
Abstract
In normal brain, neurons, astrocytes, and oligodendrocytes, the most abundant and active cells express pannexins and connexins, protein subunits of two families forming membrane channels. Most available evidence indicates that in mammals endogenously expressed pannexins form only hemichannels and connexins form both gap junction channels and hemichannels. Whereas gap junction channels connect the cytoplasm of contacting cells and coordinate electric and metabolic activity, hemichannels communicate the intra- and extracellular compartments and serve as a diffusional pathway for ions and small molecules. A subthreshold stimulation by acute pathological threatening conditions (e.g., global ischemia subthreshold for cell death) enhances neuronal Cx36 and glial Cx43 hemichannel activity, favoring ATP release and generation of preconditioning. If the stimulus is sufficiently deleterious, microglia become overactivated and release bioactive molecules that increase the activity of hemichannels and reduce gap junctional communication in astroglial networks, depriving neurons of astrocytic protective functions, and further reducing neuronal viability. Continuous glial activation triggered by low levels of anomalous proteins expressed in several neurodegenerative diseases induce glial hemichannel and gap junction channel disorders similar to those of acute inflammatory responses triggered by ischemia or infectious diseases. These changes are likely to occur in diverse cell types of the CNS and contribute to neurodegeneration during inflammatory process. Antiox. Redox Signal. 11, 369–399.
Introduction
The most sensitive CNS detectors of threatening conditions are microglia. Upon stimulation, microglial cells release high levels of pro-inflammatory cytokines, leading to dysfunctional astroglial communication (105, 150, 235, 314, 321). Direct communication between astroglial cells occurs mainly through gap junction channels (GJCs) (Fig. 1). Intercellular GJCs are believed to protect neurons through the spatial buffering of neurotoxic substrates. Moreover, reduced gap junctional communication between astroglial cells is associated with neurotoxicity (31, 271). In addition to intercellular channels, glial cells can also communicate through the release of small signaling molecules via hemichannels that open and close infrequently, but possibly enough to mediate paracrine cell interactions (Fig. 1). However, GJCs and hemichannels may exert opposite effects on neuronal viability, since overactivated glial cells show enhanced hemichannel activity (314) that very likely worsens their neurotoxic effects (352).
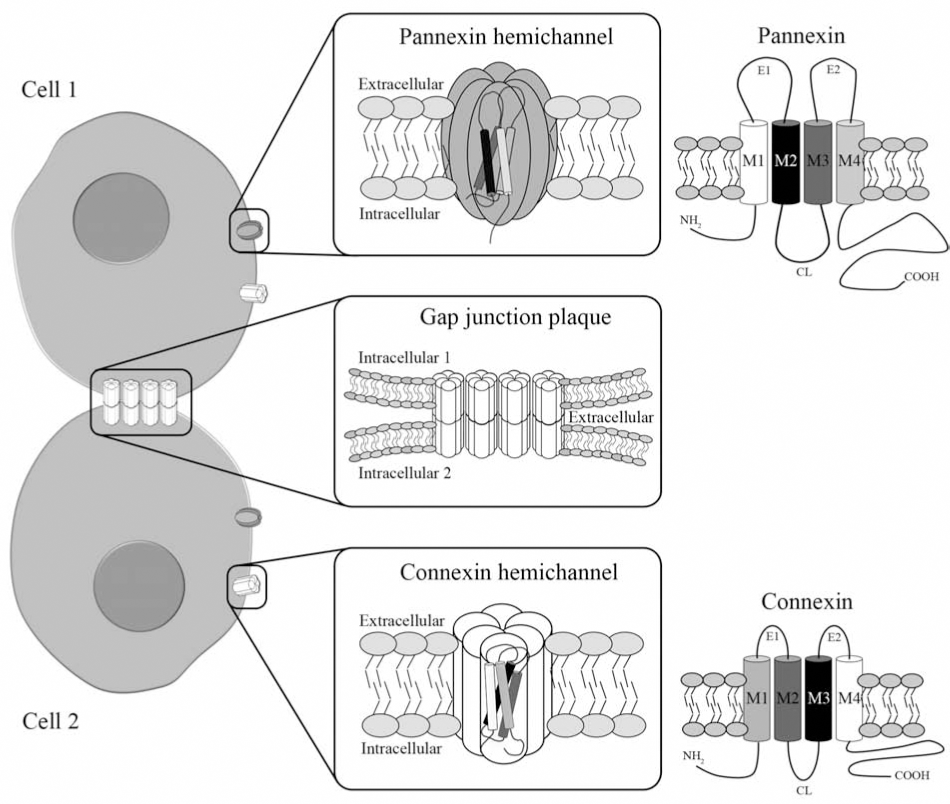
Gap junctions are plasma membrane specializations formed by the aggregation of tens to thousands intercellular channels that allow direct but selective cytoplasmic continuity between communicating cells. Each GJC spans the appositional plasma membranes of contacting cells and is formed by the serial docking of two hemichannels or connexons (Fig. 1). Each hemichannel is composed of six protein subunits termed connexins (Cxs), a family of highly conserved proteins encoded by at least 21 different genes in humans (388). Cxs are abundantly expressed in cells of the CNS, and they are named after their predicted molecular mass expressed in kDa, so that Cx43 has a molecular mass of ∼43 kDa.
Gap junctions allow the intercellular exchange of metabolites, such as ATP, ADP, glucose, glutamate, and glutathione, and second messengers including cAMP and inositol 1,4,5-triphosphate (128, 129, 170, 199, 265, 296, 328) and other molecules including antigens (236, 263). In addition, GJCs permit the intercellular spread of electrotonic potentials in excitable and nonexcitable tissues (60, 200, 319). All these functions depend on the protein subunit composition of GJCs which defines their conductance and permeability properties. While some GJCs are more permeable to anions, others show preference for cations or exhibit little charge selectivity (57, 95). Consequently, the potential physiological roles of GJCs are limited by the importance of signals and metabolites that can pass through them. Moreover, gap junctions can have an adhesive role (96, 208), and also may act through protein–protein interactions in the cytoplasmic face (125).
Recently, the presence of “free” hemichannels at the unapposed plasma membrane (Fig. 1) has been demonstrated using several experimental approaches (329). Exogenous expression systems have been used to study the electrophysiological and permeability properties of hemichannels, and identify mechanisms that control their activity. So far, all studied Cxs (Cxs 23, 26, 30, 30.2, 31.9, 35, 37, 38, 43, 44, 45, 45.6, 46, 48.5, 50, 52.6 or 56) expressed in exogenous systems, generate nonselective currents attributed to hemichannel openings (316). Most systems show little hemichannel activity in the presence of physiological [Ca2+]o (1–2 mM), but this activity can be markedly increased after removal of extracellular divalent cations. Although this regulation has been proposed to control opening of hemichannels under normal conditions (298) it might be more prominent under pathological conditions. In agreement with this possibility, an ∼10-fold reduction in extracellular concentration of divalent cations (Ca2+ and Mg2+) occurs in ischemic brain (187, 188, 210, 264). Moreover, in low [Ca2+]o, biologically relevant signaling molecules (e.g., ATP, glutamate, glutathione, NAD+, and PGE2) can be released through hemichannels (49, 72, 350, 396).
Opening of hemichannels in the presence of physiological concentrations of extracellular divalent cations (Mg2+/Ca2+) occurs in cortical astrocytes under metabolic inhibition (62), after oxygen and glucose deprivation (Orellana JA, Velarde V, Giaume C, Bennett MVL, and Sáez JC, unpublished data) or after exposure to pro-inflammatory cytokines (314). In addition, the activity of hemichannels composed of Cxs 43 or 45, but not Cx26, is enhanced by fibroblast growth factor-1 (FGF-1), mainly due to an increase in surface levels of hemichannels mediated by a rise in the [Ca2+]i (331). A parallel reduction in cell–cell communication through GJCs occurs under the latter conditions (62, 63, 65, 91, 105, 164, 204, 227, 235, 243, 253, 266, 313 –316, 331).
More recently, another gene family encoding a set of membrane proteins, named pannexins (Pxs), has been identified. Pxs have similar functional properties to Cxs and they have been found in various CNS cell types, mostly in neurons (see below). So far, three Px isoforms were discovered in mouse and human. Cxs and Pxs share similar topology, with four α-helical transmembrane domains connected by two extracellular loops and both intracellular N- and C-termini (Fig. 1). However, there is only 16% overall identity when their full-length amino acid sequences are compared.
Here we summarize the current knowledge on the functional modulation of hemichannels and GJCs in brain cells during inflammation, and we discuss its possible role in neurodegeneration.
Where are Connexins and Pannexins in the Mammalian CNS?
Pattern of connexin localization in the CNS
The expression of different Cxs varies according to the developmental stage, cell type and brain region (24, 30, 58, 84, 202, 230, 249, 251, 269, 333, 347, 369). Cx gap junctions form the most common type of electrical synapse between neurons in mammals and other vertebrates (Fig. 2A). Although early on the history dye transfer was found in crayfish septate axon, electrical synapses were first characterized by electrophysiological recordings and were later identified by electron microscopy of thin sections and freeze-fracture replicas in vertebrates (27). Despite the detection of Cxs 26, 30.2, 31.1, 32, 37, 40, 43, 47, and 57 in different neuronal types at different developmental stages (30, 58, 186, 202, 230, 249, 251, 347, 369), only Cxs 36 and 45 have been reproducibly identified at the ultrastructural level in neuronal plaques of the adult rat brain (116, 303, 305 –308). In fact, using immunogold labeling of freeze-fracture replicas, Cx36-based neuronal gap junctions were demonstrated in inferior olive, spinal cord, retina, olfactory bulb, visual cortex, suprachiasmatic nucleus, and locus coeruleus, whereas Cx45-based neuronal gap junctions have been found in the retina and olfactory bulb (116, 171, 303, 305 –308). Also, functional Cx57 GJCs are expressed between horizontal cells, and horizontal cell receptive fields are reduced in Cx57-deficient mice (152, 338). These findings are in agreement with demonstration of electrical coupling and detection of the Cx type in the corresponding brain regions (27). Cx36 knockout mice show a selective impairment of the hippocampal gamma oscillations (50). In addition, deficit in electrotonic neuronal coupling, behavioral impairments, and disrupted circadian rhythms in Cx36 knockout mice have demonstrated the functional relevance of Cx36 in adult brain (113, 154, 216).

So far, only one report has provided functional evidence supporting the existence of functional Cx36 hemichannels in neurons. In this study, cultured cerebellar granule cells expressing Cx36 show prominent ATP release after depolarization, which is blocked with Cx (but not Px) hemichannels blockers or down regulation of Cx36 with siRNA (333).
Cortical astrocytes express Cxs 26, 30, 40, 43, and 45 (81, 255, 256, 308), but only Cxs 26, 30, and 43 show colocalization at ultrastructurally-defined astroglial gap junctions (254, 308) (Fig. 2B) Cxs 30 and 43 have been detected in astrocytes of the visual cortex (318) and in astrocytic gap junctions of the CA1 and CA2 areas of the hippocampus (34). Nevertheless, single channel conductance studies of GJCs in cultured astrocytes reveal only currents consistent with those found in Cx43 transfectants (51, 82, 120, 191, 241). Cx26 is expressed by cultured astrocytes but is not detected at their cell–cell contacts areas (226). Moreover, cultured astrocytes from Cx43-deficient mice do not form functional gap junctions (261). Thus, Cx43 is thought to be the main functional Cx in cultured astrocytes. However, astrocytes co-cultured with neurons have been shown to express Cx30 (322). Gap junctional communication between astrocytes and neurons in adult mammals is still controversial (Fig. 2H). Although dye transfer and electrical coupling between astrocytes and neurons occurs in the locus coeruleus (11, 369) and in astrocyte–neuron co-cultures (114, 115, 323), there is still no evidence of ultrastructurally-defined neuroastroglial gap junctions in vivo (305). The discrepancy between the functional and the morphological observations might be related to the presence of small or less compact junctions that are difficult to identify with ultrastructural techniques.
Oligodendrocyte precursor cells become dye coupled after acquiring the commitment to the oligodendrocyte linage (381). Although there is no ultrastructural evidence of gap junctions between oligodendrocytes in the adult brain (304, 309), gap junctional communication between oligodendrocytes of the gray matter, but not in the white matter of the encephalon and spinal cord, has been consistently reported (80, 176 –178, 245, 277, 375). Oligodendrocytes of the gray matter express Cxs 29, 32, 36, 45, and 47 (9, 80, 189, 251, 274, 277, 309, 346, 347), and are extensively coupled to astrocytes via gap junctions (269). By double immunolabeling Cx32 is co-localized at heterocellular contacts with astrocytic Cxs 26, 30, 43, and Cx47 (10, 205) (Fig. 2). Recently, Orthmann–Murphy et al. (269), using electrophysiological recordings and immunolabeling, demonstrated heterotypic GJCs formed by Cx47/Cx43 and Cx32/Cx30.
In the adult rat brain, a few microglial cells express low levels of Cx43 (102), and primary cultures of resting microglia (which may be partially activated), express low levels of Cx36, Cx43 and Cx45 (88, 102, 274) (Fig. 2). After microglial activation with cytokines or peptidoglycan (PGN), Cx32 and Cx43 levels increase (102, 119, 361), which is accompanied by augmented functional gap junctions that are absent in microglia of Cx43 knockout mice, suggesting that under those conditions Cx43 is the main microglial gap junction protein (102, 119). In resting heterocellular cultures, approximately one-third of microglial cells show low-strength electrical coupling with neurons through GJCs formed by Cx36 (88). Cx36 has been proposed to form heterocellular gap junctions with neurons and might provide a new mechanism for transfer of death signals from microglia to neurons (88). Cx32 forms hemichannels through which neurotoxic glutamate concentrations can be released (361). Since Cx hemichannels are permeable to ATP (78, 172), it is possible that in microglia they provide a pathway for ATP release which stimulates microglial migration to the sites of injury (76).
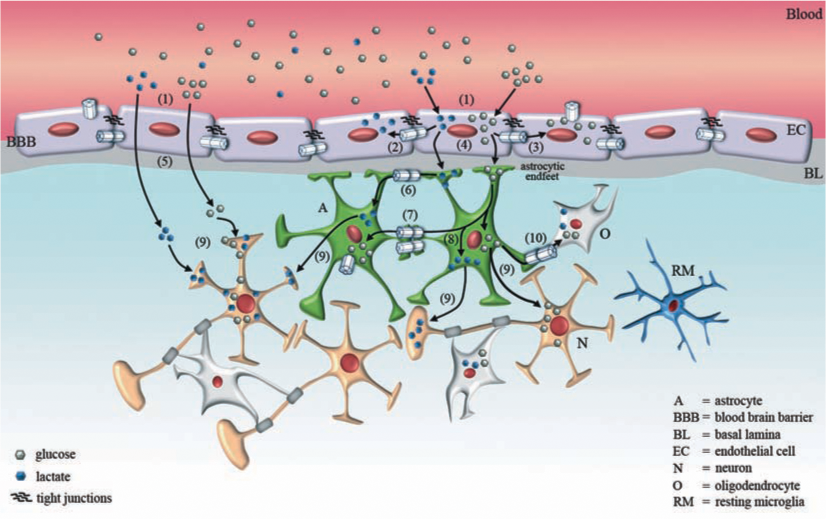
The blood-brain barrier (BBB) is another potentially important player in neurodegeneration because it contains the first cellular component affected by blood-borne pro-inflammatory agents and can also be influenced by pro-inflammatory agents generated in the brain parenchyma. In vivo, endothelial cells of the BBB express Cx43 and Cx40 (98, 211) (Fig. 2). Cell lines derived from the BBB are well coupled and intracellular photoliberation of inositol-1,4,5-trisphosphate induces Ca2+ waves that depend on extracellular ATP released through Cx hemichannels (44). Another functional interaction of Cx40 and Cx43 was reported in primary cultures of porcine BBB endothelial cells, in which these Cxs interact with tight junction proteins occludin, claudin-5, and ZO-1 (250). In this system, oleamide and β-GA, two gap junction blockers, permeabilize the barrier established by tight junctions as determined by measurements of transendothelial electrical resistance and mannitol and inulin paracellular flux (250). Cx37 and Cx43 also form myoendothelial gap junctions (137) that enable electrical coupling between muscle and endothelial layers and facilitate the synchronization of calcium waves and vasomotor activity along arterioles (137). Although, there is no in vivo evidence of direct intercellular coupling between astrocytes and endothelial cells at the capillary level, which might be prevented by the basal lamina between the two cell types, in vitro studies demonstrate the presence of weak electrical coupling between astrocytes and BBB endothelial cells, which is associated with propagation of both gap junction dependent and independent Ca2+ waves (43).
Several other brain cell types including ependymocytes, tanycytes, and leptomeningeal cells can also form gap junctions, but their possible role in brain diseases remain largely unexplored. Ependymocytes are glial cells that line the brain ventricles and the central canal of the spinal cord. They are highly coupled through gap junctions identified at the ultrastructural (304) and functional level (41, 130). Both Cx26 and Cx43 have been consistently detected in ependymocytes (84, 130, 393, 394), whereas presence of Cx30 in these cells has been controversial (190, 256). Interestingly, ependymocytes form Cx43 gap junctions with astrocytes in vivo as demonstrated in ultrastructural studies (304). Gap junctions between tanycytes, which are specialized ependymocyte, have been identified at the ultrastructural level (299) and primary cultures of these cells are dye and electrically coupled (162). To our knowledge, the Cx type expressed by tanycytes and the functional role of these junctions remain unknown. Gap junctional communication is wide spread in the developing and adult meningeal membranes, and cultured leptomeningeal cells are strongly dye coupled (83, 349). In cultured cells, dye transfer probably occurs through GJCs formed by Cx26, Cx30, and Cx43 that are expressed at high levels in these cells (237, 256). In vitro studies have also identified intercellular communication mediated by gap junctions at heterocellular meningeal cell–astrocyte contacts (132), but their functional significance remains speculative.
Pattern of pannexin localization in the CNS
Pxs are found throughout in the animal kingdom in Cnidaria as well as Bilateria. Three genes have been identified in mammals, termed Pxs 1, 2, and 3, (21, 48). They are homologous to innexins, a term used for gap junction proteins in invertebrates. Primary sequence analyses indicate a membrane topology of Pxs like that of Cxs (Fig. 1) (273). Information on the biosynthesis and assembly of Px channels is still lacking.
Px1 is widely expressed in mammalian tissues (21). In the brain, several regions including cortex, striatum, olfactory bulb, hippocampus, thalamus, inferior olive, inferior colliculus, amygdala, spinal cord, retina, and cerebellum show Px1 immunoreactivity (48, 311, 399). At the cellular level, Px1 has been localized in different neuronal types, including olfactory bulb mitral cells, Purkinje cells, and dopaminergic, cholinergic, and glutamatergic neurons (48, 311, 399). In the cerebral cortex and hippocampus, Px1 is localized in the postsynaptic cell membranes (406). Only one report showed Px1 presence in white matter (48), but others have failed to detect in vivo glial expression of Px1 mRNA (311, 379), except in the Bergmann glial cells of the cerebellum that are Px1 immunopositive (399). Px1 was also detected in cultured astrocytes, immature oligodendrocytes, and neurons under resting conditions using immunofluorescence and Western blot analysis, but it has not been found to locate in the cell surface of neurons and astrocytes (157).
Px2 is expressed exclusively in the brain (273), including the olfactory bulb, hippocampus, amygdala, superior colliculus, substantia nigra, cerebellum, hypothalamus, and spinal cord (48, 400). Under resting conditions, hippocampal astrocytes do not express Px2. However, Px2 is expressed in hippocampal astrocytes several hours after ischemia/reperfusion (400). It remains unknown whether it forms functional GJCs and/or hemichannels.
The extent to which Pxs can form functional GJCs remains unclear. Overexpression of Px1 in paired Xenopus oocytes, LNCaP, or C6 glioma cells increases intercellular coupling (48, 196, 373). However, other groups report that expression of Px1 in Xenopus oocytes, or N2A, Neuro2a, or HeLa cells does not induce intercellular communication (35, 157, 284). In addition, Px2 or Px3 do not induce intercellular currents in the oocyte expression system (48), and Px3 expression does not mediate the formation of detectable gap junctional communication in N2A cells (284). Up to now, there is no evidence of gap junction formation by endogenous Px1 in cultured neurons and astrocytes (157). However, functional Px1, Px1/Px2, and Px3 hemichannels have been demonstrated in several exogenous expression systems (20, 22, 47, 48, 215, 282, 284). Moreover, endogenous Px1 hemichannels have been reported in hippocampal neurons (364), but not in cortical astrocytes (157).
Activation of Px1 hemichannels occurs in hippocampal neurons submitted to glucose/oxygen deprivation (364). Moreover, ATP can increase Px1 hemichannels function through P2X7 or P2Y receptors transactivation in erythrocytes and macrophages (215, 282). In addition, the ATP-driven cryopyrin-mediated caspase 1 activation in response to bacterial stimuli requires Px1 activation (173). Activated Px1 hemichannels are permeable to Ca2+ and small molecules such as ATP and ethidium (281, 282), calcein (364), sulforhodamine (284, 364), and carboxyfluorescein (215). Px1 hemichannel activity is inhibited by intracellular acidification and classic Cx hemichannel blockers (such as carbenoxolone (CBX), β-GA and flufenamic acid, but they are insensitive to 1-heptanol, 1-octanol, La3+ and Gd3+ (282).
Glial Cells and Their Implication in Neurodegenerative Diseases
Microglial function and activation
Microglia, the CNS resident macrophages, represent ∼12% of total number of brain cells. Under normal conditions, they are sparse within the nervous tissue and present a resting phenotype characterized by a ramified morphology and weak expression of molecules associated with macrophagic function (32). During brain ontogenesis, microglia promote apoptosis in neuronal populations of the cerebellum (223) and cerebral cortex (368). However, they enhance neuronal survival in in vitro models (242), possibly by releasing trophic factors, for example, transforming growth factor-β (TGF-β), nerve growth factor (NGF), brain-dervied neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4). Thus, microglia are far from inactive in their resting state. Since in the adult rat brain only a few microglial cells express Cx43 at low levels (102) and primary cultures of resting microglia express low levels of Cx36, Cx43, and Cx45 (88, 102, 274), it is likely that basal microglial function is independent of Cx-based channels. Nonetheless, transient Cx expression and a role in specific cell functions cannot be conclusively ruled out. Moreover, the possible role of Px-based channels in microglial cells (339) remains unexamined.
Although microglial activation, also called microgliosis, plays a relevant role in the innate immune response that should be beneficial, their overactivation may be neurotoxic (32, 291, 351). Microglial activation occurs rapidly in response to acute brain insults, and cells acquire an amoeboid phenotype (397). In vitro, microglial activation can be induced by several stimuli, including hypoxia/reoxygenation (195), glutamate (175), lipopolysaccharide (LPS) (220, 324; Froger N, Rétamal MA, Sáez JC, and Giaume C, unpublished data), extracellular proteoglycans (52), prion peptides (23, 46), cell debris (29), amyloid-β peptide (Aβ) (59, 295), α-synuclein (402), and cytokines (244). Nevertheless, molecular mechanisms that determine whether the microglial activation is deleterious or favorable to neuronal survival are not completely understood.
Except for activation via a dectin-1-dependent pathway (337), activation of microglia is most frequently characterized by their synthesis and secretion of pro-inflammatory cytokines such as TNF-α, IL-1α/β, IL-3, IL-6, IL-8, IL-10, IL-12, IL-15, and IL-18 (142), several of which are neurotoxic (229, 363, 376). In this regard, microglia may act on neurons directly through the release of neurotoxic compounds (e.g., cytokines, oxygen- or nitrogen-derived free radicals) or indirectly by promoting astroglial overactivation and dysfunction (see below). In support of the latter, LPS-treated astrocytes are neurotoxic only in the presence of microglia (117, 124, 220, 324). A similar mechanism may operate after microglial activation by other cell insults (e.g., hypoxia/reoxygenation, ischemia/reperfusion, cytokines, Aβ, and α-synuclein). Overactivated microglia may lead to neuronal damage or even death in the normal tissue surrounding a primary affected focus, resulting in propagation of neuronal death. Local vasogenic and cytotoxic edema and damage to microvessels could generate a wave of hypoperfusion contributing to inflammatory response and cell death (see Fig. 8C). Activated microglia release glutamate through hemichannels (361) and their neurotoxic effect is blocked with glutaminase or the gap junction blocker CBX (395). Moreover, glutamate induces astrocyte edema (141, 185) and therefore contributes to the ischemic condition that propagates as a wave to the periphery of the ischemic core (see Fig. 8C). Microglia mediated neurotoxicity is known to be progressive over time (118, 158, 231) and may contribute to the progressive nature of various neurodegenerative diseases. In agreement with this notion, immunosuppression reduces delayed cell death (140).
Which cell type acts as the first threat sensor? Generally, low concentrations of pro-inflammatory molecules activate microglia but do not affect astrocytes or neurons on the same time scale. Concentrations of ∼1 μM of Aβ25–35 peptide are toxic to microglia in vitro (182), but concentrations above 20 μM Aβ25–35 are required to induce neuronal death (111, 217, 391). In primary cultures, gap junction-mediated coupling between activated microglia occurs in 30–50% of the cells, is limited to few cells (three–four), and is transient (119). A remarkably similar response has been described for Kupffer cells, the resident macrophages of the liver (103), and other cells of innate immune system such as dendritic cells (64), monocyte/macrophages (101), and granulocytes (45). The transient coupling might be a property of migratory cells and the low number of coupled microglia might be explained by their heterogeneity and various states of activation. Alternatively, the limited gap junctional communication might serve for direct transfer of signals between microglia during a brief period of time, but the functional implications could become evident long after the cells had ceased to be coupled. In other cell types, the formation of GJCs has been shown to require the expression of cell adhesion proteins (166, 248). Activated microglial are known to transiently express surface molecules, such as CD45, CD14, and intercellular adhesion molecule-1 (351), but their possible role in the formation of gap junctions between microglia remains unknown.
The role of gap junctions between activated microglia remains unclear, but studies in other macrophages and/or migratory cells suggest that they might coordinate several cellular functions, including synthesis and release of metalloproteinases (101), diapedesis (293), and antigen cross-presentation (236, 263). In addition, gap junctional communication between activated microglia might enhance the release of cytokines which in turn reduce gap junctions in parenchymal and/or mesenchymal cells. In agreement, LPS does not affect liver parenchymal cells but acts through Kupffer cells to induce downregulation of gap junctional communication between hepatocytes (131). Similarly, in microglia-free astrocyte cultures, LPS does not affect gap junctional communication, but it induces cytokine release from microglia in heterocellular cultures which reduces the astrocytic dye coupling (235, 314). Hence, block of microglial gap junctions might diminish the inflammatory response and consequently limit neurodegeneration. A major current limitation is that all available blockers would also block astroglial gap junctions which might induced adverse neuronal effects (see below). Alternatively, specific blockers of hemichannels that did not block GJCs might reduce the neurotoxic consequences of microglial activation.
Astroglial function and dysfunction
Over the last decade, the perspective on astrocytes, the most abundant brain cells, has changed from supporting cells to a cell type with multiple homeostatic properties. They do not only organize the structural architecture of the brain but also organize its communication pathways and plasticity (97, 262). An important feature of astrocytes is that they are highly coupled to each other, forming extensive intercellular networks (121) (Fig. 3 and Fig. 4). In specific brain area each tri-dimensional astroglial network is poorly coupled via gap junctions with other neighboring networks (155). Domains of resulting intercellular networks take up local extracellular K+ accumulated during episodes of high neuronal activity, and the excess of intracellular K+ is dissipated by simple diffusion towards network areas with lower [K+]o (Fig. 3). Depolarization spreads electrotonically in the coupled network; K+ ions are the major charge carrier in the cytoplasm and move in where extracellular K+ is high and out where it is low. Consequently, action potential depression is prevented in the area with high neuronal activity (201). The hippocampus of mice with coupling-deficient astrocytes retains a large capacity for K+ clearance, indicating that gap junction-dependent processes only partially account for the K+ buffering (383). The hippocampal CA1 is the most sensitive CNS region to global ischemia, and dysfunction of Cx-base channels different of gap junctions or hemichannels may play a more prominent role in this brain area.
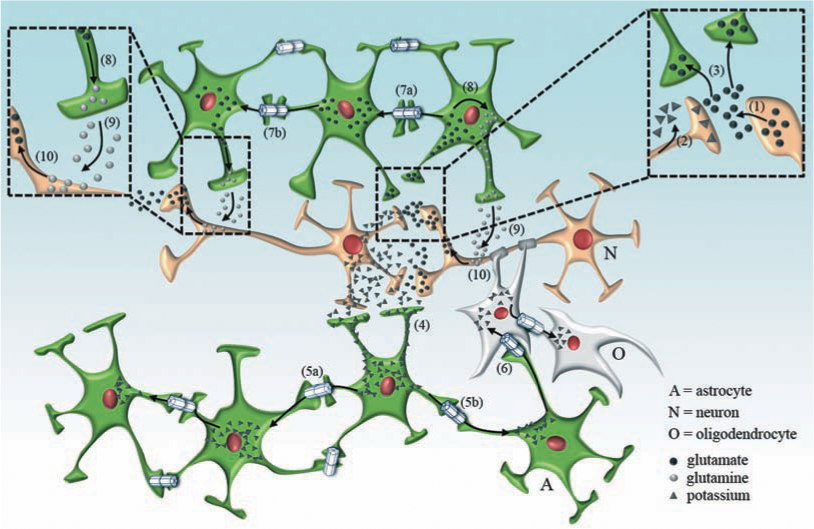
Astrocyte networks also contribute to maintaining H+ homeostasis (73) and preventing glutamate excitotoxicity in the brain (74, 334, 386) (Fig. 3). Glutamate accumulated in astrocytes is transformed to glutamine by glutamine synthase, and glutamine can be transferred to neurons to be recycled as a precursor of glutamate and/or GABA (148) (Fig. 3). In addition, glutamate and glutamine have been reported to permeate GJCs in cultured astrocytes (122). In the hippocampus, astroglial cells expressing AMPA-type glutamate receptors are not coupled via gap junctions, while those expressing glutamate transporters are very well coupled (383). A direct relationship between the expression of astroglial Cx43 based channels and expression of glutamate transporters has been recently demonstrated (107), suggesting that both glutamate transport pathways co-exist and might be under the control of the same transcription factors. Moreover, intercellular astrocyte communication is enhanced in a concentration-dependent manner after elevating extracellular K+ concentrations (77), suggesting that astrocyte depolarization might enhance gap junction-dependent neuroprotective functions (e.g., spatial buffering capacity and intercellular signaling).
Astrocytes release various neuromodulators and transmitters or so-called “gliotransmitters”, which include glutamate (276), GABA (7), ATP (297), taurine (219), adenosine (8), D-serine (240, 272, 332), and eicosanoids (247). The mechanisms underlying the gliotransmitter release are not clear, although a number of studies demonstrate release via hemichannels (350, 396) and vesicle-mediated exocytosis (42, 56, 276). Moreover, the activity of these secretory mechanisms is enhanced by elevation of [Ca2+]i (276, 294). For example, neural stimulation can increase [Ca2+]i in astrocytes via ionotropic and/or metabotropic ATP, glutamate, GABAB, and acetylcholine receptors (380). Increased [Ca2+]i might induce release of gliotransmitters from astrocytes, which increase [Ca2+]i of neighboring neurons, for example through an NMDA receptor-dependent pathway (276). Moreover, the increase in astroglial [Ca2+]i does not just occur locally but also can propagate as a Ca2+ wave through the astroglial network and affect the neural activity in distant locations (330).
A recently discovered astrocyte function is their involvement as signaling pathway in the neurovascular coupling (Fig. 4). This function is drastically reduced by inhibition of Cx-based channels, which mediate the propagation of Ca2+ waves and release of vasoactive arteriolar factors. Thus, astroglial cells have been proposed as sites for production of vasoactive factors in response to neuronal activity causing rapid and localized changes in cerebral blood flow upon changes in energy demand (239, 392, 407) (Fig. 4). Thus, astrocytes may nourish neurons by controlling the glucose availability through the regulation of blood flow as well as by generation of lactic acid in anaerobic metabolism for more efficient ATP generation in the tricarboxilic acid cycle (283) (Fig. 4). Physiopathological conditions that reduce the expression of Cx based channels in astrocytes should also have a negative impact in glucose tissue supply (Fig. 4), increasing the neuronal susceptibility to injuries.
Two mechanisms underlying Ca2+ wave propagation have received strong experimental support. One is mediation by the intercellular transfer of IP3, which spreads through GJCs and causes release of Ca2+ from intracellular stores (38, 123). The other is mediation by extracellular ATP acting as a paracrine messenger (66, 67, 123, 135, 143). Thus, ATP released from an astrocyte can activate purinergic receptors (P2X and P2Y) in surrounding cells, leading to increased [Ca2+]i (67). Release of ATP from these neighboring cells and then their neighbors allows the spread of a calcium waves to increase [Ca2+]i of distant cells. The mechanisms of ATP release from astrocytes are not well defined, but may include vesicle-mediated exocytosis (42, 56) and release through Cx43 hemichannels (62, 350). In addition, cultured astrocytes express Px1, which under resting in vitro conditions is not present in the surface membrane (157), but in particular conditions Px1 hemichannels may be inserted and provide a conduit for ATP release (20, 350). Moreover, a recent report describes the activation of an astrocytic macropore termed a maxi-anion channel in response to oxygen–glucose deprivation, which is resistant to several Px/Cx hemichannel blockers, but sensitive to Gd3+ and arachidonic acid (213). Further experimental confirmation of these findings and development of experimental paradigms to identify each channel type (formed of Cxs, Pxs, or unrelated proteins) will help to determine the contributions of each to Ca2+ wave propagation.
Cultured astrocytes express CD38, an integral membrane ectoenzyme also facing the lumen of the endoplasmic reticulum, and under resting conditions release NAD+ through hemichannels, possibly formed of Cx43 (377). CD38 may then generate cyclic ADP ribose for autocrine and paracrine signaling. It is possible that enhanced activity of hemichannels also enhances this intercellular signaling pathway.
Astrocytes are also involved in maintaining the redox potential. They produce glutathione, an important free radical scavenger, and transfer it to neurons (90, 374). This process is essential for protecting neurons from free radicals under adverse conditions such as ischemia and acute or chronic inflammation (374). Recently, hemichannels were demonstrated to act as an astrocytic pathway for release of glutathione to the extracellular milieu (302, 352). In addition, Cx43 GJCs are permeable to glutathione; thus, glutathione levels may be similar in a functional network. Nevertheless, during acute inflammation, astrocytes located within the affected area are likely to show enhanced hemichannel activity that would favor the release of glutathione to the interstitial space, and the reduced gap junctional communication will impair the replenishment of glutathione by diffusion from astrocytes located in less regions of the network. Because astrocytes perform numerous homeostatic functions (221) diverse insults that affect them can critically affect neuronal susceptibility to the same or different insults (14). Indeed, in ischemic episodes the metabolism of both neurons and astroglia is affected (138), and astrocytic functions such as glutamate uptake, dissipation of K+, and/or free radical scavenger activity are impaired contributing to the development of neuronal damage (276, 359).
In several CNS disorders, astrocytes can be activated by various inflammatory mediators, including cytokines (110, 365), lipoteichoic acid (181), LPS (233, 405), and Aβ (384). Some suggested mechanisms that underlie this effect include release of high levels of NO, peroxynitrite, glutamate, TNF-α, and IL-1β (18, 146). Astrocyte activation involves changes in cellular phenotype and gene expression which can be deleterious for neurons (18, 39, 149), Accordingly, activated astrocytes increase neuronal death in vitro (71, 156, 180).
A recent study revealed novel astrocytic gap junction functions. Uninfected astrocytes coupled through gap junctions with HIV-infected astrocytes become apoptotic (100). Gap junction blockers abolished apoptosis in uninfected astrocytes, supporting the role of these channels in amplifying cell death. Therefore, this is a novel mechanism of toxicity within the brain, triggered by low numbers of HIV-infected astrocytes and amplified by gap junctions or hemichannels that might contribute to neurodegeneration in AIDS (100). It would be of interest to study the role of Cx-based channels in HIV-infected astrocytes co-cultured with microglia or treated with inflammatory mediators that would mimic the AIDS inflammatory condition. A general cellular reaction observed after brain injury is that astrocytes adjacent to the damaged tissue undergo a process of hypertrophy and proliferation, called reactive astrogliosis (280). These astrocytes might be well coupled via gap junctions since Cx43 mRNA and protein levels are increased in the glial scar of photothrombotic ischemic lesions (145). Moreover, during the astrogliosis response observed 24 h after reperfusion, de novo synthesis of Px2 occurs in hippocampal rat astrocytes (400). The functional role of Cx43 and Px2 expression during astrogliosis remains unresolved.
Effect of Free Radicals on Gap Junction Channels and Hemichannels, and Possible Implications for Neurological Disorders
CNS cells are highly susceptible to oxidative insults, due to their high oxygen consumption, high levels of peroxidizable fatty acids, and poor antioxidant defenses (54). Oxidative stress occurs during brain invasion by pathogens, as well during partial ischemic episodes and after ischemia/reperfusion (87, 94, 133, 238). Elevated reactive nitrogen species are also neurotoxic and their generation is particularly exacerbated in cells under metabolic stress (127, 167, 403). Moreover, a large body of evidence suggests an important role of increased ROS production leading to oxidative stress as well as of increased reactive nitrogen species (RNS) in aging and neurodegenerative disorders (55, 292). The increased oxidative alterations of Aβ in Alzheimer's disease (AD), α-synuclein in Parkinson's disease (PD), and SOD1 in an animal model of amyotrophic lateral sclerosis (ALS) might result in altered protein folding and impaired protein degradation. One consequence of intracellular protein aggregation is ER dysfunction (ER stress), which leads to mitochondrial uncoupling and contributes to excessive production of ROS, thus causing or worsening oxidative stress (54). In fact, postmortem brain tissues from patients with neurodegenerative disorders, including PD, AD, and ALS, display increased indices of ROS in affected brain regions (85, 147, 279).
Membrane damage secondary to ROS-mediated lipid peroxidation can affect the activity of functional membrane proteins (5, 53, 153, 228). Moreover, NO or NO-derived compounds enhance astrocytic Cx hemichannel activity (62, 63, 313, 315) and reduce gap junctional communication (40).
Currently, all the available information on the effect of oxidant agents on gap junctions is only at the functional level. The application of H2O2 rapidly increases gap junctional communication, an effect associated with increased death of astrocytes evaluated 24 h later (320). The toxicity is not prevented by gap junction blockers. The effect on gap junctional communication is mimicked by the oxidizing agent diethylmaleate and prevented with reducing agents dithiothreitol (DTT) and N-acetyl-cysteine (320). The presence of microglia in astrocyte cultures prevents the effects of H2O2 on astrocyte gap junctions and toxicity (320), suggesting that microglial cells are better equipped with antioxidant mechanisms and might be protective under particular conditions. In contrast, treatment with the NO donor S-nitroso-N-acetylpenicillamine, the
Gap junctions between astrocytes have been proposed to be neuroprotective during oxidative stress. In agreement with this notion, a transient and pronounced reduction in astroglial dye coupling occurs during metabolic inhibition (227), a condition that induce generation of free radicals; the reduction in intercellular communication is prevented by reducing agents (227). In addition, blockade of gap junctions with 18-α-glycyrrhetinic acid (α-GA) in co-cultures of neurons and coupled astrocytes or in organotypic hippocampal slice cultures after oxidative stress induced by FeSO4 and 4-hydroxynonenal results in a markedly enhanced generation of intracellular peroxides, impairment of mitochondrial function, and neuronal death (31). In contrast, gap junctions could also be deleterious for neurons because they allow the intercellular diffusion of death signals and amplify cell injury and death in astrocytes, a phenomenon known as “bystander death” or “glial fratricide” (209). In support of this concept, pretreatment with the gap junction blockers CBX, α-GA, or endothelin, decreases the number of apoptotic neurons in the hippocampus, as compared to the contralateral hippocampus treated with saline in a model of global transient ischemia in rats. A significant reduction in lipid peroxides of the hippocampus treated with CBX suggests that the actions of gap junctional coupling during injuries may be causally related to oxidative stress (285). An alternative interpretation of these findings is that CBX reduces cell death by blocking hemichannels formed by either Cxs or Pxs. Clearly, the molecular mechanisms and biological implications of gap junction modulation in injury by free radicals are still poorly understood and may be related to differences in experimental conditions (e.g., differences in insult intensity and/or duration and differences in cell or tissue type and conditions). In addition, the studies described above did not consider the possible involvement of Cx hemichannels and the differences may be explained by the relative involvement of cell-cell channels and hemichannel.
The number of open Cx43 hemichannels is enhanced in astrocytes under metabolic inhibition (62, 63, 313), a condition that reduces the intracellular redox potential due, in part, to generation of free radicals including nitrogen- and oxygen-derived species (127, 167, 403). The increase of hemichannel activity is abolished by intracellular but not extracellular cystine reducing agents (313), suggesting the involvement of the carboxy terminal cystine residues as sensors of the redox changes. The enhanced hemichannel activity elicited by metabolic inhibition is explained in by an increase in total surface levels of hemichannels, but it is sensitive to cystine reducing agents, suggesting the involvement of a gating mechanism activated by oxidation (62, 313). Application of NO donors enhances astroglial hemichannel activity and induces nitrosylation of intracellular Cx43 cystine residues. S-nitrosylation of Cx43 hemichannels also occurs in astrocytes under metabolic inhibition (313), and thus is likely to occur during ischemia. The enhanced hemichannel activity elicited by NO is not associated with changes in levels of surface hemichannels or changes in the state of phosphorylation of surface Cx43 evaluated by Western blot analysis (313), supporting the involvement of an NO-sensitive gating mechanism in cells under metabolic inhibition. The increased hemichannel activity may compromise the capacity for handling redox changes because Cx43 hemichannels are permeable to free radical scavengers such as reduced glutathione and ascorbic acid (4, 302). Trolox and melatonin, two free radical scavengers, can prevent the increase in hemichannel activity induced by metabolic inhibition in rat astrocytes and blockade of hemichannels delays cell death (62). Activation of hemichannels has also been demonstrated in hippocampal neurons under oxygen–glucose deprivation (401). The effect is mediated by NO and is reversed with DTT (401). Because hippocampal neurons do not express Cx43 but might express Cx36 and/or Px1, it is possible that hemichannels formed by one of these protein subunits are also sensitive to redox changes
How Might Hemichannels and Gap Junction Channels Induce Neuronal Damage in Neurological Disorders?
Cell and tissue responses to injuries depend on properties if the cells (e.g., age, hormonal experience, and stage of cell cycle) and insult (e.g., duration, intensity, and quality). Moreover, tissue responses depend on interactions between their constituent cells, including chemical and electrical transmission as well as paracrine and autocrine signaling (e.g., by cytokines and ROS), possibly mediated by GJCs and hemichannels. Despite the difficulty of assigning contributions to these channels in the pathogenesis of neurodegenerative diseases, recent studies using homo and/or heterocellular cultures have provided clues. In most chronic diseases, additional mechanisms are progressively added to the primary cause and thus, complicating assignment of the contributions of each factor to the final condition. For sake of clarity, in the following sections we first describe acute pathological conditions in which the primary cause or insult is identifiable, and the cell/tissue responses can be attributed to them (e.g., stroke and infection). Then, we described four chronic and progressive neurodegenerative diseases with specific attention to the possible involvement of Cx-based channels and inflammatory responses.
Stroke
The application of a sublethal insult in many cases induces resistance to a subsequent more severe insult, a phenomenon called preconditioning response. Mechanisms of this induced resistance include the release of neuroprotective agents, but pathways for this release remain unknown. Recently, in vitro studies reveal that preconditioning reduces degradation of Cx43, and leads to a marked increase in the number of Cx43 hemichannels in the surface membrane (207). Consequently, more ATP is released which is hydrolyzed to adenosine, a potent neuroprotective molecule (207). In agreement with the possible involvement of hemichannels in preconditioning responses, Cx43 null mice were shown to be insensitive to hypoxic preconditioning but, wild-type littermate mice exhibit a prominent reduction in infarct volume after induction of preconditioning through occlusion of the middle cerebral artery (207). The same year, the involvement of Cx36 hemichannels in preconditioning response of cultured cerebellar granule neurons was demonstrated (333). Most likely these observations will be replicated in numerous cell types that manifest preconditioning and express Cxs and/or Pxs that can form hemichannels at their surface. Cx43 translocation to the mitochondria has been observed in heart during preconditioning (36), but the functional role of mitochondrial Cx43 remains speculative. To our knowledge, similar studies in brain and cultured brain cells have not been reported.
More intense or prolonged ischemia, due to partial or complete blockade of a brain artery, causes detrimental cellular consequences. Among the early cellular changes, the electrochemical gradient across the neuronal and astroglial membrane potential is lost, and both neurons and astroglial cells become depolarized (174). Voltage-gated Ca2+ channels are activated and neurotransmitters are released into the extracellular milieu (165, 192). At the same time, the energy-dependent processes such as neurotransmitter re-uptake are reduced which increases accumulation of extracellular glutamate. Consequently, the activation of NMDA and metabotropic glutamate receptors contribute to the increase in [Ca2+]i (6). It is believed that the increase in [Ca2+]i activates nuclear and cytoplasmic events that influence the development of cell damage. These changes include the activation of phospholipase A2, generation of arachidonic acid and metabolism via cyclooxygenase/lipooxygenases leading to increase in formation of free radicals, lipid peroxidation, and plasma membrane damage. All these intracellular Ca2+-dependent events occur with a concomitant reduction in [Ca2+]o, which should enhance opening of Cx43 hemichannels (298), but not Pxs hemichannels (282).
The events described above do not take place uniformly throughout the ischemic brain tissue. In the center, or ischemic core, where there is deficient blood flow, occurs a permanent anoxic depolarization since the beginning of ischemia and cells die quickly. Between the core zone and normal brain tissue is found an area with restricted blood flow and with partial energy supply termed penumbra. Afterwards, this area may undergo excitotoxicity or deleterious secondary phenomena such as propagated depolarization, postischemic inflammation, and apoptosis. After a mild ischemia or in the penumbra zone, specific neurons may die with considerable delay, while other neurons and glia survive. The involvement of hemichannels and GJCs in these events has been discussed previously (63).
In models of ischemia/reperfusion, neurons are the main cell type that dies, in part, because their metabolism is primarily aerobic. However, a prolonged ischemic episode also causes astroglial death (356), and their relative insensitivity seems to be due to their ability to switch from aerobic to anaerobic metabolism when exposed to hypoxia (288). Under this condition, ischemia is the causal condition but the response can be worsening by conditioning factors. For example, patients or experimental animals with hyperglycemia (a difference in the ischemic episode) and stroke suffer massive death in neuronal and glial cell populations (168, 246). In primary astrocyte cultures, high glucose levels also worsen the reduction in gap junctional communication and increase the enhancement in hemichannel activity (Orellana JA, Velarde V, Bennett MVL, Giaume C, and Sáez JC, unpublished data), suggesting a direct link between astroglial Cx-based channel dysfunction and reduced neuronal and glial viability. Other conditions that might aggravate stroke damage are neurodegenerative diseases such as AD. In an animal model, focal ischemia causes larger infarct volumes in the presence of Aβ (387). A possible mechanism involves Cx-based channels, since Aβ enhances the cytokine-induced reduction in gap junctional communication and increases hemichannel activity in cortical astrocytes (235) (Orellana JA, Sáez JC, and Giaume C, unpublished data).
The enhanced hemichannel activity of astroglia under metabolic inhibition accelerates cell death (62, 63). A similar scenario may occur in other in vitro models of ischemia where an increase in Cx or Px hemichannel activity has been observed (203, 316, 341, 364, 401). Demonstration of the relative importance of enhanced hemichannel activity versus reduced gap junctional communication in astroglial as well as neuronal cell viability will require new experimental approaches including drugs that specifically block Cx or Px based cell-cell channels and hemichannels. Currently available reagents are quite unsatisfactory.
In vitro gap junctional communication is only partially inhibited by ischemia or ischemia-like conditions (62, 65, 204, 227), and the remaining gap junctional communication may allow amplification of cellular damage. Ca2+ waves are a possible cell death propagation mechanism (209). The first in vivo evidence for the involvement of Cx-based channels in the spread of death signals in the CNS was that octanol, a nonselective gap junction and hemichannel blocker, reduces the infarct size after a focal ischemia (310). In addition, damage induced by global ischemia is more pronounced in the CA1 than in the CA3 area, although gap junctions are more common in CA1, and that CA1 damage is reduced by octanol (301). These observations are contradicted by findings obtained in Cx43 knockout heterozygous mouse or mouse astrocytes with the floxed Cx43 gene inactivated by Cre expression, in which focal ischemia causes larger lesions (257, 258, 344). This apparent contradiction might be explained by a differential distribution of Px1 in these neurons (406) where hemichannels could open under ischemia-like insults (364). In addition, ischemia-reperfusion induces more macrophage and microglia recruitment to the injured tissue of Cx43 knockout animals (258), suggesting that more neuronal death leads to more activation of macrophages.
In a rat model of transient global ischemia, pretreatment with compounds that block both hemichannels and GJCs (i.e., CBX, α-GA, and endothelin) reduces the number of TUNEL-positive neurons, as compared to the contralateral hippocampus treated with a saline injection (285). In agreement, the CA1 of mice deficient in Cx32 shows increased sensitivity to global ischemia (268). Cx32 forms hemichannels in activated microglia (361), and in Cx32 knockout animals microglial release of neurotoxins might be reduced. An additional complication to understanding the role of Cx-based channels in stroke is possible change in Cx expression. During ischemia expression of Cx32 and Cx26, but not Cx43, increases selectively in the hippocampal area CA1 before the onset of neuronal death (268). Moreover, the infarct penumbra in Cx43+/− mice shows an increase in Cx30 levels as compared to wild-type Cx43+/+ mice (257). The significance of these changes in Cx expression remains speculative that each Cx may play different roles at different stages of the insult and its sequellae. So far, most available in vivo information has been obtained using different models and conditions which complicate the interpretation. Future studies using more homogeneous systems and conditions should further understanding to the role of Cx based channels in stroke.
In vitro studies have also contributed to knowledge of the possible regulatory mechanisms and roles of Cx -and Px-based channels in stroke. Ischemia-like conditions induce dephosphorylation of astroglial Cx43 which is associated with a reduction in gap junctional communication in hippocampal slices and astroglial cultures (62, 65, 204) as well as with activation of Cx43 hemichannels. In primary astrocyte cultures, oxygen–glucose deprivation enhances hemichannel activity to an extent dependent on insult duration (316). In hippocampal slices, the reduction in gap junction coupling is associated with a rise in [Ca2+]i in astrocytes (65) that might activate Ca2+-dependent protein phosphatases. In agreement with the latter interpretation, inhibition of calcineurin, a Ca2+-dependent protein phosphatase, prevents the reduction in gap junctional communication (63, 204) but not the increase in hemichannel activity (63). Thus, junctional channels and hemichannels are under the control of different mechanisms possibly related to the different molecular compositions of the subcellular compartment where each one is located. It has been proposed that inhibition of astroglial gap junctions reduces neuronal vulnerability to glutamate or oxidant stress or disruption of Ca2+ homeostasis (31, 209, 271). However, in these studies the effect of gap junction blockers on hemichannel activity was not evaluated and now it is known that these conditions induce oxidant stress which enhances the activity of Cx43 and Px1 hemichannels (315, 401). In primary cultures of resting astrocytes and neurons Px1, but not Px2, is expressed (157, 400). However, Px1 is not detected at the surface of either neurons or astrocytes (157), suggesting that formation of Px1 hemichannels is hindered. So far, conditions that affect the expression and/or induce insertion of Px1 into the plasma membrane remain unknown.
Bacterial meningitis/encephalitis
Bacterial meningitis promotes inflammation of the pia, arachnoid, and subarachnoid space. Inflammation may also affect the brain parenchyma leading to encephalitis. Bacteria present in the bloodstream induce an innate immune response which produces systemic release of cytokines, mainly TNF-α (86, 317). Then, bacteria colonize and cross the inflamed BBB and components of their wall, such as LPS (19), peptidoglycan (PGN) (19), or streptococcal hemolysin/cytolysin (89), induce BBB activation and permeabilization (112) (Fig. 5). BBB activation is characterized by numerous changes including cytokine production, overexpression of cell adhesion molecules and NO synthesis (112). Changes in the BBB endothelium due to activation facilitate bacterial interaction with endothelial PAF (platelet activating factor) receptor which allows bacterial transmigration (385) into the brain parenchyma where they proliferate (Fig. 5).

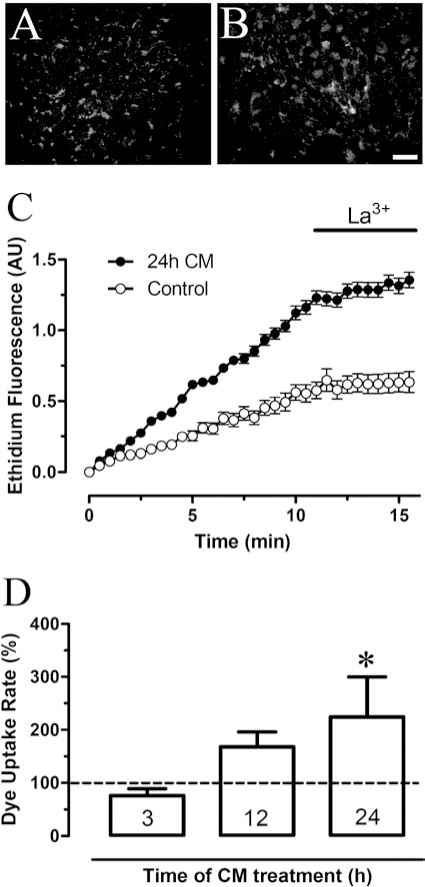
The BBB may be a target for inflammatory responses initiated in the brain parenchyma (e.g., inflammation triggered by Aβ) or in the periphery (e.g., sepsis or extensive burn of the body surface) (Fig. 5). Since the BBB restricts movement of substances into or out of the CNS, and many of its functions are likely to be coordinated, it would be of interest to learn how their hemichannels and GJCs are modified by pro-inflammatory agents or conditions. TNF-α treatment for 2 h reduces gap junction communication and increases basal ATP release in RBE4 cells (370), a cell line derived from rat brain cortical endothelium. In agreement, we found that conditioned medium of microglia activated with LPS for 24 h increases hemichannel activity of RB4 cells in a time-dependent manner (Fig. 6). In addition, 2 h of TNF-α treatment blocks ATP release induced by photoliberation of InsP3 and zero [Ca2+]o, but increases basal ATP release and hemichannel activity in a way not prevented by the gap26 mimetic peptide (370), a purported blocker of Cx43 hemichannels (104). However, gap26 does not block Cx40, which is highly expressed in brain endothelial cells (250), thus the rise in basal activity induced by TNF-α could be related to Cx40 and/or Px hemichannels. Hemichannels and GJCs have not been well studied in BBB cells, but these channels have been extensively studied in endothelium of other territories of the circulatory system (69, 108). Readers interested on Cx-based channels in endothelial cells in general are referred to reviews in this issue by Figueroa and Duling (45a) and by Brisset, Isakson, and Kwak (107a) in this issue.
Enhanced endothelial hemichannel activity could enhance release of ATP, which would recruit microglia to the injury site (76). In agreement with a role of hemichannels in ATP release during inflammatory conditions triggered by bacterial infections, Shigella infection of epithelial cells promote ATP release through Cx26 hemichannels, resulting in the activation of purinergic receptors on neighboring cells and bacterial dissemination (367). On the other hand, a reduction in gap junction communication between brain endothelial cells may impair astrocyte-endothelial cell calcium signaling (43) and increase BBB permeability by reducing tight junction formation. The latter is in agreement with the observation that block of GJCs increases transendothelial electrical resistance and enhanced paracellular flux of permeability tracers (250).
In the brain parenchyma, bacteria may undergo lysis and release pro-inflammatory and toxic factors such as PGN and LPS (353, 354), while microglia interact directly with intact bacteria (179). Bacterial-derived pro-inflammatory factors such as LPS induce neurodegeneration (295). PGN and LPS stimulate microglial Toll-like receptors (TLRs), induce translocation of nuclear factor (NF) κB (336), and activation of MAPK signaling and transcription of genes encoding inflammatory cytokines (194, 259). Moreover, PGN increases microglial Cx43 mRNA and protein expression which correlates with development of gap junction communication in vitro (119). Similarly, treatment with LPS plus IFN-γ increases Cx43 expression in rat microglia and induces gap junction communication (102). In addition, brain stab wounds induce recruitment of Cx43 immunoreactive microglia, suggesting that Cx43 is important for coordinating microglial responses (102). Expression of Cxs and formation of gap junctions, at least at homocellular contacts, might be a common feature of inflammatory macrophagic cells. Cx43 expression has been demonstrated in activated dendritic cells (64, 326), peritoneal macrophages (12, 161), foam cells of atherosclerotic lesions (290), J774 cells (12, 28), Kupffer cells (100), and polymorphonuclear cells in a tissue subjected to ischemia-reperfusion or treated with pro-inflammatory cytokines (45, 161). LPS or TNF-α also promote microglial release of neurotoxic glutamate concentrations via Cx32 hemichannels (361).
While resting microglia have a weak but significant density-dependent inhibitory effect on astroglial Cx43 expression and gap junctional coupling (105, 321), microglia activation by LPS amplifies these effects (235, 314), suggesting that they can modify signaling among astrocytes. In addition, in astrocyte cultures not devoid of microglia, LPS or pro-inflammatory cytokines reduce astroglial coupling via nitric oxide synthase (NOS) induction and a p38 kinase-dependent pathway (40, 91, 139, 164, 235, 408). Both the IL-1β and TNF-α effects on gap junctions occur as early as 2 h after application (139, 408). Because this latency is close to the half-life of Cx43 (197), the action may be due to block of synthesis or increase in degradation of Cx. In general, the reduction in astroglial coupling is associated with reduced Cx43 levels (91, 164, 235). Similar to LPS, PGN reduces astroglial coupling associated with decrease in Cx43 and Cx30 protein levels; whereas under the same conditions expression of Cx26 increases (99). In co-cultures, inhibition of astrocyte dye coupling is induced by LPS-activated microglia which is associated to a reduction in Cx43 expression (235). The effect is mimicked when microglia-free astrocyte cultures are exposed to TNF-α plus IL-1β and is potentiated by Aβ (235), suggesting that reduction of gap junctional communication can result from activation of at least two different pathways. In agreement, activation of the innate immune response by dsRNA viruses and viral replication intermediates occurs via Toll-like receptor 3 (TLR3) and activation of astrocytes with the dsRNA mimetic polyinosinic-cytidylic acid (pI:C) inhibits expression of Cx43 (404). This effect is independent of p38 MAP kinase, ERK, or JNK signaling pathways, whereas it is completely inhibited by a blocker of the PI3 kinase/Akt pathway (404). Thus, activation of innate immune response can reduce astroglial coupling through two independent pathways. It remains unknown whether their simultaneous activation elicits a greater response of hemichannels and gap junctions channels than activation of either alone.
Changes in levels of Cxs 30 and 43 have been observed in astroglial cells of mice infected with Borna disease virus (183, 184). Evaluation of the time course of the inflammatory response in those mice would help to determine whether the Cx level changes are generated by the mechanisms discussed in the present review. Similarly, studies in mixed glial cultures and astroglial cultures devoid of microglia would help to dissect changes in astroglial cells directly caused by the virus from those due to activation of microglia induced by the virus.
Beside downregulation of astrocytic coupling, the presence of a few microglia induces depolarization of the resting membrane potential (139, 150), suggesting the involvement of membrane channels located in the cell surface. Astrocytes treated with conditioned medium from microglia activated with LPS or treated with TNF-α or IL-1β show increased Cx43 hemichannel activity through a p38 MAP kinase-dependent pathway (314). Pro-inflammatory cytokines increase dye uptake mediated by hemichannels as early as 30 min after their addition (314). The astroglial hemichannel activity is sensitive to La3+ and L-NAME, and does not occur in astrocytes from Cx43 knockout mice (314). The possible consequences of increase hemichannel opening include enhanced glucose uptake, which might explain changes in the metabolic status of astrocytes under inflammatory conditions (314). Moreover, it could promote neuronal damage through astrocytic release of neurotoxic and/or inflammatory compounds such as glutamate and PGE2, respectively.
Bacterial meningitis also causes axonal damage and demyelination (260). These effects may be related to microglial cytokine release, which could promote opening of oligodendrocyte hemichannels, possibly composed of Cx32 or of Cx29, which does not form gap junctions and faces the periaxonal space (206) and, thus, promoting ion gradient imbalance and calcium overload.
Cerebrospinal fluid (CSF) normally contains leucocytes, complement factors, and antibodies at low abundance. However, severe inflammatory episodes are accompanied by high CSF cytokine and blood-derived leukocyte levels. The latter migrate to the infection zone attracted by soluble factors released by endothelial and microglial cells (362, 409). The invasion of infiltrating leucocytes often induces vasculitis which can produce thrombotic obstruction of vessels, and decreased cerebral perfusion can lead to focal ischemic lesions (224). Close to 2 decades ago, gap junction-like structures between lymphocytes and brain endothelial cells were discovered (300), suggesting that heterocellular gap junctions may be required for an inflammatory response and extravasation (see below).
Leukocytes adhere weakly to the endothelium and roll along the vessel wall at a rate ∼1% of the flow rate of nonadherent red blood cells (198). Rolling is ascribed to weak adhesive interactions between the leukocytes and endothelium surface; firmer adhesion of leukocytes to the endothelium causes cessation of rolling. The increased adhesion may result from formation of leuko-endothelial gap junctions (327). Neutrophils (161, 398) and T lymphocytes (93, 136) form gap junctions with endothelial cells that facilitate leukocyte transmigration across an endothelial cell monolayer (101, 270, 390, 398). Treatment with α-GA reduces monocyte/macrophage transmigration across a BBB model (101). Gap junctions between lymphoma cells and endothelial cells have also been demonstrated and α-GA attenuates the transmigration of lymphoma cells across an endothelial barrier (136). Interpretation of experimental results obtained using gap junction blockers or constitutive Cx knock out animals should also take in account the possible role of homocellular and heterocellular gap junctions express by inflammatory cells. In infected tissues, monocytes/macrophages play a relevant role, whereas neutrophils and lymphocytes are important protagonists in ischemia-reperfusion and autoimmune disease, respectively. This issue is particularly relevant because activated inflammatory cells generate copious free radicals which, as discussed above, can cause hemichannel and gap junction dysfunction.
It was recently shown that microglial cells cross-present antigens to naive and memory CD8+ T-cells in vitro (26). Moreover, it is conceivable that leukocytes infiltrated in the brain parenchyma might undergo antigen cross-presentation with activated microglia, and antigenic peptide domains derived from viral or cellular proteins may be transferred between inflammatory cells through gap junctions as has been demonstrated for other antigenic molecules (236, 263). Cross-presentation via GJCs might coordinate and amplify inflammatory responses. Another route of antigen presentation could arise, when one cell engulfs an area of gap junction and takes with it a bit of cytoplasm from the “donor cell”. If this vesicle contains antigens and is degraded by lysosomes, some antigenic fragments may be passed to the ER and eventually presented on the surface of the engulfing cell. Thus, skepticism on the role of gap junction in antigen cross-presentation might be reduced if the latter is ruled out.
Alzheimer's disease
Alzheimer's disease (AD) is characterized by the accumulation of the Aβ into amyloid plaques in the extracellular brain parenchyma, formation of tangles inside neurons as a result of abnormal phosphorylation of the microtubule-associated protein tau, dendritic atrophy, and changes in neurotransmission in specific brain regions (275). Aβ is generated by proteolytic cleavage of the amyloid precursor protein (APP), which plays a role in neuronal adhesion, synaptogenesis, and axonal growth (275). High concentrations of Aβ are toxic to several neuronal types (217, 275, 289). The mechanisms underlying Aβ-neurotoxicity are complex but involve activation of NMDA receptors, sustained elevations of [Ca2+]i, and oxidative stress (92, 111) which are effects common to those induced by ischemia-reperfusion but on a different time scale.
In addition to the above, the cerebral cortex of individuals with AD present activated microglia and astroglia closely associated with amyloid plaques (169, 389). Cx43 immunoreactivity is increased at amyloid plaques where gliosis occurs (252). Imbalance in brain homeostasis may explain the increase in Cx43 expression in amyloid plaques as an (ultimately failed) compensatory mechanism to ensure normal brain function (252). Alternatively, increased astroglial gap junctions may serve as a pathway for the propagation of neuronal damage, transferring death signals generated in the microenvironment of amyloid plaques to distant neurons, as death signals can propagate from C6 glioma cells injured with calcium ionophore (209, 252). In agreement with this interpretation, inhibition of gap junctions with octanol abolishes the ability of Aβ to enhance the velocity and extent of propagation of astroglial calcium waves (144). However, octanol also blocks P2X7 receptors expressed by spinal astrocytes that also show calcium waves (355). In support of P2 receptor mediation of the Aβ-induced increase of calcium wave velocity in cortical astrocytes is the fact that suramin, a P2Y and P2X receptors blocker, reduces this response (144). Nevertheless, P2X receptors as ATP release pathway in astrocytes treated with Aβ remains to be demonstrated. At least in macrophages and lymphocytes cell lines, where both P2X7 receptor and Px1 coexist, the ATP induced dye uptake occurs through Px1 hemichannels resulting from P2X7 activation (282). Although cultured astrocytes express Px1, it is not detected at the cell surface, excluding that ATP release through Px1 hemichannels mediates propagation of calcium waves (157). To be sure, the in vivo and in vitro preparations discussed may lack factors required for Px1 insertion into the plasma membrane. Studies in AD models will help to confirm or reject the above interpretations.
Overexpression of activated calcineurin induces astroglial activation, and calcineurin is upregulated in AD models (267). Activation of a calcineurin-dependent dephosphorylation pathway is associated with reduced gap junctional communication in astrocytes subjected to metabolic inhibition or glucose/oxygen deprivation followed by reoxygenation (62, 204). Hence, the Aβ-induced reduction in astroglial coupling may be in part due to dephosphorylation of Cx43 GJCs via calneurin.
Inflammatory cells inhibit astroglial coupling through gap junctions in co-cultures of resting microglia and astrocytes (321). Because microglia in primary cultures are partially activated (225) and release low levels of cytokines, the reduction in astroglial coupling in these cultures could be mediated by soluble factors instead of direct cell–cell contacts. TNF-α and IL-1β secreted by activated microglia more strongly inhibit coupling of astroglia (105, 235). This effect is potentiated by Aβ25–35, which is the neurotoxic Aβ domain (289). In addition, TNF-α and IL-1β secreted by activated microglia increase astroglial Cx43-hemichannel activity (314) (Fig. 7), which accelerates death induced by ischemia (62). Thus, neuronal death induced by Aβ-treated microglia could be mediated by cytokines released from astrocytes as well as from microglia (234, 384) that reduce gap junctional coupling and enhance Cx43 hemichannel activity (Fig. 8A). This condition would generate a positive feedback cycle because dead cells activate more microglia which release more cytokine, and the inflammatory response causes edema that reduces tissue perfusion causing partial or total ischemia (Fig. 8B and C).


The lack of neuronal protection by astroglial cells could result from astroglial death as well as dysregulation of astroglial gap junctions and hemichannels. In support of the first possibility, 0.1–10 μM Aβ25–35 induces astroglial death in vitro (15). As previously discussed, a reduction in gap junctional communication impairs the spatial buffering capacity of astroglial networks, and an increase in hemichannel activity could in turn affect the viability of other cells through a paracrine mechanism. Indeed, excessive release or deficient uptake of signaling molecules such as glutamate, arachidonic acid byproducts, PGE2 could enhance neuronal excitotoxicity (3, 238, 361, 396) (Fig. 8B). These mechanisms are likely to be enhanced by Aβ and may potentiate astroglial mediated neuronal death. The Aβ-induced rise in [Ca2+]i (384) may activate astroglial Cx43 hemichannels and promote astroglial death through at least three mechanisms: a) intracellular Ca2+ overload caused by excessive Ca2+ entry through Cx43 hemichannels leading to activation of lipases, phosphatases, and proteases, b) passive water flow generated by massive Na+ and Cl− influx through Cx43 hemichannels causing astroglial swelling, and c) Cx43 hemichannel-mediated release to the extracellular milieu of molecules required in normal metabolism (e.g., glucose and derivates, NADH, ATP, ascorbic acid, and reduced glutathione: GSH).
Aβ plaques are also commonly found in the brain of Down's syndrome patients in whom neurodegeneration is prominent and premature (218). Therefore, mechanisms involving GJCs and hemichannels as those described for AD are likely to occur in Down's syndrome.
Parkinson's disease
The neuropathological hallmarks of Parkinson's disease (PD) patients are characterized by progressive and profound loss of neuromelanin containing dopaminergic neurons in the substantia nigra pars compacta with presence of eosinophillic, intracytoplamic, proteinaceous inclusions termed as Lewy bodies, which contain α-synuclein (348).
The molecular mechanisms of PD remain elusive but important factors in the disease pathogenesis are oxidative stress (163, 212) and α-synuclein accumulation (343). Microglial activation in PD is indicated by extensive proliferation of reactive amoeboid macrophages and microglia in the substantia nigra of PD patients in postmortem studies (232). Moreover, the substantia nigra in PD presents activated glial cells expressing pro-inflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ, as well as iNOS (151, 159).
Overexpression and mutation of α-synuclein are associated with early onset PD (343), suggesting that α-synuclein dysregulation plays an important role in modulating the microglial activation state. Accordingly, fibrillary α-synuclein activates microglia in vitro (402) and microglia activation in vivo correlates with fibrillar α-synuclein deposition (68). Moreover, microglia from knockout α-synuclein mice (Scna−/−) display an increased reactive phenotype under basal conditions and an exacerbated reactive phenotype after stimulation (16), suggesting that deficient α-synuclein contribute to microglia activation. In agreement with the possible role of hemichannel and gap junction dysfunction discussed in the present review, Scna−/− microglia, after stimulation, secrete elevated levels of pro-inflammatory cytokines, including TNF-α (16).
In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned striatum, an animal model of PD, total Cx43 mRNA and astroglial Cx43-immunoreactive punctata are significantly increased, whereas astroglial coupling evaluated with dye coupling of patched cells remained unaffected (325). Moreover, FGF-2, a potent trophic factor for mesencephalic dopaminergic neurons (106) further increases astroglial density and Cx43 immunoreactivity in the vicinity of the FGF-2 source without change astroglial coupling (325). In this system, the hemichannel activity has not been evaluated and is likely to be increased due to the imminent inflammatory response described above
How can the trophic effect of FGF-2 be explained? The role of hemichannels in injury may vary with the course and severity of noxious stimuli, passing from being beneficial after sublethal insults to accelerating cell death in severely injured tissues (316). Thus, it is likely that FGF-2 promotes hemichannel uptake of energy substrates and/or release of toxic metabolites to the extracellular milieu, which may be beneficial for injured cells. In support of this view, one signaling pathway of FGF-2 depends on activation of the p38 MAP kinase (222), which is associated with an increase in astroglial Cx43 hemichannel activity induced by cytokines (314).
FGF-2 upregulates expression of Cx43 by cultured dopaminergic neurons of the midbrain floor and increases coupling (345). More importantly, block of gap junctions with oleamide abolishes the survival effect of FGF-2 on dopaminergic neurons (345). In the latter study, oleamide would block both astroglial and neuronal coupling, thus, dissection of the possible role of hemichannels and GJCs remains to be elucidated. FGF-2 may permit diffusion of ions and substances from healthy to injured neurons through gap junctions, thereby promoting neuronal survival. FGF-2 also could be beneficial by decreasing opening of neuronal hemichannels composed of Pxs and/or Cxs. Indeed, short-lasting treatment with FGF-2 or FGF-1 decreases Cx43 hemichannel activity (79). The beneficial effect of FGF-2 in PD models is likely to be related to inhibition of astroglial and/or neuronal hemichannel, which could promote both astroglial and neuronal survival.
As in the case of AD, it is probable that increased microglial, astroglial, and neuronal hemichannel activity in PD is accompanied by a reduction in gap junction coupling. Overexpression of α-synuclein inhibits gap junction communication in dopaminergic neuroblastoma cells, and this effect may increase the sensitivity of α-synuclein overexpressing cells to various toxins (357). Moreover, α-synuclein binds to Cx32 (357), a Cx expressed in substantia nigra pars compacta (345, 371) where neurons are electrically coupled (372).
Huntington's disease
Huntington's disease (HD) is a progressive and fatal neurological disorder caused by an expanded CAG repeat in the gene coding for the protein huntingtin (1). This protein accumulates and causes apoptosis of striatal neurons (278). In HD, cortico-striatal circuits are particularly affected. Degeneration of medium spiny neurons and dysfunction of the supplementary motor area and the dopamine system is particularly relevant (382). As in other neurodegenerative diseases, HD causes both microglial (358) and astroglial activation (2). Astroglia expressing mutant huntingtin enhance excitotoxic neuronal damage (340). Moreover, a mutant huntingtin fragment perturbs the microglia transcription program (126). In this view, expression of mutant proteins (e.g., Aβ, α-synuclein, and huntingtin) can perturb the normal glial response, amplifying the initial injury affecting susceptible neurons (214). Alternatively, glia expressing these mutant proteins become themselves a primary source of neurotoxicity, potentially independent of the mutation's effects in neurons at risk (214).
The distribution of Cxs 26, 32, 40, and 43 in the caudate nucleus and globus pallidus of the basal ganglia in normal and HD human brains has been studied using immunohistochemical techniques (378). Cx40, localized in vascular endothelium, is increased in HD brains, apparently due to more numerous and smaller blood vessels. Both Cx26 and Cx32 have a similar distribution in the striatum and globus pallidus of normal and HD brains, with little in the caudate nucleus but clearly detectable amounts in the globus pallidus. In the caudate nucleus the Cx43 density is increased, which correlates with enhanced GFAP expression in patches (378), indicating a reactive astrogliosis around foci of neurodegeneration.
Changes in expression of Cx36, Cx43, and Cx45 also occur in the retina of adult transgenic R6/2 mice, a model of HD (286) (The basal ganglia were not investigated in this study). In the retina, Cx36 is slightly reduced in the outer plexiform layer, whereas Cx45 is strongly reduced in the inner plexiform layer. No changes in Cx43 expression were evident in mutant HD mice. It should be determined if HD is associated with increased astroglial hemichannel activity and/or reduced gap junctional communication that could explain in part the retinal neurodegeneration as well caudate nucleus neurodegeneration (287).
Concluding Remarks
The inflammatory response is the main cause of numerous acute neuropathological processes and can progress in chronic diseases, leading to tissue dysfunction. In the brain, key cells of the inflammatory response are microglia and astrocytes that under resting conditions contribute to normal functioning of neurons. Deleterious conditions can trigger changes in both types of glial cells as well in other cells of the brain, including neurons and endothelial cells. The latter may be targets of inflammatory responses initiated within the nervous tissue as well as of inflammatory responses initiated in the periphery, such as infections, traumas, and burns. If intensity, duration, and quality of the insult are below the threshold to elicit an overactive response, release of ATP via hemichannels mediates a preconditioning response that confers tissue resistance to successive deleterious insults. If the adverse condition is excessive, microglia and astrocytes manifest rapid phenotypic changes to neutralize the threat or to adapt to the new microenvironment. In contrast, neurons do not adapt as well, and excitotoxicity and neuronal death can cause persistent glial activation and a damaging positive feedback loop. Free radicals generated during reoxygenation after an ischemic episode or cytokines and/or free radicals generated during an inflammatory response enhance hemichannel activity and reduce gap junction coupling of astrocytes. These changes tend to deprive neurons of numerous glial protective functions, and neurons become more susceptible and may undergo necrosis or apoptosis. Under acute pathological conditions, these mechanisms can be triggered by debris of necrotic cells or directly by pathogens or foreign molecules detected by microglia. In chronic diseases inflammation can be activated by anomalous molecules. The inflammation induced edema also reduces tissue perfusion worsening the inflammatory response.
The role of microglia and astrocytes in mediating inflammation of nervous tissue has been recognized previously as leading to death of neuron; these cell types can be bad neighbors for neurons (37, 360). Dysfunction of astroglial and microglial hemichannels and GJCs likely are mechanisms commonly elicited in all brain diseases associated with inflammatory responses and thus, contribute to acute and chronic neurodegeneration. Therefore, normalization of channel dysfunctions should confer tissue protection, improve quality of life, and extend survival of patients suffering acute or chronic brain inflammatory responses.
Possible changes in Cx-based channels in the brain of neonates remain unknown, and future studies might reveal interesting cues to further understanding of the role of these channels in neurodegeneration. A discussion of disease states in relation to developmental and age-dependent regulation of Cxs such as neonatal versus adult hypoxia and hypoxia-ischemia is one example. Other examples include the possible nonchannel function of Cxs and their expression at sites other than the plasma membrane.
Inflammation and neurodegeneration are closely related in other chronic and acquired neurodegenerative diseases not described in the present review, including Niemann–Pick type C disease (25), Down's syndrome (160), ALS (TBP-43) (193), experimental ALS models (75), diabetes (312), Epstein–Barr virus induced multiple sclerosis (366), Creutzfeldt–Jakob disease (17) and HIV-associated dementia (33). Elucidation of the role of hemichannel and GJCs dysfunction in these processes should be a topic of high research interest in the immediate future. How can current treatments be related to the possible role of Cx based channels in neurodegeneration? So far the most specific hemichannel blockers are mimetic peptides, but they also block GJCs (presumably by preventing formation) and most likely their chronic use will be limited by their antigenicity and susceptibility to degradation by extracellular proteases. Classical anti-inflammatory agents such as indomethacin prevent the reduction in astroglial gap junctional communication induced by arachidonic acid (226, 227). Moreover n-3 polyunsaturated fatty acids (DHA) are potent anti-inflammatory components that enhance gap junctional communication between astrocytes (70), but their effects on hemichannels remains unknown. Steroidal anti-inflammatory agents may also restore astroglial functions depressed by inflammatory responses. Corticoids do not affect gap junctions and Cx43 levels in astrocytes under basal conditions, but increase resting cytosolic free [Ca2+] levels and the extent and amplitude of Ca2+ wave propagation (342). But interestingly, corticoids prevent TNF-α and IL-1β-induced inhibition of gap junctional communication and Cx43 downregulation (Froger N. and Giaume C., unpublished data). Alternatively, anti-inflammatory cytokines (e.g., TGF-β1 and IFN-β) might be useful, since they prevent the astrocyte uncoupling response elicited by pro-inflammatory cytokines (150). A group of compounds with anti-inflammatory activity that also block hemichannels and GJCs are derived from licorice (61, 109, 134), which suggests a reduction in gap junctional communication is not so detrimental to cells as is excess hemichannel activity.
All above-mentioned compounds that are nonspecific blockers of both hemichannels and GJCs also affect other molecular targets. At this time, block of hemichannels and GJCs can be considered a side effect. From a different perspective, two common conditions known to elicit an inflammatory response, ischemia and infectious agents, reduce gap junctional communication and enhance hemichannel activity in astrocytes, suggesting that these channels could be common targets of different pathways (e.g., p38 kinase-dependent and–independent pathways) that mediate cellular responses elicited by the innate immune system. Therefore, the development or discovery of compounds with specific inhibitory action either on hemichannels or GJCs and with selectivity for Cx isoforms may offer a new generation of anti-inflammatory compounds with applicability in a much broader spectrum of disease than the compounds currently used.
Footnotes
Acknowledgment
This work was partially supported by CONICYT (24080055 to JAO), FONDECYT (1070591 to JCS) and NIH (NS37402 and NS45287 to MVLB) and INSERM/CONICYT (CG and JCS) grants.
Abbreviations
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; ASICs, acid-sensing ion channels; BBB, blood brain barrier; BDNF, brain-derived neurotrophic factor; [Ca2+]o, extracellular calcium concentration; [Ca2+]I, intracellular calcium concentration CFTR, cystic fibrosis transmembrane conductance regulator; CNS, central nervous system; CSF, cerebral spinal fluid; Cx, connexin; DTT, dithiothreitol; ER, endoplasmatic reticulum; FGF-1, fibroblast growth factor 1; α-GA, 18-α-glycyrrhetinic acid β-GA, 18β-glycyrrhetinic acid; GJC, gap junction channel; HD, Huntington's disease; IFN-γ, interferon-γ; IL-1β, interleukin-1β; LPS, lipopolysaccharide; NGF, nerve growth factor; NO, nitric oxide; NT-3, neurotrophin-3; NT-4, neurotrophin-4; PAF, platelet activating factor; PD, Parkinson's disease; PGE2, prostaglandin E2; PGN, peptidoglycan; PMCA, plasma membrane Ca2+-ATPase; Px, pannexin; RNS, reactive nitrogen species; ROS, reactive oxygen species; siRNA, small interfering ribonucleic acid; SOD1, superoxide dismutase 1; TGF-β, transforming growth factor-β; TLRs, toll-like receptors; TNF-α, tumor necrosis factor-α; TRP, transient receptor potential; ZO-1, tonula occludens protein 1.