
Abstract
In the last three decades, extensive research on human immunodeficiency virus (HIV) has highlighted its capability to exploit a variety of strategies to enter and infect immune cells. Although CD4+ T cells are well known as the major HIV target, with infection occurring through the canonical combination of the cluster of differentiation 4 (CD4) receptor and either the C-C chemokine receptor type 5 (CCR5) or C-X-C chemokine receptor type 4 (CXCR4) coreceptors, HIV has also been found to enter other important immune cell types such as macrophages, dendritic cells, Langerhans cells, B cells, and granulocytes. Interestingly, the expression of distinct cellular cofactors partially regulates the rate in which HIV infects each distinct cell type. Furthermore, HIV can benefit from the acquisition of new proteins incorporated into its envelope during budding events. While several publications have investigated details of how HIV manipulates particular cell types or subtypes, an up-to-date comprehensive review on HIV tropism for different immune cells is lacking. Therefore, this review is meant to focus on the different receptors, coreceptors, and cofactors that HIV exploits to enter particular immune cells. Additionally, prophylactic approaches that have targeted particular molecules associated with HIV entry and infection of different immune cells will be discussed. Unveiling the underlying cellular receptors and cofactors that lead to HIV preference for specific immune cell populations is crucial in identifying novel preventative/therapeutic targets for comprehensive strategies to eliminate viral infection.
Introduction
H
Of the two distinct HIV genotypes, HIV-2 is less pathogenic and predominantly found in western Africa, whereas HIV-1 accounts for 95% of HIV infections worldwide and is more virulent and treatment resistant. 8,9 As such, HIV infection generally refers to HIV-1, which will be the focus of this review.
While work on primates has provided valuable information, 10 in vitro models using infectious HIV in human cell cultures remain highly relevant for important treatment discoveries and characterizing the multiple mechanisms used by HIV to enter cells. 11 Moreover, a large number of AIDS trials have been based on research that investigated the efficacy of drugs shown to inhibit HIV binding and entry into target cells in vitro. 12,13 While this prior work has often been generalized, the specific molecules that HIV utilizes to infect different and pathologically relevant immune cell types, that is, viral tropism, is less understood, which is pertinent for the development of novel HIV therapeutic approaches.
The knowledge gained from studying HIV tropism provides researchers with unique targets to inhibit infection of specific immune cell subsets and target different stages of disease progression, and thus increase the chances of finding useful combinations of antiviral compounds. 14,15 Moreover, as resistance to current HIV-1 therapies continues to emerge, 16 inhibitors of HIV-1 attachment/entry provide a different mechanism of action than those of the current standards of care, and are potentially of great value in populations where drug resistance is more prevalent. Furthermore, during the pathogenesis of HIV infection, the virus evolves, in part, due to genetic drift of neutral mutations followed by brief episodes of natural selection during the infectious process, changing with it the cell subtype preference of the virus, which is dictated by coreceptors expressed by target cells. 17
A complete understanding of the different molecules that HIV uses to enter particular immune cells is crucial to develop effective strategies to inhibit the virus at precise stages of infection. Hence, while it is easier to focus on a single cell type or subtype for new HIV prevention strategies, an overview of HIV receptor usage on different target cells merits discussion as effective strategies will need to focus on multiple targets.
The purpose of this review is to summarize the latest information on HIV receptor usage based on in vitro studies of different human immune cells, as this clarifies not only the tropism of HIV for certain target cells but also how this may correlate with cell-to-cell transmission of HIV, and thus, how HIV spreads to different anatomical sites throughout the human body. In addition, drugs and other experimental compounds that have been used to target different HIV receptors, coreceptors, and cofactors will be discussed and are summarized in Table 1. This review will be presented in four sections based on the following HIV target cells: (1) CD4+ T cells, (2) macrophages (MΦs), (3) professional antigen-presenting cells (APCs), and (4) other immune cells. Each section will focus on primary receptor(s), coreceptor(s), and other cellular cofactors/components that are exploited by HIV to efficiently enter the aforementioned target cells, as well as discuss prophylactics/therapeutics that have been developed to target several of these mechanisms in an effort to halt infection.
CCR5, C-C chemokine receptor type 5; CD4, cluster of differentiation 4; CXCR4, C-X-C chemokine receptor type 4.
CD4+ T Cells
As the major targets for HIV, T helper cells (CD4+ T cells, Fig. 1) are the most recognized and studied immune cells in HIV research. Depletion of these important immune cells is the hallmark of HIV infection 18 and contributes to the symptomatic manifestation that characterizes AIDS 19 ; however, direct versus indirect reductions of these T cells within HIV-infected individuals remain contentious. 20,21 Nevertheless, the loss of these T cells results in compromised immunity, leading to opportunistic infections as well as cancers arising in HIV-infected individuals. These conditions include pneumonia caused by uncontrolled growth of a common airborne fungus, Kaposi's sarcoma arising from unchecked infection of Kaposi's sarcoma-associated herpesvirus, tuberculosis, and several other secondary diseases involving the nervous system and skin. 22
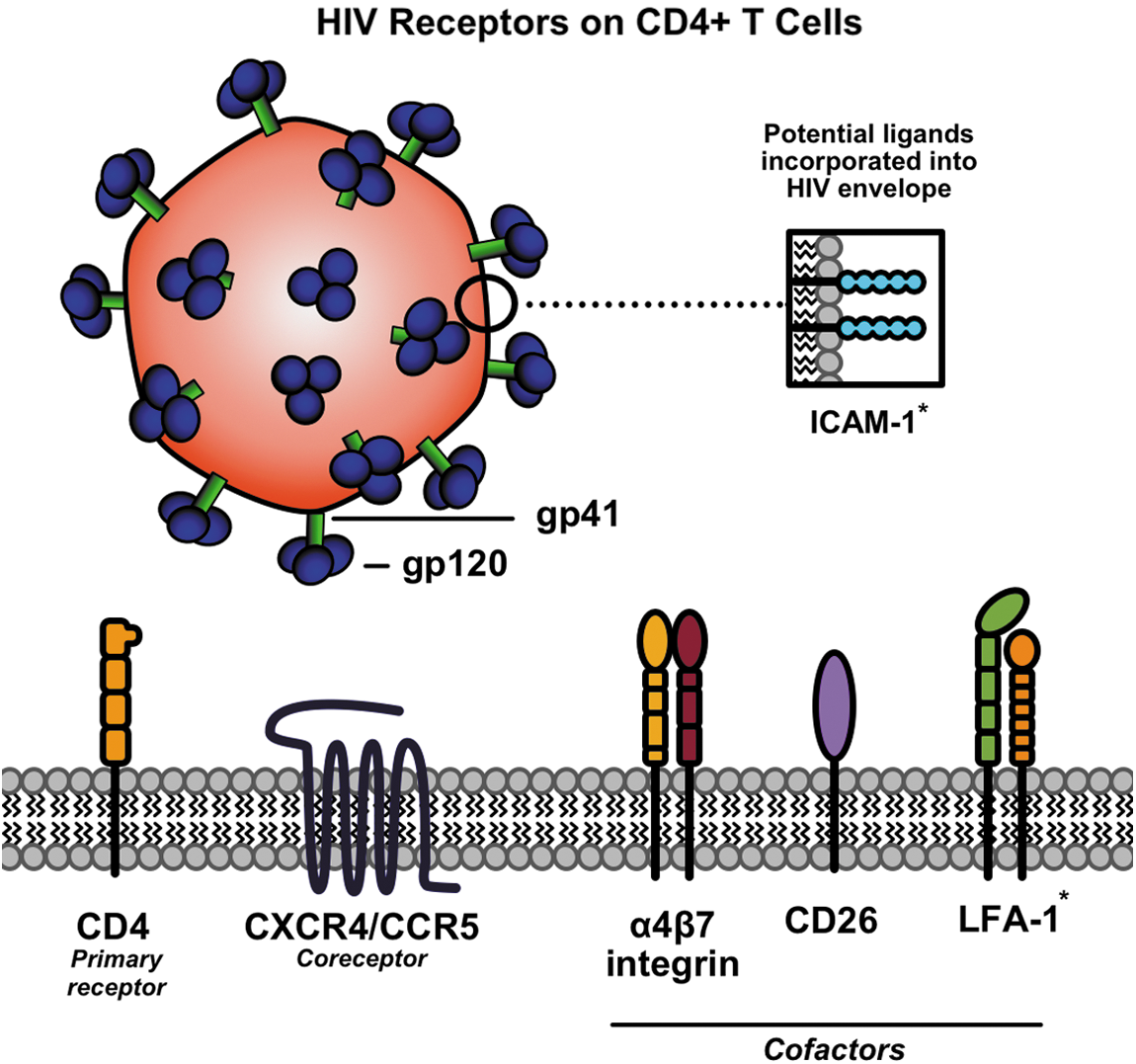
Summary of receptors used by HIV to enter CD4+ T cells. CD4 is the primary receptor for gp120, while either CXCR4 or CCR5 can act as coreceptors. On attachment to these moieties, the gp41 fusion protein is exposed and facilitates viral membrane fusion. CD26 and α4β7 have been shown to aid in catalyzing these interactions as cofactors. When ICAM-1 is incorporated into the viral envelope during budding events, it can interact with its receptor LFA-1 to enhance infectivity (interaction indicated by *). CCR5, C-C chemokine receptor type 5; CD4, cluster of differentiation 4; CXCR4, C-X-C chemokine receptor type 4; HIV, human immunodeficiency virus; ICAM-1, intercellular adhesion molecule-1; LFA-1, lymphocyte function-associated antigen 1. (Color image is available at
Primary receptor
Cluster of differentiation 4 (CD4) was demonstrated to be the principal receptor of HIV on T helper cells in 1986 when the virus was tentatively named lymphadenopathy-associated virus. 23 In this study, McDougal et al. used radiolabeling techniques in experiments that exposed CD4+ T cells to HIV and found that one of two monoclonal antibodies (mAbs) recognizing different CD4 epitopes was unable to bind HIV-treated cells. Then, through antibody–antigen complex analyses, the authors demonstrated that the CD4 molecule binds to the viral glycoprotein gp120, thus providing evidence that CD4 plays a major role in HIV infection. Other studies utilizing mAbs against CD4 have showed that HIV infection of target CD4+ T cells could effectively be blocked. 24 –26 In accordance with these results, the forced expression of CD4 through gene transfection into CD4− human cell lines conferred susceptibility to HIV infection. However, forced CD4 expression in other mammalian cells lines, such as those from mice, yielded nonproductive viral infections. 27 These conflicting results led to the conclusion that there were proteins specific to human cell lines, in addition to CD4, responsible for viral infection and propagation, and systems deficient in these factors do not support HIV replication. 28
The first successful crystal structure of a fragment of CD4 in complex with the gp120 core was solved by Kwong et al. in 1999 through X-ray diffraction. 29 This interaction has since become a key target between virus and host to block viral entry. Importantly, there are several antiviral therapies under development that impede the gp120-CD4 interaction, potentially serving as HIV entry inhibitors. For example, TNX-355, a humanized murine anti-CD4 mAb previously referred to as hu5A8, was shown to have high antiviral potency alone, synergized with antibodies targeting gp120 in vitro, and reduced HIV viral load in primates in vivo. 30,31 Furthermore, it has been shown that a single dose of TNX-355 reduced HIV plasma levels and increased T-cell counts in HIV-infected patients. 32 A natural, broadly neutralizing gp120-binding antibody, known as b12, was isolated from HIV-infected subjects, and a growing class of such antibodies is now available. 33,34 Experiments with b12 show reduced HIV binding to target cells; however, the emergence of HIV-resistant variants has limited its success. 35
Beyond antibodies, HIV attachment/entry inhibitors that interfere with gp120-CD4 interactions or stabilize conformations of gp120 that do not associate with CD4 have become an attractive strategy to prevent HIV infection. 36,37 Early candidates included soluble CD4 (sCD4) mimetics, although their efficacy has been hindered due to their slow kinetics of HIV-1 inactivation. 38,39 A more potent design that combines sCD4 with a coreceptor peptide mimetic has been shown to protect primates when delivered using adenovirus-associated virus vectors. 40 However, protein-based sCD4s lack oral availability, which is characteristic of small molecules. 41
Along these lines, two main chemotypes of small molecules have predominated in HIV entry inhibitor research, including the N-phenyl-N-piperidin-4-yl-oxalamide (NBD) analogues 42,43 and Bristol-Myers Squibb (BMS) compounds that specifically interfere with the gp120-CD4 interaction through different mechanisms. 44 One inhibitor in the latter group that is currently under development is BMS-663068. 45 BMS-663068 is a prodrug, of which activation inhibits gp120 interactions with CD4 via binding to gp120, and this is the first drug that blocks HIV-receptor interactions to undergo clinical development. 46,47 Although phase III trials are planned for BMS-663068, 47 modifications of its active form (BMS-626529) and new classes of similar compounds are currently under investigation and may allow for lower dose formulations without the need of a prodrug. 48,49
A third class of candidate anti-HIV drugs currently being examined utilizes CD4-specific designed ankyrin repeat protein (DARPin) technology, which promises a potent and stable inhibitory strategy with low production costs and high specificity to CD4 that can outcompete viral gp120 binding. 50,51 Cyclotriazadisulfonamides (CADAs) are yet another type of small molecule that target CD4 rather than envelope (Env) proteins, and work by knocking down CD4 in an mRNA-independent manner 52 ; however, long-term adverse effects on the immune system are of concern. Lignosulfonic acid, a low-cost lignin-derived polyanionic macromolecule, has also shown potency in inhibiting HIV entry by interfering with gp120 binding to the cell surface of CD4+ T cells, although the mechanism of action is not yet fully understood. 53 Most recently, thiolated pyrimidine derivatives were shown to prevent HIV entry by interacting with redox-active thiol groups on both CD4 and gp120 that are paramount for HIV entry in vitro, 54 although more work is required to establish clinical viability of these compounds.
Coreceptor(s)
Due to results that demonstrated that some non-CD4 expressing cells were susceptible to HIV infection, 28,55 scientists began to look for secondary receptors that permitted HIV infection in CD4− cells. Although several laboratories reported the sequence of a coreceptor involved in viral fusion, 56,57 it was not until 1996 when Feng et al. identified a seven-transmembrane G protein-coupled receptor that functioned as a critical coreceptor for T-cell line-tropic HIV infection. 58 The suggested name for this newly identified protein was “fusin” because of its role in HIV viral envelope fusion to T-cell membranes. Studies by Bleul et al. followed shortly thereafter and implicated the same protein, although named it with the more widely used chemokine field nomenclature as the C-X-C chemokine receptor type 4 (CXCR4). 59,60 As such, HIV strains capable of exploiting the CXCR4 coreceptor to efficiently gain entry into T cells have been dubbed X4-tropic, and the natural CXCR4 ligand stromal cell-derived factor α (SDF-α) has been shown to inhibit X4 strains. 59,61
These X4-tropic strains typically emerge during the later stages of AIDS, and a shift toward production of this HIV-subtype provides a measure of disease progression within patients. Consequently, compounds that interfere with the interaction between HIV and the CXCR4 coreceptor have been explored in a therapeutic setting. 62 Unfortunately, targeting CXCR4 has proven to be challenging. For example, a CXCR4 antagonist named plerixafor, or AMD3100, was under development for HIV treatment, but showed limited inhibitory potency in preliminary trials. 63 This result may be explained as X4-tropic viruses are commonly present together with R5-tropic viruses that utilize the C-C chemokine receptor type 5 (CCR5). 64,65
CCR5 is another major HIV coreceptor on T cells, 60 particularly on certain T-cell subsets, including T follicular helper T cells (TFH) 66 and effector memory T cells (TEM). 67 Specifically, CXCR4 is expressed on 88.5% of CD4+ T cells in resting tissues, whereas CCR5 is expressed on 10.4% of cells. 68 Thus, HIV infection inhibition via CCR5 targeting will be discussed in more detail below. Due to the concurrence of viral subtypes that have different cellular tropism, the inhibition of CXCR4 alone may not be sufficient to decrease viral load. 69 In addition, inhibition of CXCR4 also impacts its natural role in maintaining hematopoietic stem cells in their bone marrow niche. 70 While CXCR4 and CCR5 are the primary coreceptors for HIV entry into T cells, other minor coreceptors, such as CXCR6, have also been proposed for the expanding list of T-cell subsets, 71 and variations in the genes that encode for HIV-1 coreceptors and their natural ligands modify viral susceptibility and disease progression. 72
Cofactors and other components
In addition to the importance of the CD4 and the chemokine receptor coreceptors, the enzyme CD26 (also known as dipeptidyl peptidase IV) has been investigated as an HIV cofactor due to its protease activity at a specific motif within a highly conserved portion of the variable loop 3 (V3) of gp120 from different HIV isolates. 73 CD26 is preferentially expressed on memory and helper CD4 subsets and is highly upregulated on T-cell activation. 74,75 It has also been demonstrated that mAbs against CD26 are able to inhibit HIV entry, and coexpression of human CD4 and CD26 in murine NIH 3T3 fibroblasts made them permissive to HIV infection. 76 Follow-up studies supported the role of CD26 in a variety of lymphocytic functions 77 and showed that CD26 expression levels by CD4+ T cells are positively correlated with the rate of HIV infection. 78 Consequently, scientists began to strategize methods to inhibit CD26 as potentially useful treatment options. 79 However, other groups have published conflicting evidence suggesting that high CD26 expression decreases HIV infection of T cells in vitro, 80 and that high expression of CD26 confers HIV resistance in vivo. 81 Therefore, the future of targeting CD26 in therapeutic approaches remains controversial.
Several integrins have also been implicated in HIV infection. Specifically, HIV has been shown to target gut CD4+ T helper cells (Th17 cells), in part, through attachment via α4β7 integrin, 82 –84 although more recently it was suggested that α4β7 increases HIV susceptibility via an attachment-independent mechanism. 85 Another cofactor that is part of the leukocyte integrin family involved in HIV infection of CD4+ T cells is lymphocyte function-associated antigen 1 (LFA-1). 86 In its activated form, LFA-1 has a high binding affinity for its ligand, intercellular adhesion molecule-1 (ICAM-1), which can be incorporated into the HIV viral envelope during the budding process from other immune cells. 87,88 For example, CD4+ T cells deficient in LFA-1 were shown to be less susceptible to HIV-1 transmission from dendritic cells (DCs) that express ICAM-1. 89 Moreover, HIV gp120 was shown to be sufficient for LFA-1 activation, thus promoting the use of this cofactor. 90 Monoclonal and single domain antibodies against the beta subunit of LFA-1 (CD11a) efficiently blocked HIV-1 transmission both in an in vitro and in an in vivo mouse model. 91 Interestingly, a murine mAb against LFA-1 (also known as Cytolin) was shown to inhibit HIV replication by inducing the secretion of a currently unidentified soluble antiviral factor on LFA-1 engagement, providing a novel mechanism of action for targeting this cofactor in antiviral approaches. 92
As an alternative approach to block HIV infection, it may be possible to target viral envelope fusion to the host cell membrane, which occurs after initial binding and exposure of the HIV gp41 fusion peptide required for HIV entry into CD4+ T cells. 93 Therapeutic strategies aimed at preventing viral fusion are appealing, given that such approaches would likely be strain independent. Early work by Ebenbichler et al. found that three cell surface proteins on human T-cell lines in vitro bound recombinant gp41 with high affinity, 94 which may have promoted viral fusion. Thus, inhibitors of viral fusion have long been explored to prevent HIV-1 entry primarily by targeting the N- or C-terminal heptad repeats (NHR and CHR, respectively) of gp41. The most studied of such fusion inhibitors is T20 (or enfuvirtide), a peptide that mimics the CHR sequence of gp41 (aa 643–678) and competitively binds to gp41, thereby blocking the formation of the postfusion structure. While T20 has been approved for use since 2003 and remains the only approved HIV-1 fusion inhibitor, 95 it has been met with drawbacks, including costs of up to $25,000 per year, high dosage requirements due to relatively low activity, and drug resistance. 96,97 Nonetheless, efforts were made to develop new fusion inhibitors with improved pharmaceutical profiles, including sifuvirtide, 98 SC35EK, 99 and T2635, 100 although they have suffered from similar difficulties. A related fusion inhibitor, C34, has been considered one of the most promising fusion inhibitors, 101 and efforts have continued to increase its potency. 102 Moreover, C34 is currently being used in a phase I clinical trial for HIV-positive men in the United Kingdom (EudraCT No. 2014-002671-28). Most recently, a newly designed peptide (HP32) has been developed that shows cross-strain HIV antiviral activity by specifically targeting a gp41 pocket different from the T20 resistance sites. 103,104 As such, HP32 has been shown to effectively inhibit T20- and MT-SC22EK-resistant HIV-1 strains, making it an ideal candidate for clinical development. An overview of HIV receptor usage on T cells is provided in Fig. 1.
Macrophages
One of the first immune cells to encounter HIV during sexual transmission are MΦs (Fig. 2). 105 While other immune cells have been identified to be important early targets for HIV infection, 106,107 MΦs have been the most extensively studied for their roles in viral dissemination throughout the host. 108,109 Specifically, MΦs are able to promote viral dissemination due to their ability to migrate throughout the entire body and are key players in persistent HIV infection. 110 Due to their resistance to the cytopathic effects of HIV, 111 MΦs can produce virus for long periods of time and act as HIV reservoirs for viral transmission to other lymphocytes. 112 Because of their unique roles in HIV infection, MΦs not only function in initial HIV infection 113 but also play roles in HIV-associated neurodegenerative and innate immune system disorders. 114,115
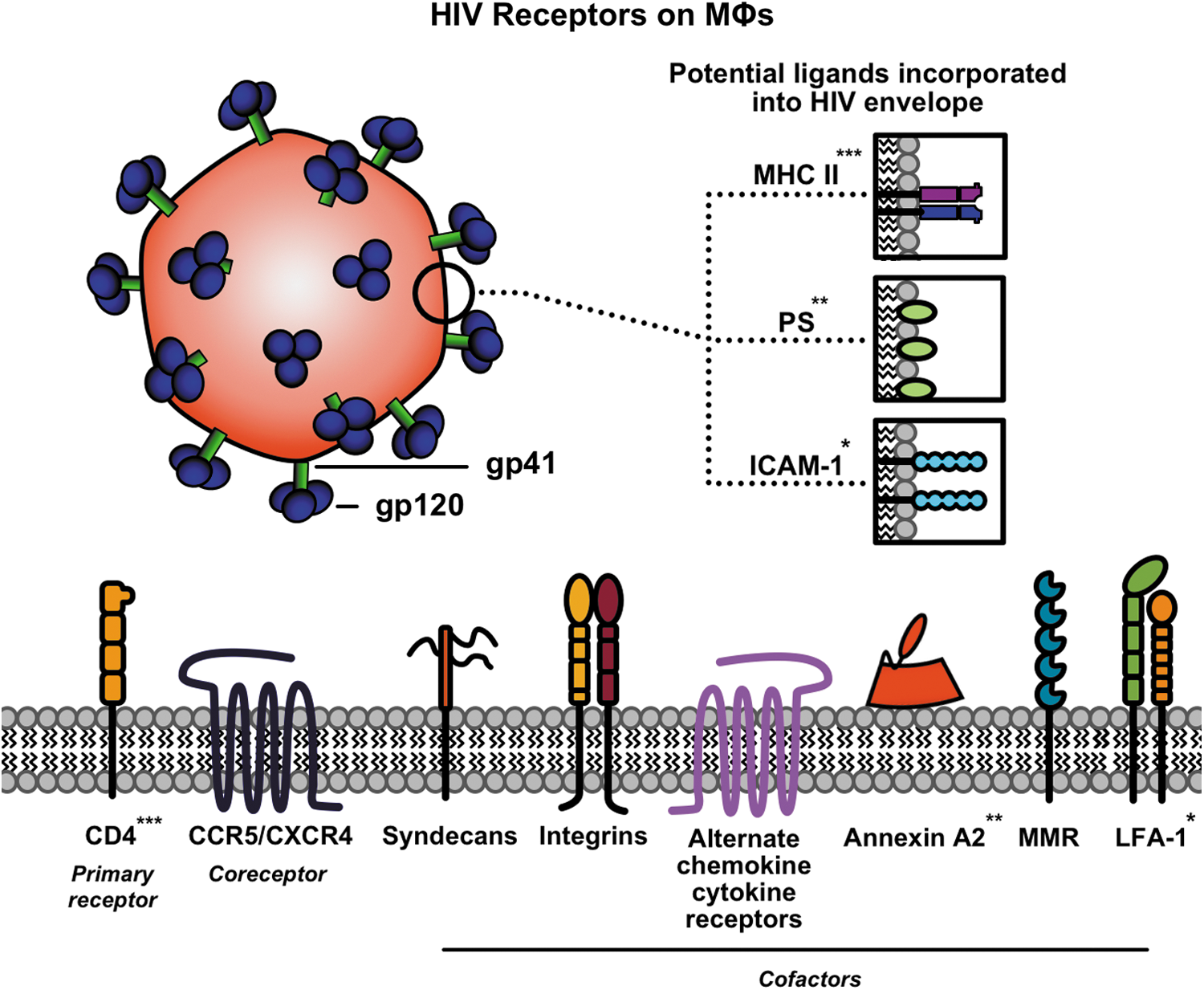
Summary of receptors used by HIV to enter MΦ. CD4 is the primary receptor for gp120, while CCR5 or CXCR4 can act as coreceptors. During viral propagation, surface proteins such as MHC II, PS, or ICAM-1 have been shown to incorporate into the viral envelope and act as ligands that enhance virus-MΦ interactions through binding to their receptors CD4, annexin A2, and LFA-1, respectively (protein interactions indicated by *, **, and ***). Syndecans, integrins, alternate chemokine/cytokine receptors, and MMR have also been shown to be cofactors that play a role in HIV infection of MΦs. CCR5, C-C chemokine receptor type 5; CD4, cluster of differentiation 4; CXCR4, C-X-C chemokine receptor type 4; HIV, human immunodeficiency virus; ICAM-1, intercellular adhesion molecule-1; LFA-1, lymphocyte function-associated antigen 1; MHC II, major histocompatibility complex class II; MMR, macrophage mannose receptor; PS, phosphatidylserine. (Color image is available at
Primary receptor
The original paradigm that HIV tropism was restricted to T cells was reevaluated once researchers demonstrated that other immune cells were permissive to HIV infection, primarily due to CD4 expression combined with yet to be identified coreceptors. 25,116 While it is now understood that HIV can exploit CD4 to bind to MΦs, 117 it stands that cells of the MΦ lineage phenotypically express lower levels of this molecule. 118 Consequently, certain HIV strains have evolved or developed adaptations to infect cells with low surface CD4 expression and are known as macrophage tropic (M-tropic). 119 These strains are able to efficiently infect MΦs, including those resident in the brain (known as microglia). 120 M-tropism arises from gp120 modifications at the CD4 binding site. Specifically, it has been demonstrated that M-tropism correlates with sensitivity to reagents that block gp120-CD4 interactions, indicating that the HIV gp120 on M-tropic strains has a higher contact affinity to CD4, enabling it to infect cells with low CD4 levels. 121
Sensitivity to gp120-CD4 inhibitors (i.e., sCD4, PRO 542, BMS-378806, b12, and human mAbs targeting conserved regions of gp120 or gp41) is characteristic of M-tropic strains, and the stronger the affinity for either target, the more effective the agent is at blocking infection. 122 –124 It remains, however, that viral replication in macrophages is limited, and in vivo infection is restricted to a minor percentage of the macrophage population. 105,125 Ultimately, observations of primary HIV isolates that only infected monocyte and MΦ populations rather than T cells promoted research focused on identifying cofactors specific for the various CD4+ MΦ subsets. 116,117
Coreceptors
MΦ infection by certain HIV isolates was shown to be inhibited by the release of Th1-associated cytokines, including RANTES and macrophage inflammatory protein 1 alpha (MIP-1α) and MIP-1β, 126 and thus, investigations were performed to identify the associated coreceptor(s) to explain these results. Interestingly, in rare cases, it was observed that individuals were resistant to sexually transmitted HIV infection even if their T cells could be readily infected by T-tropic variants in vitro. Researchers elucidated that this was due to a deletion in the CCR5 gene that conferred protection against M-tropic strains. 127,128 These observations, among others, led to the identification of CCR5 as the main coreceptor involved in HIV viral fusion to MΦs. 129 –131 Because of this, M-tropic strains that exploit CCR5 are now classified as R5-tropic, in line with the aforementioned nomenclature for X4-tropic isolates that use CXCR4, although CXCR4 can also be utilized by macrophages. 132 Since CCR5 has been shown to be essential for HIV disease progression, much effort has been invested into targeting this coreceptor for therapeutic purposes. 133 Specifically, CCR5 antagonists that impede CCR5-HIV interactions such as PRO 140, TAK-652, vicriviroc, aplaviroc, and maraviroc have been investigated. 134 –138 Among these, only maraviroc has been approved by the FDA for clinical use. 139
Coinciding with the sCD4 group of inhibitors, CCR5 coreceptor mimics have also been investigated and were shown to synergistically increase the efficacy of sCD4s to block HIV-1 infection by acting as “bait” CCR5 to the sCD4-primed virion thus catalyzing a premature discharge of HIV fusion potential. 39 More recently, a peptide that mimics a sulfated region of HIV-1 V2 (pV2alpha-Tys) was used to prevent CCR5 utilization and block HIV-1 entry, 140 and such peptidomimetics have been combined with sCD4 in a single construct to produce a more potent inhibitor. 40 MΦs can also be targeted by certain HIV isolates that are phenotypically considered T-tropic through entry via CXCR4, 141 as well as by dual-tropic strains classified as R5X4, which can utilize either CXCR4 or CCR5; however, these R5 or R5X4 strains are primarily found in advanced stages of disease. 142 –146 Interestingly, while the majority of M-tropic viruses utilize CCR5, it has become apparent that not all R5 viruses are M-tropic. 147 Therefore, while the interrelation between coreceptor specificity and HIV tropism has to be taken into consideration, it cannot be fully relied on to define certain viral isolates, and CCR5 targeting alone may not be sufficient to stop HIV infection.
Cofactors and other components
Despite the identification of CD4 and CCR5 as the primary receptor and coreceptor for MΦ infection, their binding to HIV envelope proteins represents only a small portion of the myriad of interactions that occur during viral interaction with the cell surface. Multiple other surface proteins such as integrins and syndecans are also required for efficient HIV infection of MΦs and transmission to T cells, and can influence infection rates. 148,149 In addition, other receptors can replace CCR5 in the fusion step preceding entry into MΦs. 150 These alternate chemokine/chemoattractant receptors include CCR1, CCR2b, CCR3, CCR8, CX3CR1, CXCR6, formyl peptide receptor 1, G protein-coupled receptor 1 (GPR1), GPR15, apelin receptor, and chemokine-binding protein 2 (CCBP2), 151,152 exponentially increasing the complexity of HIV infection. Also, HIV can incorporate numerous host cell surface molecules into its envelope during budding events, each of which potentially enhance subsequent infectivity. 153 Two highly relevant host-derived molecules, which have been found in the viral envelope, are ICAM-1 as previously mentioned and major histocompatibility complex class II (MHC II), which bind to the receptors LFA-1 and CD4, respectively. Integration of either of these surface proteins was shown to increase the virulence of HIV in vitro as well as its ability to replicate in target cells. 86,154
It was also shown that the efficient virological synapse-mediated transmission of HIV-1 from macrophages to T cells in vitro was facilitated by interactions between ICAM-1 and LFA-1, where antibodies against either protein decreased transfer to T cells. 155 Interestingly, forced expression of the recently identified membrane-associated RING CH 8 (MARCH8) protein, a member of the RING (really interesting new gene)-finger E3 ubiquitin ligases, was shown to block the incorporation of MHC II into the viral envelope and thus decrease viral entry, and MARCH8 knockout in macrophages increased the infectivity of progeny virions produced within them. 156 Moreover, these results highlight the novel role of MARCH8 as a potent endogenous antiviral protein that may be of interest for future research. Additional promiscuity of Env proteins has also been reported, as gp120 was shown to bind to macrophage mannose receptor (MMR), a C-type lectin receptor, and that MMR-bound HIV on MΦs could be transmitted to T cells. It is worth noting that this method of transmission does not result in productive infection of MΦs, and a similar mechanism will be discussed later that involves a C-type lectin receptor in HIV transmission by DCs. 157
Another molecule that, when incorporated into the viral envelope, can act as a significant cofactor in MΦ infection is phospholipid phosphatidylserine (PS). A notable property of PS is that it is a marker of apoptosis when present on the outer leaflet of a cell. 158 Because HIV-infected cells may express more PS on their surface due to viral-induced apoptotic events, PS can be incorporated into the HIV envelope, which then influences the infection rates and tropism of progeny virions. Experimental evidence for this was provided by in vitro studies of primary monocyte-derived MΦs and three differentiated monocytic cell lines. In this study, it was demonstrated that PS-containing vesicles blocked HIV infection without altering viral binding to MΦs, whereas phosphatidylcholine vesicles did not. 159 The ability of host-derived PS to influence HIV-1 infection led to the prediction that an unknown interacting partner on target cells facilitated viral binding and/or fusion through PS interactions leading to entry.
Also, secretory leukocyte protease inhibitor (SLPI) was shown to inhibit HIV-1 infection of MΦs independent of its antiprotease activity, and SLPI expression in the mucosa was implicated in viral susceptibility. 160 –162 Ma et al. later revealed that SLPI directly interacted with annexin A2, a PS-binding moiety, and that it disrupted interaction between annexin A2 and PS on the HIV-1 envelope in vitro. 163 Moreover, the authors demonstrated that mAbs against or RNA silencing of annexin A2 dramatically reduced HIV-1 infection of MΦs similar to that of SLPI blocking, indicating that annexin A2 was a cofactor for HIV-1 infection of MΦs. In addition, it was shown that HIV-1 produced from monocyte-derived MΦs that had been treated with annexin A2 siRNA exhibited decreased infectivity. 164 However, small-molecule inhibitors of the annexin A2 heterotetramer failed to block HIV infection of MΦs in vitro, indicating that the monomer form rather than the heterotetramer form may be active in HIV infection (unpublished data). An overview of HIV receptor usage on MΦs is provided in Fig. 2.
Professional APCs
As previously mentioned, HIV-1 is most often sexually transmitted across mucosal epithelial barriers, in which APCs (Fig. 3), particularly DCs and DC subtypes, are highly abundant. 165 –167 Depending on the localization and expression of different molecules such as C-type lectins, 168 DCs are commonly divided into subsets that include dermal DCs and epidermal Langerhans cells (LCs), which have been extensively investigated as HIV targets, reservoirs, and vectors for viral dissemination. 169,170 Therefore, the following sections will distinguish between the roles played by DCs and LCs in HIV entry and processing through different receptors and pathways, which can lead to productive infections, viral dissemination, as well as viral degradation used to limit viral spread.
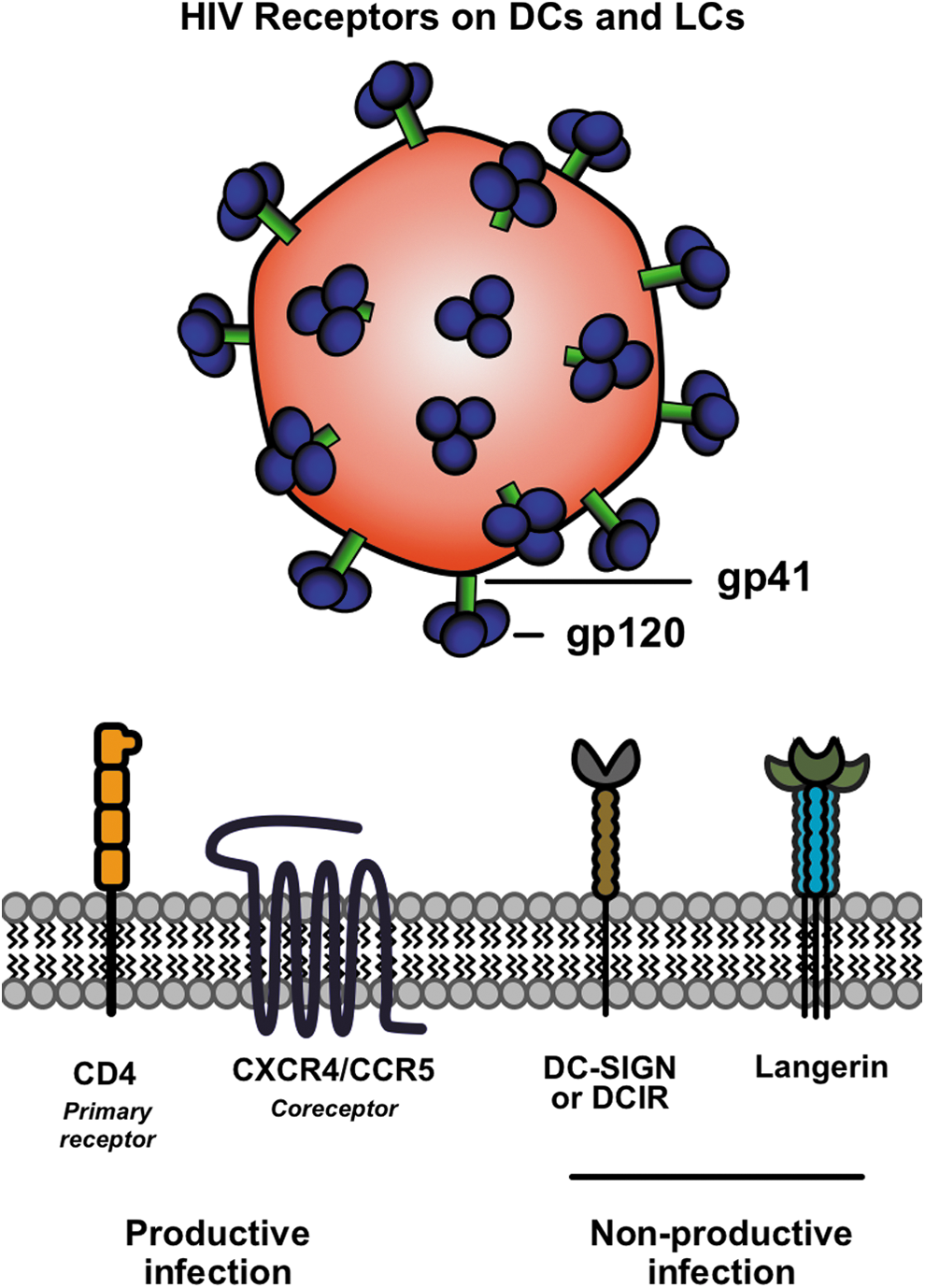
Summary of receptors used by HIV to enter professional APCs (i.e., DCs and LCs). CD4 is a primary receptor for gp120, while CCR5 or CXCR5 can act as coreceptors. Entry through these pathways can yield productive infections. Alternatively, HIV has been shown to interact with the C-type lectin receptors DC-SIGN, DCIR, and langerin at the surface of DCs and LCs. Association with these receptors can result in viral transmission to T cells via trans-infection, or potentially in viral degradation. APC, antigen-presenting cell; CCR5, C-C chemokine receptor type 5; CD4, cluster of differentiation 4; CXCR5, C-X-C chemokine receptor type 5; DCs, dendritic cells; DCIR, DC immunoreceptor; DC-SIGN, DC-specific ICAM-3-grabbing nonintegrin; LCs, Langerhans cells; HIV, human immunodeficiency virus. (Color image is available at
Dendritic cells
DCs are potent APCs found in the dermis as immature DCs (iDCs), and on infection or capture of viruses, including but not limited to HIV, can mature and migrate toward lymphoid tissues to present viral antigens to T cells. 171,172 Once bound to DCs, the fate of HIV particles depends on several factors: the particular DC subtype they are bound to, the state of DC maturation, and the receptor through which they interact with at the DC surface. Furthermore, HIV can be transmitted from DCs to T cells through so-called trans-infection via immunological synapse formation occurring independent of HIV replication. 173 –176 However, transmission to T cells can also occur by DCs productively infected with HIV in a process known as cis-infection, 177 and this difference is largely determined by the maturation state of the DC. 178,179 Although DCs express both CXCR4 and CCR5, only R5 strains were shown to efficiently replicate in DCs, thus contributing to the high proportion of R5 variants present during sexual transmission. 180 Below, different pathways of HIV entry and processing in DCs will be discussed in more detail.
CD4 and coreceptor usage in DCs
Primary peripheral blood iDCs were shown to have the highest surface expression of CCR5 among different leukocyte subsets, but had low CXCR4 expression; however, on in vitro maturation, DCs had dramatically increased CXCR4 expression, although HIV itself was not able to induce significant activation. 181 Also, it was shown that iDCs expressed the primary HIV binding receptor, CD4, at low levels, but these levels likely remained above the threshold to permit initial gp120 interaction and viral entry. Furthermore, it was demonstrated that ex vivo-isolated myeloid DCs were more susceptible to an R5 isolate, whereas donor-matched plasmacytoid DCs were more susceptible to an X4 isolate, further suggesting the differential expression of coreceptors on the surface of DC subsets. 182 Moreover, R5 isolates replicate readily in iDCs, while mature DCs transmit both R5 and X4 isolates to T cells. 183 Taken together, these results suggest that DC maturation status and resultant chemokine receptor profile have a major impact on the efficiency of infection by different HIV variants. Other chemokine coreceptors, including CCR3, CCR8, CCR9, and CXCR6, have also been implicated in HIV entry into DC. 177,184 Although the CD4 and coreceptor pathways can allow DCs to be infected by HIV via canonical membrane fusion mechanisms, several factors are able to impede HIV from efficiently replicating in these cells, potentially explaining their low infectivity compared to T cells. 185 For example, tripartite motif-containing protein 5 alpha (TRIM5α) and sterile α motif and HD domain-containing protein 1 (SAMHD1) both interfere with the HIV reverse transcription process within DCs. 186,187 Since the activation status of DCs contributes both to HIV transmission to T cells and anti-HIV immune responses, DC-targeted HIV vaccine strategies have focused on activating DCs and improving DC function to elicit anti-HIV cellular immunity. 188
DC-specific ICAM-3-grabbing nonintegrin promotes HIV trans-infection
DC-specific ICAM-3-grabbing nonintegrin (DC-SIGN) is a C-type lectin receptor highly expressed on the surface of DCs that binds to and cointernalizes with HIV-1 gp120. 189 Importantly, DC-SIGN does not lead to canonical HIV entry, but rather promotes trans-infection of T cells by trapping HIV on the surface or within endocytic vesicles where their infectivity is retained or augmented. 189,190 This facilitates viral transmission in trans from mature DCs to resting T cells in lymphoid organs through high-affinity interactions between DC-SIGN on DCs and ICAM-3 on the T-cell surface. 191 Interestingly, the three-dimensional structure of DC-SIGN revealed that the particular interaction with ICAM-3 occurs through a binding site distinct from that of the DC-SIGN-gp120 interaction site. 192 By exploiting this alternative receptor, HIV persists in a highly infective state due to stabilization mechanisms and acts as a passenger on migrating DCs to get from the initial site of exposure in the periphery to its target site of infection in lymphoid tissues. 189
In a more recent study, it was shown that DC-SIGN enhances HIV infection not only by promoting viral dissemination to T cells but also by increasing CD4-gp120 affinity. Specifically, it was shown that soluble variants of gp120 had a higher affinity to CD4 in the presence of soluble DC-SIGN via surface plasmon resonance analysis with immobilized HIV-1 gp140 molecules (soluble variants of gp120). 193 Moreover, the authors showed that the affinity of the b12 neutralizing mAb, containing an overlapping gp120 binding site that competes with CD4, 194 was enhanced by DC-SIGN, providing further evidence that this receptor increases the exposure of the CD4-gp120 binding site. Recently, lactoferrin, a protein found in colorectal mucus, was shown to bind to DC-SIGN and block HIV-1 trans-infection of both R5 and X4 strains, acting as a natural barrier of HIV-1 infection. 195 Despite these studies indicating a role for DC-SIGN in trans-infection, Burleigh et al. demonstrated that DC-SIGN-mediated HIV internalization was dispensable for trans-enhancement through the use endocytosis-defective DC-SIGN, and the authors further suggested that DC-SIGN cooperates with HIV entry receptors to facilitate cis-infection by which new viral progeny from DC contribute to HIV dissemination to T cells. 196 Therefore, future studies are needed to clarify these controversies and to determine if DC-SIGN is a viable target to control HIV during the early stages of pathogenesis.
Another C-type lectin receptor similar to DC-SIGN that can act as an attachment factor for HIV is the DC immunoreceptor (DCIR), although recently it was shown that DC-SIGN played a stronger role than DCIR in trans-infection of a broader range of isolates. 197 Moreover, DC-SIGN is downregulated upon DC activation, while trans-infection is strongly enhanced via a glycoprotein-independent capture pathway that involves sialyllactose-containing membrane gangliosides, which was recently shown to be mediated by sialic acid-binding Ig-like lectin 1 (siglec-1 or CD169) on mature DCs. 198 Hence, siglec-1 represents yet another target to block DC-mediated trans-infection of T cells. HIV-1 opsonization by complement proteins has also been shown to enhance HIV entry into DCs via DC-SIGN and complement receptor 3 (CR3). 199,200 Moreover, complement opsonized HIV-1 (C-HIV) interactions with CR3 may be part of an immune escape mechanism utilized by the virus to establish infection in the host, as CR3 engagement of C-HIV decreased inflammatory responses by DCs in vitro. 201
Langerhans cells
Since their discovery in 1868 by Paul Langerhans, 202 LCs have been extensively investigated in relation to viruses because of their proximity to pathogen entry portals of the epidermis and mucosal epithelia, including the ectocervix, vagina, and foreskin. 203,204 This makes LC likely to be the first immune cell to encounter HIV during sexual transmission, 205 and they are the only resident cell of the epithelium that harbor productive infections. 206,207 Hence, it is not surprising that LCs are readily infected via the canonical CD4 and multiple coreceptor pathways. 208,209 While it has been demonstrated that nonproductive HIV entry into LCs results in virions remaining within the cytoplasm, eventually contributing to productive infection of T cells in vivo, 210,211 LCs have also been implicated in a protective role against HIV in which the virus is degraded in LC-specific organelles known as Birbeck granules. 212 In light of these different roles, both the HIV transmission and destruction pathways will be discussed below.
CD4 and coreceptor usage in LCs
LCs are known to express both the CD4 receptor and the CCR5 coreceptor, 208,213 and are therefore vulnerable to R5 strains. 214 Although it has also been reported that some LCs express functional levels of both CXCR4 and CCR5 coreceptors and thus can be infected by both X4 and R5 strains. 209,215 Particularly, it was shown that 9% of fresh LCs isolated and purified from the epidermis expressed CXCR4 on the cell surface and 16% expressed CCR5 (95% expressed CD4), and the expression of these coreceptors was significantly increased on maturation. 209 Moreover, mature LCs were susceptible to both R5 and X4 variants. In a more recent study utilizing an ex vivo tissue model, primary LCs were infected by both R5 and X4 strains, but only R5 strains were selectively transmitted by immature LCs to T cells, which was dependent on de novo progeny in cis-infections; however, activation facilitated the transmission of both R5 and X4 variants. 215 Therefore, given the specific location of these cells, it may be advantageous to target both CCR5 and CXCR4 through topically applied microbicides to prevent LC-mediated HIV transmission to T cells.
Langerin-mediated HIV degradation
Just as DCs can be defined by their expression of the C-type lectin receptor DC-SIGN, LCs exclusively express a C-type lectin receptor known as langerin, which is also structurally involved in the formation of the aforementioned Birbeck granules. 216,217 Birbeck granules are organelles found only in LCs and are speculated to be involved in the antigen-processing pathway. 218,219 Moreover, after entry into LCs, HIV virions have been detected in Birbeck granules by immunoelectron microscopy, in what was determined to be through a degradation pathway. 212 Receptor binding assays coupled with flow cytometry analysis demonstrated that langerin interacted with soluble gp120 with a higher avidity than that of DC-SIGN, 168 and mAbs specifically designed to disrupt the langerin-gp120 interaction enhanced HIV infection and transmission to T cells. 212 A later study further corroborated the protective role of langerin in HIV infection by showing that HIV virions were taken up and destroyed in langerin-containing Birbeck granules. 220
Taken together, these results led to the conclusion that langerin-mediated entry and degradation in Birbeck granules act as a natural barrier for HIV, as this mechanism prevents transmission to T cells. However, future studies are required to resolve the inconsistencies between these results and the aforementioned studies demonstrating that LCs are readily infected by HIV and are involved in viral dissemination, although one may speculate that experimental viral load may have caused a saturation of the langerin route and allowed alternative interactions leading to productive infection. If langerin is determined to be protective, preventative strategies could be used to enhance its expression and subsequently enhance HIV degradation in the epithelium. An overview of HIV receptor usage on DCs and LCs is provided in Fig. 3.
Other Immune Cells
B cells
While it is well established that HIV-1 infection leads to the progressive depletion of CD4+ T cells, infection also negatively impacts B cells in the humoral arm of adaptive immunity (reviewed in Moir and Fauci 221 ). Although little evidence suggests that HIV can productively infect B cells, strong evidence has shown that HIV can bind to B cells in vivo through interactions with the complement receptor CD21, 222 and that this interaction promotes trans-infection of T cells in peripheral blood and lymphoid tissues. 223 A similar mechanism of HIV transmission has been suggested for follicular dendritic cells expressing CD21. 224 Interestingly, DC-SIGN expression on activated B cells was also shown to enhance trans-infection of T cells. 225 Despite the ability of CD21 to promote trans-infection, a high proportion of B cells lose CD21 expression during HIV infection, 226 and this may be associated with B-cell dysfunction, a common comorbidity during HIV infection.
Granulocytes
Granulocytes, primarily neutrophils, eosinophils, and basophils, were recently shown to express a variety of HIV-1 attachment factors. 227 Particularly, basophils expressed DC-SIGN, DCIR, heparan sulfate proteoglycan, and α4β7 integrin and exhibited the most efficient capture of HIV-1 on their cell surface among all granulocytes. Neutrophils expressed DCIR and eosinophils expressed α4β7 integrin, but showed limited and no virus-binding capacity, respectively. Accordingly, basophils, but not neutrophils and eosinophils, efficiently facilitated trans-infection of CD4+ T cells, 227 and thus, strategies designed to prevent basophil-mediated viral capture and transfer may represent a novel approach to control HIV infection.
Summary and Perspectives
In the current review, we underlined the important receptors, coreceptors, and cofactors involved in HIV tropism for different immune cells, and highlighted that the location of HIV infection plays an important role in which immune cells are targeted. In addition, different therapeutic approaches that have targeted some of these factors were discussed. A summary of the different molecules associated with HIV entry and infection of different immune cells is provided in Figs. 1 –3, and compounds that have been used to target them are provided in Table 1. In the perpetual battle against HIV, immune escape mechanisms in addition to the multitude and promiscuity of entry mechanisms, all may explain why a cure for AIDS has remained elusive. Among the immune escape mechanisms used by HIV, the high mutation rate and conformational fluctuations of gp120 stand out, as even slight modifications in its structure/conformation can modify neutralizing antibody recognition and receptor binding 228 and can allow for HIV receptor/coreceptor use in an inhibitor-resistant manner. 229 Moreover, as an enveloped virus, HIV can take advantage of cellular budding events to incorporate host molecules into its outer leaflet to enhance infectivity. 159
Finally, as the number of surface molecules that HIV exploits to efficiently enter target cells continues to grow, it becomes apparent that therapies aimed at multiple infectious pathways simultaneously with antiretroviral (ARV) drugs that inhibit reverse transcription may ultimately prove to be the best preventative/therapeutic strategy. Combinations of ARVs are currently used in highly active antiretroviral therapy for HIV+ individuals, 230 but topical preventative trials to date have been based on a single ARV. 231 –233 More recently, however, a preclinical study with tissue explants demonstrated that ARVs in combination with the CCR5 inhibitor maraviroc could be used as an effective pre-exposure prophylactic (PrEP) strategy in line with newer PrEP approaches. 234 In conclusion, elucidating the mechanisms and identifying the specific molecules by which HIV preferentially exploits different receptors, coreceptors, and cofactors on certain immune cells provide a foundation for developing novel strategies to prevent AIDS, which remains one of the deadliest diseases in the world.
Footnotes
Acknowledgments
This work was supported by Public Health Service grant R01 CA074397 from the National Cancer Institute (to W.M.K.). Contributions from the Netherlands-American Foundation, Sammie's Circle, Christine Ofiesh, Yvonne Bogdanovich, and Johannes van Tilburg are also gratefully acknowledged. W.M.K. holds the Walter A. Richter Cancer Research Chair. J.G.S. was supported by the Keck School of Medicine/USC Graduate School PhD Fellowship. Additional support for J.R.T. from the ARCS Foundation John and Edith Leonis Award is greatly appreciated. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.