
Abstract
Before the modern era of HIV/AIDS therapeutics, which enabled a cascade of early recognition of infection, prompt initiation of effective antiretroviral therapies, and close follow-up, severe forms of microvascular clotting disorders known as thrombotic microangiopathies (TMAs) were frequent in the setting of advanced HIV disease. Their incidence was as high as 7% in the period 1984–1999, but fell dramatically, to <0.5%, by 2002. This profound change was predicated on one critical development: availability of new classes of anti-HIV drugs, enabling reduction and maintenance of HIV viral loads to undetectable levels. Another development in the period 1999–2002 related to TMA therapy: with recognition of autoantibodies against the von Willebrand factor cleaving protease ADAMTS13 as the etiology of most cases of one major form of TMA, thrombotic thrombocytopenic purpura, it permitted appropriate use of life-saving interventions based on plasma exchange and immune suppression. A more recent factor in TMA therapeutics was the 2011 approval by the US FDA and European EMA of eculizumab, a humanized monoclonal antibody against complement component C5, for the treatment of atypical hemolytic uremic syndrome, another major form of TMA. Despite these milestones, life- and organ-threatening TMAs still occur in untreated HIV disease and, to a much lesser extent, in those patients with suppressed viral loads. Confusion in terms of the differential diagnosis of these TMAs also impedes use of directed treatments. This report utilizes a case study of a young woman with advanced AIDS who presented with a severe TMA, characterized by coma and renal failure, to highlight the diagnostic and therapeutic challenges raised by complex hematologic conditions occurring in the setting of HIV.
Introduction
T
In general, both aHUS and TTP can affect any organ, as microvessel thrombosis leads to widespread tissue ischemia and infarction. While TTP is historically linked to CNS manifestations, about half of aHUS patients also have severe CNS disease. 1 Twenty percent of initial aHUS presentations involve normal serum creatinine, despite the word “uremic” in its title. (Most of these patients do have some renal involvement, characterized by proteinuria, microscopic hematuria, and/or hypertension. 1 ) In contrast, classic TTP involves the kidneys in more than 50% of cases, although levels of creatinine elevation tend to be far lower than in aHUS. 1 (The one possible exception to multiorgan involvement in the TMAs is the lung, which is virtually never involved structurally in TTP, while pulmonary pathology is frequent in aHUS. 1,2 ) However, the pathophysiology of TTP and that of aHUS is distinct and demands divergent therapies.
Because of the high incidence of TMAs in the setting of untreated HIV disease, there is a particular need for those involved in the care of HIV-infected patients to be cognizant of these differences. The first case of an HIV-linked TMA was published in 1984, just 3 years after the identification of AIDS. 3 At that time, over a decade before discovery of an autoantibody against the von Willebrand factor cleaving protease ADAMTS13 as the cause of acquired TTP, and thus, the capacity to more precisely distinguish TTP from aHUS, this initial case was labeled aHUS based on the prominence of renal manifestations. Soon thereafter, the true extent of microangiopathic hemolytic anemia in the setting of HIV was recognized, including the fact that it can be the initial and only presenting clinical manifestation of HIV infection. 4,5
Pre-2000, the incidence of a TMA in the setting of clinical AIDS ranged from 0.6% to 7.1% in five studies examining 4437 individuals. 6 In a series from Miami, Florida, an early epicenter of the US AIDS epidemic, 14% of TMA patients were HIV infected. 7 Mortality was high: survival rates of <1–2 years were characteristic 7 and independent of the extent of immune suppression at the time of TMA presentation. 8 Plasma exchange (PEx) was then the only recognized treatment, whether or not the TMA was related to HIV. Of interest, the pattern and kinetics of response to PEx, in terms of normalization of platelet counts and LDH and, in many instances, resolution of neurologic and renal dysfunction, appeared to be equivalent to non-HIV TMA cases. 9 Some reports suggested that fresh frozen plasma (FFP) infusions alone were sufficient in the HIV+ cases, with resolution of at least the hematologic abnormalities occurring more rapidly than in HIV-negative cases. 10 However, TTP was generally not distinguished from aHUS and these responses were short lived; no patient survived more than 2 years from time of TMA diagnosis in one HIV+ cohort. 9
Hematologic response to intensive plasma therapy in a TMA patient cannot be used to distinguish TTP from aHUS, regardless of HIV status. FFP, given as infusions or through PEx, can correct the ADAMTS13 deficiency in more than 90% of TTP cases, leading to a complete clinical and laboratory response. 11 However, there are sufficient quantities of complement factor H (CFH) and complement factor I (CFI), the two most commonly mutated proteins in individuals susceptible to aHUS, 12,13 in FFP to normalize hematologic parameters, and occasionally improve renal function in aHUS as well. 11 The important consideration is that, unlike TTP, PEx has no long-term impact on morbidity or mortality in aHUS. 11 Indeed, the poor life expectancy of HIV+ TMA patients treated with PEx in the pre-modern antiretroviral therapy (ART) era is likely a consequence of three factors: poorly controlled HIV infection; the myriad sequelae of HIV infection itself, including immune thrombocytopenia purpura, HIV-associated nephropathy, cardiomyopathy, CNS disease, and opportunistic infections such as cytomegalovirus; and the characterization of the TMAs as undifferentiated “TTP/HUS” cases, thus failing to consider the need for differential therapy.
The modern era of HIV/AIDS therapeutics enabled a cascade of early recognition of infection, prompt initiation of effective ART, and close follow-up. 14 Uncontrolled HIV infection is thus now much less of a factor in precipitating a TMA. However, the various complications of HIV-linked inflammation, fibrosis, opportunistic infection, malignancy, and ART side effects, all of which can cause laboratory and clinical findings, especially renal dysfunction and cardiovascular disease, 15,16 reminiscent of a TMA, may obfuscate an aHUS or TTP diagnosis. In the setting of an acute exacerbation of any potentially HIV-related condition, appropriate treatments for those disorders must be initiated before suggesting a diagnosis of a primary TMA triggered or unmasked by the HIV infection. 17
Response to ART cannot be used to distinguish between a TTP-type and an aHUS-type TMA. For example, Gervasoni et al. reported the “disappearance” of all HIV-linked TMAs by 2002, with 0 cases among 347 patients in their European cohort in the modern ART era. 18 Similar data were derived from a US cohort, with only 17 of 6022 HIV+ individuals (0.27%) developing a TMA. 19 It is clear that ART must have had an impact on both TTP-type and aHUS-type TMAs, as once ADAMTS13 testing became available, it was discovered that only 40–50% of patients with an HIV-linked TMA had enzyme activities in the TTP range, that is, <5–10%. 6 That is the category of TMA patient for which PEx, with the ability to replete ADAMTS13 through multiple infusions of FFP, create space to permit intensive plasma repletion, and perhaps plasmapheresis off ADAMTS13 autoantibodies would be expected to have long-term impact.
Other clues as to the exact nature of HIV-linked TMAs came from biopsy. Renal biopsy in four of four HIV-TMA cases unresponsive to PEx revealed extensive fibrin thrombi with inflammation of microvessels, findings characteristic of aHUS. 20 These changes are a consequence of the inability to regulate activation of the alternative complement pathway in an aHUS-susceptible individual, leading to formation of the proinflammatory anaphylatoxin C5a and generation of the membrane attack complex (MAC) C5b-9, capable of activating platelets and injuring both erythrocytes and endothelium. These events also initiate a positive feedback loop, with thrombin activation leading to both fibrin deposition and cleavage of C5, 21 resulting in formation of more C5a with inflammation, and C5b to initiate C5b-9 complex formation. In contrast, TTP is characterized by platelet-rich rather than fibrin microthrombi, inflammation is usually absent, 22 and von Willebrand factor complexes rather than C5b-9 are deposited on the endothelium.
The following case report illustrates the diagnostic challenges in identifying and treating an HIV-linked TMA.
Case Report
A 24-year-old female with known HIV infection, acquired about 6 years before admission through heterosexual intercourse, was poorly adherent to her antiretroviral regimen of abacavir, lamivudine, and lopinavir. She had a history of infections and chronic kidney disease. She was admitted for evaluation of low-grade fevers, weakness and fatigue, nausea, emesis, nonbloody diarrhea, and an acute kidney injury, with a serum creatinine of 3.8, elevated from a baseline of 2. She was found to have a CD4+ T-cell count of 7/mm3 and an HIV viral load of >900,000 genome equivalents/mL plasma. There was no evidence for DIC, with INR, PTT, and fibrinogen within normal ranges. Stool cultures were negative. A TMA was diagnosed based on a platelet count of 56,000, a hemolytic anemia characterized by a hemoglobin of 7.2 g, LDH of 483, a haptoglobin <15, and a peripheral smear showing multiple schistocytes. The direct Coombs test was positive for complement. Levels of C3, C4, and CH-50 were within normal ranges. Over the ensuing week, her platelet count progressively decreased to a nadir of 22,000, her hemoglobin dropped to 5.9, and her LDH rose to 1165.
Given the clinical picture of a TMA, ADAMTS13 was drawn and, pending its result, FFP infusions initiated. This led to a drop in LDH to 520—some of which may have been dilutional—and an increase in haptoglobin from <15 to 21. Yet, she suffered a rapid decline in mental status, becoming unresponsive to verbal stimuli and then to deep pressure, and was intubated for airway protection. Cerebrospinal fluid evaluation was unrevealing. Four days after initiation of plasma infusions, her ADAMTS13 activity assay result returned and the activity level proved to be within normal limits, at 91%. The diagnosis of aHUS was established, FFP infusions were stopped, and the patient started on eculizumab. Eculizumab is a humanized mouse monoclonal antibody that binds with high affinity to C5, preventing generation of C5a and C5b and thus also blocking MAC formation. 13,23 Within 3 days, her LDH began to decline and her haptoglobin and platelet counts increased (Fig. 1A, B, D). Creatinine began to decrease by day 21, the time of the third weekly dose of eculizumab (Fig. 1C). Her mental status started improving a week after the initial dose of eculizumab; she was extubated the following week.
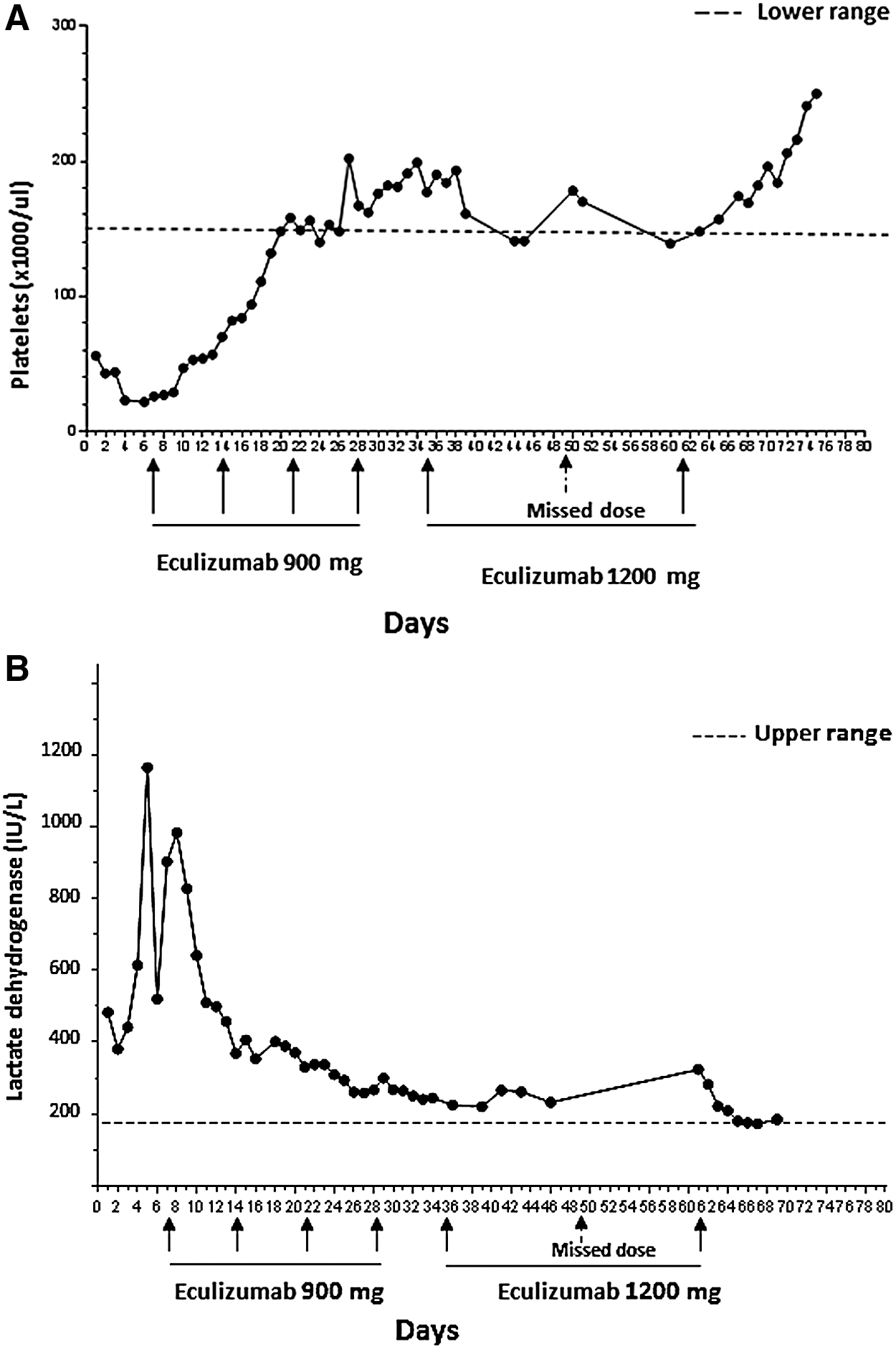
Laboratory findings consistent with a thrombotic microangiopathy in the Case Report patient were present at time of initial hospitalization and resolved with eculizumab therapy. Immediately following one missed dose of drug, these laboratory abnormalities returned, but improved following reinstitution of drug. Plasma infusion was performed daily for 4 days, just before the first dose of eculizumab. (
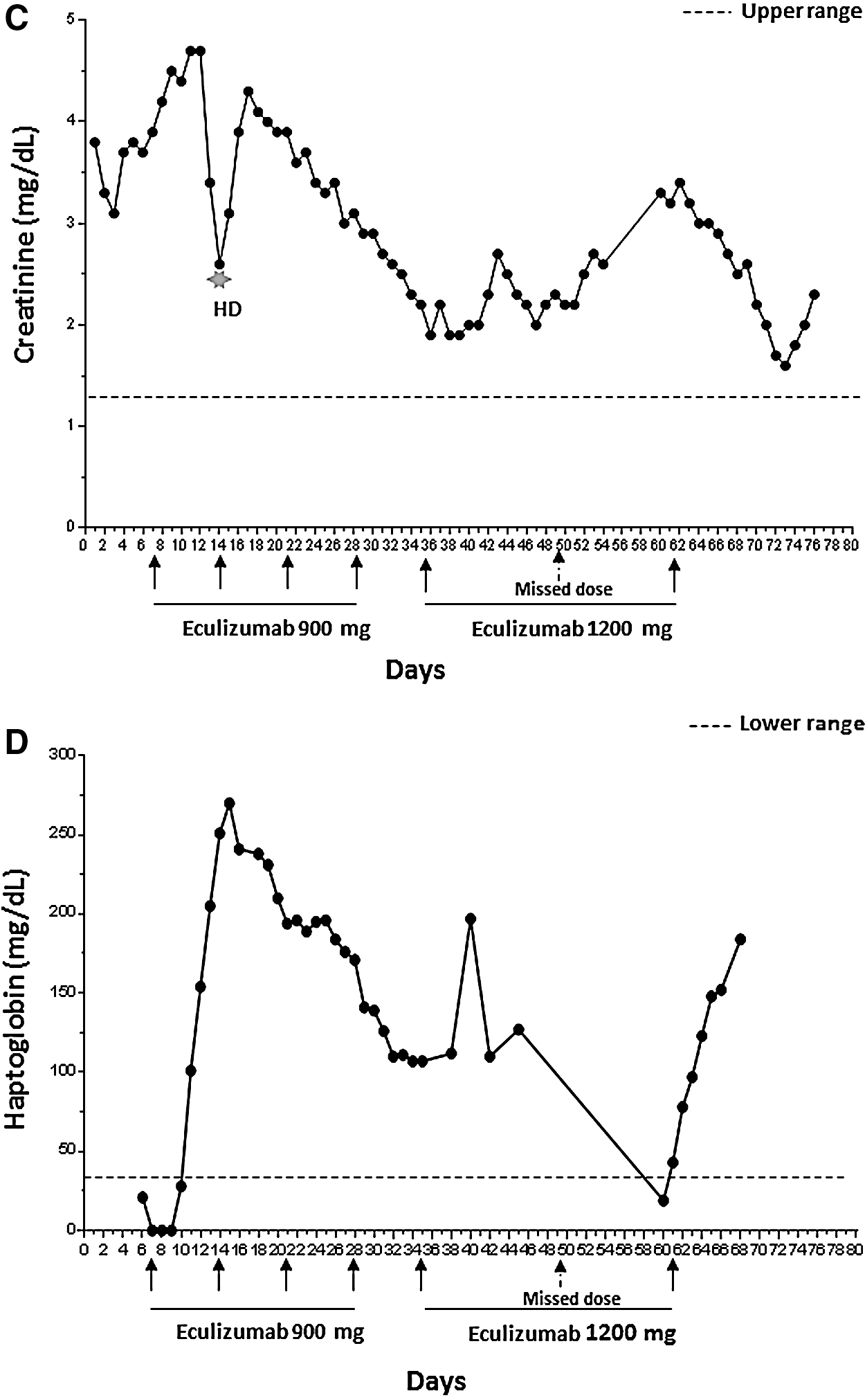
The patient was also begun on a new ART regimen of darunavir, dolutegravir, etravirine, and ritonavir, guided by HIV genotyping results. Her viral load fell from >900,000 to 29,519 on day 17, and to 5576 on day 39. On day 53, her viral load was 3203 with a CD4 count of 95. The possibility that resolution of her TMA was simply a consequence of this ART-mediated viral suppression was ruled out by the rapidity of return of TMA-related laboratory abnormalities following an unexpected eculizumab treatment interruption. As per treatment guidelines for aHUS, she received four weekly intravenous infusions of 900 mg, followed by infusions of 1200 mg every other week. (She was already on broad-spectrum antibiotics as prophylaxis for her immune deficiency. These antibiotics would also serve as prophylaxis for Neisseria infections, a side effect of eculizumab therapy.) However, the planned second biweekly dose of 1200 mg was missed, leading to a prompt rise in LDH and creatinine and a fall in platelet counts and haptoglobin—at a time when her viral load was <5000 (Fig. 1). These abnormalities persisted until that missed dose was administered 2 weeks later (Fig. 1).
Discussion
TMAs occurring in the setting of advanced, untreated HIV infection are life threatening. Unless an appropriate differential diagnosis is pursued, and thus appropriate interventions instituted, mortality rates are near 100% at 1–2 years. As in this patient, it is critical to first rule out, or identify and treat, complications of HIV infection itself, which might mimic a primary TMA. Infections and malignancies characteristic of AIDS may also initiate DIC; it is difficult, if not impossible, to diagnosis a TTP-like or an aHUS-like TMA in the setting of DIC. The underlying condition responsible for the DIC must first be resolved. If the TMA then persists and STEC-HUS has been ruled out by the absence of diarrhea or if, as in our Case, diarrhea is present, negative culture and polymerase chain reaction for shiga toxin, then complications of multiple tissue and organ system sequelae of HIV itself must be considered and ruled out or treated before finally diagnosing a primary TMA that may have been unmasked by HIV, and proceeding to characterize that TMA as TTP or aHUS.
Figure 2 illustrates such a scheme to unravel the etiology of an HIV-associated TMA. First, one must rule out possible accompanying infections or other conditions that might have exacerbated a known HIV complication. In our Case, this involved presentation with exacerbation of her chronic HIV-linked renal disease. Next, establish the existence of a microangiopathic hemolytic anemia—our patient had a markedly elevated LDH, low haptoglobin, thrombocytopenia, and schistocytosis. Third, eliminate most cases of TTP by obtaining an ADAMTS13 that is >5–10%. Finally, with aHUS now a primary consideration, one should attempt to identify a complement amplifying condition that could initiate a TMA in a genetically susceptible individual. In our Case that was simple as she presented with one: uncontrolled HIV infection. Adjuncts to our clinical diagnosis of aHUS could have included biopsy and genetic analyses. They were considered unnecessary in our Case, but are reviewed briefly below.
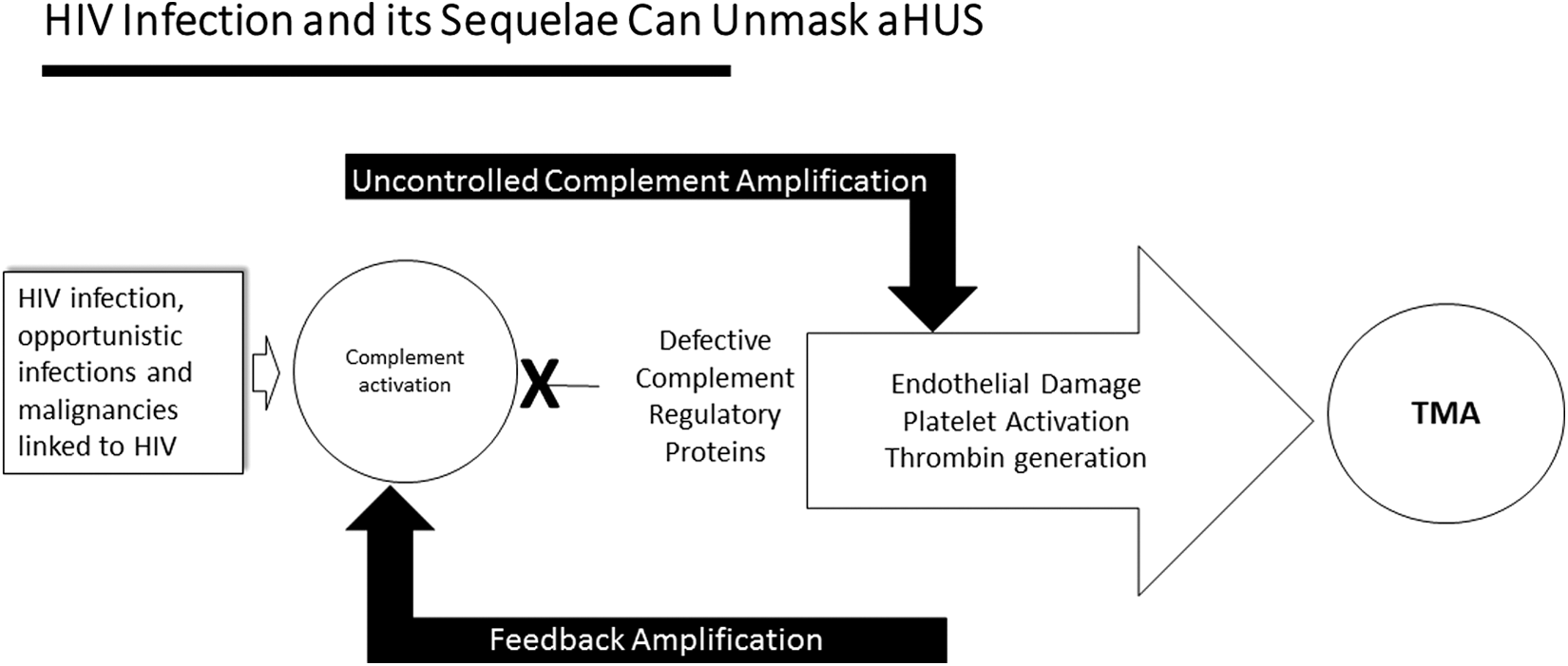
A general scheme for determination of an aHUS-type TMA in the setting of HIV infection. aHUS, atypical hemolytic uremic syndrome; TMA, thrombotic microangiopathy.
In terms of TMA treatment, while waiting results of the ADAMTS13 activity and inhibitor assays, most patients will benefit, in the short term, from FFP infusions or PEx. Our patient may have benefited, at least in terms of LDH and haptoglobin, but her clinical symptoms worsened. If her ADAMTS13 activity had been <5% of normal control levels (or <10% in some assays), the diagnosis should have been TTP. In this case, PEx would have been continued. If, after 5–9 PExs, each representing replacement of 1–1.5 plasma volumes, there had been only minor responses in laboratory and clinical parameters, other treatment modalities shown to be effective in recalcitrant TTP, including immune suppression (e.g., rituximab, mycophenolate mofetil), would have been considered. 13 In our Case, the ADAMTS13 activity was in the normal range and the diagnosis was thus aHUS. PEx was immediately stopped and specific, complement-based treatment with eculizumab instituted. Complement-based assays have been used by some groups in an attempt to confirm the diagnosis of aHUS, but they have no relationship to treatment response and are of little utility. In our Case, they were all within normal ranges.
It was noted above that the susceptibility to aHUS rests on a congenital problem: mutation in one or more complement-related genes. However, such mutations of soluble and membrane-linked complement regulatory proteins have been documented in only 60–70% of classic aHUS cases responsive to eculizumab, with or without HIV involvement, and are thus an unreliable means of excluding aHUS. 23 –25 (It is hypothesized that more extensive, genome-wide sequencing will eventually reveal complement-related abnormalities in most aHUS cases. Such analyses will be complicated by the fact that nonsynonymous mutations, involving amino acid substitutions, in CFH, the most frequently involved protein in aHUS, are present in 5% of healthy controls, with no family history of TMA. 26 ) In addition, such testing takes months and is expensive.
Biopsies may serve as a guide in difficult diagnostic situations. Sampling of any accessible, highly vascular site can help inform the differential diagnosis of a TMA, based on pathologic distinctions between TTP and aHUS that were discussed above. These differences have been recognized postmortem for a couple of decades, 20 but until recently, rarely used antemortem. Gingiva, rectum, or skin are suggested sites, regardless of whether there is an apparent lesion. 27,28 Renal tissue can also be examined, 29 although it is more difficult to obtain, especially in the setting of thrombocytopenia. The microthrombi of TTP are typically “white clots,” composed of platelets and von Willebrand factor, with only small amounts of fibrin, and vascular or perivascular inflammatory cell infiltrations are minimal or absent. 30 In contrast, biopsy of similar sites in aHUS typically reveals “red clots” in which fibrin predominates, and an inflammatory infiltrate may be seen 20,30 together with deposits of C5b-9 in the absence of immunoglobulin. 27,28 It must be emphasized, however, that the sensitivity and specificity of biopsy in distinguishing TTP from aHUS, or in classifying a TMA arising in the setting of HIV or other complement activating disorders, have not been authenticated in clinical trials.
In their 1998 review of the clinical features of HIV-associated TMAs, Dr. Viale et al. observed that these disorders “appear to have an ominously increasing incidence” in HIV/AIDS but that “there is probably no other disease in which prompt diagnosis and treatment can lead to as great a difference in clinical outcome as with TMA.” 31 Some two decades later that first statement is thankfully much outdated. However, the second comment remains highly accurate, cogently arguing for better diagnosis, and specific treatment, of TMAs occurring not only in the setting of HIV but also when unmasked by other complement-amplifying conditions, from pregnancy and malignant hypertension to allotransplantation and cancer. 1,11,32
Footnotes
Acknowledgments
This work was supported by an ongoing grant from the Angelo Donghia Foundation.
Author Disclosure Statement
J.L. is on the Speakers' Bureau of Alexion Pharmaceuticals, Inc., the manufacturer of eculizumab (Soliris®). All other authors have no conflicting financial interests.