
Abstract
Background:
Crystalline silica particles are fibrogenic agents and established carcinogens, but the mechanisms for disease initiation and progression are not well understood. Previous studies demonstrate that the tumor suppressor gene, programmed cell death 4 (PDCD4), and its upstream regulator, microRNA 21 (miR-21), may be oncogenes for novel cancer prevention or anticancer therapies.
Methods:
This study examined the alterations of miR-21-PDCD4 signaling in mouse epidermal JB6 cells after exposure to freshly fractured silica particles.
Results:
The results demonstrate that exposure to crystalline silica caused PDCD4 inhibition in JB6 cells and a significant increase in miR-21 expression. Inhibition of phosphorylated extracellular signal-regulated kinases (ERKs) or phosphorylated p38 with U0126 or SB 203580 reversed silica-induced PDCD4 inhibition. Reactive oxygen species (ROS) scavengers and N-acetyl-
Discussion:
These findings demonstrate that freshly fractured silica particles may induce miR-21 expression and PDCD4 inhibition, which may be mediated through ROS and ERK pathways. Unraveling the complex mechanisms associated with these events may provide insights into the initiation and progression of silica-induced carcinogenesis.
Introduction
Epidemiological and pathological studies have established that occupational exposure to crystalline silica is associated with the development of chronic and acute pulmonary silicosis and a higher propensity toward the risk of lung cancer.1–4 In case of acute silicosis, the exposure occurs in individuals working in areas that fracture and grind silica into fine powders by mechanical processes. The onset of acute silicosis symptoms becomes clinically apparent within 2–5 years postexposure and can result in death due to hypoxia.5–7 Chronic silicosis occurs with prolonged exposure to silica dust and becomes clinically apparent 20 years or more postexposure.5–7 The National Institute for Occupational Safety and Health has issued a recommended exposure limit (REL) of 50 µg/m3 for up to 10 hours/day during a 40-hour work week, and the Occupational Safety and Health Administration recommends a permissible exposure of 100 µg/m3 for an 8-hour work exposure. 5 Increasing evidence from epidemiological and laboratory animal studies demonstrates that the inhalation of silica has carcinogenic effects in rats, and tracheal instillation has been shown to be carcinogenic in other animal models.8–12 Based on this evidence, the International Agency for Research on Cancer (IARC) has classified silica as a Group 1 human carcinogen. 13
Although silica is a documented carcinogen, the molecular events mediating cellular responses to silica are not fully elucidated. Previous studies from our laboratory illustrate that freshly fractured silica (FFSi) causes the upregulation of activator protein-1 (AP-1). The in vitro and in vivo mechanisms for this activation are potentially mediated through extracellular signal-regulated kinases (ERKs) and p38 mitogen-activated protein kinase (MAPK) pathways.2,8 AP-1 is the transcription factor, consisting of either a Jun-Jun or Jun-Fos heterodimer that interacts with regulatory DNA sequences known as TPA (12-O-tetradecanoyl-phorbol-13-acetate) response elements and is associated with neoplastic transformation and malignancy.
13
Furthermore, it has been shown that the activation of AP-1 by crystalline silica is due, in part, to the involvement of reactive oxygen species (ROS).
14
In particular, hydrogen peroxide (H2O2) plays a significant role in this activation as demonstrated by a reduction in P-p38, P-ERK, and AP-1 in JB6 cells pretreated with N-acetyl-
Programmed cell death 4 (PDCD4) was originally isolated from a human glioma library and is homologous with the mouse PDCD4 (MA-3/TIS/A7-1) gene. 16 PDCD4 is a tumor suppressor gene that acts as a neoplastic transformation inhibitor in JB6 cells. 17 When activated, PDCD4 can inhibit the activation of AP-1-dependent transcription, skin tumorigenesis, and tumor progression in transgenic mouse models. Furthermore, the overexpression of PDCD4 results in subsequent inhibition of TPA-induced transformation on JB6 cells and tumor phenotype of transformed cells.18,19 Knockout of the PDCD4 gene in mouse models has shown an increase in the incidence of lymphomas with an increase in metastasis as well as DMBA/TPA-induced skin papilloma formation.20,21 Collectively, the results from these studies suggest that PDCD4 suppresses carcinogenesis at both the promotion and progression stages.
PDCD4 is recognized as a major and functionally significant target of microRNA 21 (miR-21). MicroRNAs (miRNA) are small, noncoding RNAs approximately 19–25 nucleotides in length. These miRNAs can bind to specific sites on the 3′ untranslated regions of target mRNAs and cause translational repression, leading to negative regulation of the target gene. Earlier studies in colon and breast cancer models demonstrated that posttranscriptional downregulation of PDCD4 by miR-21 resulted in stimulation of invasion and metastasis of these cancers.22,23 Given this observed mechanism, PDCD4 and miR-21 are emerging as potential targets for novel cancer therapies or prevention. Alterations of miRNA expression have been reported following exposure to various carcinogens. Previously, our laboratory reported that tungsten carbide–cobalt nanoparticles induced miRNA-21 expression while inhibiting PDCD4 production in JB6 cells; the mechanism by which this occurs could be mediated by the postexposure production of ROS and upregulation of MAPK pathways involving ERK.24,25
To date, silica is classified as a Group 1 carcinogen by IARC. 13 The mechanisms of involvement in silica-induced carcinogenesis pathways are not fully understood. In this study, we examined the alterations of miR-21-PDCD4 signaling in JB6 cells after exposure to FFSi particles. We demonstrate that PDCD4 signaling is repressed, and there is significantly increased miRNA-21 expression in silica-treated JB6 cells. Furthermore, the inhibition of ROS, ERK, and p38 pathways resulted in a reversal of PDCD4 repression, suggesting that this mechanism involves MAPK and ROS mediation. Last, we establish that JB6 cells and BEAS-2B human lung cells transform under chronic exposure conditions in the soft agar assay, which is indicative of carcinogenesis.
Materials and Methods
Reagents
Eagle’s minimal essential medium (EMEM) and phosphate-buffered saline were purchased from Whittaker Biosciences (Walkersville, Maryland). Fetal bovine serum (FBS), penicillin, streptomycin, and
Cell culture
Mouse epidermal JB6 P+ cells stably transfected with AP-1 (American Type Cell Culture) were maintained in 5% FBS EMEM containing
Preparation of FFSi
Crystalline Colorado silica was obtained from the Generic Center, Pennsylvania State University (State College, Pennsylvania). We used a previously described method for the preparation of FFSi.2,8,26 Briefly, crystalline silica (0.2–10 mm in diameter) was ground for 30 minutes using a ball grinder equipped with agate mortar and balls. The ground silica was further sieved through a 10-µm mesh filter for 20 minutes before use. Silica treatments were applied to the cells within a media suspension 1 hour after the preparation for each experiment.
Anchorage-independent transformation assay
The effect of FFSi on cell anchorage-independent transformation was evaluated in JB6-AP-1 and BEAS-2B cell lines using the previously described soft agar assay.28,29 The cells were seeded in six-well plates with or without TPA (20 ng/mL) or FFSi (0.025 or 0.1 µg/cm2) with six replicates per treatment per cell line. Cells were continuously exposed to FFSi at subcytotoxic concentrations and passaged weekly. Soft agar assay was performed monthly to determine cellular transformation. Cells (104) were resuspended in 1 mL of incubation medium and 0.33% Bacto agar containing 15% FBS and placed over 0.5% agar medium containing 15% FBS. As previously described by Magaye et al., 27 after one week, cells were fed a combination of enriched cell media and agar. The cultures were maintained in a humidified 37°C incubator with 5% CO2 for at least 2 weeks then analyzed using an Olympus 1X 70 inverted microscope with Simple PCI Software (Compix Imaging System). Colonies were quantified using Software Image Pro Plus (Media Cybernetics). Results are shown after 28 weeks of exposure for BEAS-2B cells and 45 weeks of exposure for JB6-AP-1 cells.
Western blot analysis
JB6 cells were plated into six-well plates with treatments repeated in duplicate on each plate. Cells were grown for 24 hours and then starved overnight in 0.1% FBS EMEM. Cells were treated with or without various concentrations of FFSi particles (0, 50, 100, 200, or 300 µg/cm2) for 0.5–4 hours. After treatment, cells were extracted with 1X SDS sample buffer. Concentration of protein was determined using the Pierce Bovine Serum Albumin protein assay (Life Technologies, Carlsbad, California).2,8,26 Equal amounts of protein were loaded and separated using Novex 10% Tris-glycine gels (Life Technologies). Immunoblots were analyzed for the expression of target proteins. Phosphorylated and nonphosphorylated proteins were assessed using the same transferred blot, and equal loading of proteins was ensured by measuring the housekeeping protein, β-tubulin. Experiments were performed in triplicate.
Quantitative real-time polymerase chain reaction
JB6 cells were plated into six-well plates with treatments repeated in duplicate on each plate. Cells were grown for 24 hours and then starved overnight in 0.1% FBS EMEM. Cells were then treated with or without various concentrations of FFSi particles (0, 100, 200, or 300 µg/cm2) for 1, 2, 3, 4, or 24 hours. TPA (20 ng/mL) was added to one cell well in the absence of particle to serve as a positive control. Total RNA was isolated and purified using the mirVana miRNA Isolation kit (Life Technologies) according to the manufacturer’s protocol. Concentration and purity of the RNA were evaluated using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technology Inc., Wilmington, Delaware). Relative miR-21 expression was determined by TaqMan real-time polymerase chain reaction (PCR). Reverse transcription reactions were performed using the following conditions: 16°C for 30 minutes, 42°C for 30 minutes, 85°C for 5 minutes, then hold at 4°C. The complementary DNA (cDNA) products were then placed into the PCR reaction with TaqMan primers. The PCR reaction conditions were as follows: 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 60°C for 60 seconds. Values obtained from untreated cells were analyzed for fold change and set to 1 using the Pfaffl equations for normalization. U6 was selected as the housekeeping gene. Experiments were performed in triplicate.
Effect of antioxidant inhibitors and ERK or p38 inhibitors
JB6 cells were pretreated with ERK inhibitor U0126 (20 µM), p38 inhibitor SB203580 (20 µM), CAT (10,000 U/mL), SOD (1000 U/mL), miRNA-21 inhibitor, NAC (10 mM), deferoxamine (2 mM), sodium formate (1 mM), or polyvinylpyridine-N-oxide (PVPNO, 50 µg/mL) for 1 hour before removal of medium and exposure to FFSi (200 µg/cm2) prepared in fresh medium (0.1% FBS supplemented EMEM). Treatments were performed in duplicate. PDCD4 was analyzed via western blot, and miR-21 expression was assayed using quantitative real-time PCR (qRT-PCR) as described earlier. Experiments were performed in triplicate.
Statistical analysis
Data were analyzed using analysis of variance and Student’s t-test in JMP Software (SAS Institute) and are presented as the means ± the standard error of the mean (SEM) of n experiments, as denoted in figure captions. Significance was set at p ≤ 0.05 for all experiments.
Results
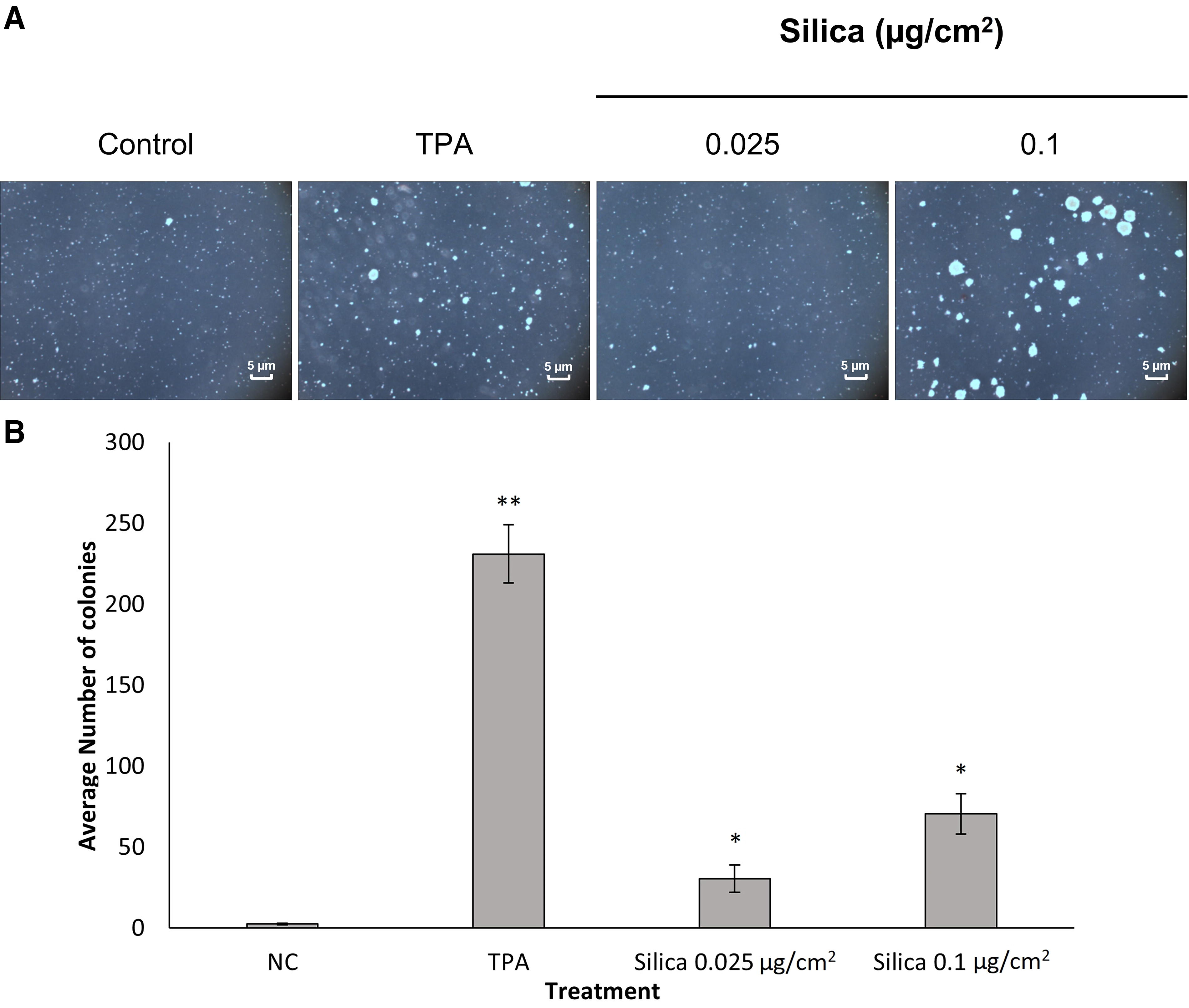
Silica exposure results in neoplastic transformation in soft agar
Previous studies8,28 showed that FFSi induces AP-1 activation through ERK and p38 MAPK pathways that are mediated through ROS production. Because of the important roles of AP-1-MAPKs signaling in tumor promotor-induced neoplastic transformation, we examined whether FFSi particles induce cell transformation. These studies were conducted on JB6 and BEAS-2B cell models using soft agar assays. Low doses for cells in this study were formulated to equate to the REL (50 µg/m3 of air in an 8-hour work shift). Briefly, cells were treated with or without low-dose (0.025–0.1 µg/cm2) FFSi particles for 6 months, then subjected to soft agar analysis. Cell colonies were scored by a computerized image analyzer and assessed based on previously published methods. 28 JB6 cells showed a significant increase in colony formation in cells treated with FFSi when compared to the negative control (Fig. 1). There was also a significant increase in colony formation of cells treated with 0.1 µg/cm2 when compared to cells treated with 0.025 µg/cm2. Similarly, BEAS-2B cells exhibited significant increases in colony formation in cells treated with FFSi compared to the negative control and in cells treated with 0.1 µg/cm2 compared to cells treated with 0.025 µg/cm2 (Fig. 2). Collectively, these results indicate that low-dose FFSi induces transformation in chronically exposed cells.

Freshly fractured silica (FFSi) induces neoplastic-like transformation of JB6 cells. The effect of FFSi on cell anchorage-independent transformation was evaluated in the JB6 cell line.
Freshly fractured silica induces neoplastic-like transformation of BEAS-2B cells. The effect of FFSi on cell anchorage-independent transformation was evaluated in the BEAS-2B cell line.
PDCD4 expression is inhibited by FFSi
PDCD4 is involved in AP-1-dependent transcription. 29 We hypothesized that PDCD4 is involved in the silica-induced cellular response. To verify this, JB6 cells were exposed to various concentrations of silica, and the PDCD4 expression was measured. The results indicate that silica caused a dose-dependent decrease in PDCD4 expression and a dose-dependent increase in phosphorylated ERKs and p38 (Fig. 3). At the concentration range of 50–300 µg/cm2, silica induced a significant decrease in PDCD4 expression compared to the untreated control.

Effects of freshly fractured silica on PDCD4, phospho-ERKs, and phospho-p38 expression in JB6 cells. JB6 cells were cultured in EMEM containing 5% FBS under standard culture conditions until 80% confluent, then cultured in 0.1% FBS overnight. Cells were then exposed to various concentrations of FFSi for 2 hours and then lysed. PDCD4, phospho-ERK, phospho-p38, and β-tubulin were analyzed using the same transferred membrane blot.
A time-course study revealed that inhibition of PDCD4 was first observed after 0.5 hours of incubation (200 µg/cm2). Thereafter, the expression of PDCD4 decreased to a maximum of 1 hour and lasted until 4 hours. Furthermore, incubations of cells (12 hours) with silica resulted in a poor response (Fig. 4A and B). Phosphorylation of ERKs increased in a time-dependent manner as expected (Fig. 4A and C). These results suggest that FFSi inhibited PDCD4 expression and activated ERKs in JB6 cells.

Time-course study of freshly fractured silica on PDCD4 and phospho-p38 expression in JB6 cells. JB6 cells were cultured in EMEM containing 5% FBS under standard culture conditions until 80% confluent, then cultured in 0.1% FBS overnight. Cells were then exposed to FFSi from 0.5 to 4 hours and then lysed. PDCD4, phospho-p38, and β-tubulin were analyzed using the same transferred membrane blot.
Effect of FFSi on miR-21 expression
PDCD4 is known to be a major, functionally significant, downstream and upstream regulator of miR-21. MicroRNA 21 expression was measured in JB6 cells after exposure to FFSi using real-time qRT-PCR. We proposed that the expression of miR-21 would be elevated following exposure to FFSi. To test this hypothesis, the cells were exposed to FFSi (100–300 µg/cm2) for 1, 2, 3, 4, and 24 hours (Fig. 5). Total RNA was purified from the respective cell pellets and analyzed by qRT-PCR for the expression of miR-21. Values obtained from the untreated cells (NC) were set to 1. TPA was utilized as the positive control. The expression of miR-21 increased significantly from 1 to 4 hours, as expected. Cells exposed to 100 µg/cm2 FFSi particles resulted in a significant increase in miR-21 expression at 1, 3, and 4 hours posttreatment as compared to the negative control. Cells exposed to 200–300 µg/cm2 FFSi demonstrated significant elevations in miR-21 expression 2, 3, and 4 hours posttreatment. There were no changes between treatments associated with dose after 24 hours postexposure.

miR-21 expression in JB6 cells postexposure to FFSi. JB6 cells were exposed to various concentrations of FFSi from 0.5 to 4 hours. Total RNA was purified. Real-time RT-PCR was utilized to analyze gene expression using the TaqMAN microRNA reverse transcription kit, FAST TaqMan Universal Master Mix, TaqMan probe, and the primers for miR-21 and housekeeping gene U6 (Applied Biosystems). Gene expression was analyzed using normalization to U6. The results are mean ± SEM, a representative of three experiments, and (*) p ≤ 0.05 versus untreated cells. miR-21, microRNA 21; RT-PCR, real-time PCR.
Effects of antioxidant reagents and MAPK inhibitors on FFSi-induced alteration in PDCD4 and miR-21
Previously, our laboratory reported that ROS play a critical role in MAPK and AP-1 activation after exposure to FFSi.8,28 To examine the role of ROS on silica-induced PDCD4 miR-21 alteration, the effects of antioxidants and MAPKs inhibitors on silica-induced PDCD4 inhibition and phosphorylation of MAPKs were investigated. JB6 cells (5 × 104) cultured in 24-well plates were pretreated for 1 hour with antioxidants or inhibitors: CAT (10,000 U/mL), SOD (500 U/mL), NAC (10 mM), deferoxamine (1 mM), sodium formate (2 mM), PVPNO (50 µg/mL), ERK inhibitor U1026 (20 µM), p38 inhibitor SB203580 (20 µM), or miR-21 inhibitor (20 µM). Then, the cells were exposed to 200 µg/cm2 FFSi in the presence of the same reagents for 2 hours, and the expression of PDCD4, p-38, and ERKs was measured using western blot. The effects of antioxidants, p38, and ERKs inhibitors on silica-induced PDCD4, P-p38, and P-ERKs are shown in Figure 6 and Figure 7.
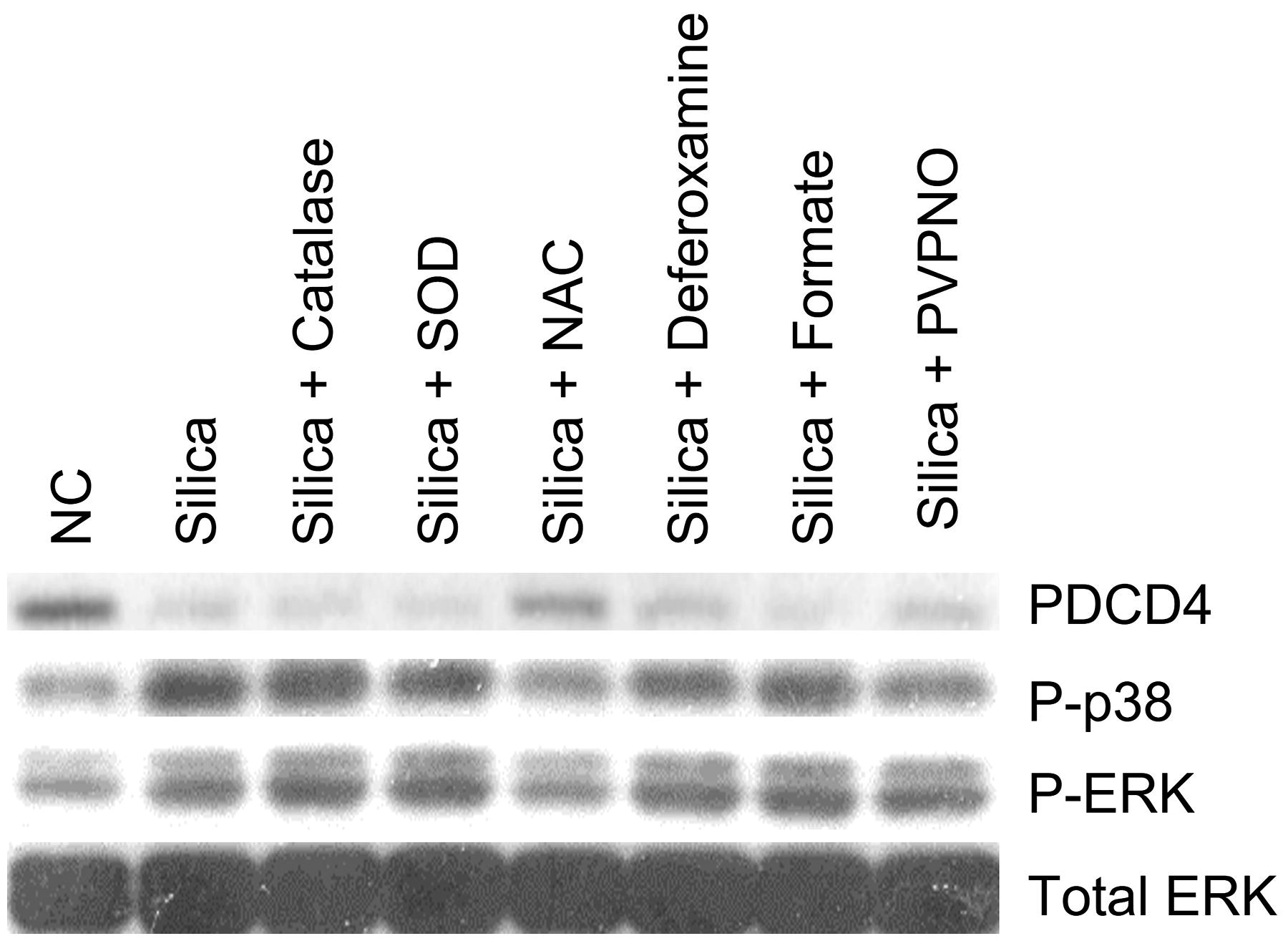
Effect of ROS scavengers on PDCD4, phospho-p38, and phospho-ERK in JB6 cells treated with FFSi. Cells were pretreated with ROS scavenger catalase (CAT, 10,000 U/mL), NAC (10 mM), SOD (500 U/mL), deferoxamine (1 mM), sodium formate (2 mM), or PVPNO (50 µg/mL) for 1 hour, then exposed to FFSi particles (200 µg/cm2) prepared in fresh medium (0.1% FBS supplemented EMEM). The expression of target proteins was analyzed using the same transferred membrane blot. NAC, N-acetyl-

Effect of inhibitors on PDCD4, phospho-p38, and phospho-ERK in JB6 cells treated with FFSi. Cells were pretreated with ERK inhibitor U1026 (20 µM), p38 inhibitor SB203580 (20 µM), or miR-21 inhibitor (20 µM). Following this pretreatment, cells were exposed to FFSi particles (200 µg/cm2) for 2 hours. Expression of target proteins was analyzed using the same transferred membrane blot.
Interestingly, the results show that cells pretreated with NAC before exposure to FFSi demonstrate the increased expression of PDCD4, suggesting a potential reversal of the effects of silica (Fig. 6). Pretreatment of the cells with SOD, PVPNO, sodium formate, CAT, or deferoxamine resulted in inhibition of PDCD4 similar to the effects of the positive control. NAC is a thiol-containing antioxidant that can reduce H2O2, a ROS intermediate, thereby protecting against the toxic effects of H2O2. These results demonstrate further that ROS, specifically H2O2, may be involved in the inhibition of PDCD4 following exposure to FFSi particles in JB6 cells. Results also demonstrate that PDCD4 expression increased in cells pretreated with ERK, p38, and miR-21 inhibitors (Fig. 7). Phosphorylated p38 expression was inhibited by U0126, SB203580, and the miR-21 inhibitor (Fig. 7). Likewise, phosphorylated ERK expression in JB6 cells was inhibited by U0126 and SB203580 pretreatment and reduced by pretreatment with the miR-21 inhibitor (Fig. 7).
The expression of miR-21 was measured using qRT-PCR. Previous findings from our laboratory demonstrate that tungsten carbide–cobalt (WC-Co) nanoparticles induced miR-21 gene expression while inhibiting PDCD4 expression. 25 In this study, miR-21 gene expression was not altered in any of the cells pretreated with antioxidants as compared to the negative control. JB6 cells pretreated with the ERK inhibitor U0126 showed a significant decrease in miR-21 expression compared to the positive control (Fig. 8). Considering that PDCD4 expression increased in the presence of NAC and inhibitors but miR-21 expression was unaltered, we suggest that alternative pathways may be governing this response to treatment with FFSi particles following pretreatment with antioxidants. A numerical decrease in miR-21 gene expression in cells treated with U0126 suggests a potential involvement of ERK signaling.
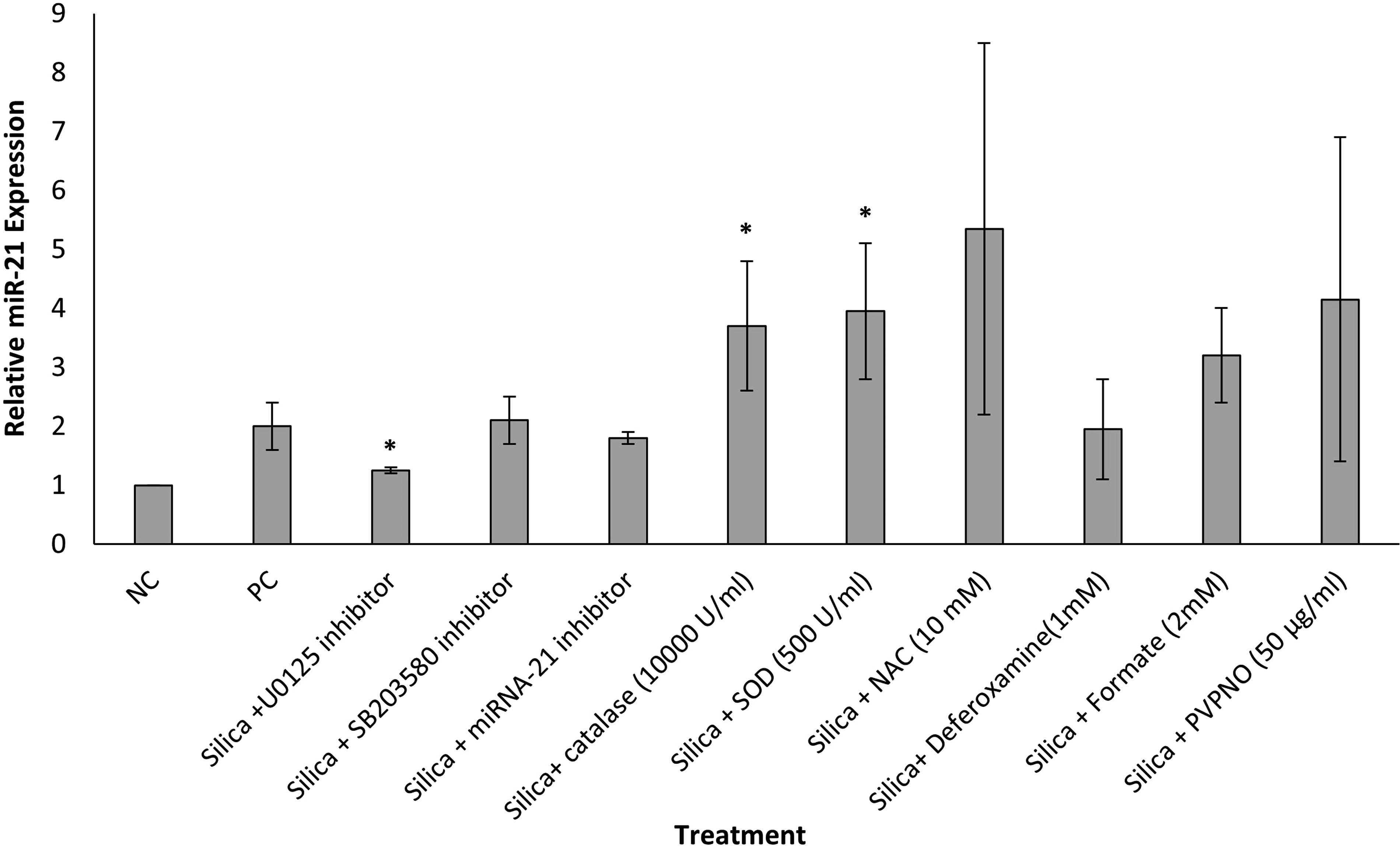
miR-21 expression in JB6 cells pretreated with ROS scavengers or inhibitors. Cells were pretreated with ROS scavengers CAT (10,000 U/mL), NAC (10 mM), SOD (500 U/mL), deferoxamine (1 mM), sodium formate (2 mM), PVPNO (50 µg/mL), ERK inhibitor U1026 (20 µM), p38 inhibitor SB203580 (20 µM), or miR-21 inhibitor (20 µM) for 1 hour, then exposed to FFSi particles (200 µg/cm2) prepared in fresh medium (0.1% FBS supplemented EMEM). Total RNA was purified. Real-time RT-PCR was utilized to analyze gene expression using the TaqMAN microRNA reverse transcription kit, FAST TaqMan Universal Master Mix, TaqMan probe, and the primers for miR-21 and housekeeping gene U6 (Applied Biosystems). Gene expression was analyzed using normalization to U6. The results are mean ± SEM, a representative of three experiments, and (*) p ≤ 0.05 versus positive control cells. CAT, catalase; NAC, N-acetyl-
Discussion
Occupational exposure to crystalline silica has been associated with the development of acute or chronic silicosis and cancer.5,28 Crystalline silica that is administered to rats via an intrapleural route was shown to induce localized malignant histiocytic lymphomas. 30 Administration of silica particles by intratracheal instillation or inhalation has been carcinogenic in rat lung tissues.9,10,31 Studies have also demonstrated that silica causes direct DNA damage and transformation of mammalian cells. 32 Many molecular mechanisms by which carcinogenesis is induced following exposure to crystalline silica particles are not fully understood. The present study examined the theory that the activation of transcription factors following crystalline particle exposure is a primary event that initiates a signal transduction cascade at the cellular level which may result in neoplastic transformation of the cells.
In this study, JB6 cells were exposed for 28 weeks to freshly fractured crystalline silica and formed colonies in soft agar in a dose-dependent fashion. Similar results were obtained for BEAS-2B cells exposed to FFSi for 45 weeks. We demonstrate that exposure of JB6 cells to FFSi resulted in an increase of miR-21 expression at 1, 2, 3, and 4 hours postexposure and caused a subsequent timewise decrease from 0.5 to 4 hours postexposure in expression of its downstream target, PDCD4. PDCD4 decreased in a dose-dependent manner following exposure to FFSi (50–300 µg/cm2). Furthermore, MAPKs p38 and ERK demonstrated increases in expression following exposure to FFSi (200–300 µg/cm2) and in a timewise fashion from 2 to 4 hours postexposure. Pretreatment of cells with NAC, a thiol-containing antioxidant, resulted in an increase in PDCD4 expression after treatment with FFSi. Taken together, these data suggest that miR-21-PDCD4 signaling and MAPK activation induced by ROS production following exposure to FFSi may play a role in cellular responses that can result in neoplastic-like transformation.
Conceptually, carcinogenesis occurs in three steps: initiation, promotion, and progression. The soft agar anchorage-independent transformation assay is a well-documented assessment of neoplastic transformation in cellular models.33–36 Previous studies involving chronic carcinogen exposure suggest that key signaling alterations leading to malignant transformation occur in a period of 4–7 months and are evidenced by colony formation in soft agar.37,38 In this study, JB6 and BEAS-2B cells exposed to FFSi at low doses for a chronic period (28 and 45 weeks, respectively) in an anchorage-independent soft agar assay showed increased colony formation in a dose-wise manner as compared to the negative control. Furthermore, the cells exposed to 0.1 µg/cm2 had a significantly higher colony count as compared to the lower dose in both cell types. In conclusion, the transformation of cells confirms the carcinogenic effects of low-dose FFSi.
Studies in various model systems have suggested an important role of AP-1 and MAPKs in preneoplastic to neoplastic transformation events in cellular as well as animal models.13,39–41 AP-1 is a transcription factor that interacts with regulatory DNA sequences in the TPA response element. Previous studies have shown that freshly fractured crystalline silica induces AP-1 through ERK and p38 MAPK. 8 It is known that AP-1 and MAPK signal transduction pathways are crucial to cell transformation and tumor promotion. 41 It is also known that stimulation with UVB and WC-Co nanoparticles activates the phosphorylation of ERK and p38, which are signaling pathways required for the activation of AP-1 JB-6 mouse cell lines.2,24,25 Furthermore, it was reported that the ERK pathways, but not p38 pathways, may be responsible for the mechanism of AP-1 activation and consequent carcinogenic properties following exposure to WC-Co nanoparticles. 25 The results from this study found that the inhibition of ERK with U0126 and p38 with SB203580 reversed the inhibitory action of FFSi suggesting that MAPK pathways may mediate PDCD4 suppression in JB6 cells.
Previous studies indicate that ROS production following exposure to crystalline silica is known to induce MAPKs and AP-1. 2 ROS are associated with all three stages of the carcinogenesis model and a reduction in antioxidant defense mechanisms. 42 Oxidative stress occurs when there is an imbalance in ROS production favoring the oxidants. Reducing oxidative stress is correlated with the suppression of tumor cell proliferation. 43 In this study, JB6 cells were pretreated with known antioxidants to determine if any of these compounds would reverse the suppression of PDCD4 following exposure to FFSi. JB6 cells pretreated with CAT and SOD did not demonstrate recovery of PDCD4 expression following treatment with FFSi. However, the effect of FFSi exposure was ameliorated in JB6 cells pretreated with NAC, showing recovery of PDCD4 expression. Studies suggest that H2O2 can mediate gene regulation and cellular injury response through PDCD4 and AP-1 pathways in vascular and cardiac tissue.44,45 Findings from these studies similarly suggest that ROS, particularly H2O2, may play a role in FFSi-induced MAPK expression.
PDCD4 plays an important role in antitumor promotion. 18 Yang et al. 19 demonstrated that PDCD4 can inhibit AP-1-dependent transcription. PDCD4 is a well-documented tumor suppressor that inhibits neoplastic transformation, tumor progression, and translation.46,47 It was first discovered to inhibit tumor promoter–induced neoplastic transformation in the JB6 murine epidermal model of neoplastic transformation. 48 PDCD4 stalls the translational machinery, decreases benign and malignant tumor progression, and controls lymphoma initiation and autoimmune inflammation.22,49,50 The expression of PDCD4 increases during apoptosis46,47 and decreases during human and mouse carcinogenesis.22,29,51 The overexpression of PDCD4 inhibits tumorigenesis and tumor progression in a transgenic mouse model and inhibits tumor cell invasion in vitro.17,46,47,49,51 PDCD4-null mice show spontaneous development of lymphomas 16 as well as enhanced sensitivity to carcinogen-induced skin cancer. 29 In this study, we observed decreased PDCD4 expression postexposure to FFSi in JB6 cells over time and in a dose-dependent manner. Like WC-Co nanoparticle exposures, 25 carcinogenesis associated with FFSi exposure may be due, in part, to the downregulation of PDCD4 expression and function.
PDCD4 has emerged as a major, functionally significant target of miR-21.16,47,52 The observed inverse correlation between PDCD4 and miR-21 expression may be a useful prognostic marker.18,47 MicroRNA-21 was first identified as an antiapoptotic factor in glioblastoma. 53 Increased miR-21 expression is observed during carcinogenesis, in tumors, and in response to chemotherapy. 54 Causal links have been established between miR-21 expression and cellular proliferation, migration, apoptosis, tumor growth, invasion, and metastasis.55,56 In recent years, multiple studies have focused on the biological functions of miR-21 and shown that it is significantly upregulated in many human malignancies, including breast cancer, glioblastoma, hepatocellular carcinoma, cholangiocarcinoma, lung cancer, tongue squamous cell carcinoma, esophageal cancer, stomach cancer, colorectal cancer, chronic myelogenous leukemia, cervical cancer, and prostate cancer.54–60 Previously, our laboratory demonstrated that JB6 cells exposed to WC-Co and UVB had an increased expression of miR-21 over time.24,25 In the present study, miR-21 expression was increased by FFSi in a timewise manner from 1 to 4 hours postexposure and in a dose-wise manner at 1, 3, and 4 hours. Additionally, pretreatment of cells with ERK inhibitor U0126 decreased miR-21 expression and increased PDCD4 expression, suggesting the involvement of MAPK signaling pathways in the overall mechanism governing carcinogenesis following FFSi exposure. However, pretreatment with antioxidant compounds did not suppress miR-21 expression, suggesting that, taken together with recovery of PDCD4 expression, other regulatory pathways involving miR-21 may be at work. These results suggest that FFSi does, in fact, stimulate proto-oncogene miR-21, which may play a role in silica-induced carcinogenesis.
In conclusion, the results obtained herein demonstrate that FFSi particles induce the transformation of JB6 mouse epithelial and BEAS-2B human lung cell lines. FFSi inhibits expression of PDCD4, a tumor suppressor gene, and induces miR-21, a proto-oncogene through MAPK signaling pathways and ROS production, particularly H2O2 (Fig. 9). This study provides important insight into the mechanisms governing silica-induced carcinogenesis. Elucidation of these mechanisms may provide future insight into understanding and potential prevention of silica-induced carcinogenesis.

Schematic of proposed mechanism of events in silica-induced carcinogenesis.
Footnotes
Authors’ Contributions
J.A.: Writing—review and editing (lead), visualization (lead), investigation (equal), formal analysis (equal), and writing—original draft (supporting). K.R.: Writing—review and editing (equal), visualization (supporting), and formal analysis (supporting). T.M.: Investigation (equal), writing—review and editing (supporting), data curation resources (supporting), and formal analysis (supporting). J.R.: Writing—review and editing (equal), validation (supporting), supervision (supporting), and formal analysis (supporting). T.B.: Conceptualization (lead), writing—original draft (lead), investigation (equal), and formal analysis (equal).
Author Disclosure Statement
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the
Funding Information
Funding for this project was provided by the Nanotechnology Research Center (NTRC) of the