
Abstract
Neovascularization is involved in normal development and wound repair as well as ischemic heart disease and peripheral artery disease. Both angiogenesis and vasculogenesis [de novo new vessel formation through mobilization of stem/progenitor cells from bone marrow (BM) and their homing to the ischemic sites] contribute to the formation of new blood vessels after tissue ischemia. Angiogenesis is dependent on cell proliferation, migration, and capillary tube formation in endothelial cells (ECs). Stem/progenitor cells have been used for cell-based therapy to promote revascularization after peripheral or myocardial ischemia. Excess amounts of reactive oxygen species (ROS) are involved in senescence and apoptosis of ECs and stem/progenitor cells, causing defective neovascularization. ROS at low levels function as signaling molecules to mediate cell proliferation, migration, differentiation, and gene expression. NADPH oxidase is one of the major sources of ROS in ECs and stem/progenitor cells, and is activated by various growth factors, cytokines, hypoxia, and ischemia. ROS derived from NADPH oxidase play an important role in redox signaling linked to angiogenesis ECs, as well as stem/progenitor cell mobilization, homing, and differentiation, thereby promoting neovascularization. Understanding these mechanisms may provide insight into NADPH oxidase and its mediators as potential therapeutic targets for ischemic heart and limb disease. Antioxid. Redox Signal. 11, 2517–2533.
Introduction
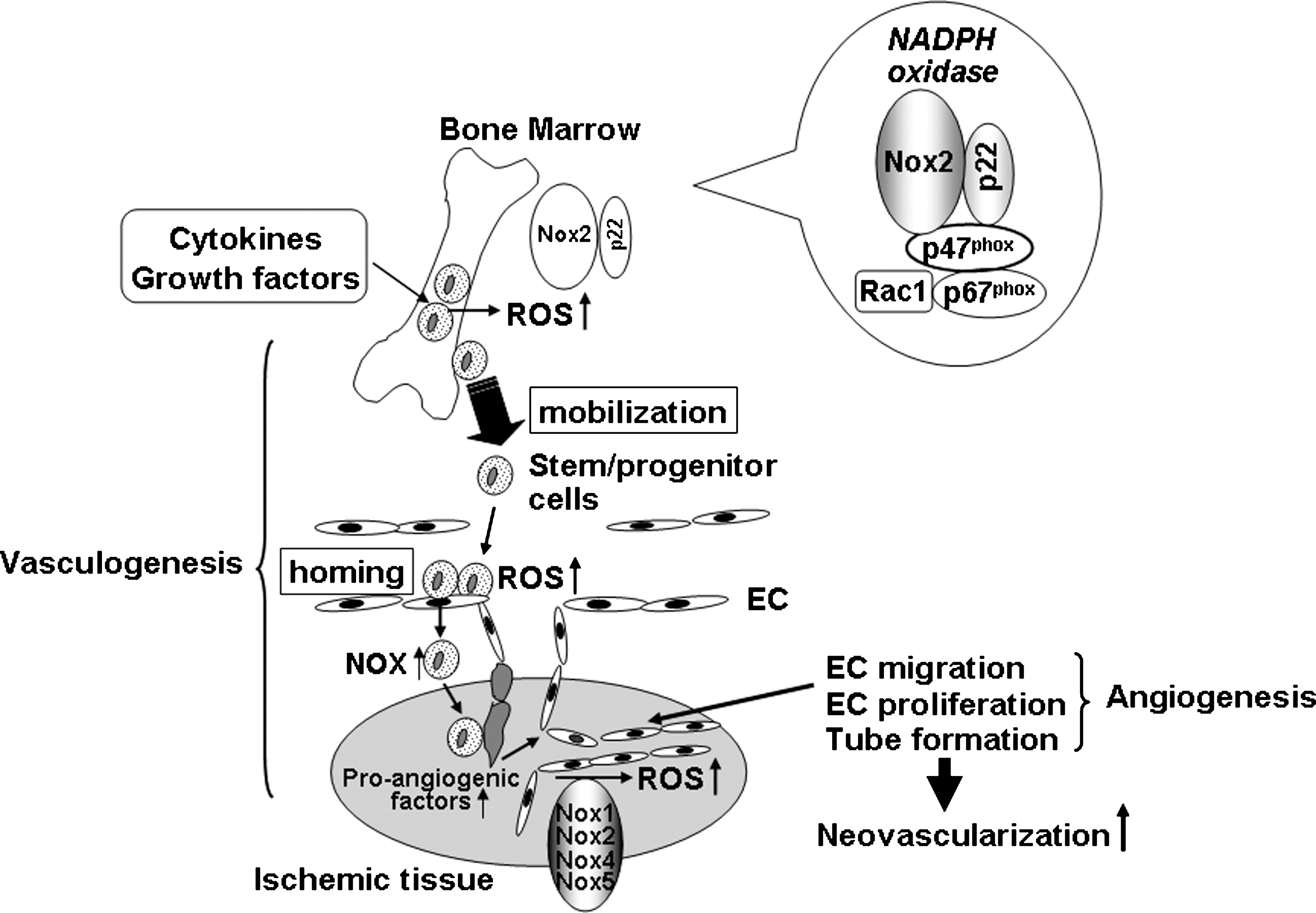
Reactive oxygen species (ROS) such as superoxide anion (O2 •−) and hydrogen peroxide (H2O2) play an important role in normal cell growth, migration, differentiation, apoptosis, and senescence (58, 70). Excess amounts of ROS are toxic and involved in EC and stem/progenitor cell senescence and apoptosis (34), thereby contributing to oxidative stress–dependent cardiovascular diseases such as hypertension, heart failure, atherosclerosis, and diabetes (233). By contrast, ROS at low levels function as signaling molecules to regulate angiogenesis and vasculogenesis (42, 135, 139, 185, 189, 194, 201, 207, 210). Signal transduction activated by ROS, “redox signaling,” has been an emerging area of investigation.
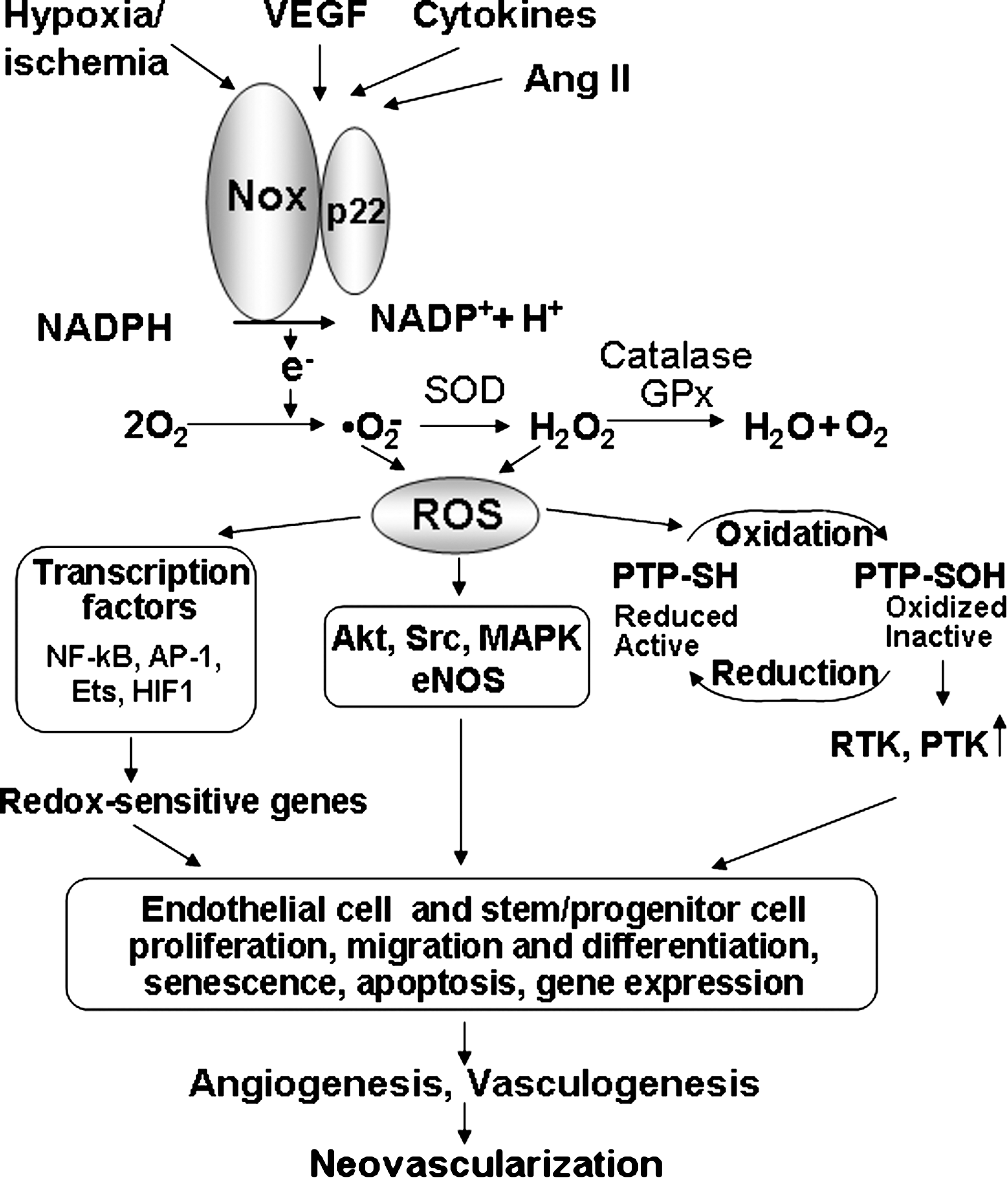
NADPH oxidase is one of major sources of ROS in ECs and stem/progenitor cells. In phagocytic cells, NADPH oxidases consist of membrane-associated cytochrome b558, comprising the catalytic gp91 phox and regulatory p22 phox subunits, and cytosolic components including p47 phox , p67 phox , p40 phox , and the small GTPase Rac1 (17) (Fig. 1). In nonphagocytic cells, several homologues of gp91 phox (also termed Nox2), including Nox1, Nox3, Nox4, and Nox5, as well as the dual oxidases (Duox)1/2, have been identified (64, 116, 120). Nox1, Nox2, Nox4, and Nox5 (only human) are expressed in ECs, whereas Nox2 and Nox4 are found in stem/progenitor cells. NADPH oxidase is activated by angiogenic growth factors and cytokines such as VEGF, angiopoietin-1, shear or mechanical stress, hypoxia and G protein–coupled receptor agonists, including angiotensin II (Ang II) in ECs and stem/progenitor cells (77, 208). NADPH oxidase–derived ROS are involved in redox signaling pathways, leading to angiogenesis in ECs, as well as various functions of stem/progenitor cells, including proliferation, migration, differentiation, and gene expression (27, 57, 77, 88, 158, 205, 210, 230), thereby regulating postnatal neovascularization (8, 201, 205) (Fig. 2). Because the role of excess amounts of ROS linked to stem/progenitor cell dysfunction has been reviewed by others (23, 34, 181, 203, 220, 233), this review briefly describes this aspect and mainly summarizes the recent progress and information on the role of NADPH oxidase and physiologic levels of ROS in angiogenesis and stem/progenitor cell function.
Role of NADPH Oxidase in Angiogenesis in ECs
Exogenous ROS increase VEGF or VEGFR2 expression (66) and stimulate EC proliferation and migration (44, 135, 234), as well as cytoskeletal reorganization (216) and tubular morphogenesis (183) of ECs. Furthermore, angiogenesis growth factors such as VEGF and angiopoietin-1, as well as Ang II or 20-hydroxyeicosatetraenoic acid (20-HETE), a product of cytochrome p450, or leptin, a circulating adipocytokine, hypoxia/reoxygenation, or adhesion of activated polymor-phonuclear leukocytes to ECs, stimulate angiogenesis-related responses via an increase in ROS (73, 77, 84, 88, 125, 210, 228, 230, 235). Natural antioxidants such as green tea catechins and vitamin E, as well as polyphenols from red wine, prevent angiogenic responses by decreasing ROS levels in ECs (195). These suggest that many of angiogenic-related responses are dependent on ROS in cultured ECs.
ROS are generated from a number of sources including the NADPH oxidase, mitochondrial electron-transport system, xanthine oxidase, cytochrome p450, uncoupled NOS, and myeloperoxidase (70, 128). Complex interactions may occur among different sources of ROS, as well as feed-back and feed-forward regulation of ROS accumulation (128). Mitochondria-derived ROS are involved in hypoxia inducible factor 1 (HIF1) stabilization (22) and angiogenesis (46); however, the role in receptor-activated signal transduction in ECs remains unclear. NADPH oxidase has emerged as a major source of ROS in nonphagocytic cells, including ECs, and is activated by numerous angiogenesis stimuli including VEGF, angiopoietin 1, cytokines, shear stress, hypoxia, and G protein–coupled receptor agonists, including Ang II (70) (Fig. 2). The prototype phagocyte NADPH oxidase consists of membrane-bound gp91 phox (now also termed Nox2) and p22 phox , which compose the flavocytochrome b 558, together with the cytosolic p47 phox , p67 phox , p40 phox , and the small GTPase Rac (Fig. 1). Nonphagocytic NADPH oxidase (Nox) consists of several homologues of Nox2, including Nox1, Nox3, Nox4, and Nox5, as well as the dual oxidases (Duoxs) (64, 65, 117, 118). In ECs, all the phagocytic NADPH oxidase subunits including Nox2, p22 phox , Rac1, p47 phox , as well as Nox1, Nox4 and Nox5, are expressed. Various Nox enzymes contribute to ROS production of different degrees, locations, and EC types.
Evidence suggests that several Nox enzymes are involved in angiogenesis in ECs. Nox2 is activated by various stimulants in ECs (61, 62, 67, 126, 210). VEGF and angiopoietin-1 stimulate EC migration via activation of Nox2-based NADPH oxidase, suggesting an essential role of Nox2 in angiogenesis in ECs (4, 45, 77, 88, 130, 210, 230). Nox1 stimulates branching morphogenesis in sinusoidal ECs (106), and Nox1 overexpression increases VEGF and VEGF-receptor expression and MMP activity, thereby promoting vascularization (11). Nox4 is involved in basal ROS production and proliferation in ECs (157). Datla et al. (49) reported that overexpression of Nox4 enhances, whereas dominant negative Nox4 inhibits VEGF-induced EC migration and proliferation. The most recent study shows that overexpression of Nox5 stimulates ROS production, proliferation, and formation of capillary-like structures, whereas Nox5 siRNA prevents thrombin-induced responses in human ECs (21). Note that Nox5 is present in most mammals but is absent in rodents, in which its function may be taken over by another Nox. The role of Rac1-derived ROS in VEGF-induced VEGF expression (51) and EC migration also has been reported (77, 110, 146, 210, 214). These suggest that various Nox homologues (Nox1, Nox2, Nox4, Nox5) play an important role in angiogenesis in ECs (Fig. 1), which may depend on species and experimental conditions.
Role of NADPH Oxidase in Angiogenesis In Vivo
ROS play an important role in physiologic and pathologic angiogenesis in vivo. ROS are increased during the reperfusion of the ischemic retina, which contributes to upregulation of VEGF mRNA (113). Short periods of ischemia/reperfusion or preconditioning induce an increase in ROS, thereby stimulating myocardial angiogenesis and arteriogenesis (48, 72, 115). Thiol antioxidant, N-acetylcysteine attenuates EC invasion and angiogenesis in a tumor model in vivo (29). Pigment epithelium-derived growth factor, which has antioxidant properties, inhibits angiogenesis and melanoma growth (1). ROS inhibitors prevent neovascularization in a mouse model of angiogenesis (160, 237). Angiopoietin1-induced angiogenesis is enhanced in catalase-knockout mice, suggesting an involvement of H2O2 in angiogenesis in vivo (105). In a pathophysiologic state, a strong correlation between ROS elevation, neovascularization, and VEGF expression has been reported in eyes of humans with diabetes (30, 55, 56) and in an animal model of atherosclerosis (171). Transgenic mice overexpressing MnSOD in ECs prevent diabetic retinopathy through reducing O2 − production and VEGF expression in vivo (68). Antiangiogenic therapy reduces plaque growth and intimal neovascularization in atherosclerosis (148). Antioxidant vitamins C and E reduce VEGF and VEGFR-2 expression in apolipoprotein-E–deficient mice (153), which may contribute to their inhibitory effects on atherosclerosis. These studies suggest that ROS are required for neovascularization in vivo.
The role of NADPH oxidase in pathologic and physiologic angiogenesis in vivo has been demonstrated. Neovascularization in response to VEGF is inhibited in Nox2−/− mice as well as in mice treated with antioxidant or NADPH oxidase inhibitor or gp91ds-tat (8, 89, 210). Hindlimb ischemia stimulates Nox2-dependent ROS production in ischemic tissues, and blood-flow recovery and angiogenesis in response to ischemia are markedly impaired in Nox2−/− mice (8, 201). Similarly, Nox2 expression and ROS production are increased in a mouse retinopathy model, which is attenuated by NADPH oxidase inhibitor or gp91ds-tat (8). With an in vivo arteriovenous loop–containing tissue engineering chamber, Jianq et al. (100) recently showed that Nox2 plays an important role in vascularization and tissue growth. Vallet et al. (211) reported that Nox4 expression is upregulated in new capillaries in brain ischemia–induced angiogenesis of mice. HMG-CoA reductase inhibitor, statins, which block Rac1 activity (122), prevent angiogenesis in vivo (221). Overexpression of Nox1 increases VEGF and VEGF-receptor expression and matrix metalloproteinase (MMP) activity through increases in ROS, thereby promoting tumor angiogenesis (11). Transgenic mice overexpressing p22 phox show enhanced VEGF expression and neovascularization of experimental atheromas (102). Myocardial ischemia/reperfusion of mice increases ROS in a p47 phox -dependent manner, which is required for VEGF expression, sprouting, and vessel outgrowth (41). Optimal levels of NADPH oxidase–derived ROS are required for p38MAPK activation and coronary collateral growth in a rat model of repetitive ischemia (168). By contrast, overproduction of Nox2-derived ROS rather contributes to impairment of postischemic neovascularization in pathologic conditions such as diabetes (53). In line with this report, extracellular SOD–deficient mice show impaired ischemia-induced neovascularization and enhanced O2 − levels, which are rescued by SOD mimetic infusion (104). These studies support a double-edged role of ROS in postischemic neovascularization; physiologic levels of ROS are required, but excess amounts of ROS are inhibitory for new blood vessel formation.
Redox Signaling in Angiogenesis
ROS can act as signaling molecules for activation of diverse signaling pathways by oxidation of reactive cysteine on the specific target molecules, including kinases, phosphatases (43, 124, 167), redox-sensitive transcription factors, AP1, Ets, NF-κB, and HIF1 (190), as well as redox-sensitive genes, VEGF, MMPs, cyclooxygenase-2 (COX-2), urokinase plasminogen activator (uPA), and plasminogen activator inhibitor-1 (PAI-1), which are involved in angiogenesis (207, 209) (Fig. 2).
NADPH oxidase is activated by numerous stimuli including VEGF, EGF, cytokines, shear stress, hypoxia, and G protein–coupled receptor agonists including Ang II in ECs (70) (Fig. 2). VEGF binds to two tyrosine kinase receptors, VEGF receptor-1 (VEGFR1, Flt-1) and VEGFR2 in ECs. The mitogenic and chemotactic effects of VEGF in ECs are mediated mainly through VEGFR2 (138), which is activated through autophosphorylation of tyrosine residues in the cytoplasmic kinase domain. This event is followed by activation of downstream signaling pathways, such as mitogen-activated protein kinases, Akt, and eNOS, which are essential for EC migration and proliferation (138) (Fig. 2). Nox2-derived ROS are involved in VEGF-induced VEGFR2 autophosphorylation in ECs (45, 88, 210, 230). Angiopoietin 1 binding to the Tie-2 receptor also stimulates Nox2-based NADPH oxidase, which is required for phosphorylation of Akt and ERK linked to EC chemotaxis (40, 77). p47 phox siRNA inhibits VEGF-stimulated phosphoinositide 3-kinase (PI3K)-Akt-forkhead as well as p38MAPK, but not ERK1/2 or JNK, in ECs (3). ROS also are important for VEGF-induced cSrc activation, phosphorylation of VE-cadherin, thereby disrupting cell-to-cell contacts of established vessels to promote angiogenesis in ECs (130, 212, 229). VEGF also induces peroxynitrite formation, which is involved in phosphorylation of VEGFR2 and cSrc as well as EC migration and tube formation (54). Thus, not only ROS but also peroxynitrite mediate VEGF angiogenic signaling in ECs.
Evidence suggests that NADPH oxidases are localized within discrete subcellular compartments, which is required for localizing ROS production and activation of specific redox-signaling pathways that mediate various cell functions (198, 206). EC migration is a key event for tissue repair in response to injury, angiogenesis, and wound healing. Rac1- and Nox2-dependent NADPH oxidase plays an important role in EC migration (2, 45, 89, 210). The PI3K-Rac pathway is involved in localized production of ROS at the membrane ruffles (155), which is required for cytoskeletal reorganization and directed cell migration (89, 147). Nox2 and its regulatory subunits, p47 phox and p67 phox are also targeted to the focal complexes or membrane ruffles in lamellipodia (89, 225, 226). Wu et al. (226) identified targeting molecules that may specify the site of ROS production at lamellipodial focal complexes during EC migration. p47 phox binds to the orphan adaptor TRAF4, which in turn binds to the focal contact scaffold Hic-5, thereby targeting p47 phox to the focal complexes. The mechanism for targeting NADPH oxidase to the lamellipodial leading edge and membrane ruffles is through the interaction of p47 phox with moesin and WAVE1, which are enriched within this specific structural compartment (47, 223). WAVE1 catalyzes actin nucleation responsible for lamellar structure in a Rac1-dependent manner, and thus p47 phox– WAVE1 complexes contain Rac1 and Rac1 effectors PAK1, which phosphorylates p47 phox . Antioxidants and inhibition of p47 phox –WAVE1 interaction block ROS production and ruffle formation (225).
Another important targeting protein is IQGAP1, an actin-binding scaffold protein and Rac1 effector that is required for Rac-mediated polarized cell migration (26, 137). IQGAP1 functions as a scaffold protein to target Nox2 and Rac1 to the leading edge to localize ROS production, which may contribute to ROS-dependent directional EC migration (89). Moreover, Nox2 and Nox4 activate distinct redox-signaling pathways in HEK293 cells transfected with Nox2 or Nox4 (9), suggesting that differential expression and localization of Nox enzymes may determine the specificity of activation of redox signaling.
The signaling properties of ROS are due, in part, to reversible oxidative inactivation of redox-sensitive target proteins, including protein tyrosine phosphatases (PTPs) (43, 124, 167) (Fig. 2). Several PTPs, including SHP-1, SHP-2, and low-molecular-weight PTP (HCPTPA), inducibly associate with VEGFR2 after VEGF stimulation (74, 85, 111). HCPTPA overexpression inhibits VEGF-induced VEGFR2 autophosphorylation (85). SHP-1 mediates a TNF-α–induced inhibitory effect on VEGFR2 phosphorylation without affecting VEGF-induced responses in ECs (151). A recent report shows that SHP-1 constitutively binds to VEGFR2, and siRNA enhances VEGFR2 autophosphorylation and EC proliferation without affecting migration (24). SHP-2 negatively regulates VEGFR2 signaling in ECs that are cultured only on type I collagen (143). PTP1B siRNA or small-molecule inhibitor enhances VEGF-induced VEGFR2 autophosphorylation and proliferation of ECs (152, 186). PTP1B inhibitor promotes neovascularization in a mouse Matrigel model (186). High cell density–enhanced PTP1 (DEP-1)/CD148 attenuates phosphorylation of VEGFR2 in contact-inhibited confluent ECs (69). Angiopoietin-1 stimulates association of SHP-2 with the phosphorylated Tie-2 receptor in ECs (86), which in turn inhibits the PI3K-dependent signaling involved in EC migration. Nox-derived ROS inactivate PTP-PEST at focal contacts, thereby promoting membrane ruffling and endothelial migration (10). Thus, it is important to determine which PTPs are reversibly oxidized and inactivated by angiogenesis growth factor–induced ROS, thereby promoting tyrosine phosphorylation–dependent redox signaling linked to angiogenesis (Fig. 2).
Lipid phosphatase and tumor suppressor, PTEN, which dephosphorylates phosphatidylinositol 3,4,5-triphosphate (PIP3), a product of the PI3K activation, is also subject to the oxidative inactivation by ROS (114). ROS regulate an EGF-induced increase in VEGF and HIF-1α expression through activation of the PI3K/Akt/p706K pathway, which is involved in angiogenesis in ovarian cancer cells (131). Although underlying mechanisms remain unclear, it is possible that ROS-dependent oxidative inactivation of PTEN (114) may be involved in this response. Similarly, MnSOD overexpression increases mitochondrial H2O2, leading to PTEN oxidation and PIP3 formation at the plasma membrane, and Akt activation, which results in VEGF expression and EC tube formation (46).
In addition to phosphatases, several serine/threonine kinases, such as cAMP dependent kinase (PKA), cGMP-dependent kinase (PKG), Akt kinase, S6 kinase, and protein kinase C (PKC), all contain a Cys-SH residue within their active-site domain. These Cys-SH residues are not required for activity, but they are fully conserved among these kinases. One may speculate that interaction with regulatory proteins may be modulated by the reversible oxidation of this Cys-SH.
Role of NADPH Oxidase in Stem/Progenitor Cell Function
NADPH oxidase and stem/progenitor cell differentiation, proliferation, and senescence
Stem cells and EPCs hold great promise for tissue repair and regenerative medicine and play a significant role in neovascularization of ischemic tissue. Expression of “stemness” genes that distinguish stem cells from mature cells, as well as stress-response genes involved in redox balance, is a characteristic feature of stem cells, suggesting that one essential attribute of the stemness is high resistance to stress (98, 166). Of note, ROS levels in endothelial progenitor cells are lower than those in mature ECs, which is due to higher expression of antioxidant enzymes (MnSOD, catalase, glutathione peroxidase) and is required for preserving stemness, such as an undifferentiated, self-renewing state (50). Hypoxia effectively maintains the biologic characteristics of CD34+ cells through keeping lower intracellular ROS levels by regulating NADPH oxidase (57). Factors promoting self-renewal cause the reduction of the redox state, whereas molecules promoting differentiation lead to excess oxidation in neural progenitor cells (154). Hyperbaric oxygen stimulates vasculogenic stem/progenitor cell growth and differentiation in vivo through an increase in physiologic levels of ROS and Nox2 (141). Thus, ROS seem to be involved in the balance between self-renewal and differentiation of stem/progenitor cells (76) (Fig. 3).
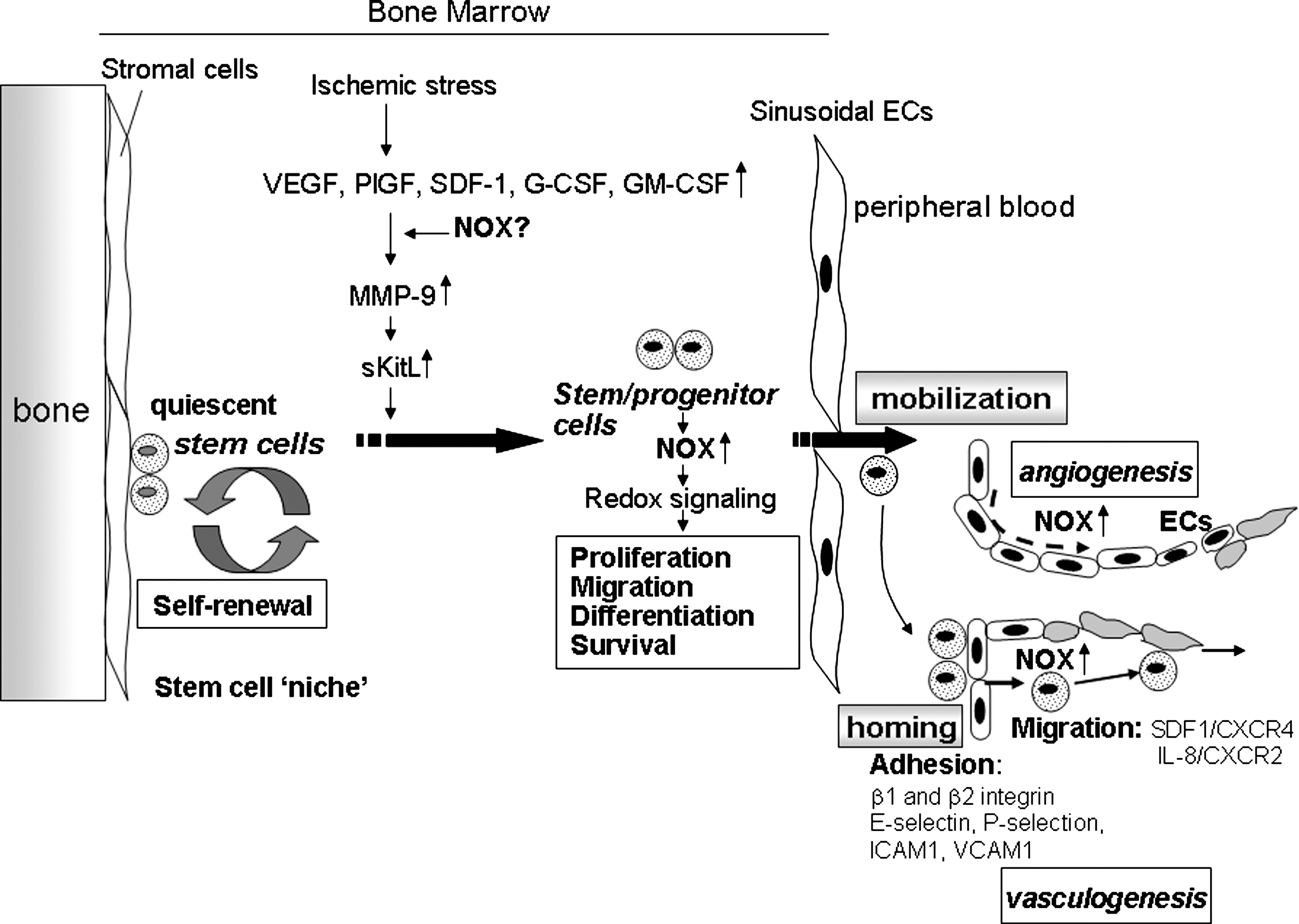
Although excess amounts of ROS are toxic to EPCs and stem/progenitor cells (233), optimal levels of ROS, which are balanced by ROS-generating and antioxidant enzymes, are involved in normal stem/progenitor cell functions, such as proliferation, differentiation, and survival (50, 63, 104). NADPH oxidase components are expressed in various stem/progenitor cells including circulating human CD34+ cells (57, 158, 159), mouse embryonic stem cells (27), skeletal muscle precursor cells (145), and rat mesenchymal stem cells (218). The high-resolution imaging of hematopoietic stem cells (HSCs) with the immunodetection of NOX indicates the presence of membrane raft–like microcompartments in which the assembly/activation of the NOX components may be functionally integrated for creating redox signaling platforms (60). Thus, NADPH oxidase–derived ROS may contribute to redox signaling involved in various function of stem/progenitor cells (Figs. 2 and 3). In undifferentiated human myeloid cell lines, Nox2, p22 phox , p47 phox , and p67 phox expression is minimal, whereas O2 −-producing capacity is increased to the level of normal neutrophils during differentiation, with a concomitant increase in p47 phox . Thus, low expression of p22 phox and Nox2 is sufficient to obtain normal O2 − production, and p47 phox might play an important role in the functional differentiation of myeloid cells (94).
H2O2 plays a role in native osteoclast differentiation (188). Stimulation of BM monocyte-macrophage lineage cells with receptor activator of NF-κB ligand (RANKL) promotes differentiation into osteoclasts through an increase in ROS via TNF receptor–associated factor (TRAF) 6/Rac1/Nox1 pathway (123). Nox2 and Nox4 are expressed in skeletal muscle precursor cells, which are involved in proliferation of these cells (145). By contrast, proliferation of EPCs is limited by the capacity to divide and the onset of a nondividing state known as senescence. Inhibition of telomerase activity with ROS, increased by oxidized low-density lipoprotein (oxy-LDL), accelerates the onset of EPC senescence, which leads to impaired proliferative capacity (93). This mechanism is through telomerase inactivation by ROS and subsequent inactivation of Akt (90). Thus, ROS-mediated proliferation and senescence in stem/progenitor cells may be determined by the amount, duration, and location of ROS generation, which activates specific redox-signaling pathways.
In ES cells, Nox2, p22 phox , p47 phox , and p67 phox of NADPH oxidase are expressed early in embryonic development, and the expression occurs in a consistent sequential fashion, indicating their involvement of ES cell differentiation (18). Mechanical strain or direct-current electrical field stimulation of mouse ES cells elevates intracellular ROS through upregulation of NADPH oxidase subunits, thereby increasing expression of HIF-1α and VEGF, which contributes to cardiovascular differentiation (176, 180). Low levels of H2O2 stimulate cardiomyogenesis of ES cells and induce proliferation of cardiomyocytes derived from ES cells. This response is associated with nuclear translocation of cyclin D1, downregulation of p27Kip1, phosphorylation of retinoblastoma (Rb), and an increase of Ki-67 expression and BrdU, which are inhibited by free radical scavengers. ROS-induced cardiomyogenesis of ES cells increases expression of NADPH oxidase components, indicating feed-forward regulation of ROS generation in cardiovascular differentiation of ES cells (27). Consistent with this, mechanical-strain stimulation of embryoid bodies grown from ES cells promotes cardiovascular differentiation through ROS elevation, which is followed by induction of Nox1 and Nox4, supporting feed-forward upregulation of ROS production (178). Similarly, cytokine cardiotrophin-1 (CT-1) stimulates proliferation of ES cell–derived cardiomyocytes through an increase in ROS and activation of redox-signaling cascades (177). CT-1 expression itself is also regulated by ROS and HIF-1α, suggesting existence of a feed-forward mechanism for promoting proliferation of ES-derived cardiomyocytes by ROS (14). Peroxisome proliferator–activated receptor alpha agonists enhance cardiomyogenesis of mouse ES cells through an increase in ROS and NADPH oxidase activity (182). The role of small GTPase Rac1 in cardiac differentiation of ES cells through NADPH oxidase is also reported (162). Nox4-derived ROS promote ES cell differentiation into smooth muscle cells (227) or cardiac cells (27, 127). Stimulation of embryoid bodies by PDGF-BB increases vascular sprouting and branching of capillary-like structures via NADPH oxidase–derived ROS and its downstream activation of ERK1/2 (119). Prenylflavonoid-induced cardiomyocyte differentiation of murine ES cells is also dependent on NADPH oxidase–derived ROS and p38MAP kinase with increased expression of Nox4 (52, 224). These reports suggest that NADPH oxidase–derived ROS are involved in cardiovascular ES cell differentiation.
NADPH oxidase and mobilization of stem/progenitor cells
Stem/progenitor cells and EPCs are mobilized from BM to the circulation in response to injury or cytokines, which contributes to postnatal neovascularization (6, 13, 192) (Figs. 1 and 3). By using Nox2-deficient mice and BM transplantation, we showed that Nox2 plays an essential for increase in ROS in BM after hindlimb ischemia, which is required for stem/progenitor cell mobilization from BM (205).
In another study, a cyclophosphamide-, G-CSF–, or IL-8–induced increase in circulating stem and progenitor numbers is inhibited in Nox2−/− mice (213). Moreover, renovascular hypertension of mice that is dependent on mechanical stress stimulates EPC mobilization through p47 phox -dependent ROS, which is associated with an increase in BM SDF-1 and MMP-9 (173). In humans, the number of circulating CD34+ cells is low in X-linked patients with chronic granulomatous disease (CGD) in which the gp91 phox (Nox2) coding gene, CYBB, is mutated (180a). These reports suggest that ischemic or mechanic stress increases circulating cytokine levels, thereby stimulating NOX-dependent ROS production in BM, which may promote reparative mobilization of stem/progenitor cells from BM and the resulting revascularization of ischemic tissues or tissue repair (Figs. 1 and 3).
HSCs are embedded in a local BM microenvironment, the so-called stem cell “niche” (31, 103, 241), and are maintained to be quiescent with low oxygen (184) and ROS levels (99) (Fig. 3). Recent reports indicate that increased ROS in BM facilitates HSCs to exit from the quiescent state, thereby stimulating proliferation and differentiation during the early steps of commitment (99, 202). Angiogenic growth factors or cytokines increase ROS in HSCs, thus stimulating cell growth (174, 175), and exit quiescent BM stem/progenitor cells to promote cell-cycle progression (87). ROS are involved in BM stem/progenitor cells proliferation, differentiation, migration, and survival in vitro (32, 36, 75, 175) and in vivo (12, 82, 97, 144, 202), which may contribute to ROS-mediated stem/progenitor cell mobilization from BM (Fig. 3). Hematopoietic cytokine G-CSF induces both serine phosphorylation and membrane translocation of p47 phox to activate NAPDH oxidase, leading to cell proliferation of BM myeloid cells (244). NADPH oxidase–derived ROS are involved in myeloid expansion (32, 244) and monocytes/macrophage survival via regulating Akt and p38MAPK (219). These suggest that Nox-dependent ROS play an important role in redox signaling involved in the mobilization of some stages of BM progenitor cells (Fig. 3).
Mobilization of stem cells from BM is a dynamic process that requires release of chemocytokines, expression of adhesion molecules, activation of proteases, and stimulation of chemotaxis of stem/progenitor cells (163, 164). Thus, ROS also may affect on some of these events, thereby promoting egress of stem/progenitor cells from the BM to the circulation. BM sinusoidal ECs are functionally and phenotypically distinct from microvascular ECs of other organs (108) and constitutively express cytokines such as SDF-1 and adhesion molecule such as E-selectin and VCAM-1 that are important for hematopoietic stem cell mobilization from the BM (15, 165). Thus, NADPH oxidase might be involved in their expression in BM sinusoidal ECs. Moreover Nox2-derived ROS are downstream mediators of CXCR4-Akt signaling, thereby stimulating migration of stem/progenitor cells (205). VEGF also is known to induce proangiogenic cell mobilization including VEGFR2+ EPCs (101) and VEGFR1+ hematopoietic cells (164) Placental growth factor also promotes recruitment of VEGFR1+ HSCs from a quiescent to a proliferative microenvironment within the BM, favoring differentiation, mobilization, and reconstitution of hematopoiesis (163). VEGF induced by acute ischemia causes MMP-9 upregulation in the BM microenvironment, resulting in the disruption of SCF-cKit bond between stem/progenitor cells and niche cells (79). In addition, activation of MMP-9 and subsequent release of soluble Kit ligand (sKitL) and mobilization of proangiogenic cells are impaired in eNOS−/− mice, suggesting a role for the eNOS-MMP9-sKitL pathway in BM EPC mobilization (5). Note that H2O2 causes endothelial NO release by phosphorylating eNOS serine 1179 (28) and that antioxidant and NADPH oxidase inhibitor attenuates eNOS activity in EC (161). In line with this, exercise training, known to increase both endothelial shear stress and H2O2, increases eNOS expression (121). Thus, NADPH oxidase–derived ROS may regulate eNOS-MMP-9 signaling, thereby promoting BM stem/progenitor cell mobilization (Fig. 3).
NAPDH oxidase and homing of stem/progenitor cells
BM-derived stem/progenitor cells homing and angiogenic capacity play in important role in postnatal neovascularization by directly incorporating into endothelial lining, structurally supporting the neovasculature, or secreting proangiogenic factor in ischemic tissues (96, 109, 134) (Figs. 1 and 3). This homing is mediated through highly concerted mechanisms, which involve adhesion, chemotaxis, and invasion, followed by integration of cells into vascular structures (38). Understanding the molecular mechanisms of homing is essential for enhancing cell-based therapy. Recent studies suggest that adhesion molecules on the endothelial surface plays an important role in the homing of progenitor/stem cells to sites of neovascularization, as demonstrated for leukocytes recruitment to the sites of inflammation. The homing capacity of EPCs and stem/progenitor cells is mediated by various adhesion molecules, including E-selectin, ICAM-1 and VCAM-1 (38), P-selectin (59), β1 integrin (Duan et al., 2006), and β2 integrin (37) (Fig. 3). NADPH oxidase–derived ROS are involved in expression of E-selectin in diabetes (80, 240) during ischemia/reperfusion (156, 172) and cytokine stimulation (35, 71, 156), as well as expression of ICAM-1, VCAM-1 (142), and P-selectin (7, 95, 193) on the endothelial surface. A recent study shows that stimulation of Rap1 through cAMP-mediated Epac1 activation increases integrin activity and integrin-dependent homing of progenitor cells, thereby enhancing their therapeutic potential in vivo (33). Given that Rap1A is involved in regulating NADPH oxidase (25, 129), NADPH oxidase might be involved in integrin-dependent homing functions of progenitor cells. β2 integrin and PI3Kγ are involved in homing capacity of EPCs and BM progenitor cells by regulating adhesion of EPCs on fibronectin and ICAM-1 (38). However, adhesion of BM cells to fibronectin and ICAM-1 is not affected in Nox2−/− cells (205), suggesting that β2 integrin– and PI3Kγ-mediated EPC adhesion capacity is not dependent on Nox2-derived ROS. Whether other Noxs are involved in the adhesion capacity of BM stem/progenitor cells remains unknown.
Intravenous infusion of Nox2−/− BM cells in a mouse hindlimb ischemia model shows that Nox2-derived ROS are required for the homing and angiogenic capacity of BM cells, thereby promoting neovascularization of ischemic tissues in vivo (205) (Fig. 1). Short-term pretreatment with low-dose H2O2 enhances the efficacy of BM cells for neovascularization in a mouse hindlimb ischemia model (112). Similarly, pretreatment with H2O2 rescues the impaired homing and migration capacity of Nox2-deficient BM cells (205), suggesting an important role of ROS in stem/progenitor cell function. Mechanistically, Nox2-derived ROS are selectively involved in actin polymerization as well as the pathways linking SDF-1/CXCR4 to Akt, thereby promoting migration of stem/progenitor cells (205) (Fig. 3). Similarly, receptor tyrosine kinase activation by hematopoietic cytokines increases generation of ROS that are involved in BM stem cells proliferation (87). IL-8 is an inflammatory chemokine that is able to stimulate angiogenesis (149) and provides a chemoattractant gradient for BM-derived endothelial progenitors or angioblasts. This chemokine-mediated homing of BM progenitors to the ischemic heart regulates their ability to induce myocardial neovascularization, protection against apoptosis, and functional cardiac recovery (107). IL-8 stimulation via CXCR2 causes NADPH oxidase activation by Rac translocation to membrane (242), and blocking CXCR2 inhibits the incorporation of human EPCs expressing CXCR2 at sites of arterial injury (83). Thus, IL-8/CXCR2-mediated activation of NADPH oxidase may be involved in EPCs and BM progenitor/stem cell homing, thereby promoting angiogenesis and repair in response to injury (Fig. 3).
NADPH oxidase and dysfunction of stem/progenitor cells in pathophysiologies
Although physiologic levels for ROS are required for normal function, excess amounts of ROS produced in various pathophysiologies contribute to dysfunction of EPC and stem/progenitor cells (20, 81, 132, 181, 215). Evidence suggests that number and functional capacity of EPCs are reduced in cardiovascular diseases, including hypertension, atherosclerosis, diabetes, coronary artery disease, and heart failure, as well as comorbid risk factors such as aging, hypercholesterolemia, and cigarette smoking (23, 34, 181, 203, 220, 233), all of which are associated with oxidative stress.
The renin–angiotensin system increases vascular oxidative stress, leading to reduction in the bioavailability of NO, thereby contributing to hypertension, diabetes, atherosclerosis, and heart failure. Ang II accelerates the senescence of EPCs by increasing oxidative stress via upregulation of Nox2, which is mediated through AT1Rs (91). Hypercholesterolemia and in vitro exposure to LDLs are associated with increased AT1R expression in CD34+ cell, and enhance AT1R-mediated HSC differentiation into proatherogenic CD11b+ monocytes (191). Impaired BM stem/progenitor cell function and postischemic revascularization in spontaneously hypertensive rats (SHRs) are rescued by cotreatment with Ang II converting enzyme (ACE) inhibitor, AT1Rs blocker, or antioxidants (238). Moreover, AT1Rs blockers or ACE inhibitors improve EPC dysfunction through antioxidative mechanisms in hypertension (239). Thus, AT1R signaling is involved in stem/progenitor cell dysfunctions leading to hypertensive vascular injury. In line with these findings, antioxidative β1-adrenoceptor blocker celiprolol decreases oxidative stress in hypertension and improves EPC numbers and function (232). Aldosterone, which increases oxidative stress, inhibits the formation of EPCs, at least in part by attenuating VEGFR2 expression and subsequent Akt signaling (136).
By contrast, Ang II at low concentrations functions as a positive regulator for EPC and stem/progenitor function. Ang II potentiates VEGF-induced human EPCs proliferation and network formation through the upregulation of VEGFR2 (92). It also promotes NO production, inhibits apoptosis and enhances adhesion potential of BM-derived EPCs via the PI3-kinase/Akt pathway (236). Ang II stimulates proliferation of hematopoietic progenitor cells as well as differentiation of dendritic cells (150). It also promotes hematopoietic recovery in multiple cellular lineages after chemotherapy through an increase in the number of early hematopoietic progenitors (169). In two-kidney, one-clip or Ang II–infused hypertensive mice, hypertension-induced mechanical stretch of the vessel wall stimulates the mobilization of EPCs from BM, in a p47 phox -dependent manner (173). This response seems to be a compensatory vascular adaptation.
Increased oxidative stress plays a role in EPC dysfunction in type 1 and type 2 diabetes (133, 179). In type 1 diabetic mice, overproduction of Nox2-derived ROS contributes to impaired angiogenesis and EPC dysfunction, which is rescued in Nox2-knockout mice (53). Uncoupled eNOS in diabetic BM, glucose-treated EPCs, and EPCs from diabetes patients produce overproduction of O2 •−, which results in impaired EPC mobilization and function (200). Human EPCs from type 2 diabetes, which are associated with oxidative stress, exhibit the mobilization of functionally defective EPCs with impaired proliferation, adhesion, and incorporation into existing vasculature (197). BM cells obtained from diabetic db/db mice are dysfunctional because of oxidative stress–mediated apoptosis, which is restored by the antioxidant ebselen (39). Diabetes mellitus impairs CD133+ progenitor cell function after myocardial infarction because of a lower resistance of CD133+ cells to oxidative stress (217). This explains the delayed postischemic vascular healing and myocardial recovery in diabetes patients. Diabetes patients show an increase in NADPH oxidase expression in circulating lymphomonocytes, membrane-associated PKCβ2 activity in monocytes, and a decrease in number of EPCs (16). The in vivo reendothelialization capacity of EPCs from patients with type 2 diabetes is impaired because of increased NADPH oxidase–dependent O2 •− production and subsequent reduced NO bioavailability (187). AT1R antagonists increase the number of regenerative EPCs in patients with type 2 diabetes mellitus (19), presumably through inhibiting NADPH oxidase.
In myocardial infarction model of rats, ROS elevation as well as reduced ERK phosphorylation and MMP-9 activity in BM contribute to impaired EPC mobilization, which is rescued by ACE or HMG-CoA inhibitors (199). In patients with coronary artery disease, peroxisome proliferator–activated receptor-γ (PPAR gamma) agonists, which are used for the treatment of diabetes, increase EPC number and migration by inhibiting NADPH oxidase activity (222). A study using p47 phox knockout mice and BM transplantation demonstrated that NADPH oxidase–derived ROS in BM contribute to lung ischemia/reperfusion injury (231). Oxidized LDL plays a key role in the pathogenesis of atherosclerosis by accelerating the onset of EPC senescence (93). In diabetes, oxidized LDL contributes to EPC dysfunction via a p53-mediated signaling pathway (170). By contrast, high-density lipoprotein, which is atheroprotective by antioxidant and antiinflammatory properties, promotes progenitor cell–mediated endothelial repair in apoE−/− mice (204). Homocysteine, which increases excess production of ROS, accelerates senescence and reduces proliferation of EPCs (243).
The availability and function of EPCs are also affected by other risk factors including cigarette smoking (140) and aging (78), which are associated with oxidative stress. ROS and the natural by-products of oxidative metabolism are known to cause DNA damage closely associated with aging. ATM (ataxia telangiectasia-mutated) is important for stem cell function in regulating ROS levels. Hematopoietic stem cells from ATM-deficient mice show enhanced ROS levels and impaired stem cell functions, rescued by the treatment with the antioxidant N-acetyl-
Understanding the molecular mechanisms by which oxidative stress is involved in reducing the number and functional activity of EPCs is potentially important, as it could create novel therapeutic strategies for treatment of cardiovascular diseases (203, 233). Inhibition of NADPH oxidase of progenitor cells may lead to improvement of endothelial repair and ischemia-induced neovascularization in various oxidative stress–dependent pathophysiologies (23).
Conclusion and Future Directions
NADPH oxidase–derived ROS play an important role in physiologic and pathologic postnatal angiogenesis and vasculogenesis. The effects of ROS derived from ECs and stem/progenitor cells during neovascularization are tightly regulated by the balance of prooxidant and antioxidant enzyme activity. ROS at low levels function as signaling molecules to mediate cell proliferation, migration, differentiation, survival, and angiogenic gene expression in ECs and stem/progenitor cells, which contributes to reparative neovascularization in response to injury. Moreover, NADPH oxidase expression is increased in BM after ischemic injury, which is required for BM stem/progenitor mobilization, homing, and angiogenic capacity at the site of new-vessel formation. Recent studies show the crucial role of NADPH oxidase in cardiovascular differentiation of ES cells. By contrast, excess amounts of ROS (oxidative stress) involved in various pathologies including hypertension, atherosclerosis, aging, and diabetes contribute to dysfunction of ECs and stem/progenitor cells, thereby resulting in impaired angiogenesis and vasculogenesis. Cultured EPCs express higher levels of antioxidant enzymes MnSOD and catalase than do mature ECs, thereby being resistant to oxidative stress. Significant work remains to be performed (a) to define the role of each Nox and its regulatory subunits in angiogenesis and vasculogenesis; (b) to determine the activation mechanisms of NADPH oxidase by various angiogenesis factors and cytokines in ECs and stem/progenitor cells; (c) to identify molecular targets of NADPH oxidase–derived ROS in signaling pathways involved in angiogenesis in ECs, as well as stem/progenitor cell function; and (d) to determine the contribution of ROS derived from other cell types such as inflammatory cells, platelets, and T cells in postnatal neovascularization in vivo. Understanding these mechanisms will lead to development of new therapeutic strategies for many angiogenesis- and vasculogenesis-dependent cardiovascular diseases such as ischemic heart and limb diseases and hypertension, diabetes, and atherosclerosis.
Footnotes
Acknowledgments
This work was supported by NIH R01 HL077524, AHA Grant-In-Aid 0555308B, and AHA National Innovative Research grant 0970336N.