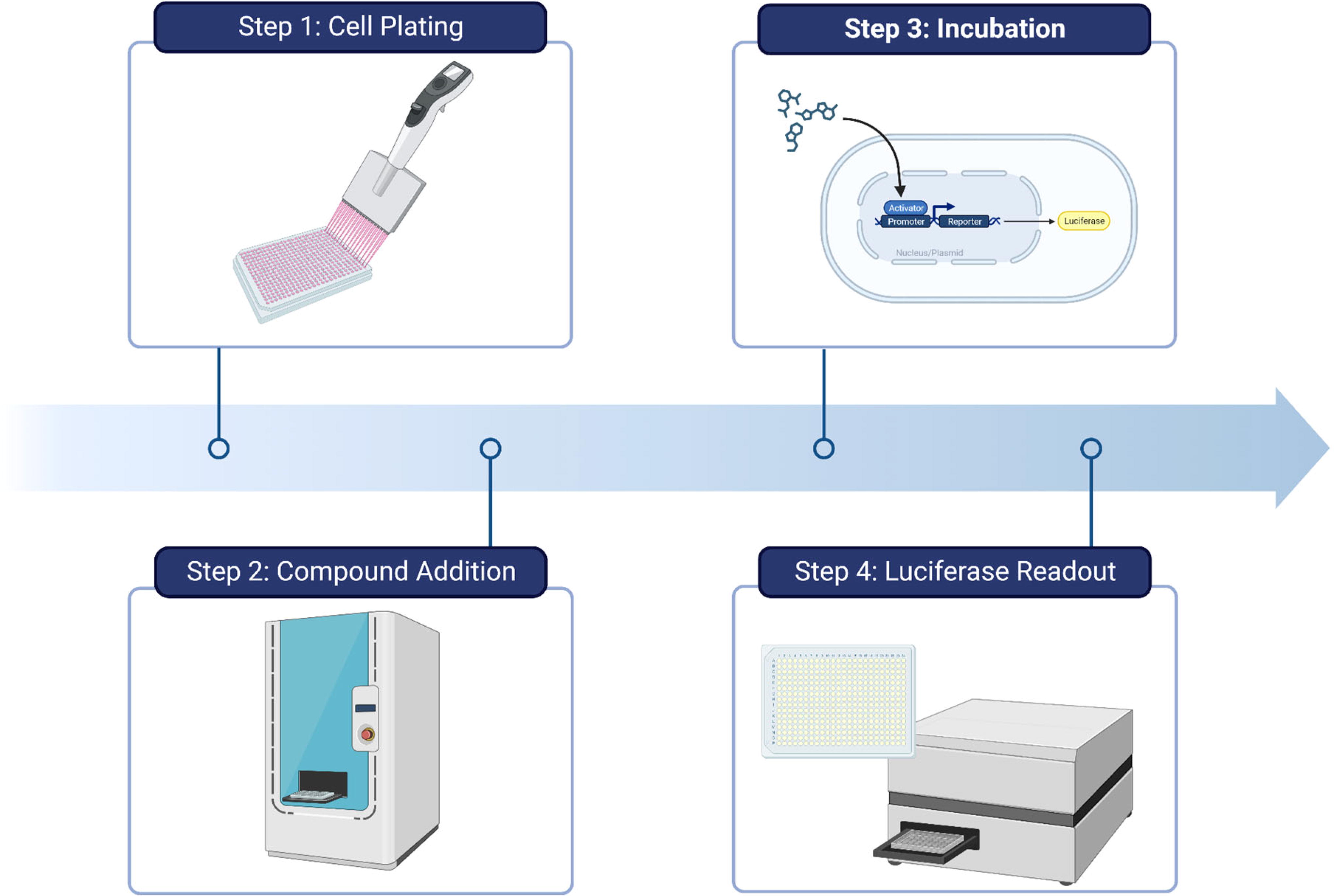
Workflow for high-throughput luciferase assays.
It is established that the high level of circulating glucose and lipids, persistent over the course of T2D, induces beta-cell apoptosis. 14 Recent studies developed a cell-based GLT model to facilitate screening for protective molecules. 15,16 In this innovative assay, immortalized beta-cell lines are exposed to high concentrations of glucose exceeding 25 mM and simultaneously challenged with palmitate, a saturated fatty acid. Conveniently, palmitate can be precomplexed with fatty acid-free bovine serum albumin at various ratios for seamless addition during screening. The primary readouts for this assay were cell viability, using either an XTT assay or CellTiter-Glo. Miniaturization to a 1,536-well format has permitted the screening of over 300,000 compounds. These phenotypic screens identified several molecular targets and pathways that protect against GLT by decreased cytosolic calcium influx in islet cells and reduction of stress-induced apoptosis by MAP4K4 inhibition offering new druggable targets to halt the progression of beta-cell loss.
In the context of type I diabetes (T1D), a condition characterized by beta-cell loss through an autoimmune-mediated pathway, a similar approach was applied to establish an HTS model. A cytokine-induced beta-cell apoptosis model was developed using an immortalized rat insulinoma cell line, exposed to a cocktail of cytokines. These cells were exposed to a cocktail composed of 10 ng/mL interleukin-1, 50 ng/mL interferon-gamma, and 25 ng/mL tumor necrosis factor alpha, a mixture known to induce beta-cell death. Following the addition of compounds and a 48-hour incubation period, beta-cell viability was measured using CellTiter-Glo. 17 This pilot screen, comprising 2,240 compounds, led to the understanding that GSK-3B inhibitors were also effective at preventing beta-cell apoptosis.
A more targeted approach for luciferase screening involves the creation of genetically encoded promoter–reporter systems. This system, with the luciferase sequence alongside a target gene promoter, can be integrated into cells. When these cells express the target gene, they concurrently synthesize luciferase. The subsequent light emission observed when luciferin is added to these cells allows for the measurement of gene expression level. Two examples that have been used in this fashion for HTS in the context of diabetes are MYC and Nkx6.1.
The MYC proteins have long been recognized as crucial drivers of cell growth. HepG2 cells, a hepatocyte line, were transfected with a luciferase reporter under the control of the human MYC promoter. Although HepG2 cells are not an endocrine cell line, they were chosen because they yielded the most robust luciferase responses. Among 100,000 compounds screened, 86 were selected for testing in primary rat islets. Ultimately, Harmine, a DYRK1A inhibitor that has since gained prominence in the field, was the sole compound from the screen to confer efficacy in stimulating islet beta-cell regeneration. 18
A cDNA screen conducted in a hamster insulinoma cell line unveiled the impactful role of homeobox genes on beta-cell function. 19 Further investigation efforts focused on the homeobox domain protein Nkx6.1, revealing its role in beta-cell replication and its regulation of glucose-stimulated insulin secretion (GSIS). In addition, it was found to promote beta-cell survival via the production of the VGF peptide. 20 –22 Leveraging this promising target, a screen of 630,000 compounds was executed, using a luciferase reporter for the VGF promoter in the 832/13 rat insulinoma cell line. 23 This extensive screening process yielded three lead candidate molecules, each containing the ability to induce beta-cell proliferation, enhance GSIS, and protect against apoptosis.
Although luciferase-based screening is cost-effective and efficient for HTS, it is widely acknowledged for its susceptibility to assay interference. One study investigating 360,000 compounds in PubChem found that 12% of screening compounds inhibited FLuc. 24 Likewise, compounds dubbed “luciferase stabilizers” can increase the luminescence signal through the formation of stable adducts with ATP, leading to false positives. 25 Small molecules that have linear, planar structures have the highest likelihood of causing assay interference. In light of this phenomenon, newer analogs of FLuc, such as NLuc, RLuc, or TLuc, exhibit reduced interference. It is also worth noting that cytosolic ATP levels have been observed to increase during apoptosis, introducing another potential source of interference in luciferase viability assays. 26 To mitigate such challenges, carefully choosing orthogonal assays that do not rely on a luciferase readout is key, as they provide an effective method to filter out assay interference compounds. Alternatively, using the same screening method but with the utilization of ATP-independent RLuc can be sufficient to filter out both false positives and negatives in expression assays, thus enhancing the efficacy and accuracy of the screening process. 27 In fact, the Dual-Luciferase Reporter Assay uses sequential reads from FLuc and RLuc in the same cell lysate to overcome potential assay interference. 28 The development of NanoLuc has provided a luciferase that is smaller in size, drastically brighter, and less prone to compound interference. 29 A summary of luciferase-based diabetes bioassays is shown Table 1.
Summary of Luciferase-Based Assays in Diabetes Drug Screening
ATP, adenosine triphosphate.
GSIS
The transition from healthy to diabetic involves the deterioration of several beta-cell functions, one of which is the first-phase insulin release following postprandial increase in blood glucose. 30 This was underscored by two longitudinal studies that identified changes in acute insulin response as the primary determinant of glucose tolerance. 31,32 However, until recently, large-scale screening for molecules that can modulate this process has been exceptionally difficult. A typical GSIS assay requires a starvation period before introducing glucose. This, coupled with multiple wash steps to remove previously secreted insulin, makes this assay system cumbersome for HTS. After glucose stimulation, insulin levels in the supernatant are measured; however, there are a limited number of cell lines that are glucose-responsive, many of which lose that ability over time/passages that pose an added difficulty. 8 Historically, the quantification of insulin relied on enzyme-linked immunosorbent assay (ELISA). Although highly sensitive, they are time-consuming, expensive, and can have issues with high variability. Nevertheless, recent advances in proximity luminescence and fluorescence technologies have led to the emergence of high-throughput, cost-effective assay kits to enable high-throughput interrogation of insulin production and secretion. Another challenge in performing robust GSIS assays is the avoidance of cell disruption/lysis during sampling of the media for insulin quantitation. Beta cells contain large quantities of insulin and secrete only a small fraction; therefore, any cell disruption can cause significant increases in insulin content. Therefore, a slow and careful aspiration of media from cell plates, ideally using liquid handling robotics, can greatly reduce noise in the insulin readout by avoiding cell disruption.
An improvement in assay technologies is the use of homogeneous “mix and read” assays where reagents are added, and the signals are detected directly as shown in Figure 2. Homogeneous proximity biosensor assays have become a prominent tool for insulin detection in GSIS experiments. One notable tool that has emerged is a luminescence-based assay kit known as AlphaLisa (Perkin Elmer). With this assay technology, donor and acceptor beads are coated with anti-analyte antibodies. Upon simultaneous binding of both beads to the analyte, in this case, surrounding insulin, they are brought into proximity. The subsequent activation of the donor beads using a 680 nm wavelength triggers a singlet oxygen reaction sequence with the nearby acceptor bead, which produces photon emission at 620 nm. Notably, this system can be miniaturized to require only 1 uL of analyte and is compatible with a 1,536-well format. Despite its remarkable sensitivity, the AlphaLisa encounters certain limitations. The primary limitation is its irreversible chemical reaction that leads to diminished signal upon sequential reads and may suffer interference from ambient light. 33

General workflow for GSIS assays. Cells are plated in 384-well format and are allowed to recover, generally overnight. Compounds are added, and the cells incubate for an optimized amount of time. Cells are then starved of glucose before being stimulated with a buffer containing a high concentration of glucose. The buffer is removed, and various methods can be used to quantify the amount of insulin released during glucose stimulation. GSIS, glucose-stimulated insulin secretion; MALDI-TOF, Matrix Assisted Laser Desorption/Ionization-Time of Flight.
An alternative approach uses time-resolved fluorescence resonance energy transfer assays. With execution and cost similar to AlphaLisa, these assays implement a proximity-based fluorescence readout, as opposed to luminescence. The adoption of long-lived fluorophores minimizes background noise stemming from buffers, proteins, or chemical compounds present in the sample mixture. Several commercially available kits offer this technology such as Perkin Elmer’s Lance, Invtrogen’s Lanthascreen, and CisBio’s Homogeneous Time-Resolved Fluorescence (HTRF) platform. Particularly intriguing is the potential to use the HTRF assay system in a 3,456-well format, paving the path for ultra-HTS campaigns. 34 However, it is important to consider that these technologies are not entirely free from assay interference. Assay interference in this context is primarily caused by a reduction in assay signal because of mechanisms including absorbance, quenching, light capture, and disruption of affinity components. Overcoming these may require deploying strategies such as product spiking or orthogonal assays to mitigate interferences. 35
In a concerted effort to drive down the cost of insulin secretion assays, several groups have pioneered similar luciferase-based insulin secretion assays. The defining feature of these systems is the fusion of Gaussia luciferase and the C-peptide portion of proinsulin, based on the previous work where green fluorescent protein (GFP) was fused with the proinsulin protein to facilitate fluorescence quantitation. 36 Intrinsic to the insulin secretion process, the C-peptide of mouse proinsulin is cleaved within beta-cell vesicles. It is then co-released with insulin during exocytosis. Since C-peptide and insulin are released concurrently, a luminescence readout can be achieved using the supernatant. Cell lines from both mouse (MIN6) and rat (INS-1E) that stably expressed the reporter were developed, with rigorous validation efforts confirming a strong correlation between luciferase activity and insulin content. 37,38 Expanding the reach of this method, the system was successfully miniaturized to the 1,536-well format. This was achieved by performing the glucose starvation and wash steps while the cells were suspended, prior to plating. One example of the implementation of this system is the screening of more than 1,100 plant extracts for their ability to modulate insulin secretion. 39
The recent work of Delannoy et al. presents a novel approach for quantitative high-throughput GSIS, using the Matrix Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF) mass spectroscopy. 40 This innovative method requires samples to be spiked with an internal standard of bovine insulin to quantitate and normalize insulin content. Following dilution and spiking, small sample quantities are dried onto a 384 steel MALDI target and subsequently analyzed. The advantage of this method lies in its ability to directly detect insulin, thereby circumventing potential complications such as incomplete antigen binding or interference from compounds within the detection assay. Interestingly, experimental results from both ELISA and MALDI-TOF were nearly identical. Although several preparatory steps are required before mass spectrometry analysis, these stages are brief, and overall assay efforts are comparable with those required for luminescence or fluorescence proximity assays. Such innovations underscore the potential for efficient, high-throughput interrogation of GSIS dynamics. However, it is noteworthy that these assays continue to be single endpoint assays instead of multiplexed assays, even though the cells hold potential for imaging or other readouts after the GSIS is complete.
HIGH-CONTENT SCREENING
Multiplexed imaging-based phenotypic cell screening, also known as “high-content screening” (HCS), is designed to extract a wealth of information from cells using fluorescent probes and automated fluorescence microscopy as shown in Figure 3. With significant advancements in the field, this approach has evolved where screens can be conducted in a completely automated and quantitative manner, providing robust results capable of measuring multiple end points, including biomarker intensity, spatial distribution of signals, and morphologic end points. 41 The primary goal of this approach is to extract as much information as possible from cell populations within each experiment. Although earlier HCS efforts include one or two image-based features, the advent of cell painting and the availability of open-source image/data analysis software, such as CellProfiler, CellPose and Knime, have expanded the information content in screening to encompass hundreds to thousands of measured cellular features. 42,43 This technique is particularly advantageous for targeting cell subpopulations to enable image cytometry. Assays focusing on cell cycle, infection, and differentiation have shown robust results, leading to their adoption within the diabetes research field.
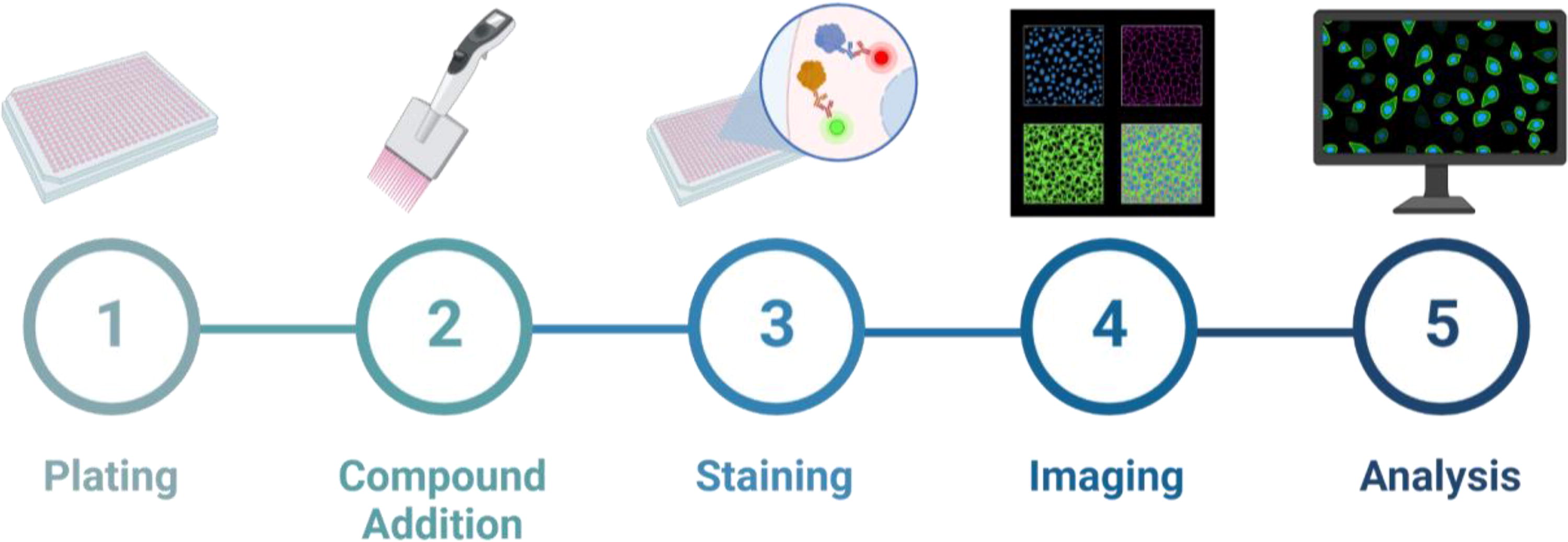
General workflow for HCS.
In conducting an HCS, it is critical to ascertain the reliability and robustness of the assay. This is typically assessed through a statistical coefficient called Z-prime (Z′). 44 The Z′ offers a measure to evaluate the quality of a biological assay factoring in both the assay window and variability of the data. This includes consideration of background and positive controls. The mathematical formulation of Z′ equals one minus the absolute value of ratio of the difference between the standard deviations of the controls and the difference of their means. It is important to note that the accuracy of this metric degrades if hit rates exceed 20% of the samples. Meanwhile, the B-score plays a crucial role as a normalization method in high-throughput experiments. 45 This method is designed to handle spatial bias and plate/batch effects. For instance, a common plate effect can be the evaporation of media in the outer wells, leading to a systematic error in determining efficacy. Normalization is achieved by comparing the measurement of each well to the median of its neighboring wells within the same plate. 46 One benefit of high-content multivariate assay end points is the ability to generate machine learning (ML) models that can significantly enhance the robustness of the assay. These methods generally rely on training ML random forest models against the positive and negative controls in the plate, enabling scoring of compound-treated wells for robust quantitation of efficacy with simultaneous detection of cytotoxicity. We have previously described a pragmatic approach to these general methods in the National Institutes of Health Assay Guidance Manual as a resource to help guide the implementation of ML models in the context of HCS. 47 This approach also streamlines the integration of high-dimensional screening data into conventional HTS data analysis paradigms intended for univariate plate reader-based compound screening.
An early application of imaging in diabetes drug discovery was in the search for hepatocyte nuclear factor (HNF) 4α agonists. This screen took advantage of T6PNE cells, a line originating from human fetal islets and engineered to express Pdx1, NeuroD1, and E47. 48,49 The cells were further adapted for HTS through the incorporation of a lentiviral vector. This vector was designed to express GFP under the control of the human insulin promoter. In this screen, tamoxifen was used to impair insulin expression, and the objective was to identify compounds that rescued insulin production. Following a 2-day incubation, the cells were imaged using a high-content imaging system. The primary parameter analyzed from the images was the intensity of the GFP; however, this assay ultimately suffered low Z′-scores because of variability in GFP expression. 49
Through the deployment of a dual reporter system of Ins1/GFP and Pdx1/red fluorescent protein (RFP) in an HCS, a link was identified between sodium channel inhibition and increases in insulin transcription where the multiplexed readout delineates the “maturation” of the beta cells. Cells expressing high Pdx1 but low Ins1 were “immature,” whereas cells expressing high Pdx1 and high Ins1 were deemed “mature.” 50 This screen was carried out in 96-well plates using a MIN-6 cell line. Cells were imaged live on a Zeiss 200M microscope. Variability was observed in the outer rows, which was corrected through B-score normalization. 51 The study took advantage of a library of 1,120 compounds that contained many off-patent drugs. The well-documented mechanism of action of these known drugs enhances hypothesis generation when used in HCS. 52 Regrettably, owing to excessive variability, the assay proved incompatible with primary human islets.
A proposed strategy to restore beta-cell mass involves the conversion of other endocrine cells to beta cells. Such a process has been confirmed through lineage tracing in mouse models following beta-cell loss. 5,6 Particularly, when examining alpha cells, it was found that simultaneous loss of DNMT1 and ARX genes led to the expression of insulin, Pdx1, and Nkx6.1 genes within those same cells. 53 To identify compounds capable of inducing this transformation, a MIN-6 cell line was manipulated to overexpress Myc-tagged ARX, which leads to the repression of their normal beta-cell genes. Cells were plated in 384-well format, and compounds were introduced with the goal of repressing ARX expression and stimulating insulin production. Outcomes were determined by the fluorescent staining of insulin and Myc-ARX, followed by high-content imaging. 54
The scarcity of human-derived beta-cell lines has necessitated the use of cadaveric human islets as a model for diabetes drug discovery because of their physiological relevance. It has been well established that islets sourced from donors remain viable in culture and are capable of demonstrating islet cell proliferation and normal insulin secretion dynamics. 55 Yet, their three-dimensional (3D) structure poses challenges for rapid imaging and analysis. The most common approach begins with the dispersal of isolated cadaveric islets into a single-cell suspension; however, they exhibit high viability under specific culture conditions. 56,57 To enable the use of these cells in the 384-well screening environment, plates were initially coated with an extracellular matrix formed by HTB-9 cells, a bladder carcinoma line. After these cells secreted matrix factors, they were removed using ammonium hydroxide, and dissociated human islet cells were plated on top of the matrix. 58 Without this extracellular matrix, dispersed islet cells have very low attachment strength and tend to detach over the course of the protocol. 59 In addition, the matrix allowed dispersed islet cells to be cultured for up to 7 days. The cell populations were examined via imaging, applying a combination of glucagon, C-peptide, and Ki67 staining. A library of 1,280 compounds was screened in cells from three different donors. 60 However, prominent variability in the beta-cell response was observed between donors, a factor that should be considered for optimization in large-scale screening endeavors. A summary of two-dimensional HCS is shown in Table 2.
Summary of 2D High-Content Screening Methods
2D, two-dimensional; GFP, green fluorescent protein; HNF, hepatocyte nuclear factor.
IN VIVO SCREENING
The zebrafish has emerged as an interesting model for whole organismal in vivo phenotypic screening, bridging the gap between the simplicity of in vitro cellular models and the expense of in vivo mouse models. Owing to their optical transparency, ability to absorb compounds added in their environment, and compatibility with 96-well plates, zebrafish embryos are ideally suited for HTS. 61 Several studies have used zebrafish in screens to identify pathways leading to the induction of beta-cell proliferation. In one study, the use of yellow fluorescent protein expression, driven by the insulin promoter, and RFP expression, driven by the somatostatin 2 promoter, allowed for beta-cell production to be observed. Moreover, the inclusion of RFP could facilitate the identification of compounds that selectively increase beta-cell mass. 62 To further explore beta-cell dynamics, two other studies leveraged variations of fluorescent ubiquitination-based cell cycle indicator (FUCCI) technology. The core of FUCCI features of a plasmid containing GFP and RFP fused to proteins unique to growth 1 or synthesis/growth 2/mitosis phases. 63 In one approach, this system was placed under the control of the insulin promoter, thus allowing the visualization of proliferating beta cells within the zebrafish pancreas. 64 The alternative approach replaced the fluorescent proteins with NLuc for a luminescence-based readout upon addition of the appropriate substrate. 65 Both these techniques deploy a few larvae per well of a 96-well plate for compound exposure. The zebrafish can be anesthetized with Tricaine, allowing for high-throughput imaging and image analysis-based quantitation. 66
MICROTISSUES AND PRIMARY ISLETS
Although immortalized or primary cell monolayers have significantly contributed to diabetes drug discovery, they fall short in replicating the complexity of the intact pancreatic islet. Moreover, the widespread use of rodent-derived cell lines has led to a more comprehensive understanding of the intracellular signaling pathways steering rodent beta-cell proliferation than that of human beta cells. 67 Human islets are small, 3D structures composed of five main endocrine cell types, interspersed in the islet along with other nonendocrine cell types. 68 Their 3D structure makes drug penetration, as well as imaging and analysis, a considerable challenge. Microscopy of intact microtissues often involved mounting them on glass slides and imaging on low-throughput confocal microscopes, requiring significant time and effort. Nonetheless, recent advances in confocal microscopy have facilitated high-content imaging and analysis of single cells in 3D structures such as pseudoislets or intact primary islets.
One strategy to address low drug penetration involves dissociating islets to single cells prior to compound or transgene vector treatment. These cells are then reaggregated into islet-like structures often referred to as “pseudoislets.” 69,70 Pseudoislets reestablish gap junctions and maintain GSIS similar to their native counterparts. They also bring the added advantage for screening by reducing variability in size and enhancing uniformity and cellular composition. 71,72 Originally, pseudoislets were formed in bulk using the “hanging-drop” method, a labor-intensive process that involved suspending 30 uL drops, containing dispersed endocrine cells, upside down in culture dishes. 73 However, a modern adaptation of this method uses round-bottom, ultra-low-attachment 384-well plates to accomplish reaggregation. 74 This results in pseudoislets that can be maintained in culture for up to 4 weeks. The utilization of round-bottom plates ensures uniformity in pseudoislets’ position across all wells, thereby facilitating rapid high-resolution imaging. Staining using DAPI, Nkx6.1, and EdU allows for the assessment of beta-cell proliferation. Interestingly, this method can be multiplexed with GSIS before fixation, accommodating a multi-endpoint assay. 75 Although this system provides a more physiologically relevant model than with monolayer cultures, the generation of only one islet per well significantly hampers throughput necessitating multiple wells to interrogate the effects of a single compound. In addition, because the pseudoislets are not attached to the microwell plate, fixation and staining that require multiple additions and wash steps can lead to the loss of pseudoislets, further hampering screening efforts.
Although the uniformity of pancreatic pseudoislets reduces variability, it reduces the physiological relevance. The creation of these microtissues destroys the original size, ratio, and organization of cells within islets. Although the heterogeneity of islets presents some challenges, the primary hurdle is their small size relative to the surface area at the bottom of the microplate wells. High-resolution imaging of an entire well for detailed images of islets would require a tremendous amount of time, and a majority of images would be empty. Conversely, low-resolution imaging to identify areas that contain islets would expedite the process but sacrifice detail within the islets. A solution to this problem was addressed by the advent of a two-pass high-content imaging technique. In this method, each well is imaged at low resolution, the location of each islet is determined via image analysis, and the location data are fed back to the microscope to facilitate high-resolution second-pass imaging where a single islet is centered within a field. Furthermore, coupling the multiplexed staining of Nkx6.1, glucagon, and Ki-67 with ML analysis enabled the detection of islet cell proliferation and the analysis of islet spatial morphometrics at the single-cell level. 76
Recent advances in modeling of pancreatic islets using induced pluripotent stem cells (iPSCs) have enabled the generation of islet-like cells and have gained utility in biomedical research. Although differentiating iPSCs into pancreatic beta cells can be a complex, expensive, and time-consuming process, requiring complex combinations of growth hormones and signaling molecules, it offers advantages over cadaveric islets. 77 Specifically, it ensures greater control over genetic diversity, age, and overall health of the islets. 78 Furthermore, iPSC-derived islets generated in either a planar or a 3D scaffold are notably similar to native islets; they are glucose-responsive and display a mix of insulin and glucagon-expressing cells. 79 However, challenges remain in ensuring the efficient differentiation of iPSCs into fully mature and functional islet cells that accurately represent native pancreatic islets. Ongoing research efforts are focused on optimizing differentiation protocols and improving the functional maturity of iPSC-derived islet cells to enhance their utility in drug discovery. iPSC-derived pancreatic islets represent a powerful tool in drug discovery, offering novel opportunities for disease modeling, drug screening, and the development of personalized medicine approaches for diabetes drug discovery.
SMALL MOLECULES IN CLINICAL DEVELOPMENT FOR DIABETES
Using methods described previously, small molecules that modulate beta-cell function have been successfully identified. Luciferase-based screening has enabled the evaluation of millions of compounds for their ability to modulate beta cells. For instance, Harmine was initially discovered in HepG2 cells and was observed to promote islet cell proliferation through DYRK1A inhibition. 18 The aminopyrazine GNF-4877 similarly promotes beta-cell proliferation through the inhibition of DYRK1A and GSK-3B. 13 In addition, both Alsterpaullone and Ro 31-8220 were found to protect beta cells against inflammatory-based apoptosis through GSK-3B inhibition. 17 Several compounds were confirmed to reduce fatty acid-induced apoptosis in beta cells (lipotoxicity) through either an MAP4K4 kinase or a calcium channel-mediated pathway. 15,16 In contrast, compounds including Milrinone, Forskolin, and Anagrelide were found to enhance GSIS through a luminescent reporter of insulin secretion. 37 These results were corroborated, with the addition of repaglinide, in a MALDI-TOF GSIS screen. 40 An HCS discovered the importance of HNF4α in the modulation of insulin promoter expression. 49 Another HCS effort found that Artemisinins cause alpha cells to take on beta-cell characteristics. 54 Zebrafish screening studies revealed retinoic acids, glucocorticoids, and several other drug classes that can increase beta-cell mass. 62,64 The usage of cadaveric human islets in recently developed screening assays holds exciting potential for the additional detection of beta-cell modulating compounds. 75,76
Despite the discovery of numerous small molecule modulators of beta-cell function, there are many barriers to clinical translation for diabetes indications. However, several existing therapeutics have undergone or are undergoing clinical trials to investigate their effectiveness in affecting beta-cell modulation to regenerate or preserve beta-cell mass. For instance, Lansoprazole (Prevacid) and Sitagliptin (Januvia) were studied in combination with Dyamide to protect and replenish beta-cell mass in new-onset T1D. 80 Another therapy, Verapamil, demonstrated potential in inhibiting beta-cell expression of proapoptotic thioredoxin-interacting protein and is currently being studied for its ability to prevent functional beta-cell loss in T1D. 81 –83 Though Venetoclax exhibited promise in treating T1D in mouse models, it has yet to advance to clinical trials. 84 The liraglutide and beta-cell repair (LIBRA) study in 2011–2014 was a Phase 3 trial to study the ability of Liraglutide, a glucagon-like peptide 1 agonist, to preserve beta-cell function. 85 The current Phase 4 trial hopes to discern if Liraglutide can protect beta-cell mass by diminishing endoplasmic reticulum stress in T2D patients. 86 On another front, Fingolimod (Fingoland) has displayed positive outcomes against T1D and is presently in a Phase 4 trial to evaluate its efficacy in countering T2D. 87 Although the repurposing of current therapies holds promise, the discovery of various small molecule modulators presents an exciting frontier in the pursuit to find new therapies for T2D.
CONCLUSIONS
This review of bioassay technologies summarizes the common modalities used in pancreatic islet-based drug discovery approaches toward the identification/development of first-in-class pharmacotherapies for diabetes that target pancreatic islet cells. Despite the persistent and significant challenges in islet discovery, the diabetes drug screening holds great promise to identify efficacious small molecule therapies and novel molecular mechanisms influencing diabetes. Advancements in primary islet screening platforms, imaging hardware, and ML analysis have significantly advanced the field enabling high-throughput phenotypic and targeted drug discovery that were hitherto inaccessible. One significant aspect lies in the continued integration of iPSC-derived islets into the screening processes. iPSC islets offer a more physiologically relevant model compared to traditional cell lines, as they closely mimic the behavior of native pancreatic islet cells. As iPSC technology continues to improve and become more accessible, we can anticipate even greater adoption of this approach in drug discovery efforts.
Moreover, the incorporation of multi-endpoint assays represents a paradigm shift in screening methodologies. By simultaneously measuring multiple parameters and end points within a single assay, such as GSIS and cell type-specific replication, researchers can gain a more comprehensive understanding of drug effects and mechanisms of action. This holistic approach enables the identification of compounds with multifaceted therapeutic effects and can uncover synergistic interactions that may not be apparent when focusing on single end points alone.
The convergence of HTS with advanced imaging techniques, artificial intelligence (AI)/ML, and integration of spatially resolved transcriptomics methods holds tremendous potential. These technologies enable the rapid analysis of large datasets and the extraction of valuable insights that can inform drug discovery efforts. For example, AI/ML methods can help identify patterns in complex biological data, predict drug responses, and even suggest novel therapeutic targets.
Overall, the future of diabetes drug screening is characterized by an integration of cutting-edge technologies, a shift toward more physiologically relevant models, and a focus on comprehensive, multidimensional analyses. These advancements hold promise to accelerate the discovery and development of novel therapeutics for diabetes, ultimately improving outcomes for patients worldwide.
Footnotes
ACKNOWLEDGMENTS
Figures were generated using Biorender.com.
AUTHORS’ CONTRIBUTIONS
S.M.M. contributed to the methodology and writing of the original draft. S.M.M., J.Z.S., and M.C.C. contributed to the conceptualization, reviewing, and editing. J.Z.S. and M.C.C. contributed to resources, supervision, and funding acquisition.
DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
We acknowledge funding from the University of Michigan Institute for Clinical and Health Research (MICHR; National Center for Advancing Translational Science Grant UL1TR002240) and the U–M Center for Drug Repurposing. J.Z.S. is supported by the National Institute of Diabetes and Kidney Diseases (R01DK120623).