
Abstract
The cellular oxidation and reduction (redox) environment is influenced by the production and removal of reactive oxygen species (ROS). In recent years, several reports support the hypothesis that cellular ROS levels could function as “second messengers” regulating numerous cellular processes, including proliferation. Periodic oscillations in the cellular redox environment, a redox cycle, regulate cell-cycle progression from quiescence (G0) to proliferation (G1, S, G2, and M) and back to quiescence. A loss in the redox control of the cell cycle could lead to aberrant proliferation, a hallmark of various human pathologies. This review discusses the literature that supports the concept of a redox cycle controlling the mammalian cell cycle, with an emphasis on how this control relates to proliferative disorders including cancer, wound healing, fibrosis, cardiovascular diseases, diabetes, and neurodegenerative diseases. We hypothesize that reestablishing the redox control of the cell cycle by manipulating the cellular redox environment could improve many aspects of the proliferative disorders. Antioxid. Redox Signal. 11, 2985–3011.
I. Introduction
A. A redox cycle within the cell cycle
Consistent with these observations, we have reported that the cellular redox environment fluctuates during the cell cycle. HeLa (human adenocarcinoma) cells synchronized by mitotic shake-off were replated and then harvested at different times after plating for flow-cytometry measurements of the cellular redox environment. The fluorescence of a prooxidant-sensitive dye (DCFH2-DA) was three- to fourfold higher in mitotic cells compared with cells in the G1 phase. The cellular redox environment increased gradually toward a more-oxidizing environment as G1 cells moved through the cell cycle (111). These results suggest that a redox control of the cell cycle regulates progression from one cell-cycle phase to the next. This hypothesis is also supported by a recent report demonstrating significantly higher GSH content in the G2 and M phases compared with G1; S-phase cells showed an intermediate redox state (64). Furthermore, pharmacologic and genetic manipulations of the cellular redox environment perturb normal cell-cycle progression (200 –202, 276, 277).
Overall, these results support the hypothesis that a redox cycle within the cell cycle represents a regulatory link between the oxidative metabolic processes and cell-cycle functions. A defect in this regulation could lead to aberrant proliferation. Aberrant proliferation is central to a variety of human pathologic conditions, such as cancer, wound healing, fibrosis, cardiovascular diseases, diabetes, and neurodegenerative diseases. It is hypothesized that reestablishing the redox control of the cell cycle by manipulating the cellular antioxidant pathways could be an innovative approach to prevent, reverse, or suppress (or a combination of these) many aspects of aberrant cellular proliferation.
Proliferation depends both on cell division and cell death. Cell division drives proliferation, and cell death prevents damaged cells from propagating damaged cellular macromolecules to daughter generations. Reproductive death, apoptosis, and necrosis are the three major modes of cell death. This review article focuses on literature reports demonstrating a redox control of cellular proliferation. The readers are referred to excellent recent reviews discussing the possible role of cellular redox environment and apoptosis in various pathologic conditions (15, 190, 233, 245, 306).
B. Reactive oxygen species
ROS are oxygen-containing molecules that are highly reactive in redox reactions. The partial reduction of molecular oxygen results in the production of superoxide (O2 •−) and hydrogen peroxide (H2O2) (120). O2 •− and H2O2 react with transition metal ions (e.g., cuprous and ferrous ions) through Fenton and Haber–Weiss chemistry, generating the highly reactive hydroxyl radical (HO•) (121).
ROS are primarily produced intracellularly by two metabolic sources: the mitochondrial electron-transport chain and oxygen-metabolizing enzymatic reactions such as xanthine oxidases, the cytochrome P450 system, NADPH oxidases, myeloperoxidase, and nitric oxide synthase (27, 30, 151, 189, 278, 284, 355). ROS levels also are dependent on oxygen concentrations. Most eukaryotic organisms require oxygen to survive. Oxygen is the terminal electron acceptor during energy production. It accepts an additional electron to create superoxide, a more reactive form of oxygen. Superoxide can be converted to hydrogen peroxide (H2O2) spontaneously.
ROS were traditionally thought of as toxic byproducts of living in an aerobic environment because they are known to damage cellular macromolecules (Fig. 1), which could subsequently lead to cell death (296). However, in recent years, several studies have shown that ROS can function as signaling molecules that regulate numerous cellular processes, including proliferation (9, 13, 19, 38, 39, 200 –202, 262, 276, 277, 315).

The second-messenger properties of ROS are believed to activate signaling pathways by activating tyrosine kinases, tyrosine phosphatases, MAP kinases, or ion channels (235). Furthermore, interactions between specific receptor-ligands also are known to generate ROS (76). This dual function of ROS, as signaling molecules or toxins, could result from the differences in their concentrations, pulse duration, and subcellular localization. The concentration-dependent effects of ROS regulating different cellular processes are clearly evident in a recent report by Laurent et al. (165). NIH 3T3 fibroblasts treated with 0.02–0.13 μM H2O2 enhanced proliferation, whereas treatment with 0.25–2 μM H2O2 resulted in cell death. Prostate cancer DU-145 cells treated with low concentrations of H2O2 (100 nM to 1 μM) enhanced c-Fos expression, which was associated with an increase in cell proliferation, whereas a higher concentration of H2O2 (200 μM) decreased c-Fos expression and induced cell-cycle arrest (341). Therefore, although higher levels of ROS can be toxic, low levels of ROS may serve as signaling molecules regulating numerous cellular processes, including proliferation (Fig. 1).
C. Antioxidant enzymes and small-molecular-weight antioxidants
Cellular ROS levels are maintained both by the production of ROS and by their neutralization by antioxidant enzymes and small-molecular-weight antioxidants. In addition to spontaneous conversion, superoxide is converted to hydrogen peroxide by superoxide dismutase enzymes (MnSOD, CuZnSOD, and EcSOD). Catalase (CAT) and glutathione peroxidase (GPx) neutralize H2O2 to water (Fig. 1). MnSOD, a nuclear-encoded and mitochondria-localized homotetrameric enzyme, is the primary defense against mitochondrially generated ROS (196). CuZnSOD is in both the cytoplasm and the nucleus. EcSOD is present in the plasma membrane and extracellular space (98). CAT is found primarily in the peroxisomes, and different isozymes of GPx are found in most subcellular compartments (225, 350).
Hydroperoxides also are neutralized by thioredoxin/thioredoxin reductase, glutaredoxin/glutaredoxin reductase, and the six-member family of peroxiredoxins (93, 262). Peroxiredoxins (Prxs) are a family of peroxidases that reduce H2O2 and alkyl hydroperoxides to water and alcohol. Prxs include both the 2-cys (Prx I-IV) and 1-cys (Prx V and VI) family of oxidoreductase proteins: Prx I, II, and VI are present in the cytosol; Prx III and V (short and long forms) are localized in the mitochondrion; and Prx IV is present in the endoplasmic reticulum and extracellular space (261). Thioredoxin is a small, 12-kDa ubiquitous protein. It reduces protein disulfides and itself is oxidized during this redox reaction. Oxidized thioredoxin is reduced by thioredoxin reductase, a seleno-cysteine protein, in the presence of NADPH (218). Glutaredoxins (Grx) are a GSH-dependent oxidoreductase family of 1-Cys and 2-Cys proteins of low molecular mass, 9–14 kDa. The human Grx family includes three members: Grx1 is present in the cytosol and nucleus, and Grx2 and Grx5 are present in mitochondria. Grx catalyzes the formation and reduction of the protein–mixed-disulfide forms in presence of the GSH/GSSG-redox couple and NADPH (130). Additional intracellular small-molecular-weight antioxidants include cysteine, vitamin C (ascorbic acid), and vitamin E (α-tocopherol) (77). Therefore, changes in the antioxidant enzyme activities or small-molecular-weight antioxidant levels or both could perturb the cellular redox environment, which in turn could affect the redox regulation of the cell-cycle progression.
D. Redox regulation of cell-cycle progression
The mammalian cell cycle has five distinct phases; quiescence is G0, whereas the proliferative state encompasses the G1, S, G2, and M phases. In response to mitogenic stimuli, quiescent cells enter the proliferative cycle and may transit back to the quiescent state. Reentry into quiescence is essential to prevent aberrant proliferation as well as to protect the cellular life span. The quiescent state is frequently incorrectly referred to as cellular senescence or differentiated states. Unlike differentiation and cellular senescence, quiescence is a reversible process that protects the proliferative capacity of cells essential for cell and tissue renewal. One of the best examples of the quiescent state in vivo is stem cells that retain the capacity to proliferate.
As mentioned previously, the redox regulation of cell-cycle progression was first reported in the cell cycle of sea urchin eggs (255). In mammalian cells, a transient increase in cellular prooxidant levels in G1 is required for entry into S phase (202). Inhibition of this prooxidant event with an antioxidant like N-acetyl-
Furthermore, in cultured hamster fibroblasts, sublethal doses of ROS added exogenously stimulated proliferation (39 –41). Likewise, H2O2 in nanomolar concentrations generated from growth factor receptor–ligand binding is known to facilitate cell proliferation (37). NADPH oxidases, such as Nox1 and Nox4, are required for growth factor–mediated production of H2O2, which subsequently activates multiple signaling pathways including the SOS-RAS-Raf-Erk and PI3K/AKT pathways (69, 259, 260). NADPH oxidase is a multi-subunit membrane-bound oxidase composed of p22 phox , p47 phox , p40 phox , p67 phox , and Nox2 (or any of its homologues: Nox1, Nox2, Nox3, etc.). The enzyme consists of two membrane-spanning subunits: p22 phox , which serves as a stabilizing and regulatory subunit for the superoxide-producing subunit Nox (174). The cytoplasmic components include p47 phox , p67 phox , p40 phox , and Rac, which helps to regulate the assembly of the functional oxidase and its activity (263). NADPH oxidase is found in various cell types, including neutrophils, smooth muscle cells, endothelial cardiac myocytes, and vascular and cardiac fibroblasts. Manipulations of cellular redox environment, by using NAC, inhibited proliferation in mouse embryonic fibroblasts, hepatic stellate cells, and vascular smooth muscle cells (147, 160, 202). The redox potential in proliferating cells is reported to be −240 mV, and necrotic cells exhibit the highest oxidizing state (−150 mV); the redox potential for the quiescent cells is in between proliferation and differentiation states, whereas apoptotic cells exhibit a redox potential of −170 mV (Fig. 1) (134, 282).
The hypothesis of a redox cycle regulating the cell cycle is also evident in other organisms (59, 328). The yeast metabolic cycle (YMC) in budding yeast oscillates between glycolytic and respiratory metabolism. Yeast cell-division cycle is restricted to the reductive phase of the YMC when oxygen consumption is minimal. The level of NADPH that provides a reducing equivalent to numerous enzymes peaks during the reductive phase of the YMC. Furthermore, the YMC in budding yeast coordinates with periods of gene expression regulating essential cellular and metabolic events (328). Mutations in the metabolic genes and cell-cycle checkpoint genes disrupt the communication between the YMC and cell-cycle progression (59). The literature discussed earlier overwhelmingly supports the hypothesis that a redox cycle within the cell cycle regulates progression through different cell-cycle phases.
E. Redox regulation of cell-cycle proteins
Progression through the cell-cycle phases is orchestrated by sequential and periodic activation of positive regulators, cyclins, and cyclin-dependent kinases (CDKs) (Fig. 2). Progression from G0/G1 to S is largely regulated by the D-type cyclins (cyclin D1 and D2) in association with CDK4-6 (112, 299, 300). CDK4-6 kinase activity in early G1 is low primarily because of lower levels of cyclin D1. After mitogenic stimulation, cyclin D1 peaks in mid-to-late G1, coinciding with higher levels of CDK4-6 kinase activity. Cyclin D1 expression is regulated at the transcriptional, posttranscriptional, translational, and posttranslational levels. A recent report indicates that cyclin D1 is transcriptionally downregulated by forkhead box O (FoxO3a) transcription factor, which subsequently inhibits cell-cycle progression (283). The FoxO-family of transcription factors are known to be phosphorylated by the mitogenic-signaling pathway, phosphatidylinositol-3 kinase (PI3K)/protein kinase B (AKT). Phosphorylated FoxO is excluded from the nucleus, thereby relieving FoxO-mediated gene repression (33). NAD-dependent deacetylases, sirtuin 1 and 2, also are known to activate FoxO transcription factor activity (35, 339).
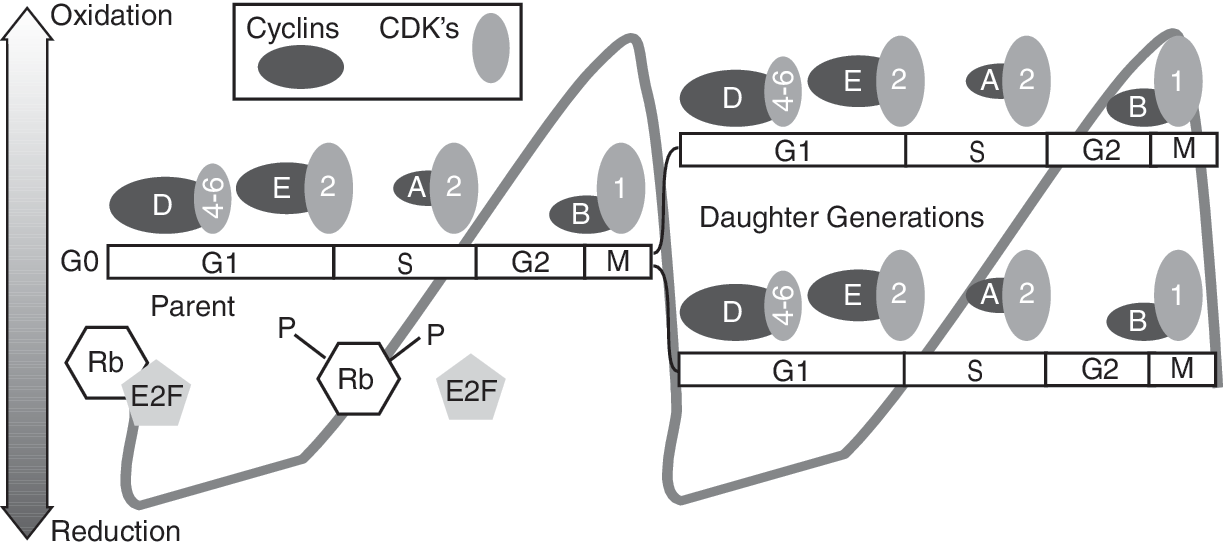
The cyclin D1/CDK4-6 kinase complex partially phosphorylates the retinoblastoma (Rb) protein, causing a conformational change that releases the E2F family of transcription factors (Fig. 2). Cyclin E/CDK2 kinase is activated in late G1 to early S and facilitates further Rb phosphorylation (112, 299). E2F activates the expression of multiple S-phase–specific genes that are required for DNA replication and progression through the S phase (214). The G1 phase of the cell cycle is critical in deciding whether proliferation will be arrested or continued. Temin (318) first proposed the presence of a “decision point” in the G1 phase beyond which cells become committed to progress through the cell cycle and divide. In 1974 Pardee (236) renamed the decision point as “restriction point” and defined it as the time in G1 after which a cell is committed to enter the S phase, more or less independent of external conditions. Although the mechanisms regulating the “restriction point” are not completely understood, it was observed that a change in the cellular redox environment toward a more-oxidizing environment is required for entry into S phase (202). Thus, the “restriction point” could represent a redox threshold necessary for progression from G0/G1 to S.
The cyclin A/CDK2 kinase complex regulates progression through S and G2 phases. The cyclin B1/CDK1 kinase complex, along with CDC25C phosphatase, regulates progression from G2 to M phase. Earlier it was believed that the functions of individual cyclins and CDKs are specific to a specific cell-cycle phase. However, recent reports demonstrate redundancy in these cell-cycle–regulatory protein functions. For example, cell-cycle progression is unaffected in CDK2, CDK4, and CDK6 knockout mouse embryos, suggesting that CDK1 can substitute for other CDKs. Cyclin D1 knockout mice are viable, possibly because of redundancy from cyclin E function. However, knockout of cyclin A and cyclin B are lethal in mice (5, 128). Cyclins are the positive regulators of cell-cycle progression, and cyclin-dependent kinase inhibitors (CKIs) are the negative regulators. The INK family of CKIs (INK4B, p15; INK4A, p16; INK4C, p18; INK4D, p19) specifically inhibit cyclin D/CDK4-6 kinase complexes. The KIP family of CKIs (p27 and p57) inhibits mainly cyclin E/CDK2 kinase complexes. The inhibitory effect of p21 is ubiquitous, and it can inhibit all cyclin/CDK kinase activities. p21 and p27 also are known to facilitate the assembly of cyclin/CDK complexes (60, 161).
Redox regulation of cell-cycle proteins p21, Rb, cyclin D1/CDK4-6 kinase, and CDC25 phosphatase is observed in NAC-treated mouse and human fibroblasts (37, 175, 279, 288, 340, 351). NAC treatments shift the cellular redox environment toward a more-reducing environment. This change is associated with a decrease in cyclin D1, an increase in p27, and Rb hypophosphorylation (199, 201, 202). The decrease in cyclin D1 is inversely correlated with MnSOD activity (201). Considering that MnSOD is mitochondrial and cyclin D1 is a nuclear protein, this inverse correlation is intriguing. One possible mechanism of this interorganelle crosstalk could be due to the FoxO-mediated transcriptional control of cyclin D1 and MnSOD expression. FoxO3a is known to activate MnSOD transcription, while inhibiting cyclin D1 transcription (153, 283). FoxO3a-mediated induction in MnSOD transcription is associated with the quiescence state. During the quiescence state, FoxO3a has been shown transcriptionally to upregulate p27 expression (198). Inhibition of FoxO3a activity is anticipated to relieve cyclin D1 from transcriptional repression, which in turn is anticipated to support cellular proliferation.
Alternatively, the redox sensitivity in cyclin D1 expression could also be regulated by posttranslational mechanisms. NIH3T3 mouse fibroblasts carrying the Thr286A cyclin D1 mutation suppressed NAC-induced cyclin D1 degradation. This suggests that redox-sensitive phosphorylation of Thr286 could influence cyclin D1 protein levels (201). Furthermore, the redox sensitivity in cyclin D1 accumulation could also be regulated by the thiol-redox reactions of critical cysteine residues. Consistent with this hypothesis, mutations of specific cysteines were found significantly to decrease cyclin D1 protein levels compared with wild-type cyclin D1 (unpublished observations).
In contrast to the effect of NAC on cyclin D1, H2O2 inhibits cyclin D1 protein degradation in Her14 fibroblasts, resulting in cyclin D1 accumulation (188). Overall, this literature supports the hypothesis that redox regulation of the cell cycle could be mediated via the redox-sensitive regulation of the cell-cycle–regulatory protein function.
The significance of the relation between cyclin D1 expression and the cellular redox environment is clearly evident from a recent report by Sakamaki et al. (274). Physiologic levels of cyclin D1 decreased aerobic glycolysis and mitochondria size and function in vivo. Mitochondria activity was enhanced by genetic deletion of cyclin D1. Subsequent study by Wang et al. (338) showed that cyclin D1/CDK4-6 phosphorylates nuclear respiratory factor 1 (NRF1) at Ser47, suppressing its transcriptional activation of nuclear-encoded mitochondrial gene expression (338). Likewise, dephosphorylation of NRF1 in the absence of cyclin D1 promotes expression of nuclear-encoded mitochondrial genes. These results provide strong evidence for cyclin D1 coordinating cellular metabolism and cell-cycle progression.
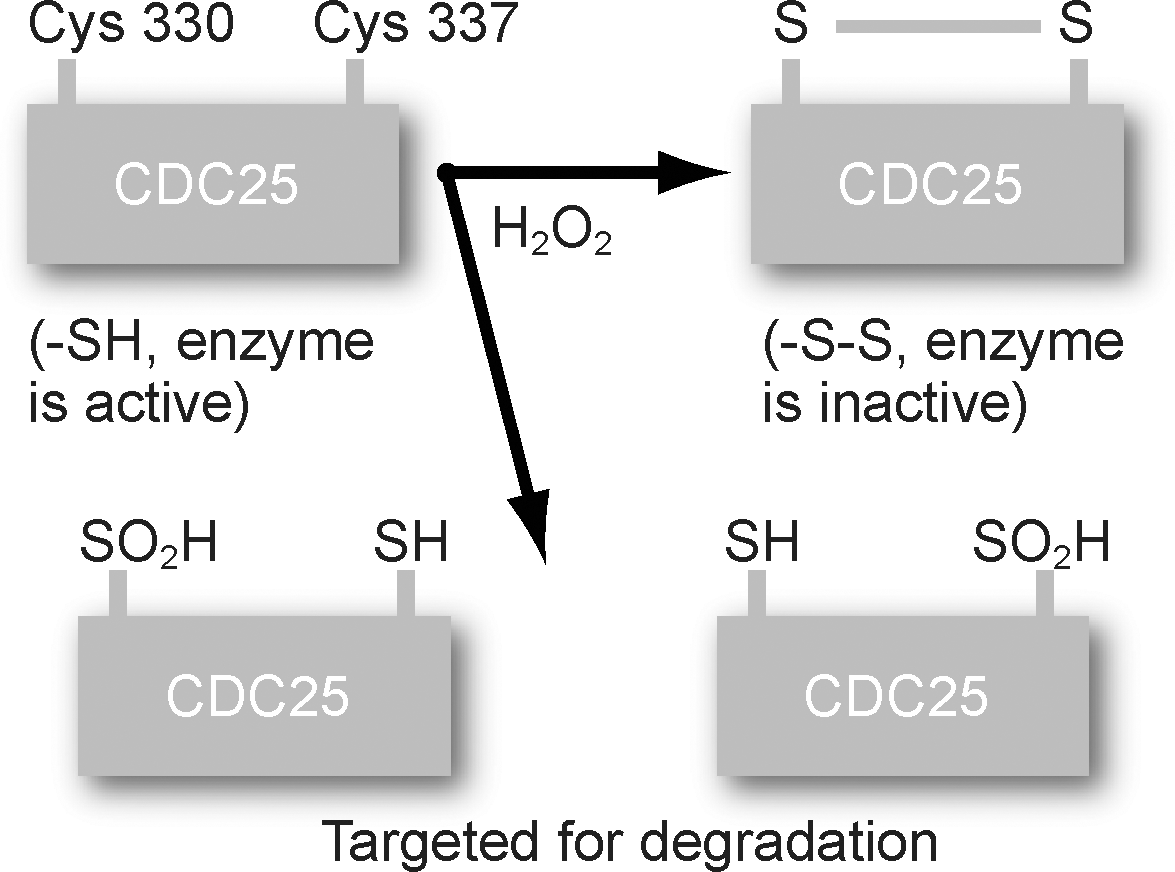
Another cell-cycle–regulatory protein that exhibits redox sensitivity in its function is CDC25 phosphatase. CDC25 phosphatases are a family of dual specific phosphatases that dephosphorylate pThr14 and pTyr15 on CDKs and activate the cyclin-CDK kinase activity (287). Dunphy and Kumagai (84) showed in vitro that the phosphatase activity of CDC25 can be inhibited by using N-ethylmaleimide, a thiol-alkylating agent, or mutating a single conserved cysteine residue. Recently, Savitsky and Finkel (279) showed that the H2O2 treatment of HeLa cells induces an intramolecular disulfide bond between two critical site cysteines, Cys377 and Cys330, of CDC25 (Fig. 3). This thiol-disulfide redox reaction is associated with an inhibition in CDC25 phosphatase activity. CDC25 harboring the double mutant of the cysteines was resistant to the H2O2-induced inhibition in its phosphatase activity. These observations are consistent with earlier reports suggesting that ROS could reversibly modify the redox state of specific cysteine residues in phosphatases (protein tyrosine phosphatases and dual-specificity phosphatases), inactivating their activities, which could favor Ser/Thr phosphorylation-dependent signal pathways initiating proliferation (95, 205, 260, 314, 320).
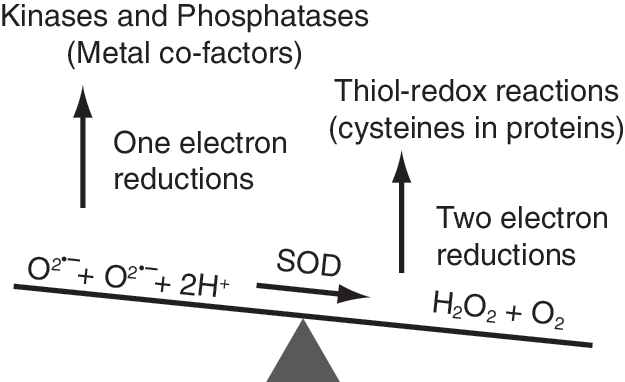
Hydrogen peroxide could influence the redox state of protein thiols, two-electron reactions (36) (Fig. 4). The reduced form of cysteine in protein (RSH) can be oxidized to sulfenic acid (RSOH), which can be further oxidized to sulfinic (RSO2H) and sulfonic (RSO3H) acids. The sulfinic and sulfonic forms of proteins are believed to be targeted for degradation; the sulfenic form can react with another RSH to form the disulfide, RSSR. RSSR can be then reduced back to RSH by cellular antioxidant machinery. This thiol-disulfide exchange reaction can regulate many of the cell-cycle–regulatory protein functions during the redox regulation of the cell cycle. In addition, superoxide can initiate one-electron reactions that can alter the redox state of metal cofactors (e.g., Fe and Zn) (Fig. 4) present in many kinases and phosphatases, thereby affecting their activities. Thus, both one- and two-electron reactions can participate in the redox regulation of cell-cycle proteins during progression from one cell-cycle phase to the next.
F. RNS signaling and cell-cycle progression
Reactive nitrogen species (RNS) are molecules derived primarily from the reactions of nitric oxide. Nitric oxide (NO) is a short-lived and highly reactive diffusible free radical that is known to regulate various biologic processes. NO is produced from
NO can also mediate important biologic effects via the activation of specific cell-signaling pathways. Lower concentrations of NO are known to activate NF-κB transcription factor, possibly by S-nitrosation of p21 and activation of IκB. In contrast, higher concentrations of NO inhibit the DNA-binding activity of the SP-1 transcription factor (79, 167). Higher levels of NO have been shown to accelerate S-phase entry basally, and facilitate entry into mitosis apically in developing chick neuroepithelium (227). Low-molecular-weight S-nitrosothiol, S-nitroso-N-acetylpenicillamine promotes the nitrosation of p21 Ras and the activation of the Ras-ERK 1/2-MAP kinase signaling pathway. This activation in the ERK-signaling pathway leads to cell-cycle progression in rabbit aortic endothelial cells (17). NO inhibits proliferation in vascular smooth muscle cells, resulting in G1 delay. NO-induced G1 delay is accompanied with a decrease in cyclin A/CDK2 activity and an increase in p21 protein levels (324). Results from these studies indicate that the cell-type specificity and signaling pathway(s) could significantly influence the mitogenic and cytostatic properties of NO.
G. Summary
This literature clearly supports the hypothesis that periodic oscillations in metabolic redox reactions, a redox cycle, within the cell cycle represent a fundamental mechanism linking oxidative metabolic processes to the cell-cycle–regulatory processes. The periodicity in cellular redox environment is maintained by a delicate balance between the production of ROS, RNS, and their removal by nonenzymatic and enzymatic antioxidants. Redox regulation of the cell-cycle–regulatory proteins could be influenced by the presence of redox-sensitive motifs, such as cysteine residues or metal cofactors in kinases and phosphatases. The literature presented in the next section integrates information supporting the concept that perturbations in the redox control of the cell cycle could lead to proliferative disorders. It is hypothesized that reestablishing the redox cycle by manipulating the cellular antioxidant pathways could reverse, suppress, and/or prevent many aspects of proliferative disorders.
II. Redox Control of the Cell Cycle and Proliferative Disorders
A. Development
Development in living organisms involves two distinctive criteria: proliferation and differentiation. Cells proliferate in low oxygen concentrations throughout the embryonic stage and in higher oxygen concentrations during neonatal life. This environmental transition from low to high oxygen during development creates a gradient of ROS that may have direct and indirect effects on cellular proliferation. ROS signaling is known to regulate many of the transcription factors that influence development (e.g., NF-κB, AP-1, and HIF-1) (162).
The role of antioxidants during development is well documented. For example, homozygous MnSOD-knockout mice survive the embryonic stage of development. However, these mice die after birth of lactic acidemia, cardiomyopathy, and degeneration of the basal ganglia (138, 325, 326). Developmental defects in MnSOD-knockout mice are associated with damage to mitochondrial aconitase, complex I, and succinate dehydrogenase. In comparison to control mice, defects in MnSOD-knockout mice were very pronounced after oxygen exposure, with a subsequent increase in ROS production (166, 171). MnSOD overexpression is known to promote differentiation (62, 168, 310).
CuZnSOD-knockout mice are viable (194), but slowly develop neuronal axonopathy, which is not as pronounced at birth in comparison to mice lacking MnSOD (166, 171). Another example of SOD-dependent developmental defects relates to CuZnSOD overexpression in Down syndrome patients; aberrant placental formation is common in these patients (10, 99, 100). Furthermore, a lack of selenium-containing protein antioxidants, such as thioredoxin reductase and phospholipid hydroperoxide glutathione peroxidase (GPx-4), is lethal in early gestation (191, 353). Interestingly, another selenium-containing protein, antioxidant selenoprotein W, was found to be highly expressed in proliferating cells during the development of the heart, skeletal muscle, and the nervous system in mice. However, as cells exit from the cell cycle, the expression of this protein is decreased (140, 177).
The changes in the cellular redox environment during development could affect both proliferation and differentiation. Although proliferation is the major event in development, both positive (cyclin/CDK) and negative (CKI) regulators of the cell cycle are involved. Results from knockout mice demonstrated that a lack of individual cyclins and CDKs is not lethal to the organism (63, 89, 184, 231, 249, 303, 304), with the exception of cyclin B1 and cyclin A2, which are fatal in early gestation (28, 208, 209). In general, these mice have a normal developmental life, suggesting that redundancy in cyclin and CDK functions could compensate for the absence of an individual member of the cyclin and CDK family of proteins. It also is interesting to note that whereas individual cyclin (or CDK)-knockout embryos develop normally to adults, they are susceptible to proliferative disorders later in life. An additional complication of this regulation is that these impaired proliferative activities are tissue specific. Cyclin-dependent kinase inhibitor (INK4 and CIP/KIP) homozygous knockout mice showed viable embryos and normal neonatal development. However, these animals did develop an increase in tumor incidence later in life (75, 150, 155, 164, 295). p57-Knockout mice were found to be neonatal fatal with severe developmental defects in the gastrointestinal tract and abnormal cell proliferation in placenta, cartilage, and eye lenses (352, 356).
This literature suggests that the cellular redox environment and cell-cycle–regulatory proteins might collectively regulate development. Although individual cell cycle–regulatory proteins may not affect the development process because of redundancy, their presence as a family is essential for the normal developmental process.
B. Aging and cancer
One of the most well-stated definitions of aging was offered by Caleb Finch (94), who defines aging as “a nondescript colloquialism that can mean any change over time, whether during development, young adult life, or senescence. Aging changes may be good (acquisition of wisdom); of no consequence to vitality or mortality risk (male pattern baldness); or adverse (arteriosclerosis).” The numerous theories of aging can be broadly classified into two major categories: error and program theories. According to the program theory, aging occurs because of a preexisting external or internal program. The program theory of aging includes both the “Hayflick limit” and the telomere-shortening phenomenon. The error theory considers aging to be a cumulative damage process (289). The free radical theory of aging, originally proposed by Harman (123), is an example of the error theory of aging. ROS-mediated damage to critical cellular macromolecules is believed to accumulate as a function of age and to lead to deleterious effects associated with degenerative diseases of aging, senescence, and carcinogenesis (96). A recent study clearly demonstrated that mitochondria-targeted overexpression of human catalase extends the median and maximal life span of mice by 20% (285). Mitochondrial overexpression of catalase delayed age-associated loss in mitochondrial DNA deletions, cardiac pathology, and cataracts in mice (285). Previous studies reported that overexpression of MnSOD and CuZnSOD extended Drosophila life span (230).
In 1961, Hayflick (reviewed in ref. 49) showed that human diploid fibroblasts cultured in vitro divide a finite number of times (Hayflick limit) before irreversible growth arrest, also known as senescence. Cellular senescence is associated with specific changes in cell morphology that include increased cell volume, expression of neutral senescence-associated β-galactosidase activity, and increased production of extracellular matrix degradative enzymes such as collagenase and stromelysin. Cellular senescence is believed to be caused by telomere shortening. Telomeres are repetitive DNA sequences (TTAGGG in vertebrates) present at the ends of chromosomes and are essential for maintaining chromosomal integrity. During DNA replication, 50–200 bp of telomeric DNA are not replicated at the end of the S phase of the cell cycle. Because telomerase, the enzyme that synthesizes telomeric DNA de novo, is not expressed by most human cells, telomeres shorten with each cell cycle. When the telomeres shorten from the maximum size of 10–15 kb to an average size of 4–6 kb, human cells irreversibly arrest in growth, producing a characteristic phenotype, defined as senescence (48, 308). These observations are known now as the telomere hypothesis of the Hayflick limit.
It is important to mention that a majority of cell-culture experiments are performed at nonphysiologic oxygen concentrations of 21% compared with physiologic concentrations of 4%. Because ROS are byproducts of oxygen metabolism, it is anticipated that oxygen concentrations will significantly affect cellular ROS levels: 21% oxygen environment is known to reduce population doublings (PDs) in cultured cells (294, 348, 349). Furthermore, human and mouse fibroblasts cultured at 21% oxygen concentrations exhibit significantly reduced replicative life span despite mouse cells having long telomeres (43, 237). Consistent with these previous reports, our unpublished results show that normal quiescent human skin fibroblasts cultured at 4% vs. 21% oxygen concentrations protect these cells from age-related loss in proliferative capacity. Interestingly, the protection of proliferative capacity at 4% vs. 21% oxygen environment correlated with the preservation of mitochondrial morphology. These results suggest that oxygen concentrations could significantly influence the redox biology of the cell cycle.
Results from several studies suggest that the redox regulation of the cell cycle could be influenced by the redox-controlled maintenance of telomeres. Overexpression of EcSOD in human fibroblasts decreased the intracellular peroxide content, slowed the telomere shortening rate, and elongated the life span of these cells under normoxia and hyperoxia (293). Minamino et al. (203) showed that hypoxia extends the life span of vascular smooth muscle cells through activation of telomerase activity. Furthermore, ectopic expression of telomerase increased MnSOD expression by more than sevenfold (298). Telomerase-immortalized cells have higher levels of the p21 cell-cycle–regulatory protein (49, 83, 110, 334). Thus, the redox regulation of the cell cycle could protect replicative senescence via a redox-sensitive regulation of the telomeres and telomerase activity.
Redox regulation of the cell cycle could also influence the chronologic life span of aging. Chronologic life span is characterized as the capacity of quiescent (G0) cells to reenter the proliferative cycle (124). The mechanisms regulating the chronologic life span are poorly understood. In response to mitogenic stimuli, quiescent cells enter the proliferative cycle and subsequently transit back to the quiescent state. This reversible property of cellular quiescence is highly essential to protect the chronologic life span and avoid aberrant proliferation. MnSOD activity protects the chronologic life span of normal human skin fibroblasts from age-dependent loss (276). Quiescent normal human skin fibroblasts cultured for 40 to 60 days were unable to reenter the proliferative cycle after replating. This inhibition of reentry was associated with a significant accumulation of p16 and a decrease in p21 cyclin-dependent kinase inhibitor protein levels. Interestingly, MnSOD overexpression suppressed age-associated increase in p16 accumulation, maintained p21 at a higher level, and restored the ability of quiescent fibroblasts to reenter the proliferative cycle (Fig. 5). Furthermore, MnSOD activity has been shown to regulate a ROS switch favoring a superoxide signal regulating the proliferative cycle and a hydrogen peroxide signal supporting quiescent growth. Higher levels of MnSOD activity were associated with quiescence, whereas lower levels support proliferation. MnSOD activity–regulated transitions between quiescent and proliferative growth was associated with changes in cyclin D1 and cyclin B1 protein levels (277). These results support the hypothesis that MnSOD activity could maintain a redox-balance protecting the chronologic life span.

Cellular ROS levels and the protection of proliferative capacity are also apparent in hematopoietic stem cells (322). FoxO transcription factor–deficient hematopoietic stem cells (HSCs) showed reduced ability to repopulate (321). Because FoxO is known to regulate antioxidant enzyme (MnSOD and catalase) and cell-cycle (cyclin D1 and p27) genes transcription (154), it has been suggested that ROS could mediate the proliferative capacity of HSCs. Consistent with this notion, small-molecular-weight antioxidant (NAC) treatment of FoxO-deficient HSCs protected the proliferative capacity of HSCs (322). Overall, this literature supports the hypothesis that a loss in the redox control of the cell cycle during transitions between quiescent (G0) and proliferative (G1, S, G2 and M) cycles could severely affect the proliferative capacity of cells.
Cancer is a disease manifesting late in life, suggesting that the very biology of aging contributes to its exponential increase in the older population. Cancer risk is elevated with aging, which could be due to an increase in ROS production or decrease in ROS removal or both (157, 331). Carcinogenesis can be divided into three distinct stages: initiation, promotion, and progression. Initiation can occur because of mutations in one or more genes, which result in loss or gain of function. Promotion is the functional enhancement and alteration of the pathway induced by initiation. Progression is the continuing change of the unstable karyotype, often leading to aberrant proliferation. Aberrant proliferation in cancer cells could be due to a loss in the redox regulation of the cell cycle. Oberley and Buettner (224) were first to report that cancer cells exhibit lower levels of antioxidant enzyme activities compared with their respective normal cells, in particular MnSOD. Other studies suggest that oxidative stress could significantly contribute to cancer progression, possibly by perturbing the redox control of the cell cycle (103, 118, 129, 239, 240, 292). Redox potential in normal cells correlates with Rb phosphorylation status during the cell cycle, suggesting that perturbations in cellular redox potential could significantly affect the function of a tumor-suppressor gene (129). Furthermore, it is hypothesized that the metabolic redox-signaling pathways could initiate as well as promote carcinogenesis (108). This hypothesis is based on numerous studies demonstrating a regulatory role of MnSOD activity in cancer cell growth in both cell-culture and tumor xenograft animal model systems (62, 223, 346, 347, 357). These results suggest that reestablishing the redox control of the cell cycle by manipulating the expression of ROS-removal enzymes (e.g., MnSOD) could suppress or inhibit (or both) carcinogenesis.
Recent reports enlighten an additional aspect of carcinogenesis that relates to the tumor microenvironment. Several studies report that aged normal human breast and prostate fibroblasts support epithelial malignancies (11, 16, 50, 81, 157, 228, 271). The majority of human cancers are carcinomas originating from epithelial cells. Fibroblasts are the primary component of the stroma that supports these epithelial tissues. Campisi et al. (47, 50, 51) showed that senescent human fibroblasts enhanced cellular proliferation in premalignant and malignant epithelial cells in vitro, and tumor growth and metastasis in mice in vivo. Prostate epithelial cells from tissue with aged stroma can become tumorigenic when co-cultured with tumor-burden fibroblasts (228). Likewise, exposure of mammary gland stroma to irradiation or carcinogens stimulates nonmalignant epithelial cell proliferation and promotes tumor formation (11). Although the mechanisms regulating this phenomenon are not completely understood, the secreted growth factors, cytokines, and extracellular matrix proteins from aged fibroblasts are believed to enhance premalignant and malignant epithelial cell proliferation. Because many of the growth factors and cytokines are known to generate ROS, it is hypothesized that ROS signaling derived from aged fibroblasts could provide mitogenic stimuli to premalignant and malignant epithelial cells.
C. Wound healing
Wounds are an inevitable part of life. They could arise both from internal and external injuries. Wound healing is a complex process that can be grouped into three major overlapping stages: inflammation, proliferation, and maturation or closure (Fig. 6). ROS are involved in all three stages of wound healing (31, 226, 256, 270, 302, 311). The inflammation stage of the wound-healing process is one of the most widely studied areas for ROS production, and it is the stage during which most ROS are produced (270). The production of ROS during the inflammation stage is believed to protect cells from pathogens and regulate angiogenesis (290). Hydrogen peroxide levels are higher in the early stages of wound repair compared with late stages; superoxide is also detected in the leading edge of the wound area (270). MnSOD, CuZnSOD, catalase, and glutathione peroxidase levels increases during the normal wound-healing process (311). Likewise, treatment with SODs and administration by hydrogels containing CuZnSOD or transgenic expression of MnSOD resulted in better wound healing in mouse models (56, 61, 291). In contrast, lower levels of antioxidants (e.g., glutathione) were associated with improper wound healing (207, 256).
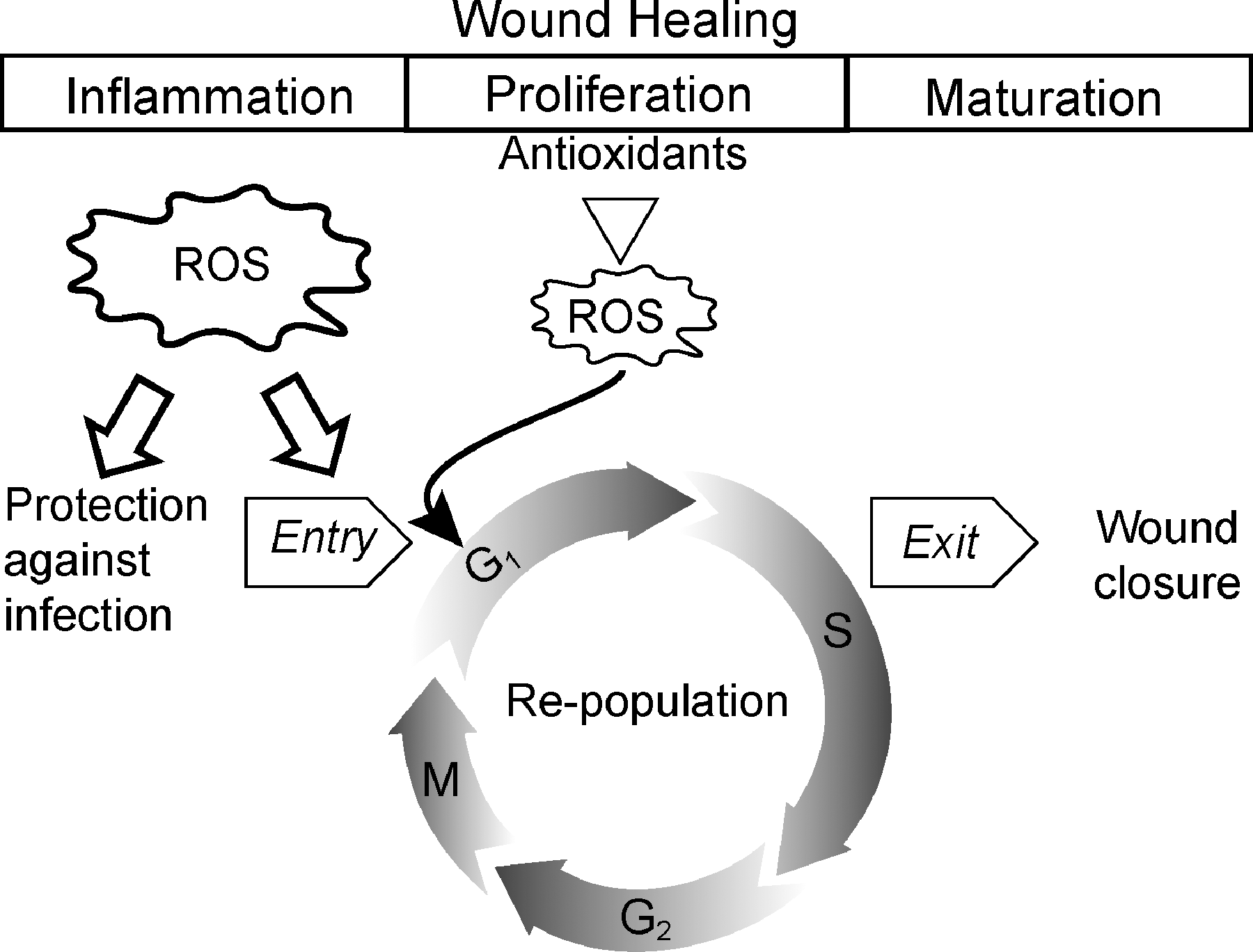
Although inflammation is the initial stage, the wound-healing process is mainly dependent on the proliferative stage. Cells repopulate during the proliferative phase and prepare the wound for closure and healing. The repopulation process depends on the ability of quiescent cells to reenter and subsequently exit the proliferative cycle (Fig. 6). Quiescent normal human skin fibroblast entry into and exit from the proliferative cycle is dependent on cellular superoxide and hydrogen peroxide levels regulated by MnSOD (277). Redox-sensitive regulation of cyclin D1 and cyclin B1 protein expression correlated with the quiescent fibroblast entry into and exit from the proliferative cycle (277). Cyclin E and Ki-67 protein levels are known to increase during the early stages of the wound-healing process (359). Increased levels of cyclin D1/CDK4-6 correlate with better wound closure (332). Furthermore, ROS are also known to regulate cellular migration and proliferation during different physiologic responses (217, 252). The decrease in cyclin levels at later stages of the wound-healing process was associated with an increase in p21 and p27 (12, 359, 360, 364). p21 and p27 were upregulated in the migrating epithelial cells on the leading edge of the wound, whereas the basal cells showed an increase in cyclin A and Ki-67 protein levels (12). Redox-sensitive control of the proliferative stage of wound healing is critical for normal wound healing.
Loss of the redox-sensitive control in the inflammatory or proliferative stages, including migration and reentry into the proliferative cycle, could lead to higher accumulations of collagen, elastin, fibronectin, and proteoglycan that are hallmarks of keloids and hypertrophic scars (1, 44, 178). These results support the hypothesis that tight redox control of the cell cycle is necessary for proper wound healing.
D. Fibrosis
Fibrosis is defined as the formation of fibrous tissue as a reaction or repair process due to disease, treatment, or exposure to chemicals. Fibrosis involves the overgrowth, hardening, and often scarring of tissue due to excess collagen. Fibrosis is most common in the lung, heart, peritoneum, and liver.
1. Radiation-induced fibrosis
Radiation-induced fibrosis (RIF) is a serious and common complication of radiation therapy that causes chronic pain, neuropathy, swelling of lymph nodes, and limited motion in the joints. It occurs most commonly in the head, neck, breast, and connective tissues. Risk factors for developing RIF include high-dose radiation, large tissue volume exposed to radiation, and radiation combined with surgery or chemotherapy.
The biologic effects of ionizing radiation begin with the generation of both early and late ROS accumulation (105). ROS signaling could activate quiescent fibroblasts to differentiate into myofibroblasts, which have the phenotype of smooth muscle cells. Myofibroblasts appear during the initial inflammatory phase, and they are present during the constitutive fibrotic phase. Myofibroblasts are characterized by increased proliferation and reduced production of extracellular matrix metalloproteinases (187). The persistent excess of myofibroblasts is believed to be responsible for the areas of hypercellular fibrosis and the clinical observation of radiation-induced fibrous swellings (74). Numerous biochemical compounds, such as cysteine, pentoxifylline, and tocopherol, are used to minimize radiation-induced damage to normal tissue (74).
EcSOD-overexpressing mice exhibit decreased fibrosis, which correlated with a decrease in TGF-β and Smad3 protein levels (144, 243). Likewise, liposomal delivery of CuZnSOD in a 3D-culture of myofibroblasts decreased TGF-β levels and facilitated myofibroblast reversal to normal fibroblasts (335). Although in general, the effect of TGF-β is growth inhibition, TGF-β is also known to promote proliferation of fibroblasts. TGF-β has been shown to decrease p21 and p27 protein levels in WI38 human lung fibroblasts and NIH3T3 mouse fibroblasts, respectively (80, 257). This decrease in cyclin-dependent kinase inhibitors was accompanied by a significant increase in cyclin E/CDK2 kinase activity. A decrease in cyclin D2 expression in fibroblasts isolated from breast cancer patients correlated with a low risk for the development of RIF (264). TGF-β remains elevated long after the radiation treatment (6). The increase in TGF-β can result in prolonged inhibition of p21 and p27, which could support continued proliferation facilitating the development of RIF.
2. Lung fibrosis
The most common types of lung fibrosis include idiopathic pulmonary fibrosis (IPF), chronic obstructive pulmonary disease (COPD), and bleomycin-, asbestos-, or cigarette smoke-induced fibrosis (137, 148). The lung is subject to the highest exposure to oxygen, which makes it susceptible to ROS- and RNS-induced abnormalities. In addition to the ETC and NADPH oxidases, ROS in lung can be generated by myeloperoxidase, eosinophil peroxidase, and xanthine oxidase (149). Xenobiotics or pathologic conditions can overcome the detoxification enzyme system (104, 149, 246, 337). GSH levels were found to be low in the epithelial lining fluid of IPF patients (52). In bleomycin-induced lung fibrosis, increased oxidation of cysteine (Cys) to its oxidized form, cystine (CySS), and a decrease in the GSH pool were observed (137). IPF patients also exhaled more NO as compared with healthy individuals (265). Failure to cope with the oxidant insult could be the major factor causing fibrosis. This hypothesis is supported by a recent report of aerosolized administration of NAC attenuating bleomycin-induced lung fibrosis (117).
The oxidant insult could lead to a loss of regulation of the fibroblast cell cycle, causing the resulting fibrosis. Fibroblasts derived from areas of fibrosis proliferate faster than cells derived from histologically normal areas of lung tissue (141). Patients with IPF have elevated levels of nitric oxide, which could stimulate proliferation of human lung fibroblasts, possibly via NF-κB–mediated activation of cyclin D1 expression and progression from G0/G1 to S phase (127, 186, 265). NO-induced proliferation in a normal human fetal lung fibroblast cell line, MRC-5, is associated with inhibition of p21 and p27, activation of cyclin/CDK complexes (cyclin D1/CDK4-6 and cyclin E/CDK2), and hyperphosphorylation of Rb (Fig. 7) (57).

As in RIF, oxidants and TGF-β may interact to enhance fibrosis in patients with IPF (149). Active TGF-β has been detected in patients with pulmonary fibrosis, in contrast to healthy individuals in whom TGF-β is present mostly in the latent form (23). TGF-β has been shown to activate NADPH oxidase in human fibroblasts, which is associated with an increase in ROS levels (319). TGF-β–induced increase in ROS levels inhibits cell proliferation by inducing a G1 arrest, decreases cyclin D1/CDK4-6 kinase activities, and increases p21, p27, and p16 protein levels (71, 206, 323). Consistent with these results, p21 overexpression demonstrates an antiapoptotic and antifibrotic effect in attenuating bleomycin-induced pulmonary fibrosis in mice (135). It is unclear how TGF-β could promote proliferation in RIF and inhibit proliferation in pulmonary fibrosis.
3. Cardiac fibrosis
Cardiac fibrosis refers to the thickening of heart valves due to increased proliferation of cardiac fibroblasts and subsequent collagen accumulation. The perturbation in cardiac fibroblast proliferation is believed to be regulated by ROS signaling generated from the membrane-bound NADPH oxidase, a major source of superoxide in the heart. An increase in Nox2, also known as gp91 phox , has been observed in the perivascular space and at sites of fibrosis in both the right and left ventricles (358). Cardiovascular NOX is thought to release low levels of superoxide intracellularly. These lower amounts of ROS could serve as second messengers initiating cellular signaling pathways that control many cellular processes, including proliferation.
Both the renin–angiotensin system and TGF-β play important roles in the development of cardiac fibrosis. Angiotensin II (Ang II) is an effector hormone of the circulating renin–angiotensin system, which has endocrine functions in maintaining cardiovascular homeostasis. Ang II-dependent induction of TGF-β expression induces cardiac fibroblast proliferation and phenotypic conversion to myofibroblasts (266). Ang II increase in cell proliferation is associated with a significant increase in cyclin D and cyclin A expression in neonatal rat cardiac myocytes (272). Furthermore, Ang II–induced phosphorylation of Rb on serine 480, a mitosis-specific event, suggests that Ang II promotes cell division (272). Overexpression of p21 and p16 inhibited Ang II–induced cardiac myocyte hypertrophy (220). These results support the hypothesis that a loss in the redox regulation of the cell cycle could contribute to the genesis of cardiac fibrosis (Fig. 8).
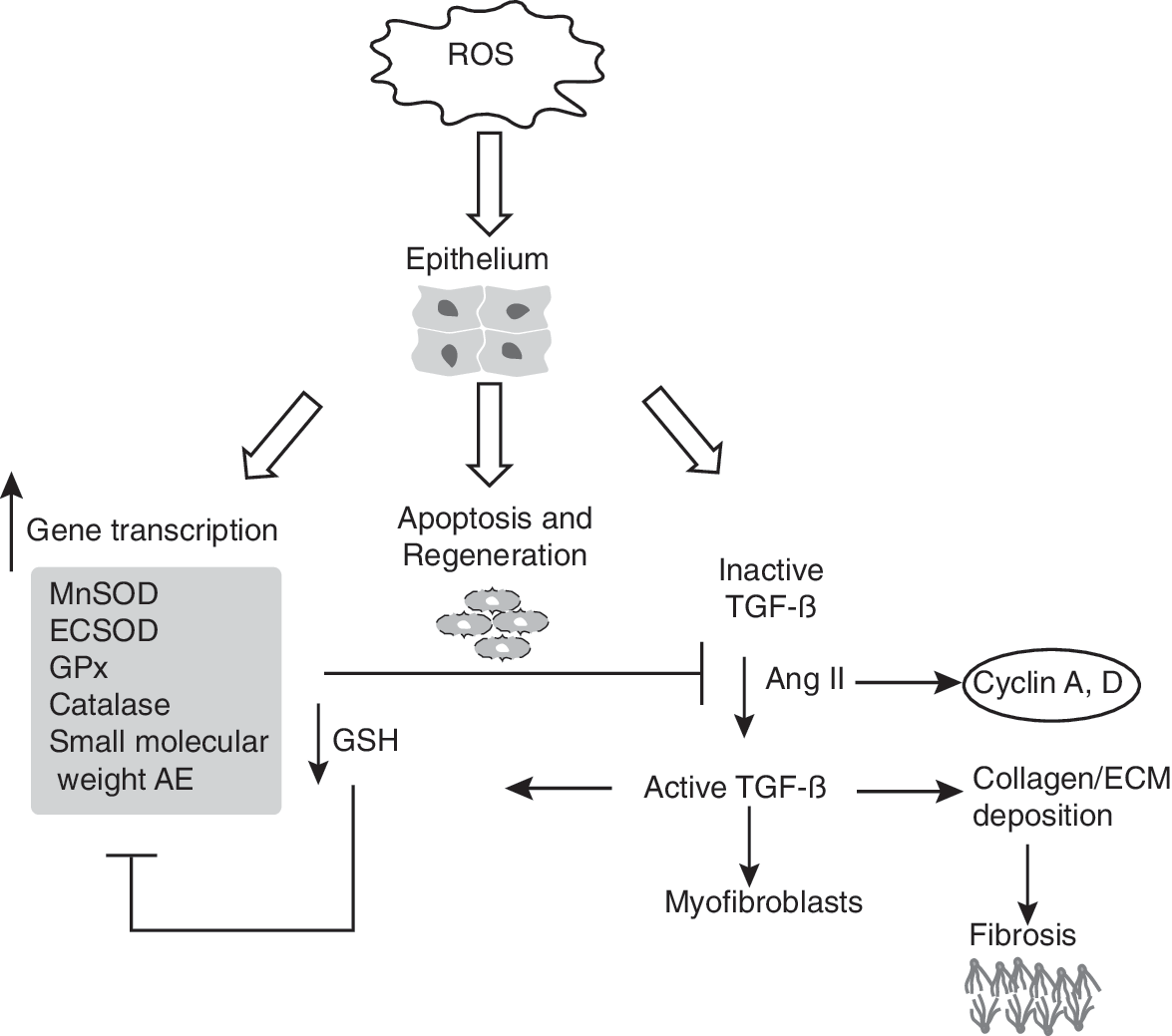
4. Liver fibrosis
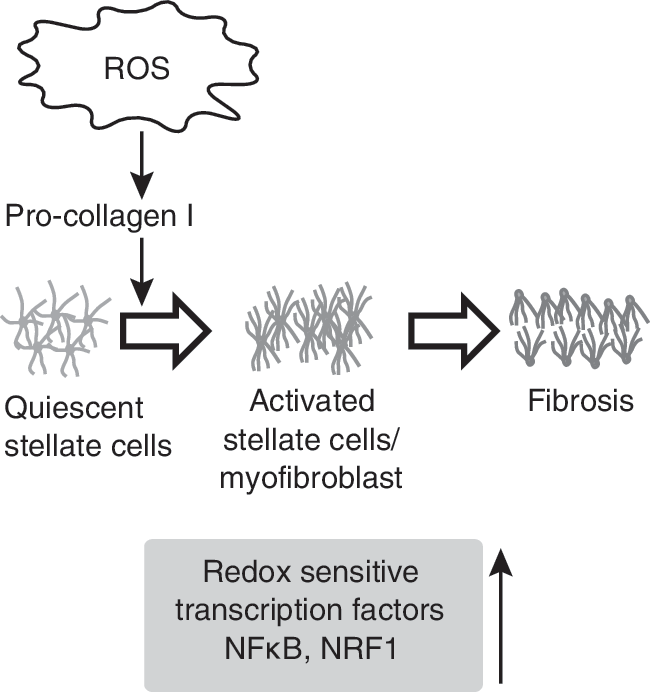
The adult human liver is the largest internal organ, and it plays an important role in the metabolism and the clearance of body toxins. The parenchymal cells of the liver contain most of the hepatic antioxidant enzymes, but Kupffer cells, hepatic stellate cells (HSCs), and endothelial cells are more exposed and sensitive to oxidative stress. Chronic liver injury is associated with accumulation of matrix proteins, causing fibrosis (336). After liver injury, parenchymal cells regenerate and try to replace necrotic or apoptotic cells. This is usually accompanied by an inflammatory response and the deposition of extracellular matrix (ECM). The excess accumulation of extracellular matrix in hepatic fibrosis is regulated mostly by HSCs. After liver injury, HSCs undergo “activation” that is accompanied with the transition from quiescent to proliferative growth (101). In the healthy liver, the HSCs are in a quiescent state and function to store vitamin A. In response to liver damage, quiescent HSCs lose vitamin A and differentiate into a myofibroblast phenotype expressing α-smooth muscle actin (101). This phenomenon is a hallmark of cellular response to liver injury. MnSOD activity is known to regulate transitions between quiescent and proliferative growth states (277), suggesting that quiescent HSC entry into the proliferative cycle could be regulated by the cellular redox environment.
The cellular redox environment in hepatocytes and Kupffer cells may be regulated by ROS produced from the NADP/NADPH oxidase system or leakage of electrons from the ETC (or both), followed by univalent reduction of oxygen to superoxide anion (72, 172). Substrates like ethanol, polyunsaturated fatty acids, and iron may enhance ROS production. Noncytotoxic levels of superoxide in human HSCs have been shown to enhance procollagen type I expression through the antioxidant-sensitive pathway Ras/ERK, which stimulates HSC migration and the profibrogenic response (219). ROS signaling could be influenced by the activities of various antioxidant enzymes, the expression of which is regulated by a number of redox-sensitive transcription factors, including NF-κB and NRF1. Quiescent HSCs lack NF-κB in contrast to activated HSCs, suggesting that a redox-sensitive activation of NF-κB could regulate expression of NF-κB–targeted genes providing an appropriate cellular redox threshold for quiescent HSC entry into the proliferative cycle (Fig. 9). Consistent with this hypothesis, inhibition in NF-κB activity by using pyrrolidine dithiocarbamate is known to protect rats from the development of hepatic fibrosis (34). Inhibition in NRF1 could alter the expression of antioxidant response element (ARE)-responsive antioxidant enzymes that could perturb the cellular redox environment and subsequently affect proliferation, cell death, and increased collagen synthesis in HSCs that collectively results in liver fibrosis (195).
E. Cardiovascular diseases
Redox signaling has been implicated in the pathogenesis of all major diseases, including those of the cardiovascular system. Cardiovascular disease is the number one cause of death in the United States, accounting for ∼35.3% of all deaths in 2005 (2). Oxidative stress in cardiovascular biology was once considered only in terms of injury, damage, and dysfunction. However, an accumulating body of literature suggests that low to moderate concentrations of ROS may act as secondary messengers and signaling molecules regulating redox-sensitive processes during the vascular smooth muscle cell (VSMC) and cardiac myocyte cell-cycle progression.
Coronary heart disease is caused by atherosclerosis, the narrowing of vessels due to a buildup of plaque, which may lead to chest pains and heart attack. VSMCs are known to play an important role in the formation of fibrous plaques in atherosclerosis and initial thickening after angioplasty (267, 286). Focal accumulations of monoclonal VSMCs are precursors to atherosclerotic lesions (286). In these accumulations, VSMCs cause vessel-wall inflammation, lipoprotein retention, and fibrous cap formation that stabilizes plaques. Proliferation of VSMCs in atherosclerosis has been linked to inflammation, apoptosis, and matrix alterations (85). Additionally, VSMC proliferation has been identified as the primary mechanism of pathogenesis in restenosis, transplant vasculopathy, and vein bypass graft failure (29). Hypertension and diabetes are also associated with VSMCs growth (235).
In general, an injury to vessels initiates vascular proliferative disorders. This injury can cause endothelial denudation/dysfunction, inflammation, and VSMC activation and proliferation (85). ROS generated during VSMC proliferation could originate from the NADPH oxidase or growth factors or both. Nox4 is highly expressed in vascular wall cells, and Nox2 is predominantly expressed in VSMCs (179). Treatment of VSMCs with Nox1 antisense inhibits superoxide production and ROS-dependent signaling pathways (163). ROS generated from growth factors, such as platelet-derived growth factor (PDGF), are associated with increases in smooth muscle cell proliferation (248). Increased expression of PDGF and its receptors has been found in lesions of atherosclerosis (248). Overexpression of catalase inhibited VSMC proliferation, indicating a causal link between ROS signaling and cell proliferation in VSMCs (315). Furthermore, TGF-β, angiotensin II, epidermal growth factor, insulin-like growth factor, and basic fibroblast growth factor could also initiate medial proliferation of VSMCs (86, 114, 173, 183, 210). Angiotensin II–induced VSMC proliferation is regulated at least in part via the NADPH oxidase–dependent generation of H2O2-sensitive signaling pathways (115).
Extracellular signal-regulated kinases (ERKs) of the mitogen-activated protein kinase (MAPKs) family form one of the H2O2-sensitive signaling pathways that could contribute to VMSC proliferation (235). Exogenous addition of H2O2 induced VSMC proliferation via tyrosine phosphorylation of MAPKs (8, 253, 254, 315). ERK 1/2 activation is known to increase cyclin D1 (4, 102). PDGF can increase PIP3 levels, which in turn mediate p70S6K and AKT activation. AKT is known to phosphorylate GSK-3β and thus negatively affect its kinase activity. Inactivation of GSK-3β kinase activity inhibits cyclin D1 Thr286 phosphorylation, thereby stabilizing cyclin D1. An increase in cyclin D1 facilitates cells progression from G1 to S phase (68). AKT-mediated inactivation of GSK-3β and stabilization of cyclin D1 have been proposed recently for enhanced proliferation of VSMCs after treatment with betacellulin and amphiregulin (Fig. 10) (301).
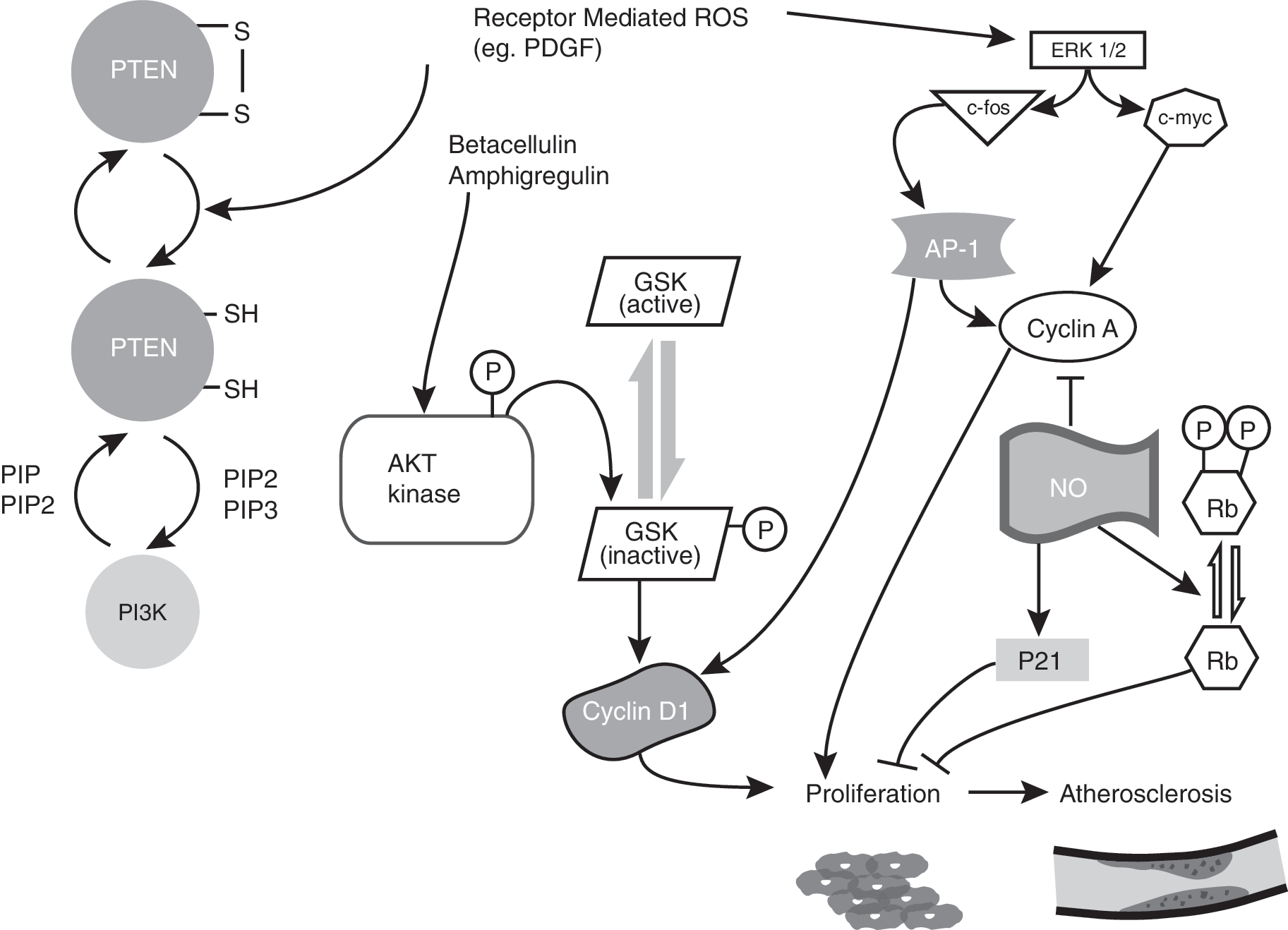
Furthermore, PDGF is known to activate redox factor 1 (Ref-1) by altering its redox status, enhance AP-1 activity, and increase cell-cycle–regulatory protein expression, facilitating progression from G0/G1 to S phase in VSMCs (125). CDK2, cyclin E, cyclin A, and PCNA protein levels were low in uninjured rat carotid arteries. However, a significant increase in these protein levels occurs within 2 days of balloon angioplasty. This increase in cell-cycle–regulatory proteins was present even after 10 days after injury, suggesting a continuous activation of proliferation during this period (169). Abundant expression of cell-cycle proteins has been observed in regions of human restenotic lesions, which also showed increased VSMCs proliferation (345). Disruptions of the E2F-Rb complex and inhibition of p53 have also been shown to stimulate proliferation of VSMCs (22).
Hydrogen peroxide is known to increase c-myc and c-fos mRNA levels, which can influence cell-cycle progression (253). Studies have shown the proliferative effects of c-myc in VSMCs (21, 213). Diez et al. (78) demonstrated an association between enhanced expression of c-myc and increased expression of cyclin A in VSMCs. c-Fos, conversely, promotes AP-1 transcription factor activity, which could activate cyclin A expression via binding to the AP1-consensus promoter sequence (317). Figure 10 summarizes the possible pathways of the redox control of cellular proliferation leading to the development of atherosclerosis.
In contrast to ROS activating VSMC proliferation, nitric oxide (NO) is a potent mitogenic repressor. NO suppresses the promoter activity for cyclin A gene transcription, resulting in decreased cyclin A mRNA and protein levels, which was associated with cell-cycle arrest (116). Additionally, NO-induced inhibition of CDK2 activity is associated with Rb hypophosphorylation, increased p21 expression, and inhibition in cell-cycle progression from G1 to S (136).
As atherosclerosis progresses and vessels are continually narrowed, myocardial infarction is a likely result. Cardiac myocytes rapidly proliferate during fetal life, but are terminally differentiated soon after birth. This limits the ability of the heart to restore function after injury. However, recent evidence suggests that cardiac myocytes may retain some proliferative potential (20, 142, 176). Additionally, cardiac progenitor cells have been identified that give rise to cardiac myocyte–like cells (3). Ki-67, a common indicator of proliferation, was found to be positive in 4% of myocytes near infarction sites and in 1% in distant myocytes in human tissue sections. Mitotic spindles, contractile rings, karyokinesis, and cytokinesis were also identified in these tissue sections (20). These results suggest that cardiac myocytes could be recruited to the proliferative cycle, at least under ischemia/reperfusion conditions.
Cardiac tissue contains many of the same growth factors seen in VSMCs, such as basic fibroblast growth factor and insulin-like growth factor (145). ROS are known to be generated in the ischemic myocardium, especially after reperfusion during acute myocardial infarction (131). Superoxide derived HO• and R• from reperfusion (366) may account for up to 50% of the final size of the myocardial infarction (354). The major sources of ROS in ischemic reperfused myocardium are from mitochondria, xanthine oxidase, and phagocyte NADPH oxidase (82, 281, 344). Although high levels of these ROS can cause significant damage, lower levels of ROS could stimulate myocyte proliferation for repair. H2O2, added to ventricular myocytes, activated the ERK pathway, leading to myocytes proliferation (159). Myocardial infarction is associated with enhanced expression of cyclin E, cyclin A, cyclin B, CDK2, and CDK1 in the remaining viable ventricular cardiac myocytes (258). In both acute and end-stage heart failure, the levels of p21, p27, and p57 reverted to a pattern similar to that observed in human fetal heart; p21 and p27 declined, whereas p57 expression was significantly increased (42). Although it appears that some ROS may activate proliferation, nitric oxide may have a different effect.
Recent evidence showed NO protects the myocardium from ischemia/reperfusion injury, possibly by scavenging superoxide (40). NO-induced increase in p21 and inhibition of cylin A/CDK2 activity prevented apoptosis in reperfused cardiomyocytes (181).
This literature supports the hypothesis that the cellular redox environment could influence VSMCs and cardiac myocytes proliferation in cardiovascular diseases. Interestingly, one of the recent strategies to improve cardiac function has been aimed at increasing the number of viable cardiac myocytes by manipulating cell-cycle–regulatory protein expression (307). The application of antioxidants could be a viable redox-based therapy for preserving the redox control of the cell cycle in VSMCs and cardiomyocytes. Additionally, some drugs already in use may have previously unknown redox function. Statins have been used to lower cholesterol to prevent heart disease by inhibiting 3-hydroxyl-3-methylglutaryl-CoA reductase and increasing LDL. Recently they have been shown also to have an antioxidant effect (313). Statins inhibit isoprenylation, resulting in decreased translocation of Rac-1 to the membrane; Rac1 is required for NADPH oxidase activity (180, 342, 343). Statins also decreased mRNA expression of NADPH oxidase subunits (343). In addition to preventing oxidant production, statins have been shown to increase catalase in liver and aortic vascular smooth muscle cells (139, 343). These effects also may serve to reduce the oxidant burden to mitogenic levels to allow repair in vasculature.
F. Diabetes
Diabetes is one of the earliest recorded diseases, found in the documents of ancient Greek and Hindu cultures (250, 268). Diabetes affects almost 8% of the United States population, and it was the seventh leading cause of death in 2006 (54). Type I, or insulin-dependent, diabetes mellitus, is usually diagnosed early in life and consists of 5–10% of the general diabetes diagnoses. Type I diabetes is denoted by a defect in insulin production and is most successfully treated with glucose monitoring and insulin administration. Type II, or non–insulin-dependent diabetes mellitus, is usually diagnosed later in life and is associated with obesity. Type II diabetes represents 90–95% of all diabetes diagnoses. Type II diabetes can be controlled with diet, medication, and glucose monitoring. Some common complications of diabetes are heart disease, stroke, high blood pressure, blindness, kidney disease, nervous system disease, delayed wound healing, amputation, and dental disease (54).
Although the exact cause of diabetes is not completely understood, one hypothesis is that the cellular redox environment and control of the cell cycle could significantly contribute to the development of this disease. A recent study by Houstis et al. (133), in an experimental model of insulin resistance, suggests that increased ROS activate insulin resistance in TNF-α and glucocorticoid dexamethasone–treated 3T3-L1 adipocytes. This resistance can be suppressed by prior treatments with small-molecular-weight antioxidants, N-acetyl cysteine (NAC), and manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) (133). The authors showed that 3T3-L1 adipocytes overexpressing CuZnSOD, MnSOD, and cytoplasmic and mitochondrial-targeted catalase were able to prevent insulin resistance in this experimental model system of diabetes. Furthermore, results from gene-expression analysis showed six of the ROS-biology–related genes (metallothionein, cytochrome P450, xanthine dehydrogenase, haptoglobin, and ceruloplasmin) increased by two- to sevenfold. Interestingly, these results also showed approximately a two- to fivefold decrease in the G0/G1 switch gene that is believed to regulate quiescent cell transition into the proliferative cycle (133). Hydrogen peroxide treatment of muscle cells (L6), human embryonic kidney fibroblasts, and mouse fibroblasts conferred insulin resistance (24, 122). Results from several studies showed that the increase in ROS levels precedes the hyperglycemia and insulin resistance, suggesting a causal role of ROS in the disease process (24, 122, 133).
Hyperinsulinemia also has been linked to diabetes as a cause for insulin resistance demonstrated in vivo and in vitro (73, 297). Several different types of cells, including adipose, insulin-sensitive hepatoma, and human skin fibroblasts, have shown evidence of ROS generation after insulin stimulation (55, 182). Prolonged insulin treatment of 3T3-L1 adipocytes inhibited insulin signaling and glucose uptake, while producing ROS (106). The reader is referred to two of the recent reviews discussing a possible link between insulin/IGF-1 and ROS/RNS signaling pathways (14, 234).
The cellular redox environment also is suggested to have a role in the late complications of diabetes, such as atherosclerosis, β-cell dysfunction, and nephropathy. Similarly, evidence of a preceding antioxidant imbalance has been linked to abnormal glucose levels (25, 275). Perturbation in the cellular redox environment also is known to alter free fatty acid levels (FFAs). FFA produced during the process of lipolysis has been shown to be higher in obese and diabetic individuals (26). Elevated FFA levels are known to uncouple oxidative phosphorylation, generate ROS, and reduce glutathione levels (88). Glucose autooxidation, NADPH oxidase, NOS, and superoxide generated from mitochondrial complexes I, II, and III are believed to be some of the sources of ROS production that could contribute to the development of hyperglycemia (13, 170, 216, 330, 365). Intermittent and stable hyperglycemia has been shown to cause ROS and eventually impaired cellular functions in pancreatic β cells (132, 242).
β-Cell dysfunction in the pancreas is one of the earliest events in the progression of type II diabetes (330). A majority of diabetes research focuses on the differentiation, function, and maintenance of pancreatic β-cell mass. In type II diabetes, the β cells exhibit defective proliferation and growth, whereas in type I diabetes, β cells are depleted by an autoimmune reaction (250). A further distinction of β cells is their low levels of antioxidant enzymes and thus their subsequent sensitivity to ROS and the eventual damage to cellular macromolecules (113). β Cells have a short cell-cycle duration compared with other cells in the body, but not all β cells retain the ability to reenter the cell cycle (316). The low percentage of proliferating β cells has been linked to lower CDK1 serine/threonine kinase and cyclin B1 mRNA levels (185). Some other cell-cycle proteins linked to diabetes are p27, CDK4, cyclin D1, and cyclin D2 (158). p27-Knockout mice showed increased β-cell proliferation and suppressed hyperglycemia, whereas p27 overexpression resulted in severe diabetes (329). CDK4-knockout mice showed poor β-cell proliferation and insulin-deficient diabetes (249). INK4a (p16) inhibits CDK4, which is necessary for β-cell proliferation (327). Transgenic overexpression of p16 exhibits decreased pancreatic islet proliferation (156). Mice expressing a mutant form of CDK4 that is unable to bind p16 develop pancreatic hyperplasia (249). Human and rat pancreatic β cells overexpressing CDK4 and cyclin D1 via adenovirus-mediated gene delivery enhanced Rb phosphorylation and increased proliferation by approximately two- to 10-fold (66). Cyclin D2–knockout mice develop severe diabetes by 12 weeks, because of defective β-cell replication, and these β cells also are unable to increase cyclin D1 or D3 expression until 2 weeks after birth (107). A cyclin D2–knockout mouse cross-bred with a heterozygote cyclin D1 mouse ablated β-cell proliferation, suggesting that cyclin D1 could also contribute to β-cell proliferation in these mice (158). AKT has been shown to regulate β-cell proliferation by cyclin D1, cyclin D2, and p21, through increased CDK4 activity (91). These results provide compelling evidence in support of the hypothesis that perturbations in the redox control of cell-cycle proteins could significantly affect β-cell proliferation and the development of diabetes.
G. Neurodegenerative diseases
Neurodegeneration is a pathologic condition affecting the nerves of the brain and spinal cord. Neuronal cell death occurs over the course of many years. In addition, distinct populations of neurons are targeted in different diseases; in Alzheimer's disease, 40% of the superior temporal sulcus is lost over a 10-year period, whereas 45% of the caudal substantia niagra is lost in Parkinson's disease within the same period of time (92, 109, 221). Throughout development, neuronal precursors proliferate to make more neurons than necessary. Apoptosis removes this excess, and the remaining neurons are terminally differentiated (152).
Alzheimer's disease is the most common form of dementia (87). The hallmark histopathology for this disease includes senile plaques, which are β-amyloid aggregates, and neurofibrillary tangles, which are tau protein aggregates. β-Amyloid, when bound to copper or iron, has been implicated as a major source of oxidative stress in Alzheimer's disease (18, 269, 280). Nunomura et al. (222) demonstrated that oxidative stress is highest early in Alzheimer's disease and lower later in the disease process. Transgenic mice overexpressing β-amyloid precursors, presenilin1 and amyloid precursor protein, showed an induction of oxidative stress (192). Presenilin 1 also has been shown to inhibit phosphorylation of Rb, suggesting a possible correlation between the cell cycle and oxidative stress in Alzheimer's disease (241). Activated microglia, upregulated in Alzheimer's disease, and surrounding tangles and plaques can be additional sources of NO and O2 •− (53, 119, 193).
Parkinson's disease is characterized by rigidity, resting tremors, and bradykinesia (65, 90). This disease is caused by selective degeneration of neuromelanin-containing neurons, resulting in a significant decrease in the neurotransmitter dopamine in the substantia niagra. Affected cells histologically contain Lewy bodies and cytoplasmic inclusions of α-synuclein protein.
Amyotrophic lateral sclerosis (ALS) is another progressive neurodegenerative disease that has a direct link to ROS. Mutations in CuZnSOD account for ∼20% of all familial cases of ALS (273, 305). More than 100 known mutations are found in CuZnSOD, all of which are dominant, and most of which confer a toxic gain of function (244, 305).
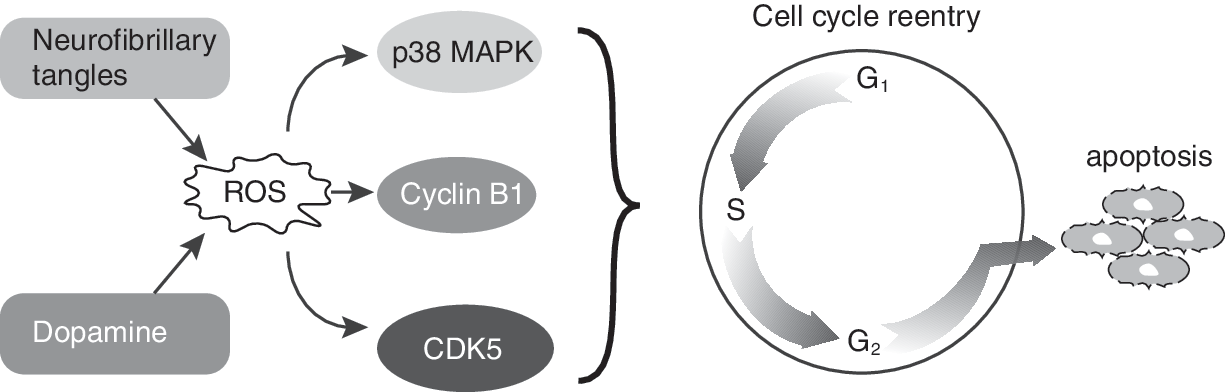
Whereas higher levels of ROS (O2 •− and H2O2) could result in oxidative stress and neuronal cell death, lower levels of ROS could be mitogenic. Rb hyperphosphorylation, increased levels of cyclin D, and E2F-1 redistribution to the cytoplasm have been observed in motor neurons and glia of ALS patients (251). CDK4 is highly abundant in mice overexpressing CuZnSOD mutants (215). Neurofibrillary tangles, present in many neurodegenerative diseases, may be an ideal source for mitogenic levels of H2O2. MAPK p38 expression has been localized to these tangles, providing a link between oxidative stress and cell-cycle reentry (7, 152, 362). Consistent with this hypothesis, cells treated with dopamine, a drug known to have an oxidative metabolism with the ability to generate ROS (152), activated a neuronal expression of cyclin B1 and CDK5 (303). Treatment of these cells with antioxidants blunted the activation of the cell-cycle protein expression, suggesting that the ROS signaling could trigger a mitogenic response in neuronal cells.
Many classic markers of proliferating cells, such as cyclin D1, CDK4, and Ki67, have been detected in degenerating neurons (197, 211, 212, 247, 309, 361). The presence of cyclin E and CDK2 in degenerating neurons suggests that the mitogenic properties of ROS facilitate neuronal cell progression from G0 to the G1/S border (212). The identification of cyclin B1, CDK1, and tau proteins as well as binucleated cells in neuronal tissues of patients diagnosed with neurodegenerative diseases suggest that terminally differentiated neuronal cells could be susceptible to unscheduled entry into the proliferative cycle (333, 361, 363). However, the absence of mitotic structures in these neurons suggests that the ROS levels that facilitate neuronal cell unscheduled entry into the cell cycle might not be high enough to stimulate neuronal cell entry into mitosis (152). This premise is consistent with a previous report demonstrating a gradual increase in cellular ROS levels as cells progress through the cell cycle; cells in M phase exhibit the highest oxidative state (111). The literature discussed earlier supports the hypothesis that the absence of an appropriate redox control of the neuronal cell cycle after reentry could activate the cell-death pathways, resulting in neuronal cell loss and the subsequent pathology of various neurodegenerative diseases (Fig. 11).
III. Summary and Future Directions
Not too long ago, ROS, diverse and abundant in biologic systems, were thought of as toxic byproducts of living in an aerobic environment. ROS are known to cause damage to cellular macromolecules, including both nuclear and mitochondrial genomes, proteins, and lipids, resulting in apoptosis or necrosis. However, recent evidence suggests ROS could be beneficial and necessary for many of the cellular processes, including proliferation and growth arrest. The literature discussed in this review indicates that a periodic oscillation in metabolic redox reactions represents a fundamental mechanism linking oxidative metabolic processes to the cell-cycle–regulatory processes. The periodicity in intracellular redox state can be regulated by a delicate balance between production of ROS and subsequent removal by nonenzymatic and enzymatic antioxidants. It is important to note that specific species of ROS can be a determining factor that drives cellular proliferation and ultimately cellular responses in health and disease. An “ROS switch” exists in which superoxide signaling promotes proliferation, and hydrogen peroxide signaling supports quiescence (277).
Future studies are necessary to decipher how the same ROS could regulate necessary biologic processes but also be toxic to cells. It is possible that this dual function of ROS could be due to the difference in their concentrations (threshold), pulse duration (flux), subcellular localization, and cell types. We believe that advances in the quantitative redox biology research may resolve many of these intriguing questions in the very near future.
Another future direction would be to determine whether the activation of the redox-sensitive signaling pathways is specific to a particular source of ROS generation. Must all cellular processes be under the control of specific ROS-sensitive signaling pathways? Can the redox regulation of the cell cycle be controlled by direct modifications of redox-sensitive motifs (cysteine residues, metal cofactors) present in cyclins/CDKs, CKIs, and phosphatases? It has been reported that critical thiol-disulfide exchange reactions between specific cysteine residues significantly affect CDC25 phosphatase activity (279). Our unpublished observations showed that specific cysteines in cyclin D1 could regulate its protein levels. These results suggest that many of the redox-sensitive processes regulating cell-cycle progression could be directly regulated by thiol-redox reactions in specific cell-cycle–regulatory proteins.
As demonstrated, ROS appear to play a critical role in many human diseases, especially in relation to the cell-cycle control (Fig. 12). However, it is interesting to note that many clinical trials using antioxidants to treat or prevent cancer (126, 229), coronary disease (32), and other diseases have been inconclusive or have yielded negative results. These results reemphasize the complexity of the redox biology and warrant that any clinical trials must take into consideration the redox threshold, flux, subcellular localization, and cell types. For example, ascorbate was first hypothesized to be an effective anticancer agent in 1972 by Ewan Cameron and Linus Pauling (45, 46, 232). However, early clinical trials by Moertel (67, 204) showed that ascorbate is ineffective in treating advanced cancers. Although the research community was quick to lose interest in ascorbate as a cancer-therapy agent, it is important to note that the oral delivery of ascorbate was insufficient to achieve a therapeutic dose level in the plasma. Recent evidence suggests that intravascular delivery of ascorbate indeed showed a much higher plasma concentration of ascorbate that exhibited a positive correlation with inhibition in tumor growth in human, rat, and murine tumor xenografts (58). Additionally, antioxidants are often chosen as therapy agents based on their availability and ease of delivery (312). Many studies have used vitamin E, which has both antioxidant and prooxidant effects (238). This dual effect of certain antioxidants warrants that extra care must be taken in selecting antioxidants for therapeutic purposes. It is believed that a more-careful development of targeted antioxidant-based therapy could be more rewarding.
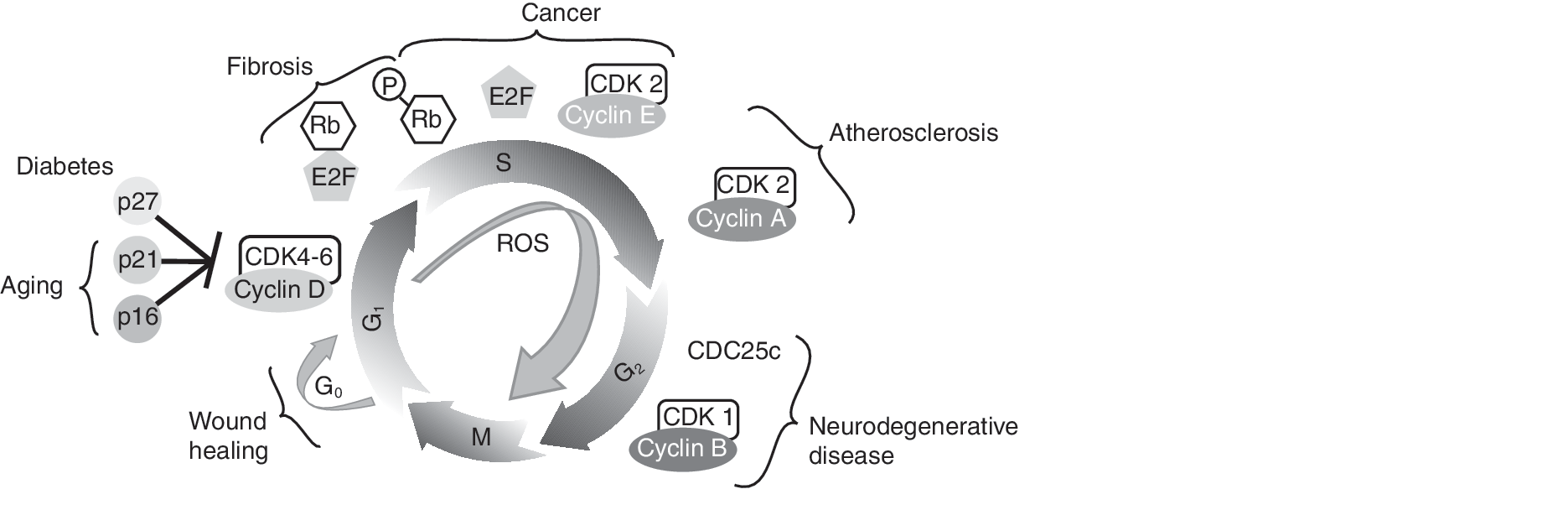
We believe the literature discussed in this review article will foster an innovative research frontier focusing on redox control of the cell cycle in health and disease. Newer and more-effective antioxidants targeted at the redox control of the cell cycle could provide additional therapy options to treat proliferative disorders.
Footnotes
Acknowledgments
The first two authors contributed equally to this work. We thank Drs. Jennifer Nickerson-Gass and Venkatasubbaiah A. Venkatesha for critical reading of the manuscript and Mr. Gareth Smith for help with illustrations. Funding from NIH CA 111365 and McCord Research foundation supported this work.