
Abstract
Little information exists on the frequency, severity, and timing of first-line anti-tuberculosis drug-related adverse events (TB-AEs) in HIV-tuberculosis coinfected (HIV-TB) patients in the antiretroviral therapy (ART) era. This matched-cohort study included HIV-TB patients as cases and HIV-uninfected tuberculosis (non-HIV-TB) patients as controls. Tuberculosis was culture-confirmed in both groups. Cases were matched to controls in a 1:4 ratio on age, sex, and year of diagnosis. TB-AEs were defined as Grade 2 or higher requiring drug discontinuation/regimen change. From 2003 to 2015, 94 cases and 376 controls were analyzed (95% men, 98% Asians). Standard four-drug combination therapy was initiated in 91% of cases and 89% of controls (p = 0.45). Cases had a higher frequency of TB-AE [51% (48/94) vs. 10% (39/376), p < 0.001]. Their major TB-AEs were fever (19%), rash (11%), and neutropenia (11%). TB-AEs were more severe in cases [Grade 3 or higher: cases (71%, 34/48) vs. controls (49%, 19/39), p < 0.001]. The time from treatment initiation to TB-AE was shorter in cases [median 18 (interquartile range 12–28) vs. 27 (15–57) days, p = 0.027], and 73% of TB-AEs in cases occurred within 4 weeks of starting anti-tuberculosis treatment. HIV infection was an independent risk factor for TB-AEs in the multivariate Cox analysis [adjusted HR (aHR): 6.96; 95% confidence interval: 3.93–12.3]. TB-AEs occurred more frequently in HIV-TB than in non-HIV-TB patients, and were more severe. The majority of TB-AEs occurred within 4 weeks of initiating anti-tuberculosis treatment. Because TB-AEs may delay ART initiation, careful monitoring during this period is warranted in coinfected patients.
Introduction
T
One of the most severe anti-tuberculosis drug-related adverse reactions is liver injury, 2 and risk factors for hepatotoxicity have been rigorously investigated. 7,8 However, little evidence is available regarding the frequency, severity, and timing of adverse reactions in HIV-TB patients. 6,9,10 Hence, the aim of this study was to evaluate the frequency and severity of each adverse reaction related to first-line anti-tuberculosis drugs, and the time to occurrence of such reactions, in HIV-TB patients in the ART era, using a well-matched cohort of HIV/TB patients and HIV-uninfected tuberculosis (non-HIV-TB) patients.
Materials and Methods
Study design and setting
This matched-cohort study was designed to compare the frequency, severity, and timing of each adverse reaction to first-line anti-tuberculosis drugs in HIV-TB and non-HIV-TB patients. This study was conducted at the National Center for Global Health and Medicine (NCGM), a tertiary care hospital with 781 inpatient beds in Tokyo, Japan. NCGM has an AIDS Clinical Center, one of the largest referral centers for HIV care in Japan, and a tuberculosis isolation ward with 40 beds. The data for this study were obtained through a chart review.
This study was approved by the Institutional Review Board at the NCGM (NCGM-G-001933-00) and conducted according to the principles expressed in the Declaration of Helsinki. The need for informed consent was waived because this study only used the data gained from clinical practice.
Study population
Cases
We included all HIV-TB patients managed at the NCGM between January 2003 and October 2015. HIV-1 infection was confirmed either with HIV-1 RNA polymerase chain reaction or HIV-1 Western Blot assay. Tuberculosis was defined as isolation of M. tuberculosis from culture specimens from sites of infection (see detailed microbiology test information in the Supplementary Data; Supplementary Tables S1–S4 are available online at
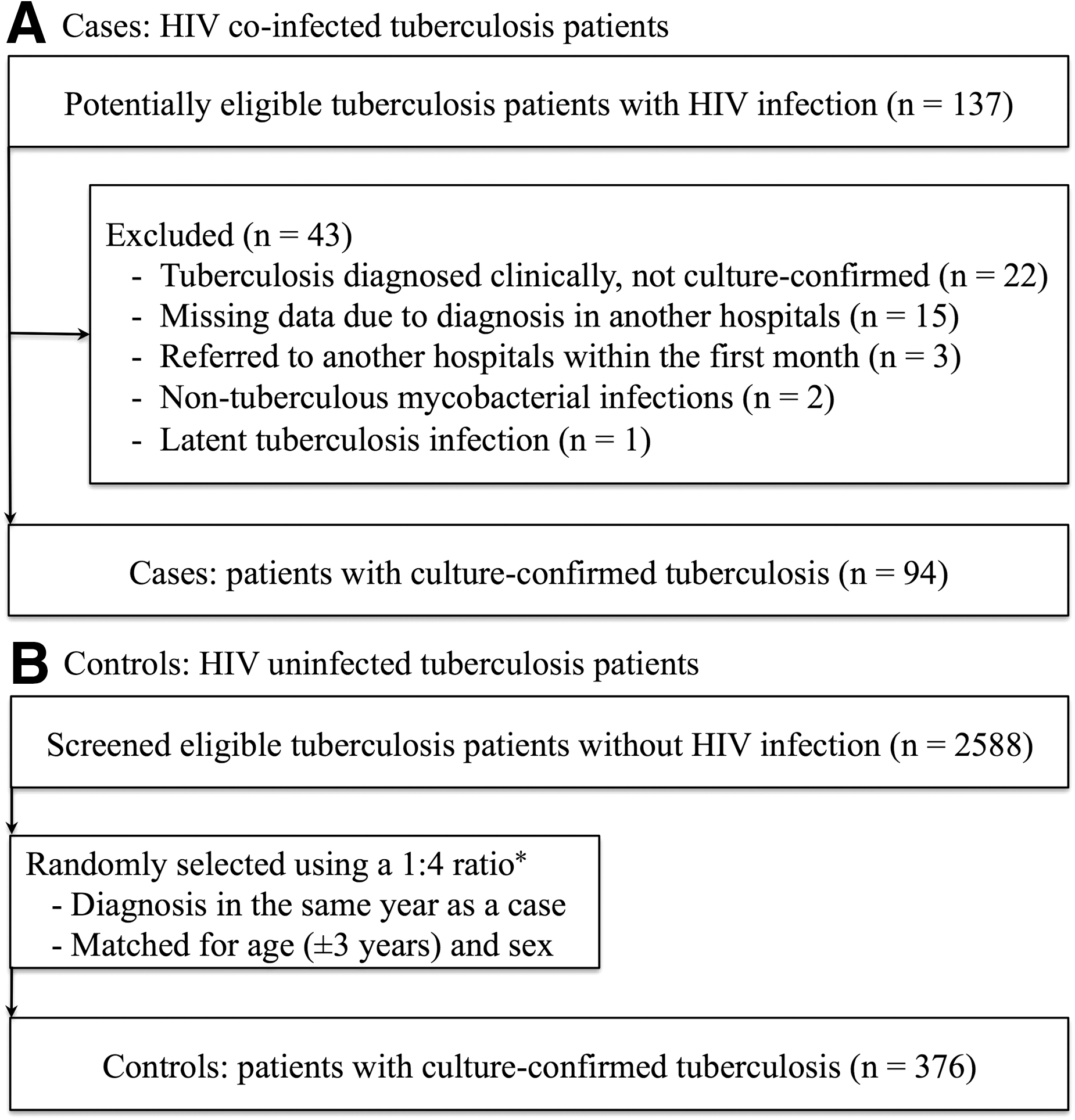
Flow diagram of
Controls
Tuberculosis patients without HIV infection during the study period were selected as potentially eligible controls (n = 2588). HIV negativity was confirmed by either third-generation HIV antibody assay or fourth-generation HIV antigen/antibody assay. The controls, matched to the cases for age (±3 years), sex, and the year of diagnosis, were randomly selected from the set of 2588 patients using the RAND function of Microsoft Excel, Office for Mac 2011 (Microsoft, WA), in a ratio of 4 controls to 1 case. The potentially matched controls were assigned a random patient number. We then confirmed whether they satisfied the inclusion and exclusion criteria, following an ascending order of the random numbers until the number of matched controls reached four to each case. Those who were not included as a control were returned to the set of potentially eligible controls to become a control candidate for the next case. In the process of control selection, 57 patients were excluded and 376 culture-confirmed tuberculosis patients without HIV-1 infection were included in the control group for analysis (Fig. 1B).
Measurements and definitions
Baseline characteristics were collected at the start of anti-tuberculosis treatment, which generally coincided with the date that the diagnosis of tuberculosis was confirmed. Baseline characteristics included age; sex; ethnicity; body mass index (BMI); serum albumin level; CD4 count and the use of ART (for cases); homelessness; history of previous tuberculosis treatment; and medications such as steroids, immunosuppressants, and chemotherapy. Comorbidities included chronic hepatitis B or C virus (HBV/HCV) infection, defined by detectable serum HBV surface antigen or HCV RNA, respectively. Alcoholic liver dysfunction was based on the diagnosis made by hepatologists. Diabetes mellitus and chronic kidney disease were defined based on the guidelines. 11,12 Multidrug-resistant (MDR) tuberculosis and extensively drug-resistant (XDR) tuberculosis were based on standard definitions. 13 A paradoxical reaction was defined as worsening of tuberculosis during effective anti-tuberculosis treatment without ART initiation. Tuberculosis-associated immune reconstitution inflammatory syndrome (TB-IRIS) was defined based on the guidelines. 4
Anti-tuberculosis drug-related adverse reactions, including symptoms or abnormal laboratory data, which resolved with discontinuation and/or relapsed after readministration of the drug(s), were extracted from the medical chart. Severity of the adverse reactions was classified in accordance with the Common Terminology Criteria for Adverse Events Version 4.0 (CTCAE). 14 An adverse event due to anti-tuberculosis drugs was defined as an adverse reaction of Grade 2 or higher, which required anti-tuberculosis drug discontinuation or regimen change. 15 Regarding hepatotoxicity, to distinguish adverse reactions from hepatic adaptation, 2 asymptomatic Grade 2 hepatotoxicity events were excluded, in accordance with the guideline's recommendation. 4 Paradoxical reactions and TB-IRIS were not regarded as adverse events. All adverse events were analyzed; however, only the initial event for each patient was used in the analysis of the time to occurrence of adverse events. Patients were censored at the time of loss-to-follow-up (such as treatment interruption by themselves or referral to other hospitals), tuberculosis-related or tuberculosis-nonrelated death, and drug discontinuation/regimen change with Grade 1 adverse events or regimen change due to detection of a resistant strain.
Treatment and follow-up
A once-daily regimen comprising the following anti-tuberculosis drugs was administered during the initial 2-month phase of treatment: isoniazid 5 mg/kg/day, rifampicin 10 mg/kg/day or rifabutin 5 mg/kg/day, ethambutol 15 mg/kg/day, and pyrazinamide 25 mg/kg/day. Isoniazid and rifampicin or rifabutin were used for the continuation phase of treatment, which lasted 4–10 months. In general, patients with pulmonary tuberculosis were treated for 6–9 months and patients with central nervous system, disseminated, or bone infection were treated for 12 months. All sputum smear-positive pulmonary tuberculosis patients were hospitalized in the tuberculosis isolation ward until three consecutive sputum smears, examined at 1-week intervals, were negative after at least 2 weeks of anti-tuberculosis treatment. For all inpatients, 100% adherence to medication was guaranteed; all outpatients underwent directly observed therapy at a public health center until the end of the treatment. Adverse reactions, paradoxical reactions, and TB-IRIS were screened for with routine clinical, laboratory, and chest X-ray monitoring every 1–2 weeks for inpatients, and at least every 1–2 months for outpatients. Treatment outcomes were observed at least every 6 months until 2-year post-treatment at the NCGM or public health center.
Statistical analyses
Baseline characteristics were compared between cases and controls using chi-square (χ 2 ) or Fisher's exact tests for nominal variables and Mann–Whitney U tests for continuous variables. The Kaplan-Meier method was used to evaluate the cumulative occurrence of adverse events in the two groups, and the log-rank test was used to determine statistical significance. Multivariate Cox proportional hazards regression was used to estimate the impact of HIV-1 infection on anti-tuberculosis drug-related adverse events. The model was adjusted for age >60 years and sex, because age >60 years and female sex were identified as risk factors for anti-tuberculosis drug adverse reactions in previous studies. 9 The model was also adjusted for a BMI <18.5 kg/m2 (identified as a confounder for liver injury), 7,16 chronic HBV infection (a risk factor for hepatotoxicity), 7 and extrapulmonary tuberculosis (that was detected significantly more frequently among cases). A two-tailed p value <0.05 was regarded statistically significant, and the hazard ratio (HR) and 95% confidence interval (95% CI) were used. All analyses were performed using SPSS for Windows, Version 21 (IBM Corp., Armonk, NY).
Results
Demographics and clinical characteristics of cases and controls
For the 94 HIV-TB cases, the median age was 39 years, 98% were men, and 16% were taking ART. The median CD4 count was 117 [interquartile range (IQR) 51–226] cells × 109/L and HIV viral load was 5.08 (IQR 4.49–5.51) log10 copies/mL (Table 1). Age and sex were not significantly different between the cases and controls; however, the cases had a higher frequency of chronic HBV infection (p < 0.001) than the controls, whereas the controls had a higher frequency of Asian ethnicity (p = 0.031) and homelessness (p = 0.009). Six cases had a malignancy (4 Kaposi's sarcoma, 1 Burkitt lymphoma, and 1 HCV-related hepatocellular carcinoma); all 10 controls with malignancy had a solid cancer. Standard four-drug combination treatment for tuberculosis (aforementioned in the methods) was initiated in 91% of cases and in 89% of controls (p = 0.45). Due to pregnancy or unstable/advanced liver dysfunction, 6% of cases and 5% of controls were treated with isoniazid, rifampin/rifabutin, and ethambutol without pyrazinamide. MDR tuberculosis was more frequently seen in cases than controls (4% vs. 1%, p = 0.016). Extrapulmonary tuberculosis was more common among the cases (67% vs. 28%, p < 0.001). In addition, tuberculous lymphadenitis (31% vs. 2%, p < 0.001) and miliary tuberculosis (22% vs. 2%, p < 0.001) were more frequent in cases, whereas tuberculous pleuritis was more common in controls (6% vs. 11%, p < 0.001) (Table 2).
ART, antiretroviral therapy; BMI, body mass index; HBV, hepatitis B virus; HCV, hepatitis C virus; HREZ/HBEZ, isoniazid, rifampicin/rifabutin, ethambutol, and pyrazinamide; IQR, interquartile range; MDR, multidrug resistant; NA, not applicable; TB, tuberculosis; XDR, extensively drug resistant.
All infection sites are included in Table 2 (duplicate count).
Subcutaneous, iliopsoas, or spleen TB were included in the category “abscess.”
Three and eight patients with vertebral osteomyelitis were included in HIV-infected and HIV-uninfected groups, respectively.
Prostate, renal, or epididymis TB was included in the category “urinary tract TB.”
TB, tuberculosis.
Drug-related adverse events
Main analyses: frequency, severity, and timing of adverse events
Anti-tuberculosis drug-related adverse events occurred more frequently in cases than in controls [51% (48/94) vs. 10% (39/376); p < 0.001], although a greater proportion of cases were treated with steroids (13% vs. 3%, p < 0.001), which might mask adverse reactions such as fever and rash (Table 3). 17,18 Major adverse events observed in cases were fever (19%), rash (11%), and neutropenia (11%), while major events observed in controls were hepatobiliary dysfunction (6%) and rash (3%). Further, the adverse events were more severe in cases; Grade 3 or higher adverse events were more frequently observed in cases than in controls [71% (34/48) vs. 49% (19/39), respectively, p < 0.001].
Adverse event was defined as regimen change or anti-tuberculosis drug discontinuation due to adverse reactions with Grade 2 or higher. One case and 10 controls were censored due to drug discontinuation or regimen change following Grade 1 adverse events, and 2 further censored as they had asymptomatic Grade 2 hepatotoxicity.
Reasons for steroid use in cases were TB-associated immune reconstitution inflammatory syndrome (n = 7), tuberculous meningitis (n = 2), pneumocystis pneumonia (n = 2), and Burkitt lymphoma (n = 1).
IQR, interquartile range; TB, tuberculosis.
Moreover, the time from anti-tuberculosis drug initiation to the occurrence of adverse events was shorter in cases than in controls [median 18 days (IQR 12–28) vs. 27 (15–57) days; p = 0.027], as displayed in the Kaplan-Meier curve (log-rank test, p < 0.001) (Fig. 2). Among patients with adverse events (48 cases and 39 controls), in 50% (24/48) cases and 18% (7/39) controls, the events occurred within 2 weeks of anti-tuberculosis drugs initiation (p < 0.001), and in 73% (35/48) cases and 54% (21/39) controls within 4 weeks (p < 0.001). Further adverse events occurred in 85% (41/48) cases and 77% (30/39) controls within 8 weeks (p = 0.218).
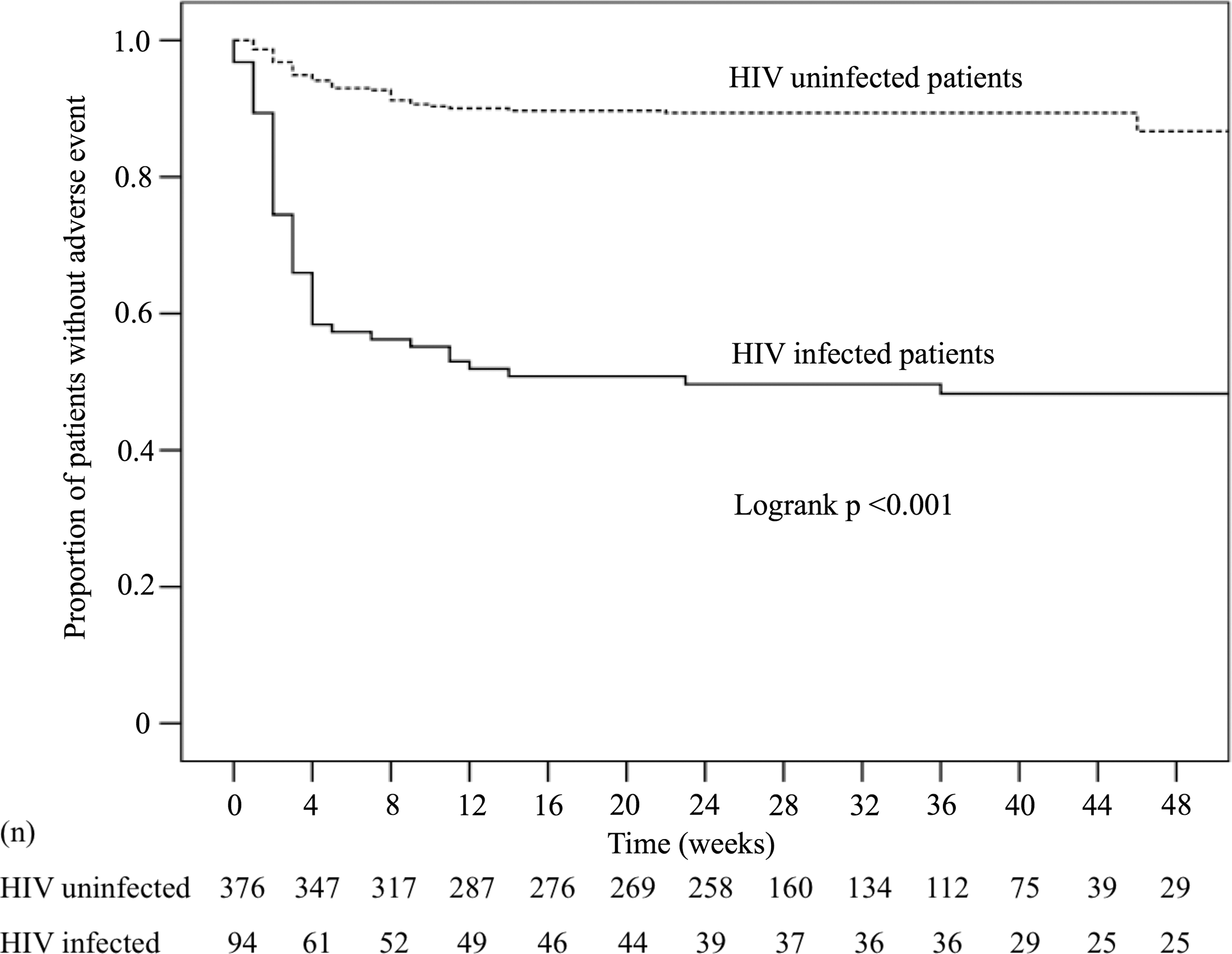
Kaplan-Meier curve of the time to the occurrence of moderate-to-severe anti-tuberculosis drug-related adverse events. The median observation period of the adverse events in cases and controls was 26 (IQR 9–39) weeks. Frequency of adverse events in HIV-infected tuberculosis patients was significantly higher than that in HIV-uninfected tuberculosis patients (p < 0.001, log-rank test). IQR, interquartile range.
Main analyses: risk factor of adverse events
Univariate analysis showed that HIV-1 infection (HR: 9.02; 95% CI: 5.35–15.2, p < 0.001), HBV infection, and extrapulmonary tuberculosis were significantly associated with the occurrence of adverse events (Table 4). HIV-1 infection remained an independent risk factor for adverse events even after adjustment [adjusted HR (aHR): 6.96; 95% CI: 3.93–12.3, p < 0.001].
CNS involvement included meningitis and brain tuberculoma.
BMI, body mass index; CI, confidence interval; CNS, central nervous system; HBV, hepatitis B virus; HCV, hepatitis C virus; HR, hazard ratio; TB, tuberculosis.
Subgroup analyses: characteristics of HIV-TB cases with adverse events
Of 48 HIV-TB cases with adverse events, 41 (85%) cases were ART naive at the time of adverse event, and 6 (13%) were taking ART before the time of tuberculosis diagnosis and continued ART at the time of adverse event. The remaining one case experienced isoniazid-induced neuropathy 21 days after ART initiation (96 days after anti-tuberculosis drugs initiation). HIV viral load was higher in cases with Grade 3 or higher adverse event than those with Grade 2 [median 5.34 (IQR 4.87–6.09) vs. 4.84 (IQR 4.13–5.14) log10 copies/mL, p = 0.018] (Supplementary Table S1). Extrapulmonary tuberculosis was more frequent in cases with Grade 3 or higher adverse event than those with Grade 2 [82% (28/34) vs. 50% (7/14), p = 0.029]. There was no relationship between timing [≤2 weeks (n = 24) vs. >2 weeks (n = 24)] or nature [fever and/or rash (n = 24) vs. others (n = 24)] of the adverse event and variables (BMI, CD4 count, HIV viral load, HBV coinfection, extrapulmonary tuberculosis, and ART use) (Supplementary Tables S2 and S3).
Other clinical outcomes
Seventy-five cases initiated ART within a median of 13 (IQR 10–23) weeks of anti-tuberculosis drug initiation. TB-IRIS occurred in 18 (24%) of these patients (Supplementary Table S4). One (1%) patient started ART within 2 weeks and 33 (44%) started between 2 and 12 weeks after anti-tuberculosis drugs initiation. Intergroup differences for all-cause mortality were insignificant (3% vs. 2%; p = 0.44), and no tuberculosis-related death occurred in the cases. The causes of death for three cases were Burkitt lymphoma, hepatocellular carcinoma, and chronic heart failure. In these three cases who died, ART was started 1, 8, and 8 weeks after anti-tuberculosis drugs initiation, respectively.
Discussion
In this matched-cohort study, we evaluated the frequency, severity, and time to occurrence of first-line anti-tuberculosis drug-related adverse events in HIV-TB patients compared with non-HIV-TB patients in the established ART era. Our study revealed that adverse events occurred more frequently among HIV-TB cases (51%) than non-HIV-TB controls (10%) and adverse events were more severe in the cases. Adverse events also occurred earlier in cases than controls, as 73% and 54% of adverse events, respectively, occurred within 4 weeks of initiation of anti-tuberculosis treatment. These study findings warrant the careful monitoring of HIV-TB patients after initiation of anti-tuberculosis treatment, especially for the first 4 weeks, and provide useful information on distinguishing adverse reactions of anti-tuberculosis drugs from these of ART, and on the timing of ART initiation in HIV-TB patients.
This study has three important strengths. First, to our knowledge, this is the first study that investigated the frequency, severity, and time to the occurrence of adverse reactions of first-line anti-tuberculosis drugs among HIV-TB patients in the established ART era. Two previous studies investigated this issue during the pre-ART or early-ART era (1996–1999 and 1997–2003), but it might be difficult to apply their findings to current clinical practice, given the difference in antiretrovirals used then (compared with now) and their likeliness to cause adverse reactions (e.g., didanosine-induced peripheral neuropathy or indinavir-induced nephrotoxicity). 6,19 Further, although one study from Rwanda in the ART era (2008–2009) also reported on anti-tuberculosis drug-related adverse events in HIV-TB patients, they included both culture-positive and clinically diagnosed tuberculosis cases, and, importantly, their definition of adverse events included not only adverse reactions but also concurrent neoplasms/infections and paradoxical reactions/TB-IRIS. 16
Second, our study is the first to use a matched-cohort design to compare the characteristics of adverse reactions between patients with HIV-TB and non-HIV-TB. This study matched the controls to the cases on age, sex, and the year of tuberculosis diagnosis in a 4:1 ratio; however, the previous studies only compared HIV-TB and non-HIV-TB patients in a cohort. 6,20 As a result, our study design enabled us to show that adverse events were not only more frequent in HIV-TB than non-HIV-TB patients (51% vs. 10%) but were also more severe, although the mechanisms by which adverse events were observed more frequently in HIV-TB patients are still under discussion.
Third, our study compared the frequency of each adverse event between HIV-TB and non-HIV-TB patients and showed that major adverse events in cases and controls were different. Among the cases, fever (19%), rash (11%), and neutropenia (11%) were major adverse events, whereas in the controls, hepatobiliary dysfunction (6%) and rash (3%) predominated. The results of previous studies reporting the frequency of each adverse event related to anti-tuberculosis drugs in HIV-TB patients differ. A study from Namibia reported that abdominal pain was the most common adverse event, 21 whereas liver dysfunction and neuropathy were most common in a Brazilian study 10 and British study, 6 respectively. Interestingly in our study, even though a greater proportion of cases than controls received steroid treatment, which might mask adverse events such as fever and rash, 17,18 fever and rash were the two most frequent adverse events experienced by the cases. Fever and rash were also the most common adverse events in a Canadian study of mostly non-HIV-TB patients, and that study showed that Asian-born patients were more likely to experience serious adverse events than patients of other ethnicities. 9 The frequency and susceptibility of anti-tuberculosis drug-related adverse reactions might vary according to genetic background, considering studies that have reported the association between single-nucleotide polymorphisms in genes such as N-acetyltransferase 2, cytochrome P450 2E1, and glutathione S-transferases, and isoniazid-induced hepatotoxicity. 22 –24 Further, one Korean study showed that a variant in adenosine triphosphate-binding cassette transporter might be a risk factor for anti-tuberculosis drug-induced rash in Asian populations. 25
This study has several limitations. First, this was a single-center study that consisted mostly of Asian men. Thus, further studies might be needed to confirm whether the findings are applicable to females and people of other ethnicities. However, considering the scarcity of data on this topic, it is noteworthy that the results of our study were derived from a well-designed matched-cohort study with a large number of tuberculosis patients. Importantly, this study was conducted in a resource-rich setting where monitoring of drug adherence and adverse events were strictly conducted as described in the Materials and Methods section. Second, we used the following definition for adverse events: symptoms/signs or laboratory data of Grade 2 or higher, which resolved on discontinuation of the drug and/or relapsed after readministration of the drug. This definition, including Grade 2, might have overestimated the frequency of adverse events. However, even if we analyzed only events of Grade 3 or higher, the results were the same; adverse events occurred more frequently in cases than in controls [34 of 94 (36%) vs. 19 of 376 (5%), respectively; p < 0.001], and among the cases, 82% of the events occurred within 4 weeks of initiation of anti-tuberculosis drugs. Further, our definition of the adverse events required drug discontinuation or regimen change due to adverse events, and we believe this definition enabled our study findings to be representative of actual clinical experience.
This matched-cohort study showed that moderate-to-severe anti-tuberculosis drug-related adverse reactions, requiring drug discontinuation or regimen change, occurred fivefold more frequently in HIV-TB patients (51%) than in non-HIV-TB patients (10%) and the adverse events were more severe in HIV-TB cases. In HIV-TB patients, 73% of such adverse events occurred within 4 weeks of initiation of anti-tuberculosis treatment. Hence, careful monitoring and appropriate management of such coinfected patients after initiation of anti-tuberculosis treatment are warranted because such adverse events can delay the initiation of ART.
Footnotes
Acknowledgments
The authors thank the physicians, nurses, and other staff at the AIDS Clinical Center and Department of Respiratory Medicine, NCGM, for their excellent work. We also thank Tetsuya Mizoue and the staff at the Department of Epidemiology and Prevention, Center for Clinical Science, NCGM, for advice regarding our study design. Funding: This study was supported, in part, by research grants from the Japanese Agency for Medical Research and Development, AMED. The funder had no role in this study such as study design, data collection, or decision to publish.
Authors' Contributions
T.M. devised the original idea for the study and collected the data. T.M. and T.N. led the analysis and drafted the original article. All authors contributed to the study design, interpretation of the data, and revision of the article for intellectual content. All authors have read and approved the final version.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.