
Abstract
The enzymatic complexes of the mitochondrial respiratory chain have been extensively investigated in their structural and functional properties. A clear distinction is possible today between three complexes in which the difference in redox potential allows proton translocation (complexes I, III, and IV) and those having the mere function to convey electrons to the respiratory chain. We also have a clearer understanding of the structure and function of most respiratory complexes, of their biogenesis and regulation, and of their capacity to generate reactive oxygen species. Past investigations led to the conclusion that the complexes are randomly dispersed and functionally connected by diffusion of smaller redox components, coenzyme Q and cytochrome c. More-recent investigations by native gel electrophoresis and single-particle image processing showed the existence of supramolecular associations. Flux-control analysis demonstrated that complexes I and III in mammals and I, III, and IV in plants kinetically behave as single units, suggesting the existence of substrate channeling. This review discusses conditions affecting the formation of supercomplexes that, besides kinetic advantage, have a role in the stability and assembly of the individual complexes and in preventing excess oxygen radical formation. Disruption of supercomplex organization may lead to functional derangements responsible for pathologic changes. Antioxid. Redox Signal. 11, 961–1008.
I. The Enzymes and Complexes of the Respiratory Chain
The concentration values are expressed as nmol/mg protein. na = not assayed. Mean molar ratios of the individual components are shown in round brackets; numbers were calculated by comparison with the content of complex III, the amount of which, as obtained from the different authors, was normalized to a value of 3.
Obtained from inhibitor-binding studies.
Estimated from electron paramagnetic resonance (EPR) and from antibody titration.
Based on flavin mononucleotide (FMN) content.
Determined by electrophoretic–densitometric approach.
Based on 0.19 nmol heme a/mg protein and stoichiometry, as indicated in parenthesis.
Determined by spectral analysis.
Based on the content of acid-nonextractable flavin adenine dinucleotide (FAD) content.
Based on half the amount of cytochrome b content, as spectrophotometrically determined.
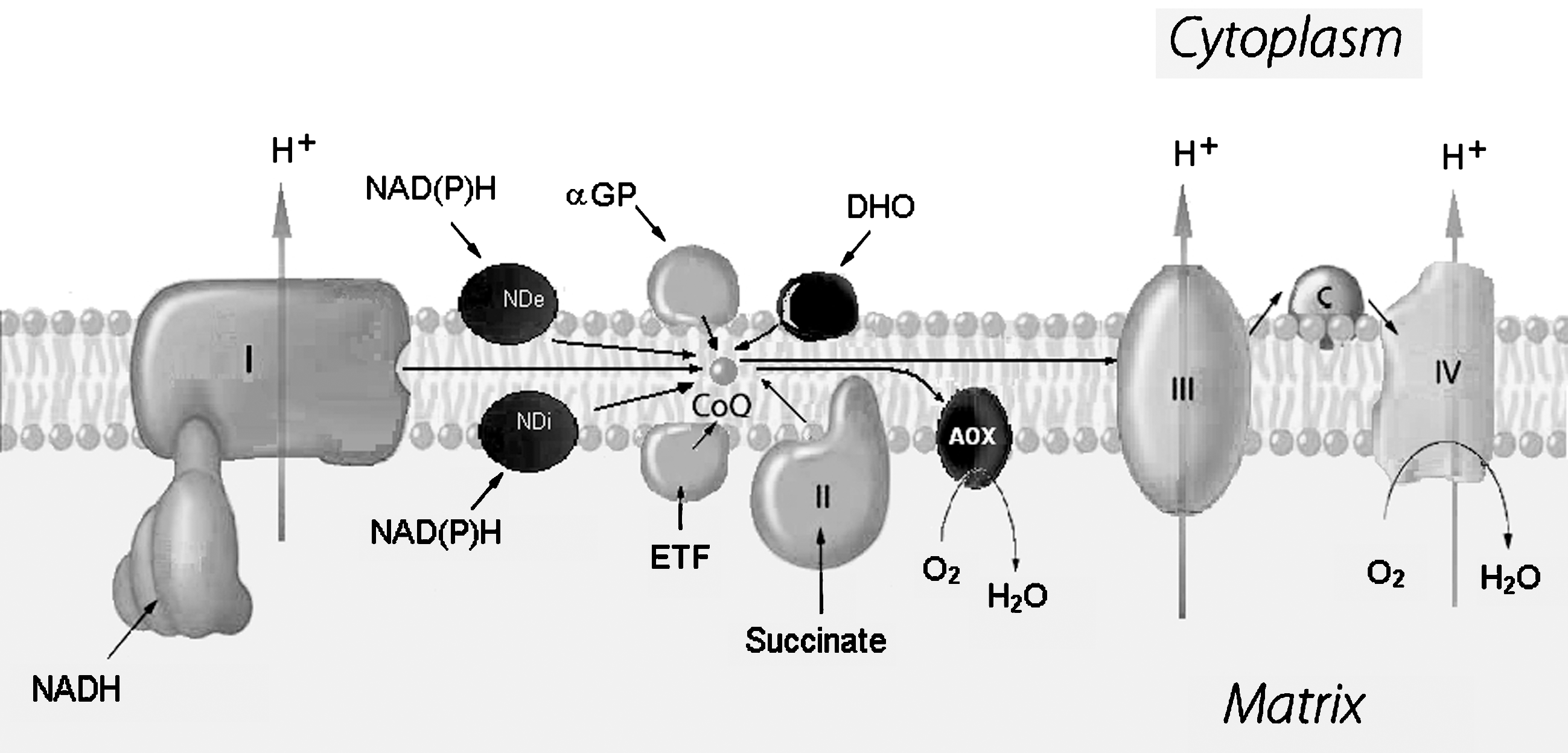
The inner membrane contains, in smaller amounts, other proteins having electron-transfer activity; among these there are electron-transfer flavoproteins capable of feeding electrons to the respiratory chain by pathways not involving complex I or NAD or both (i.e., glycerol-3-phosphate dehydrogenase, electron-transfer flavoprotein (ETF)-ubiquinone oxidoreductase, dihydroorotate dehydrogenase, choline dehydrogenase), besides alternative NADH dehydrogenases in mitochondria from several organisms, especially plants and fungi. Moreover, alternative or branched pathways of electron transfer also occur, departing from CoQ: these are the alternative ubiquinol oxidases from bacteria and plant and fungi mitochondria.
A. The “core” proton-translocating complexes
The traditional description considers the four complexes originally described by Green (107, 108) to be the structural core of the respiratory chain; a collection of other accessory enzymes feeding electrons to the chain was subsequently added to this nucleus. Nevertheless, a substantial difference exists between three of the original complexes (I, III, and IV) and complex II, because the latter shares some important properties of the accessory enzymes (i.e., they give electrons to coenzyme Q without creation of a transmembrane proton gradient; in other words, they are required for OXPHOS but do not participate directly in energy production). Moreover, contrary to the “core” complexes, they do not have subunits encoded by mitochondrial DNA. Furthermore, we provide evidence in this review that their supramolecular organization is probably different, because the three “core” complexes are associated together in a supramolecular assembly, whereas the remaining accessory enzymes appear to be free in a random organization.
1. Complex I
Complex I (NADH-coenzyme Q reductase, E.C. 1.6.5.3) catalyzes the first step of the electron-transport chain of mitochondria and several bacteria (32). The reaction is accompanied by translocation of four protons from the matrix to the intermembrane space.
Notably, some organisms lack complex I, including the most-investigated yeast species, Saccharomyces cerevisiae (92) and some other yeasts, as S. carlsbergensis and Kluyveromyces lactis (143), and the malarial parasite Plasmodium yoelii yoelii (313).
a. Structure and mechanism
The bovine enzyme is a heteromultimer consisting of 45 subunits for a molecular mass of ∼1,000 kDa, making complex I by far the largest enzyme of the respiratory chain. Seven subunits are the products of the mitochondrial genome and correspond to hydrophobic components named ND1-ND6 and ND4L. The minimal active form of the enzyme is that found in bacteria, composed of 14 “core” subunits, all of which are homologous to their mitochondrial counterparts, whereas all other “accessory” subunits still have an undefined role. From structural and phylogenetic considerations, the enzyme is envisaged to consist of three different sectors: a dehydrogenase unit and a hydrogenase-like unit, constituting the peripheral arm protruding into the matrix, and a transporter unit deeply embedded in the membrane and involved in proton translocation (91). The dehydrogenase domain contains the NADH oxidizing site, whereas the hydrogenase domain binds and reduces CoQ; both domains contain prosthetic groups, whereas the transporter unit appears to be devoid of cofactors.
Trypanosomes have a complex I slightly larger than that of bacteria, but lacking some of the core subunits: four subunits normally encoded by the mitochondrial genome. This deficiency results into loss of proton translocation, presumably an adaptation to parasitic life (231). Besides the classic NADH-ubiquinone oxidoreductase reaction performed by all mitochondrial complex I (see later), additional enzymatic activities have been found or proposed to be associated with complex I from plants, bovines, or fungi. However, the physiological significance of these activities, displayed by unique noncore subunits like γ-carbonic anhydrase,
Figure 1 is a general scheme illustrating the electron-transfer pathway from NADH to CoQ; major sites for reduction of artificial acceptors [ferricyanide, 2,4-dichloro-phenol-indophenol (DCIP)] are shown in the figure.
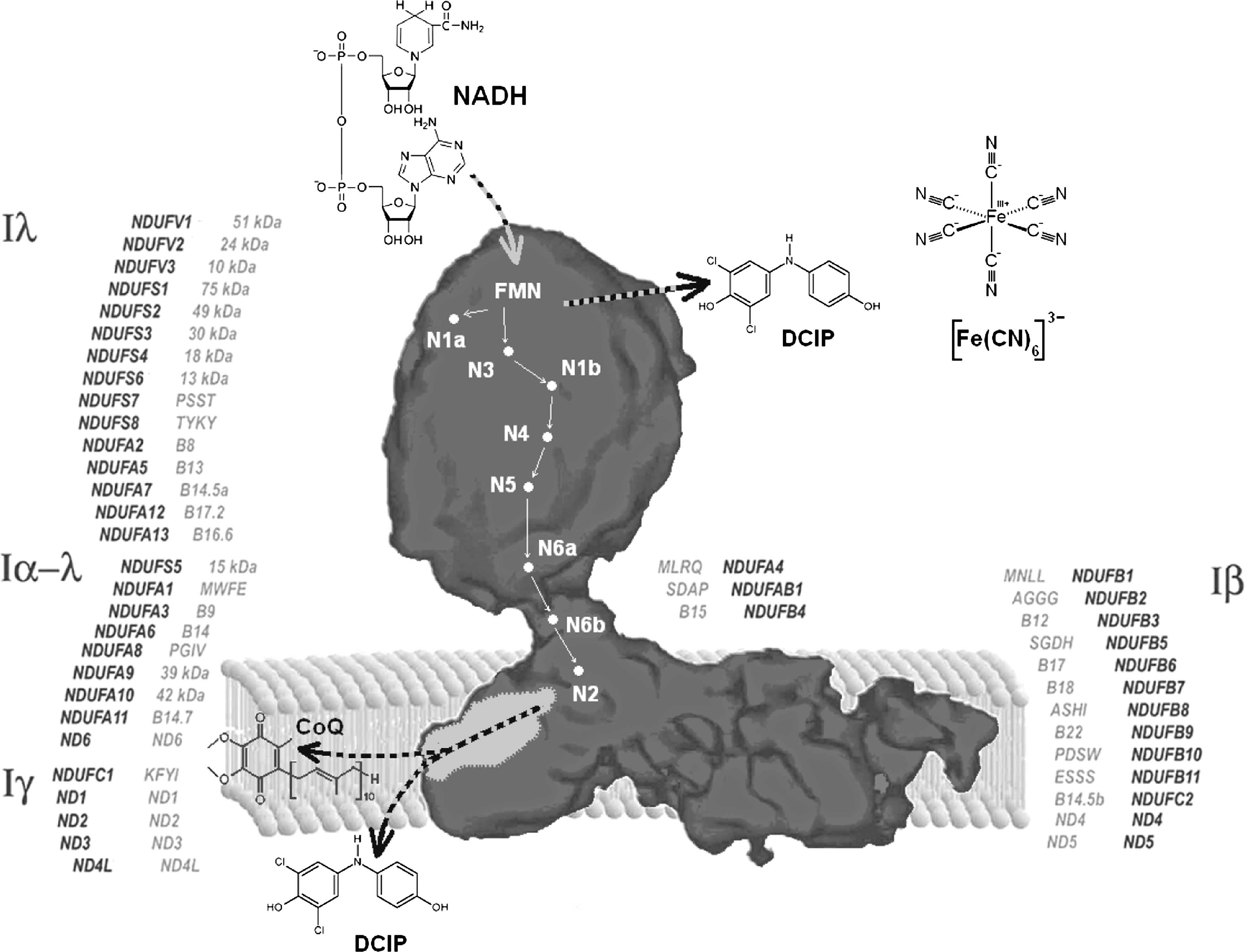
Several prosthetic groups contribute to electron transfer within the enzyme: FMN is the entry point for electrons that are then transferred to a series of iron–sulfur clusters. Two clusters present different characteristics: N1a, of the type Fe2S2, has the lowest midpoint potential (Em = −370 mV), whereas N2, that is of the type Fe4S4 and resides at the interface between the PSST and the 49-kDa subunits, has the highest midpoint potential (Em between −150 mV and −50 mV), presenting EPR magnetic interactions with the ubisemiquinone radicals; for these reasons, it is considered to be the direct electron donor to ubiquinone. N2 iron–sulfur cluster is most likely located in the connection between the peripheral and the membrane arm. The magnetic interaction with the semiquinone radical, corresponding to a distance of ∼10 Å (194), suggests that the ubiquinone headgroup could somehow reach up into the peripheral arm, as assumed by Brandt et al. (33), who hypothesized an amphipathic “ramp” guiding ubiquinone into the catalytic site. The arrangement of iron–sulfur clusters in the hydrophilic domain of complex I from Thermus thermophilus has been determined by x-ray crystallography, showing a linear chain of all clusters except N1a and N7 (275).
Complex I is inhibited by more than 60 different families of compounds from rotenone, the prototype of this series, to a number of synthetic insecticides/acaricides (192), which have been grouped into three classes (57). It is commonly accepted that they share the same hydrophobic large pocket in the enzyme (81) (Table 2).
Data are taken from ref. 57.
The pathway of electrons is now well defined (178). The primary acceptor of electrons from NADH is FMN bound to the 51-kDa subunit; because iron–sulfur cluster N1a has a very negative potential and is situated too far from the other iron–sulfur clusters, it is not likely to reside in the main pathway of electrons. Thus, electrons would flow from FMN to N3 in the same 51-kDa subunit, and to N4 and N5 in the 75-kDa subunit, and then to N6a and N6b in the TYKY subunit and to N2 in the PSST subunit shared with the 49-kDa subunit. N2 is the direct electron donor to bound ubiquinone, and probably this step is linked to proton translocation, although the mechanism is still debated (32). Because all redox groups in the enzyme appear to be located in the hydrophilic arm or at least at the interface with the hydrophobic arm, direct coupling mechanisms appear unlikely; this implies that the driving force for proton translocation must be transduced over a considerable distance to the actual pumping process in the membrane arm via conformational coupling (32, 341). Because a mutation completely abolishing the pH dependence of cluster N2 redox potential has no effect on proton translocation, it is likely that the conformational change driving proton translocation is linked exclusively to ubiquinone reduction.
The mechanism of CoQ reduction is particularly intriguing, because more than one bound quinone species has been assigned to the enzyme; three ubisemiquinone signals are detectable in the enzyme (186). The findings in our laboratory that two different classes of inhibitors have opposite effects on oxygen reduction to superoxide during forward electron transfer (see also Section III.B), together with other observations, allowed us to draw a tentative scheme of electron transfer in complex I (78) (Fig. 2). In the bifurcated scheme shown in the figure, an iron–sulfur cluster located upstream of the N2 center might act as a “switch” for electron delivery.
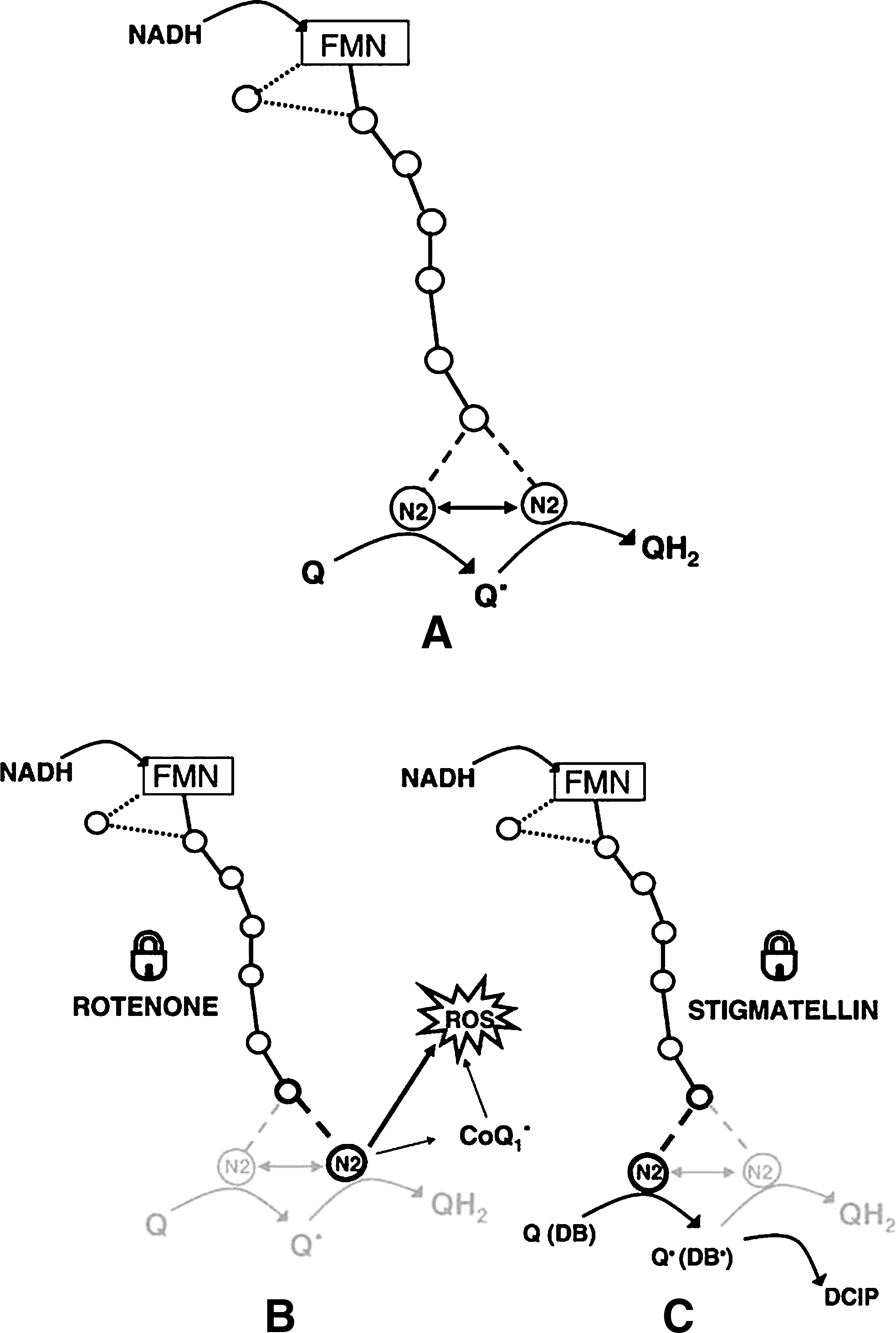
The findings of Fato et al. (78) have to be reconciled with the linear pathway of electrons along the series of iron–sulfur clusters, as demonstrated by the crystallographic study of Sazanov (276); our interpretation is not in contrast with the existence of a linear pathway, because the two electrons delivered to CoQ for its complete reduction could be provided by the same cluster (N2) consecutively, if a suitable conformational change occurs after the first electron delivery to provide a gating mechanism for the second electron.
b. Substrates and kinetics
The physiological activity of complex I is the electron transfer from NADH to ubiquinone or, in some cases, to menaquinone. Despite a difference in the substrate redox potentials of ∼400 mV, the reaction is fully reversible; it was demonstrated a long time ago that, in the presence of a proton-motive force, mitochondria can transfer electrons from succinate onto NAD+ (41). The K m value of complex I for NADH is in the micromolar range, and weak product inhibition at millimolar concentrations of NAD+ can be observed (321). In contrast, the K m of NAD+ for the reverse reaction is in the micromolar range.
Being natural ubiquinones, extremely hydrophobic molecules unsuitable as electron acceptors in vitro, a series of homologues and analogues having shorter chains in the six position are used as substrates for complex I assays. These quinones have finite membrane/water partition coefficients (80) that must be taken into account in any consideration concerning their specificity and kinetics of interaction (175). It is assumed that these compounds interact with the physiological site(s), in place of the endogenous CoQ, by first partitioning from the water phase to the membrane and that exogenous quinones are reduced directly by complex I, without the mediation of the ubiquinone pool. The discovery that the enzyme contains bound ubiquinone essential for its activity reopens the question of the mode of interaction of exogenous quinones with the acceptor site(s). Among the quinone acceptors used are the homologue series from CoQ0 up (175), including less frequently long isoprenoid chain homologues; despite their insolubility in water, the tetramethyl benzoquinone analogue, duroquinone (DQ), and analogues having straight saturated chains, such as 6-pentyl, 6-decyl, and 6-undecyl ubiquinones (usually abbreviated as PB, DB, and UBQ, respectively). These acceptors are used in the presence of endogenous ubiquinone. The suitability of many of these commonly used acceptors has been questioned (73); the main reason has been the observation that NADH-CoQ reductase activity, as experimentally determined, is often paradoxically found to be lower than NADH-cytochrome c reductase or NADH oxidase. The reasons for underevaluating the oxidation of NADH by exogenous quinones may be summarized as follows (175). The water solubility of the quinones with respect to their K
m is fundamental for assessing that kinetic saturation is reached during assay; Some quinones are complex I inhibitors. The inhibitory action of CoQ2 and other short-chain isoprenoid homologues (but not of CoQ1), well documented in beef-heart mitochondria (80), also was observed in human lymphoblast mitochondria (195); moreover, CoQ2 was shown to inhibit cell growth in culture (151). The clinically used analogue idebenone (hydroxydecyl-ubiquinone) also inhibits complex I (107). It also has been proved that some quinols, being the product of the electron-transfer activity of complex I, can potently inhibit the enzyme complex; for example, whereas decylubiquinone (oxidized form) acts as a potent acceptor for complex I electrons, its reduced form, decylubiquinol, severely impedes complex I activity (19). Despite that, measuring the initial rate of complex I activity avoids product inhibition and allows high complex I activity (80); and A further reason, not considered in earlier reports, may be in the supramolecular assembly of complex I in the native membrane (see later) in which it is strictly linked to complex III, possibly hiding the CoQ-acceptor site to exogenous quinones.
The steady-state kinetics of complex I has been investigated by Fato et al. (80) by using different quinones as acceptors; considering their partition coefficients and their real concentrations in the membrane, the best acceptors were found to be CoQ1 and DB. The kinetic pattern was shown to follow a ping-pong mechanism; however, a further study (223) in the purified enzyme suggested a sequential mechanism. The K m for CoQ1 is in the range of 20 μM but is reversibly increased to 60 μM by extraction of the endogenous CoQ10 (80). The increased K m in CoQ10-depleted membranes indicates that endogenous ubiquinone not only does not exert significant product inhibition but rather is required for the appropriate structure of the acceptor site.
2. Complex III
The cytochrome bc1 complex or complex III (ubiquinol-cytochrome c oxidoreductase, E.C. 1.10.2.2) from mitochondria of several species has been crystallized, and its structure solved to atomic resolution (139). The mechanism of the enzyme is generally well understood, although some questions remain.
The enzyme represents a confluence point for reducing equivalents from various dehydrogenases: it can catalyze the transfer of electrons from hydroxyquinones (ubiquinol, reduced CoQ) to a water-soluble c-type cytochrome, and it can, concomitantly, link this redox reaction to translocation of protons across the membrane (52).
All cytochrome bc1 complexes contain three protein subunits with redox prosthetic groups, a di-heme cytochrome b containing a relatively high-potential b H (or b566) heme and a lower potential b L (or b562) heme, cytochrome c1 and an iron–sulfur protein (Rieske protein) with a 2Fe-2S cluster (21). As many as seven or eight supernumerary subunits also are present in the mitochondrial enzymes. These nonredox subunits are not required for electron-transfer and proton-translocation activities of the enzyme; their possible functions include structural stability and regulation of coordinated activity of the dimeric enzyme, and docking sites for ternary complex formation with the dehydrogenase and oxidase complexes (277). Table 3 shows the subunit composition of mammalian complex III.
Gene name; in parenthesis, accession number in UniProtKB (
Number of aminoacidic residues; numbers in parenthesis refer to presequences, other numbers, to the mature sequences.
Mitochondrial gene.
Mitochondrial targeting presequence cleaved from UQCRFS1.
The mitochondrial complex III is a symmetrical, oligomeric dimer; it has been demonstrated that the iron–sulfur protein spans the dimer structure because it is anchored in one monomer, whereas its peripheral domain is located in the other monomer, where it forms part of the ubiquinol oxidation site. Evidence exists that the dimer behaves as a functional monomer, on the basis of the stoichiometry of inhibitor action on enzyme activity (51). Striking evidence exists that the peripheral domain of the Rieske protein moves back and forth between positions close to cytochrome b and cytochrome c1 (225), facilitating electron transfer within the enzyme. Crystal structures established the location of the ubiquinol oxidation and ubiquinone reduction at topographically separated sites within each monomer and demonstrated the transmembrane disposition of the b hemes. These structural details provide a final confirmatory evidence of the proton-motive Q-cycle mechanism of the enzyme, with protons being carried across the inner mitochondrial membrane, whereas electrons from ubiquinol are transferred through the bc1 complex (211, 232) (Fig. 3).
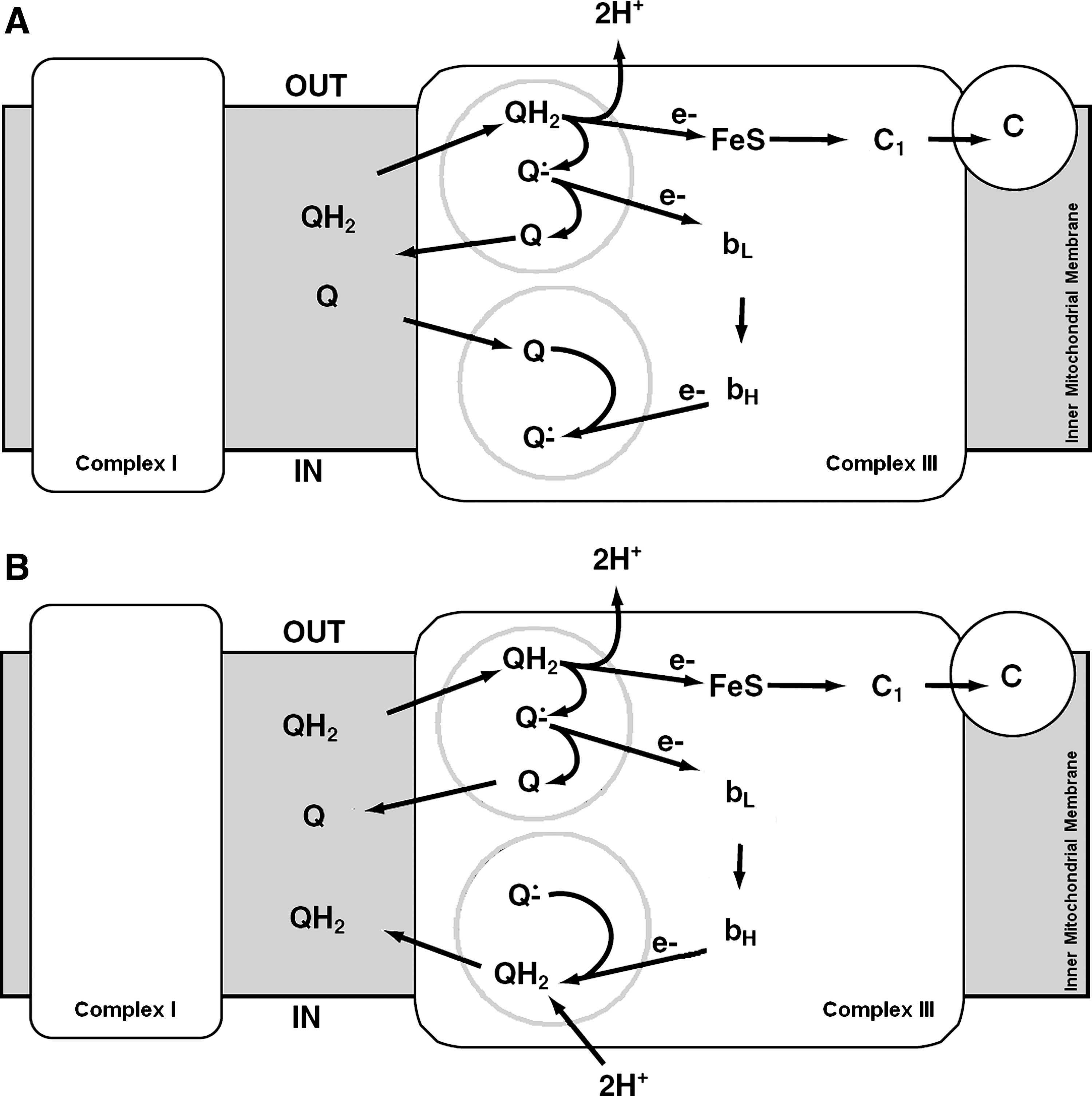
A detailed survey of the effects of complex III inhibitors has been the basis for establishing the Q-cycle as the molecular mechanism of electron transfer and proton translocation. Two major classes of inhibitors have been individuated, acting at two separate sites, denominated site or center i (inner) or N (negative), and site or center o (outer) or P (positive); site o inhibitors have been subdivided into two further subclasses of compounds (proximal and distal inhibitors, with reference to the block of the first or of the second electron released from ubiquinol (Table 4).
Data are taken from ref. 71.
Class P contains two subgroups that are distinct in their ability to induce mobile (Pm) or fixed (Pf ) conformation of iron–sulphur protein.
According to ref. 188: Class I inhibitors bind to the QP pocket and are further divided into three subclasses (Ia, Ib, Ic) based on their chemical characteristics and their ability to change biophysical and spectral properties of the heme bL and the 2Fe2S cluster in ISP; Class II inhibitors bind to the QN pocket.
Being water insoluble, reduced CoQ10 as well as other long isoprenoid chain ubiquinols cannot be used in the assay of complex III; widely used short-chain homologues are CoQ1 and CoQ2 and analogues such as duroquinol or decyl-ubiquinol (100); the hydroxyl derivative of decyl ubiquinone, idebenone, in its reduced form, is a good electron donor to complex III (58). The donor substrates interact with the enzyme by previously partitioning in the membrane lipids, so that their partition coefficients must be taken into consideration for determining the true kinetic constants of the enzyme (80). Weiss and Wingfield (327) studied the enzymology of complex II and of the bc1 complex embedded in detergent micelles by using CoQ10/ubiquinol10 as the connecting substrate, and found that the transfer of the reduced quinone from one micelle to another was the rate-limiting step of the integrated activity.
Steady-state kinetic analysis by two-substrate titrations indicates, for complex III, a two-site ping-pong mechanism; the kinetic analysis suggests that the enzyme is not controlled by ubiquinol diffusion to the active reduction site, but may be controlled by cytochrome c diffusion to the oxidation site.
3. Complex IV
Complex IV (cytochrome c oxidase, EC. 1.9.3.1) belongs to the heme-copper oxygen reductase superfamily whose members catalyze the complete reduction of dioxygen to water and promote proton translocation across the mitochondrial or periplasmic membrane, further contributing to the difference in electrochemical potential. These enzymes have in common the same general structural fold of the catalytic subunit (subunit I), which contains a low-spin heme and a bimetallic center, composed of a high-spin heme and a copper iron (CuB), retained in the protein by ligation with histidine residues. Dioxygen reduction takes place at this binuclear site. Stoichiometries for the redox-driven proton pumping are variable among the different members of the superfamily, and even the same enzyme has no fixed stoichiometry in all conditions, as generally considered in the literature (13). The maximal stoichiometry of the mitochondrial oxidase is translocation of 2H+/2e−.
Heme-copper enzymes are classified according to the amino acid residues of their proton-conducting channels. The mitochondrial enzyme is a member of the type A1 family (Fig. 4), having Asp-124 (D124, amino acid numbering as in Paracoccus denitrificans, after which the D-channel proton pathway is called) close to the negative side of the membrane and besides hydrophilic amino acid residues (Asn-199, Asn-113, Asn-131, Tyr-35, Ser-134, Ser-193) ending at Glu-278, considered a key residue for the operating mechanism of the enzyme. The residues Lys-354 (K), Thr-351, Ser-291, and Tyr-280 are part of a second proton pathway (K,channel) and have also been demonstrated to play a crucial role in the catalytic cycle (187).
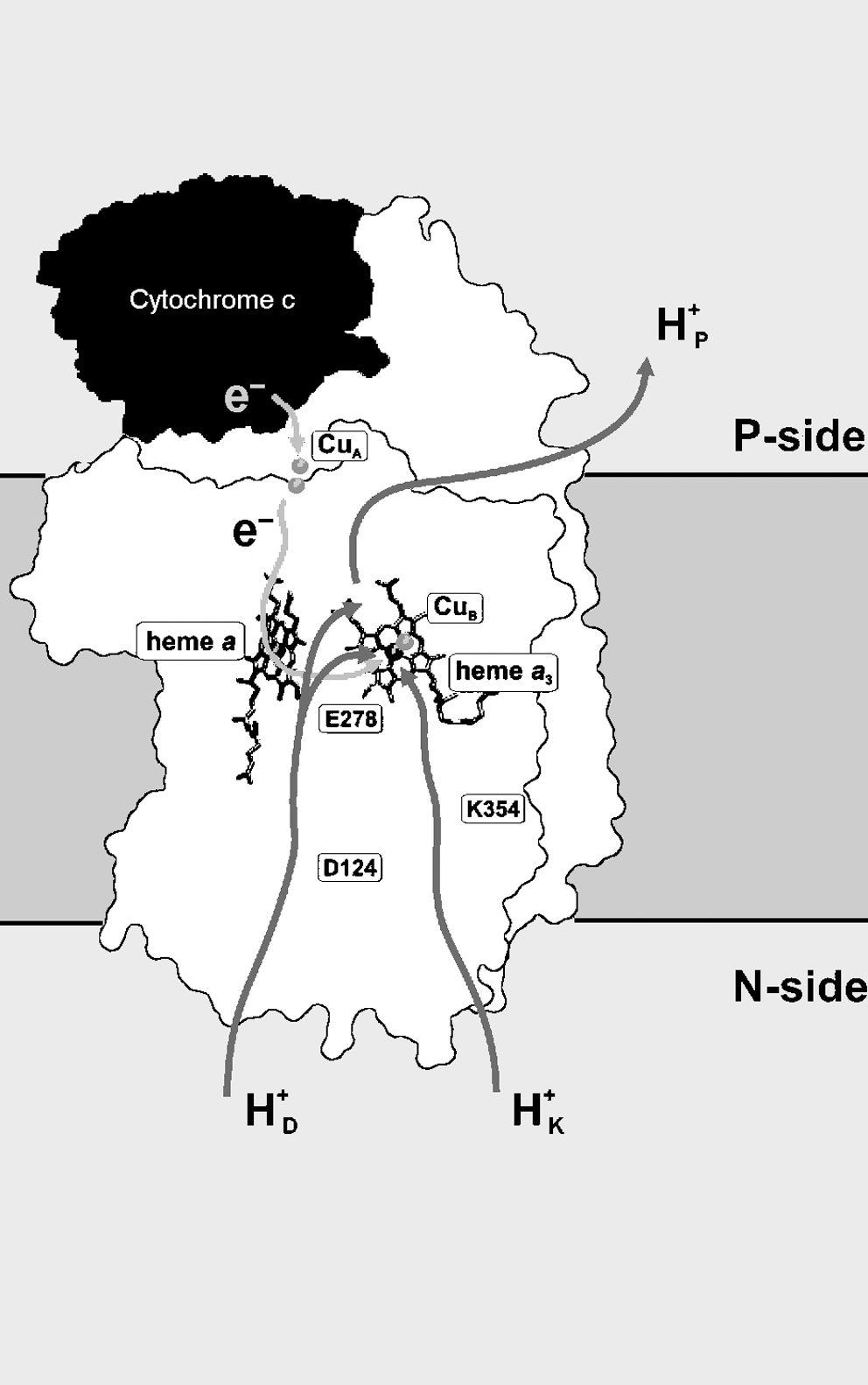
Electron transfer through complex IV occurs from ferrocytochrome c to the CuA center (which acts as a single-electron receptor), then to heme a onto the heme a3 /CuB center, and finally to oxygen bound to heme a3 . H+/e− cooperative linkage at Fe(a3 )/CuB is envisaged to be involved in proton-pump mechanisms confined to the binuclear center (330). Models have also been proposed that involve a role in proton pumping of cooperative H+/e− linkage at heme a/CuA (205, 243).
Cytochrome c oxidase is potently inhibited by cyanide, azide, and carbon monoxide, which bind at the oxygen-binding site (48). The molecular mechanism of inhibition by NO is more complex: (a) a major component is competitive with oxygen and, presumably, requires the presence of electrons in the binuclear center, but (b) an additional interaction occurs at the oxidized enzyme by binding to CuB 2+ (103). In the latter case, the enzyme becomes reduced (CuB +), and NO is oxidized to nitrite (NO2 −). Kinetically, this results in uncompetitive inhibition with respect to the oxygen kinetics. Recent kinetic models have successfully incorporated both modes of inhibition (49); therefore, depending on the balance of the two effects, the activity of cytochrome oxidase toward NO may result in the strong inhibition of cell respiration or in the removal of NO from the cell.
As extensively reviewed in a recent article by Belevich and Verkhovsky (14), a real breakthrough in the understanding of the function of cytochrome oxidase was achieved when the first crystallographic structures of the enzyme were resolved, in both oxidized and reduced states, but solving the structures of all intermediates in the catalytic cycle is still a difficult task, as those intermediates are quite unstable. At present, five x-ray crystallographic structures of heme-copper oxygen reductases have been determined, with resolution up to 1.8 Å (cf. ref. 27 for a detailed list of references), shedding light also on the structure of additional protein subunits that can compose the functional unit of cytochrome oxidase, besides the catalytic subunit I. The mammalian cytochrome oxidase has a molecular mass of ∼200 kDa and consists of 13 subunits originating both from nuclear and mitochondrial DNA, whereas the bacterial enzyme is simpler in structure because it contains three core subunits, whose sequence homology highly corresponds to the three subunits that, in the majority of eukaryotes, are encoded in the mitochondrial DNA (COXI, COXII, COXIII); one extra subunit is present in P. denitrificans only (Table 5).
MW, molecular masses (kDa) according to UniProtKB (
Because none of the nuclear-encoded subunits is associated with the active site, it was formerly assumed that they were not important in the functional mechanism of the enzyme. However, it is now demonstrated that some of those additional subunits are involved in the stabilization of a dimer state of the oxidase (88) and might participate in the interaction of complex IV with its partner complexes within a respiratory supercomplex, or they are suggested to regulate complex IV activity either by chemical modification like glycosylation and phosphorylation (120) or by binding effectors, such as ADP/ATP or protein kinase A (18). Lee and colleagues (169) proposed that the physiological meaning of such feedback regulation of the respiratory chain by its end product, ATP, is in keeping with the low membrane potential and, consequently, reduces ROS production of mitochondria. The allosteric ATP-inhibition is lost when the enzyme is dephosphorylated.
B. The auxiliary enzymes of the respiratory chain
This survey includes not only those enzymes that reduce CoQ, bypassing NAD and complex I, but also the alternative oxidases that deliver electrons from CoQ to oxygen, bypassing complex III and cytochrome oxidase. All these enzymes are characterized by lack of energy-conserving proton-translocation mechanisms.
1. Complex II
Besides its functional role as succinate dehydrogenase in the Krebs cycle, the enzyme (EC 1.3.5.1) is involved in aerobic metabolism by the respiratory chain because it can couple the two-electron oxidation of succinate to fumarate with the electron transfer directly to the quinone pool; hence complex II is more precisely termed succinate:quinone oxidoreductase (SQR) (164).
Mammalian complex II is part of a class of ubiquinone-reducing enzymes containing a single b heme and anchored to the inner mitochondrial membrane by two hydrophobic subunits, SdhC (14.2 kDa) and SdhD (12.8 kDa). Although the membrane anchor domain shows low sequence identity (less than 20%) and varies in composition between organisms, the primary sequence of the soluble domain of complex II is highly conserved (30 to 50% sequence identity) and consists of a flavoprotein subunit (SdhA, Fp, 64 to 79 kDa) containing covalently linked FAD and an iron–sulfur protein subunit (SdhB, Ip, 27 to 31 kDa), both located on the matrix side of the membrane (for a review, see ref. 40). One feature of the complex II structure is a linear electron-transport chain that extends from the flavin and Fe-S redox cofactors in the extrinsic domain to the quinone and heme b cofactors in the membrane domain (40). The interaction of quinones with complex II is an area located at the fringe of a hydrophobic pocket comprising residues from subunits SdhB, SdhC, and SdhD (132). Besides two electrons from the oxidation of succinate, the full reduction of the quinone in SQR would require two protons to be donated by the protein environment of the Q-site followed by re-protonation of the site after catalytic turnover. In the native structure of SQR from Escherichia coli, Horsefield et al. (132) identified a proton-uptake pathway suitable for such purpose that crosses the membrane anchor arriving at the Q-site. The high homology between the SQR Q-sites in E. coli and in mammalians, based on absolute conservation of amino acids in contact with ubiquinone, suggests the same mechanism for electron transfer to ubiquinone, thus making E. coli an excellent model system for mitochondrial complex II research (301). This is of particular interest in humans, because mutations in complex II result in various physiological disorders (269). The structure of complex II is shown in Fig. 5.
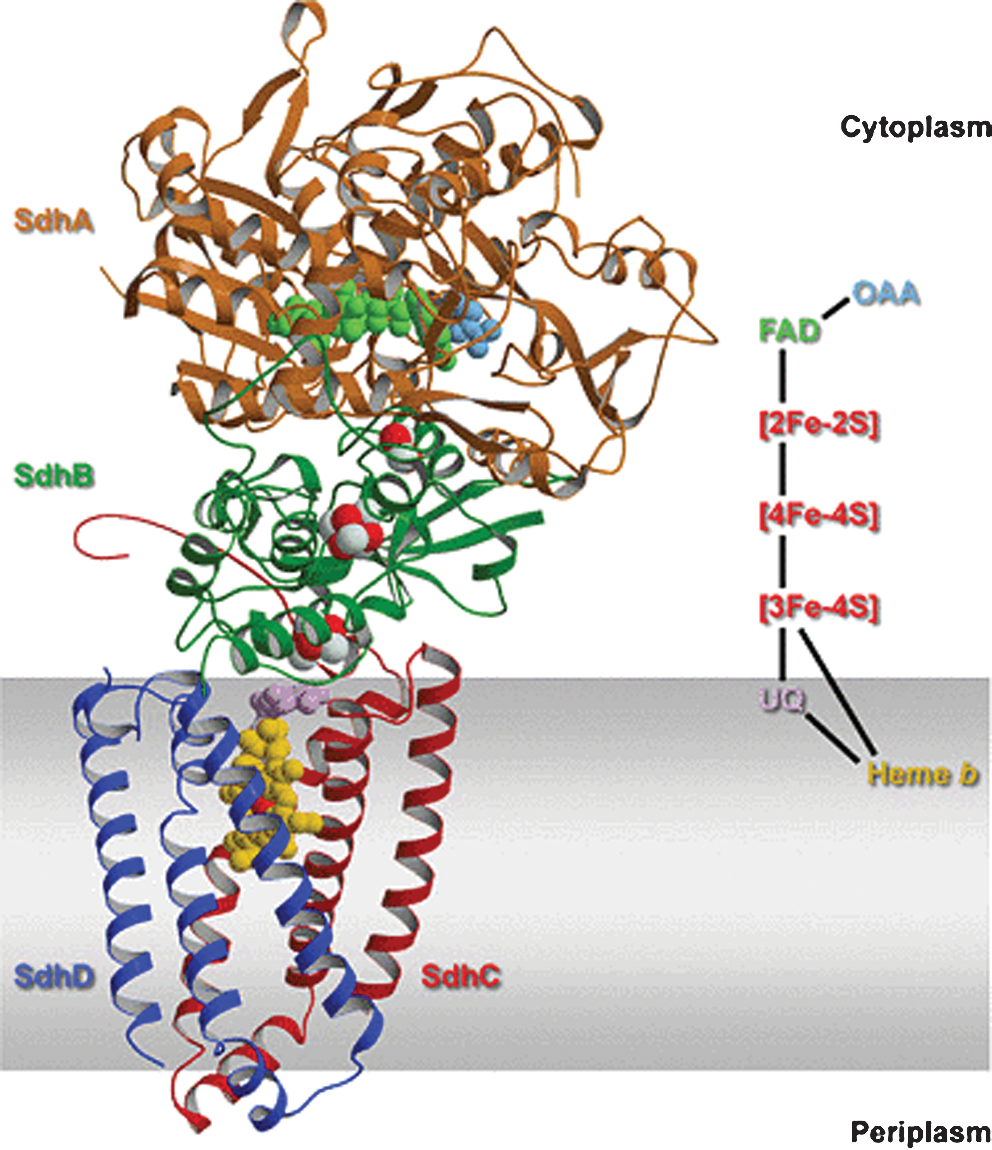
Although the natural acceptor of complex II is hydrophobic CoQ in the membrane, the enzyme is not usually assayed with short-chain quinones as acceptor substrates, but by using 2,6-dichlorophenol-indophenol, which has a higher midpoint redox potential and accepts electrons directly from endogenous CoQ (87); alternatively, especially in studies from muscle biopsies in the case of mitochondrial genetics diseases, the activity of complex II+III is measured cumulatively as succinate cytochrome c reductase (e.g., in ref. 9). “Soluble” succinate dehydrogenase is assayed by using as acceptor phenazine methosulfate, which accepts electrons upstream from the physiologic site (87). Complex II is typically inhibited by thenoyl trifluoroacetone and carboxin (5,6-dihydro-2-methyl-1,4-oxathin-3-carboxanilide) that bind to the same binding site situated in the SdhB iron–sulfur protein subunit (201).
The competitive inhibition by malonate reflects a physiologic inhibition by oxaloacetate (4): this is the reason that succinate dehydrogenase and related activities are usually low in isolated mitochondria, and incubation with succinate is needed to remove tightly bound oxaloacetate. In mitochondria from rat brain, but not heart or liver, the oxaloacetate inhibition is released by allowing mitochondria to oxidize pyruvate plus glutamate and malate, probably by removing the inhibitor through metabolic reactions (241).
Recent research has shown that the mitochondrial complex II plays an important role in the anerobic energy metabolism of parasites: often, the parasite uses aerobic metabolism during the free-living stage outside the host, but adapts to hypoxic environments and uses systems other than oxidative phosphorylation for ATP synthesis when inhabiting host mammals. Many adult parasites perform fumarate respiration by expressing a stage-specific isoform of complex II that catalyzes the reduction of fumarate (quinol-fumarate reductase, QFR), which is the reverse of the reaction catalyzed by SQR (152).
2. Mitochondrial glycerol-3-phosphate dehydrogenase
Glycerol-3-phosphate dehydrogenase (mtGPDH, EC 1.1.99.5) shuttles reducing equivalents from cytosol through the respiratory chain to molecular oxygen. This metabolic shuttle was first discovered in insect flight muscle (337) and in brown adipose tissue (133), where the enzyme has highest activity. In pancreatic islet β cells, many studies support the significant participation of the shuttle in the events signaling the release of insulin in response to increased glucose (196). The mtGPDH is a very hydrophobic protein of the inner membrane; its catalytic center is accessible from the outer surface of the inner membrane. Besides containing FAD as a prosthetic group, the presence of an iron–sulfur cluster has been suggested on the basis of ascorbic acid stimulation and inhibition by di-iron metallo-enzymes inhibitors (144); however, the putative center has not been characterized. The activity of mtGPDH is inhibited by acyl-CoA esters and free fatty acids (257) and induced by hormones (299). The enzyme is calcium dependent: because its glycerol phosphate-binding site faces the outer surface of the inner membrane, it is exposed to fluctuations in cytoplasmic calcium concentrations (226); the binding site is part of the polypeptide chain of the enzyme, contrary to other calcium-sensitive mitochondrial dehydrogenases in which calcium appears to bind separate subunits (226). A specific activation by short-chain CoQ homologues and by the CoQ analogue idebenone was related to the release of the inhibitory effect of free fatty acids. This competition suggested that the inhibitory effect of free fatty acids is exerted by occupying the CoQ-reducing site of the enzyme, thus preventing transfer of reducing equivalents to the CoQ pool. The mechanism of CoQ reduction by mtGPDH is not well understood; progress in this direction has derived from studies of ROS production by the enzyme (section III.E). Glycerol-3-phosphate dehydrogenase appears to interact directly with the CoQ pool, therefore not forming a supramolecular aggregate with complex III, as suggested by the convex hyperbolic curve of inhibition of glycerol phosphate cytochrome c reductase by myxothiazol, indicating the existence of a mobile intermediate between mtGPDH and complex III (258).
3. ETF-ubiquinone oxidoreductase
The electron-transfer flavoprotein (ETF)-ubiquinone oxidoreductase (EC 1.5.5.1) is a globular protein located on the matrix surface of the inner mitochondrial membrane. The enzyme can accept reducing equivalents from a variety of dehydrogenases (12), including those involved in fatty acid oxidation, in amino acid oxidation, and in choline catabolism (dimethylglycine dehydrogenase and sarcosine dehydrogenase), and is oxidized by ubiquinone.
Crystal structures of the enzyme (338) indicate that the molecule forms a single structural domain where three closely packed functional regions bind FAD, the 4Fe4S cluster, and ubiquinone. The ubiquinone molecule penetrates deep into its binding pocket, which consists mainly of hydrophobic residues. Only five units of the 10 isoprenes in the flexible tail of CoQ could be seen in the structure of the ubiquinone-containing protein (338). Studies of site-directed mutagenesis in Rhodobacter sphaeroides indicated that FAD is involved in electron transfer to ubiquinone but not in electron transfer from ETF, demonstrating that the iron–sulfur cluster is the immediate acceptor from ETF.
4. Choline dehydrogenase
Choline dehydrogenase (EC 1.1.99.1) catalyzes the oxidation of choline to betaine aldehyde. The enzyme is localized at the matrix side of the inner mitochondrial membrane; because its oxidation through the respiratory chain was shown to yield a P/O ratio approaching 2, it was suggested that it feeds electrons to CoQ at a similar position to that of respiratory complex II (134).
The enzyme contains FAD and an iron–sulfur cluster. The sequence predicted by computer analysis from the rat liver gene sequence in the NCBI database was confirmed by cloning a full-length cDNA; expression of the recombinant gene in S. cerevisiae led to enrichment of the active target protein in the inner mitochondrial membrane (134).
5. Dihydroorotate dehydrogenase
Dihydroorotate dehydrogenase (DHODH, EC. 1.3.3.1) is an iron-containing 43-kDa flavoprotein (FMN) that catalyzes the oxidation of dihydroorotate to orotate, the fourth step in de novo pyrimidine biosynthesis (76).
Biochemical and microscopic studies (128, 209) showed that the mammalian DHODH and that isolated from Neurospora crassa (class 2 enzymes) are integral membrane proteins localized in the inner mitochondrial membrane (325) with the active site facing the intermembrane space. The enzyme is functionally linked to the electron-transport system of the respiratory chain because it uses ubiquinone as co-substrate electron acceptor (209); thus, it is classified as a dihydroorotate:ubiquinone oxidoreductase. According to the catalytic properties described by Hines and Johnston (127), it seems reasonable that FMN functions as the proximal electron acceptor, experiencing two-electron reduction concomitant with dihydroorotate oxidation. Reduced flavin would then become reoxidized by passing electrons, perhaps one at a time, to a putative iron–sulfur cluster that, in turn, would be exposed to the quinone. Crystallographic studies, however, failed to detect iron–sulfur clusters in the enzyme (325). In situ, the reduced ubiquinone would be expected to equilibrate with the membrane CoQ-pool and to be reoxidized by complex III. By contrast, the rat liver DHODH lacks flavin, contains iron and zinc as the two apparent redox-active cofactors, and, like the cytosolic enzymes isolated from parasitic protozoa (class 1 enzymes) (245), delivers electrons directly to molecular oxygen (89).
The high-resolution crystal structure of human DHODH (189) shows a small domain that forms the opening of a tunnel leading to the bound FMN and that provides access to ubiquinone, whereas it is unlikely that orotate may enter via the same tunnel. Insight into the structure of the enzyme has been useful to design drugs active against protozoan parasites like Plasmodium falciparum and also in human diseases (7).
The role of quinone reductases in mammalian metabolism is depicted schematically in Fig. 6.

6. Alternative NADH dehydrogenases
Alternative NADH dehydrogenases (NDs) designate a family of proteins located in the inner membrane of eukaryotic mitochondria, which catalyze oxidation of NAD(P)H from either the cytosol (external enzymes) or the mitochondrial matrix (internal enzymes) and enable quinone reduction. The greatest functional difference from complex I is that their oxidoreductase activity is rotenone insensitive and is not coupled to proton pumping.
The number and specificity of alternative NADH dehydrogenases vary considerably when comparing different organisms: none was described in humans, whereas plants may have up to four proteins (two in each side of the membrane, but they were not yet conclusively identified), suggesting that they may have organism-specific roles.
Alternative dehydrogenases are present in bacteria as well as in the mitochondria of fungi. In Neurospora crassa mitochondria, the presence of both internal and external rotenone-insensitive alternative NADH dehydrogenases has been reported since the early 1970s (38). In the yeast S. cerevisiae, which lacks complex I, an internal and two external enzymes have been quite well characterized: NDI1, NDE1, and NDE2, respectively (234). Another yeast, Yarrowia lipolytica, contains only one external enzyme in the inner mitochondrial membrane in addition to complex I (147).
Alternative NADH dehydrogenases are encoded by a single nuclear gene and have a mature peptide molecular mass of 50 to 60 kDa. The only prosthetic group is FAD, by contrast with the FMN and multiple FeS centers of complex I. Gene cloning has established that NDA and NDB in potato mitochondria are markedly similar to the yeast NDI1, with sequence identity of ∼30–40% (256). Both NDA and NDB have NADH- and FAD-binding motifs, whereas neither has any indication of membrane-spanning α-helices. Both proteins bind to the inner mitochondrial membrane, but their different targeting leads to locations on opposite sides, as shown in Fig. 7.

The cellular role and need for alternative NADH dehydrogenases remains mostly unclear; it was suggested that they provide the organisms with plasticity to adapt to different environmental conditions. A pivotal question to be elucidated is how these enzymes interact with other mitochondrial dehydrogenases. The alternative NDs and the proton-pumping complex I have overlapping roles in oxidoreductase reactions and, in some cases, it was demonstrated that alternative NDs are not essential proteins, given the fact that mutants are viable (38). Evidence also suggests that alternative NADH dehydrogenases can complement complex I defects in different situations. Disruption of complex I genes in Paracoccus denitrificans was possible only after introduction in the organism of the NDH-2 gene of E. coli (86). Likewise, the segregation of complex I mutants in Y. lipolytica required the previous targeting of its external single alternative NADH dehydrogenase to the matrix face of the inner mitochondrial membrane (147). Moreover, the complementation of complex I defects in mammalian cells with the NDI1 gene of S. cerevisiae is quite amazing and points to a possible strategy for gene therapy in human mitochondrial diseases (333, 247).
In the yeast S. cerevisiae, NDE1 and NDE2 were described to be associated in a membrane-bound supramolecular complex with both other known intermembrane space- and matrix-facing dehydrogenases (glycerol-3-phosphate dehydrogenase,
Because alternative NADH dehydrogenases do not pump protons, they may be useful to keep reducing equivalents at physiologic levels and to avoid the production of reactive oxygen species associated with complex I. Moreover, data on the characterization of the expression and activity regulation of these enzymes are emerging: alternative components respond to factors ranging from oxidative stress to the stage of fungal development. For instance, their direct involvement in oxidative stress in yeast (55) or in development and light responses in plants (303) was described. Their variability among species is a sign that they accomplish specific requirements of the different organisms.
7. Malate-quinone oxidoreductase
Bacteria possess a malate dehydrogenase (EC 1.1.99.16) catalyzing the oxidation of malate to oxaloacetate by the respiratory chain without using NAD as the intermediate acceptor; the enzyme is a membrane-associated protein containing FAD as a prosthetic group and donates electrons directly to coenzyme Q (214).
The enzyme, however, was found also in mitochondria of some eukaryotes. Genes encoding for a malate quinone oxidoreductase have been detected in the genomes of P. falciparum and P. yoelii; moreover, malate was shown to stimulate rotenone-insensitive respiration and ADP phosphorylation in the parasites Toxoplasma gondii and Plasmodium yoelii (313).
8. Alternative quinol oxidases
Although complex III is the only ubiquinol-oxidizing enzyme in mammalian mitochondria, most plants and some yeasts and fungi posses a cyanide- and antimycin-insensitive alternative oxidase (AOX) that catalyzes the aerobic oxidation of ubiquinol in addition to the cytochrome pathway (155). The enzyme is nonprotonmotive and its activity does not contribute to the conservation of energy that can therefore be dissipated as heat (217). However, the ubiquitous presence of AOX in plants, including nonthermogenic species, suggested a more general physiologic role of the enzyme as an overflow mechanism. It has been predicted that AOX allows Krebs-cycle turnover when the energy state of the cell is high and that it protects against oxidative stress. In transgenic tobacco cells, the antisense suppression of AOX resulted in cells with a significantly higher level of ROS compared with wild-type cells, whereas the overexpression of AOX resulted in cells with lower ROS abundance (202). Conversely, in a long-lived respiration mutant of the fungus Podospora anserina overexpression of AOX enhanced ROS production (190); in an analysis of supercomplex arrangement of the respiratory chain in wild-type and long-lived mutants of P. anserina overexpressing AOX, Krause et al. (159) found two different types of supramolecular organization of complex I and complex III and concluded that it is the supramolecular arrangement of the respiratory chain to dictate the overall properties of the respiratory system (see section VI.A.1).
Despite the difficulty of purifying the enzyme to homogeneity in a stable, active form, recent models considered AOX as a homodimeric interfacial protein, the functional unit being a single polypeptide of around 32 kDa, peripherically associated with the matrix side of the inner mitochondrial membrane (289). Few studies addressed the problem of possible protein–protein-specific interactions of AOX with respiratory chain complexes or supercomplexes (cf. section V.A).
The structure of the active site of the oxidase comprises a nonheme di-iron center that is reduced by two electrons delivered from ubiquinol. Moore and Albury (216) proposed a model for the ubiquinol-binding site in AOX, which identifies a hydrophobic pocket, between helices II and III, leading from a membrane-binding domain to the catalytic domain; this crevice could act as a channel through which the substrate gains entry to the active site (Fig. 8).

A significant engagement of the alternative pathway is not apparent until the reduction level of the CoQ pool reaches 40%; to explain this kinetic characteristic, Siedow and Moore (288) proposed a detailed model, based on CoQ pool behavior, that predicts the changing affinity for oxygen with changes in CoQ-pool reduction. Various regulatory phenomena that affect the amount and activity of the alternative oxidase have been reported in the literature (288). For example, induction of AOX in N. crassa occurs only when mutations or chemicals inhibit the cytochrome pathway (60), and no active AOX is present under normal growth conditions, whereas the conventional and alternative respiratory pathway can operate simultaneously in other fungi (121).
Moreover, not only the amount of alternative oxidase, but also its kinetic characteristics vary with tissue conditions; a clear example is afforded by mitochondria isolated from young and mature spadices of Arum maculatum, in which the plot of AOX activity rate versus Q-pool reduction changes from nonlinear to nearly linear as a function of tissue-growth stage. Biochemical regulation is known to occur at the highly conserved cysteine residue CysI located in the structurally undefined N-terminus (Fig. 8). When the CysI residues of the AOX dimer interact with α-keto acids, perhaps forming a thiohemiacetal, the enzyme becomes activated through a charge-induced conformational change. When this conformational change is prevented, either by oxidation of CysI residues in the native homodimer to form an intermolecular disulfide bond or by substitution of CysI with a hydrophobic amino acid residue, an inactive enzyme results. These regulatory features allow the plant AOX activity to be influenced by intermediates of carbohydrate metabolism and cellular redox state, consistent with its hypothesized functions listed earlier (312).
AOX expression is well tolerated in cultured mammalian cells; cotransforming rho-0 cells with the NADH dehydrogenase of S. cerevisiae and the alternative oxidase of Emericella nidulans, NDI1 and AOX recovered full NADH oxidation without proton pumping (247). Furthermore, the ectopic expression of the alternative oxidase from Ciona intestinalis was able to complement cytochrome oxidase defects in Drosophila (82). These studies highlight the potential use of AOX for gene therapy of respiratory chain deficiencies.
9. Sulfite oxidase
Sulfite oxidase (EC 1.8.3.1) is the only enzyme, besides complex III, to be able to deliver electrons at the level of cytochrome c. The enzyme catalyzes the reduction of cytochrome c by sulfite and is involved in liver in the final steps of degradation of the sulfur-containing amino acids cysteine and methionine and in detoxification of sulfite from environmental sources (142).
Sulfite oxidase is localized in the intermembrane space; it is a homodimer composed of a large molybdenum domain linked to a small heme b domain. The model of ionic interaction between cytochrome c and its reaction partners predicts a cluster of specifically oriented carboxyl groups; such a cluster has indeed been found for other partners but not for sulfite oxidase (263).
A schematic drawing of the respiratory chain complexes and their relation with the inner mitochondrial membrane is represented in Fig. 9.
C. The small connecting molecules (“mobile components”) of the respiratory chain
1. Coenzyme Q
The natural coenzyme Q (CoQ, ubiquinone) is 2,3-dimethoxy-5-methyl-6-polyprenyl-1,4-benzoquinone, in which the polyprenylated side chain is six to 10 units long, depending on the species. Within mammals, only CoQ9 and CoQ10 are found, with CoQ9 distributed only among rodents.
Mitochondria from very few eukaryotes (Tetrahymena, Euglena) have CoQ8, whereas this homologue is present in many bacteria including E. coli (255); plant mitochondria may have either CoQ9 or CoQ10. Saccharomyces cerevisiae, among other peculiarities, has CoQ6 as the only ubiquinone species.
Parasite mitochondria may contain redox-active quinones not present in mitochondria from other animals. The reduction of fumarate to succinate, representing an adaptation to anaerobic conditions, is the opposite reaction to succinate oxidation catalyzed by complex II; prokaryotes contain two distinct enzymes and two different quinones, menaquinone and ubiquinone, for fumarate reductase and succinate CoQ reductase. Likewise, mitochondria from parasitic helminths and some marine organisms adapted to low oxygen tension also use two different quinones, rhodoquinone (in which an amino group substitutes the methoxy group in the 3-position) and ubiquinone, for fumarate reduction and succinate oxidation, respectively (315). In the widely investigated nematode Caenorhabditis elegans, not a parasite, in addition to CoQ8 taken up from the diet consisting of E. coli bacteria, contains as a major species both ubiquinone-9 and rhodoquinone-9 (306). The organic structural specificity of CoQ homologues and analogues was investigated in beef heart mitochondria after pentane extraction and reconstitution. As widely discussed by Lenaz (175), the homologue specificity based on the number of isoprenoid units in the 6-position is critical for the reduction of the quinone ring in the active pocket in complex I.
Because of its extreme hydrophobicity, natural CoQ can be present in three physical states only: forming micellar aggregates, dissolved in lipid bilayers, and bound to proteins. The former state is very important working with CoQ in cell-free systems (77); however, in the living cell, CoQ is distributed among the other two states.
The extent to which CoQ is bound to mitochondrial proteins is an important parameter in relation to its function. If we consider bound CoQ in a 1:1 stoichiometry with the complexes interacting with the quinone (CI, CII, CIII), in beef heart mitochondria, we come up to ∼0.5 nmol/mg protein, that would increase to ∼0.8 nmol, assuming more than one site to be fully occupied in complex I and complex III. Because the total CoQ content is higher than 3 nmol/mg (37, 80), we must assume that most CoQ (more than 75%) is free in the bilayer. A direct study (166) of the amount of CoQ bound to mitochondrial proteins in five different mammalian species showed that the protein-bound aliquot ranges between 10 and 32% of total CoQ.
It has been assumed for long time that the shape of the CoQ molecule is linear, with some possibility of rotation allowed for the long isoprenoid tail. Bending of the molecule is required in a model proposed by us (174), on the basis of previous evidence and of theoretic considerations, and confirmed by linear dichroism studies (274) of the location of CoQ10 in the hydrophobic midplane of the lipid bilayer, with the polar head oscillating about the third isoprene unit between the midplane (wholly linear shape) and the polar heads of the phospholipids (maximal bending of 90 degrees).
Contrary to these predictions, a computer-simulation study of the molecular dynamics of CoQ homologues in the vacuum starting from different initial configurations showed that the conformation with the lowest energy level is a folded one, in which the polar head is in tight contact with the last isoprenoid unit of the hydrophobic tail (61). Within the series of homologues, the cut-off for the folded conformation is four isoprenoid units.
Important implications of a folded structure exist. First, the similar size of short and long homologues would explain the similar high rates of lateral diffusion for all quinone homologues (61, 77). In addition, protein binding during electron transfer may require unfolding, contributing to the high activation energy and low collision efficiency observed for electron transfer (e.g., 80).
The cyclohexane/water partition coefficients of different quinones are good parameters of their hydrophobicities and are known from the literature (260). The membrane/water partition coefficients of CoQ1 and pentyl-ubiquinone (PB), determined by fluorescence quenching, agree with the cyclohexane/water corresponding values, but more hydrophobic quinones are underevaluated because their partition from water to the membrane competes with their micellization in water (77). An additional consequence of the high hydrophobicity of ubiquinones, related to their partition coefficients, is their extent of solubility in monomeric state (77); only quinones with very short chains (as CoQ1 or PB) are monomeric in the concentration ranges used in complex I assays, whereas CoQ2 and decyl-ubiquinone (DB) form micelles at or below micromolar concentrations in the assay medium. If the micelle-to-monomer transition is rate limiting with respect to the enzymatic kinetic steps, then any rate determination would become meaningless.
Water insolubility is a particularly serious phenomenon for oxidized quinones, as in complex I activity determination, because the hydroquinone forms used in complex III activity determination are significantly less hydrophobic (58).
The lateral diffusion of quinones in lipid bilayers has received particular attention in relation to their role in the electron-transfer processes in the mitochondrial respiratory chain; according to the “random collision model” of the electron transfer proposed by Hackenbrock et al. (114), all components of the mitochondrial respiratory chain are randomly distributed in the plane of the membrane and undergo independent lateral diffusion. The mobility of the smaller components, such as coenzyme Q (CoQ) and cytochrome c, is faster than that of the macromolecular complexes and assures electron transfer by random collisions with the latter. In addition, Hackenbrock and co-workers (114) suggested that CoQ diffusion in the mitochondrial membrane is the rate-limiting step in the whole electron-transfer process.
We discuss in the following sections that the random collision model is, at least in part, invalidated by the findings of direct electron channeling between complexes I and III. For the fraction of CoQ that is mobile in the mitochondrial inner membrane and is required for electron transfer between other complexes, it is likely that high diffusion rates make CoQ diffusion not rate limiting for electron transfer, as amply discussed in previous publications (174, 179).
2. Cytochrome c
Cytochrome c is a water-soluble ∼12-kDa heme-containing protein, encoded by the nuclear genome, that first forms as apocytochrome c in the cytosol. The basic assumption that cytochrome c binds to the mitochondrial inner membrane through electrostatic attraction to the phospholipid head groups has been challenged by experimental findings that demonstrate the presence of hydrophobic interactions between cytochrome c and phospholipid acyl chains that extend outward from the lipid bilayer (146). A fraction of total cytochrome c (10%) persists as membrane-bound molecules, even after treatment of mitochondria with digitonin (a mild nonionic detergent) (50), thus supporting the idea of a spatial and functional repartition of cytochrome c in the IMS between (a) a soluble, loosely bound pool that is sensitive to electrostatic alterations, such as ionic strength and surface charge density; and (b) a pool that binds more strongly to the inner membrane and, possibly, is in closer contact with the complexes of the respiratory chain. It is well known that cardiolipin is functionally relevant for the energy-transduction process. It may increase the surface concentration of cytochrome c close to the respiratory complexes so that the binding of cytochrome c to cytochrome oxidase may be facilitated (1); moreover, it modulates the catalytic activity of the major proteins of the mitochondrial OXPHOS apparatus (68, 93, 104, 264). In addition, cardiolipin was found to be required for the organization of the respiratory chain into supramolecular assemblies (340). A mechanistic model for the lipid anchorage of cytochrome c to cardiolipin-containing membranes was proposed by Kalanxhi and Wallace (146), who identified a crevice in the protein structure as a route of entry for one pivoting acyl chain of cardiolipin. The same authors indicated the electrostatic association of cytochrome c with the membrane surface as a necessary prerequisite step (146).
In the late 1980s, the modes and rates of cytochrome c diffusion were extensively investigated in both purified inner membranes and intact mitochondria, showing that the highest rate of diffusion is measured at physiologic ionic strength (100 to 150 mM), where the diffusion mode is three-dimensional and cytochrome c has the lowest affinity (concentration near the surface) for the inner membrane while it mediates the highest rate of electron transport through maximum collision efficiency with its redox partners, complex III and complex IV (110). Although largely debated, the existence of different physical pools of cytochrome c is consistent with the demonstration of a physical sub-compartmentalization of the mitochondrial interior and supports the notion of a “molecular reservoir” that could influence the modalities of respiratory-chain substrate use at different energy states, as suggested by Benard et al. (17).
II. Regulation of the Mitochondrial Respiratory Chain
Until recently, the only form of control of mitochondrial respiration was considered to be that exerted by the thermodynamic pressure of ΔμH +, created by the proton-translocating complexes, equilibrating with the transport of electrons in the respiratory chain.
Although the thermodynamic control exerted in vivo by the ATP/ADP ratios and by moderate uncoupling has received great attention in the balance of energy expenditure (261), also in relation to the generation of ROS, the kinetic control exerted by substrates and substrate-like molecules has received less attention until recently. In particular, the very low K m for O2 of cytochrome oxidase has represented a reason for considering the O2 concentration as never rate limiting. Contrary to this assumption, however, is the physiological observation that in many tissues, the O2 concentration gradient from the capillaries and the vessel endothelia to the mitochondria of parenchymal cells may be so steep as to make the O2 concentration at the site of use in the micromolar range, close to the K m of cytochrome oxidase for O2 (305). In addition, nitric oxide (NO) is a physiologic effector behaving both as a competitive inhibitor of cytochrome oxidase with respect to O2, decreasing the apparent affinity of the enzyme for oxygen, and as a substrate that is oxidized to nitrite, thus behaving as an uncompetitive inhibitor (48).
It is reasonable to consider that oxygen concentration in many tissues at the level of mitochondria may be such as to contribute to the effective rate of respiration: this may be particularly important under pathologic conditions paradoxically elevating the risk of generation of ROS (see section III).
A. Rate-limiting steps: flux-control analysis
The possibility of control of respiration at the level of individual enzymes has received attention from the clarification of the rate-limiting steps by metabolic flux-control analysis (MCA). The discovery that control is exerted at different levels of the OXPHOS apparatus subsequently prompted the possibility that other forms of regulation exist, such as allosteric and covalent control.
MCA predicts that if a metabolic pathway is composed of distinct enzymes freely diffusible in a dynamic organization, the extent to which each enzyme is rate controlling may be different, and the sum of all the flux-control coefficients for the different enzymes should be equal to unity (145, 218).
The flux-control coefficient (Ci) of a step in a metabolic pathway is defined as the fractional change in the global flux through the pathway induced by a fractional change in the enzyme under consideration, and it can be expressed in mathematical terms (145) as the ratio between the change over the metabolic flux rate (dJ/dI)I→0 and the corresponding infinitesimally small change of enzyme activity (dvi/dI)I→0 induced by a specific inhibitor. The case of a tight metabolic control is described by a flux-control coefficient approaching unity, whereas the low Ci value associated with a non–rate-limiting step indicates the characteristic phenomenon known as “biochemical threshold effect,” by which the decrease in the single enzyme activity has to exceed a critical value before a decrease in the global flux can be observed.
Flux-control analysis in intact mitochondria under phosphorylating or uncoupled conditions usually exhibits low flux-control coefficients for respiratory complexes in mitochondria isolated from various tissues (cf. 54, 136, 219, 267, 319) because the control is distributed among other components, besides the respiratory complexes, such as the adenine nucleotide carrier, the ATP synthase, and presumably the substrate carriers and the NAD-linked dehydrogenases.
Few studies have dealt with flux control analysis of respiration in intact cells: Villani and Attardi (320) measured, in human cell lines, the control of respiration by cytochrome oxidase and found a smaller reserve capacity than expected from studies in isolated mitochondria, indicating that the enzyme activity is only in slight excess for supporting maximal respiration. Likewise, in saponin-permeabilized muscle fibers, the flux control was shared by different OXPHOS enzymes; however, the flux control by cytochrome oxidase was higher than expected and was ascribed to the low oxygen tension occurring in the fiber lattice (329). Indeed, it should be noted that not all tissues appear to behave in the same way: Kudin et al. (162) found that the flux-control coefficient of cytochrome oxidase was lower and the excess capacity higher in digitonin-treated parahippocampal homogenates than in saponin-treated muscle fibers. Moreover, the distribution of the control over mitochondrial respiration depends closely on the steady state under consideration, and limitations of the flux-control analysis could be related to the type of respiratory substrate; therefore, it might be questioned whether the interpretations should be restricted in the limited experimental conditions rather than being extended to physiologic conditions. For instance, the measurement of the control by COX in living cells could be modified by the combined use of complex I– and complex II–dependant endogenous substrates or by the use of uncouplers in the assay. Moreover, limitations due to the use of cancer cell lines, which may possess abnormal bioenergetic properties, should be stated.
Because metabolic flux-control analysis has been used in our laboratory to differentiate between randomly distributed respiratory complexes and stoichiometric supercomplexes, we return to this issue in a further section (V.C.1.).
B. Covalent modification: subunit phosphorylation
It is now established that some of the mitochondrial complexes are subjected to reversible phosphorylation and dephosphorylation; mitochondria contain protein kinases and phosphatases, and phosphorylated proteins are found in mitochondria. Both serine/threonine phosphorylation and tyrosine phosphorylation of mitochondrial proteins occur and are important in regulation of activity of these organelles.
1. Phosphorylation of complex I
Early evidences on the phosphorylation of complex I subunits showed cAMP-dependent phosphorylation of 18- and 42-kDa proteins associated with complex I from bovine heart mitochondria. Newly developed procedures based on BN-PAGE, and complemented by in-gel digestion, phosphopeptide enrichment by titanium dioxide (TiO2), and phosphopeptide-directed triple MS analysis, besides confirming serine phosphorylation of the 42-kDa ESSS and B14.5a subunits, also revealed threonine phosphorylation of the B14.5b (human gene NDUFC2) and B16.6 (GRIM-19) subunits of bovine complex I (43, 238). The mass spectrometric analyses detected both the phosphorylated and nonphosphorylated peptides from these subunits, indicating that they may undergo a dynamic condition of phosphorylation/dephosphorylation.
Investigation of mammalian and human cell cultures has provided evidence that modulation of subunit phosphorylation by intramitochondrial protein kinase A and phosphoprotein phosphatase contributes to the stability of complex I and regulates its functional activity (59). Moreover, activation of the cAMP-dependent protein kinase prevents the formation of oxygen free radicals in mitochondria by affecting the ROS-generating capacity of complex I (244, 254).
Direct evidence of the potential effects of phosphorylation of the MWFE and ESSS subunits on complex I activity and assembly was recently reported by Yadava et al. (332) by mutational analysis of the phosphorylation sites in two Chinese hamster respiratory-deficient cell lines showing, respectively, null mutations in the NDUFA1 and NDUFB11 genes encoding the MWFE and ESSS subunits, respectively: complementation with mutant cDNAs appeared to cause low levels of mature protein or a complete failure for complex I to assemble. These observations strongly support the rationale that electron transport and oxidative phosphorylation might be regulated by a mechanism involving the covalent modification by phosphorylation of the respiratory complexes and clearly indicate that if phosphorylation occurs in vivo, the effects on complex I activity are significant.
2. Phosphorylation of complex IV
Lee et al. (170) examined the cAMP-dependent phosphorylation of mitochondrial complex IV isolated from fresh bovine liver and heart in the presence of theophylline, a phosphodiesterase inhibitor that induces high cellular cAMP levels. Under the conditions applied, they were able to identify Tyr-304 of subunit I (COX-I) as the target site of phosphorylation and to demonstrate that such phosphorylation leads to strong decrease of Vmax in the isolated enzyme while decreasing the K m for cytochrome c. In particular, the phosphorylated enzyme shows sigmoidal behavior such that up to 12 μM cytochrome c (i.e., in the range of physiologic content for heart muscle mitochondria) the activity is less than 20%, compared with saturating substrate concentrations, both in normal and in ADP-stimulated preparations. The same authors (334) indicated that cytochrome c is also targeted for phosphorylation in vivo and that it produces enhanced sigmoidal kinetics with cytochrome c oxidase. Other treatments that would increase phosphorylation (e.g., elevation of cAMP levels by glucagon addition or by forskolin activation of adenylyl cyclase) were tested in a cell-culture system and found to have the same effect on cytochrome oxidase kinetics as predicted by the theophylline results: Complex IV is switched off when it is tyrosine-phosphorylated in the catalytic subunit I.
Phosphorylation at COX-I appears to be rapidly lost during storage at −80°C or during freezing and thawing of the bovine heart, or both, and is obtained only under certain isolation conditions of the enzyme, including the use of protein phosphatase inhibitors in the medium, whereas more-stable phosphorylation sites are detectable at tyrosine, serine, and threonine in subunits II and III, as well as at specific amino acid residues in subunits IV, Vab, VIabc, VIIabc, and VIII (120). However, in the latter cases, the identified phosphorylated amino acids are located at the matrix side of complex IV and are apparently not related to the allosteric kinetics of the enzyme in the presence of ADP and ATP, which appears to involve phosphorylation of subunit I. Their function could be to change the binding affinity of complex IV to specific proteins [i.e., EGFR-pY845, the viral protein HBx, PKCe, NO synthase, subunit RIa of PKA, the androgen receptor; for a review, see Vogt et al. (323)]; it seems worthwhile to test whether phosphorylation at those sites may also modify supercomplex formation by cytochrome oxidase.
C. Regulation by mitochondrial dynamics
In living cells, mitochondria undergo continuous shape transformations, including isovolumetric reshaping, fission/fusion rearrangements, and contraction–swelling volume changes. The transmembrane potential brings about specific contributions to the bulk mechanical parameters (i.e., lateral tension, bending-rigidity constant, spontaneous curvature) that affect the structure and the elastic properties of the membrane. Interestingly, the theory's prediction by Chvanov (45) of a transient contraction and folding of the inner mitochondrial membrane during a state 4 to state 3 transition agrees well with experimental reports in which the increase in the ADP concentration promotes matrix folding, so that a decrease in the diameters of the cristae junctions will reduce the availability of ADP (197). In addition, it was proposed that metabolic-driven variation of such mechanical properties of the inner mitochondrial membrane can promote membrane remodeling between the principal geometric shapes and serve as a negative feedback in control of the oxidative phosphorylation (15, 45). In their analysis of the cross-talk between mitochondrial bioenergetics and organelle-network organization, Benard and colleagues (15) demonstrated a strong reduction in the endogenous rate of coupled respiration, in association with a strong inhibition of mitochondrial energy production, in DRP1-depleted HeLa cells in which the mitochondrial network is deprived of the ability to fragment and results in abnormal connectivities. These observations about the possible regulation of the respiratory-chain activity by mitochondrial dynamics that triggers subsequent changes in membrane physicochemical properties might also contribute to explaining the etiology of several diseases in which mitochondrial fusion or fission is altered.
III. The Respiratory Chain as a Source of Reactive Oxygen Species
A. General features
The cumulative term reactive oxygen species (ROS) includes all the compounds arising from partial (fewer than four electrons) reduction of oxygen. In most cellular sources, including the respiratory chain, the initial product is the anionic superoxide radical, although many enzymes directly generate hydrogen peroxide by bivalent oxygen reduction.
Within a cell, mitochondria largely contribute to the production of ROS via the respiratory chain (176); however, some flavin enzymes such as monoamine oxidase in the outer membrane and dihydrolipoamide dehydrogenase in the pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes in the matrix also may represent abundant sources of ROS under certain conditions.
The major sites of superoxide formation within the respiratory chain are linked to respiratory complexes I and III. Further sites, however, may have importance and physiologic relevance.
B. Superoxide generation by complex I
Besides its well-known redox role in the electron-transport chain, complex I is considered one of the main sites of production of ROS: early experiments already proved its involvement in the production of lipid peroxides (307) through the initial production of the superoxide anion (310); more recent studies confirmed that electron leak at complex I can release single electrons to oxygen and give rise to the superoxide anion in several types of mitochondria.
The superoxide production by complex I is higher during the reverse electron transport from succinate to NAD+(141), whereas during the forward electron transport, it is much lower. Reverse electron transfer–supported superoxide production requires high membrane potential and is inhibited by uncouplers and by processes dissipating membrane potential. Rotenone enhances ROS formation during forward electron transfer (101, 124) and inhibits it during reverse electron transfer (230).
The identification of the oxygen-reducing site has been the subject of extensive investigation, and several prosthetic groups in the enzyme have been suggested to be the direct reductants of oxygen. These include FMN, ubisemiquinone, and iron–sulfur cluster N2 (178, 180). In isolated complex I, FMN is considered the major electron donor to oxygen to form superoxide anion (72, 95) also mediated by short-chain hydrophilic quinones (150). In mitochondrial membranes, however, most studies identify the quinone-binding site as the site of oxygen reduction (101, 124, 230). A possible explanation is that two sites for oxygen reduction exist in the complex, represented by both flavin and an iron–sulfur cluster; the latter site would be predominant in membrane particles, whereas the former might be available after complex I isolation. Hirst et al. (129) admit the possible presence of two oxygen-reacting sites at the two ends of the cofactor chain, ascribing the distal one to superoxide generation during reverse electron transfer. We see in section VI.A.3. a possible different interpretation involving the supramolecular assembly of complex I.
The electron donor to the first molecule of bound ubiquinone in the complex is most probably FeS cluster N2. It is proposed (78) that this center is also the electron donor to oxygen, both directly and via one-electron reduction of several exogenous quinones (cf. Fig. 2 for the proposed mechanism). Studies in CoQ-depleted and reconstituted mitochondria indicated that endogenous CoQ is not required for superoxide generation (101).
Complex I also is involved in redox cycling of redox-active compounds (i.e., cocaine, other abused drugs, catecholamines, and several other compounds) that, through interference with physiologic electron-transfer reactions, regenerates the parent compound and releases superoxide. A typical example is doxorubicin (Adriamycin), the anticancer agent that is endowed with severe cardiotoxicity; the toxic effect is distinct from the anticancer mechanism and involves ROS formation, as is also suggested by the protective effect of overexpressing antioxidant enzymes in transgenic animals (302).
The mechanism of redox cycling involves an initial reduction to a semiquinone radical by a one-electron transfer, and a subsequent reaction with oxygen, releasing superoxide and regenerating doxorubicin (273). The mechanism of cardiotoxicity, however, implies long-term exposure, and is ascribed to secondary damage induced by ROS to complex I and other mitochondrial complexes, as well as to mitochondrial DNA, leading to permanent loss of the OXPHOS machinery. The activation pathway followed by carcinogenic polycyclic aromatic hydrocarbons may involve catalytic one-electron redox cycling, through reduction by either microsomal cytochrome P450 and cytochrome b5 reductase or mitochondrial complex I and subsequent autooxidation, establishing futile redox cycles in which superoxide generation is amplified multiple times (191).
A similar mechanism has been suggested for benzene: initial metabolism occurs in the liver by cytochrome P450, with resulting formation, among other compounds, of benzene dihydrodiol, which is oxidized to cathecol and then to benzoquinone (287), which undergoes redox cycling with superoxide production. For this purpose, p-benzoquinone derivatives, such as short-chain CoQ homologues and analogues, undergo redox cycling with oxygen at the level of mitochondrial complex I (79).
Moreover, adrenaline may undergo oxidation and cyclization to adrenochrome in a multistep process in which the main oxidant under physiologic conditions is the superoxide anion (24); conversely, adrenochrome can be reduced to the corresponding semiquinone by NADPH in liver microsomes and by mitochondrial complex I in bovine heart; a redox cycle is then established in which the semiquinone reacts with O2, producing superoxide and regenerating adrenochrome (97). Because adrenochrome reduction to the semiquinone is totally insensitive to either rotenone (which acts at the level of FeS center N2) or p-hydroxymercuribenzoate (which inhibits at the start of the iron–sulfur chain), the site of electron delivery to adrenochrome is presumably FMN. Similar events also may occur with other catecholamines, such as dopamine and nor-adrenaline.
C. Superoxide generation by complex III
The formation of superoxide in complex III depends on this peculiar mechanism of electron transfer. Because the electron transfer from cytochrome bL to bH occurs against the electrical gradient (from the positive to the negative side, cf. Fig. 3), it is strongly retarded when the electrochemical potential is high, as in the controlled state of mitochondrial respiration (state 4); this retardation prolongs the lifetime of Qo and allows reaction of the semiquinone with O2, forming superoxide (141).
Antimycin A (AA) is known not to inhibit completely the electron flow from ubiquinol to cytochrome c: according to the Q-cycle, AA blocks ubiquinone reduction by cytochrome bH at center i, at the inner or negative side of the membrane (cf. Fig. 3), thus enhancing the production of O2 ·− that mediates the reduction of cytochrome c. Because the antimycin-stimulated production of ROS is inhibited by the inhibitors acting at center o (at the outer or positive side), we may locate the site of one-electron reduction of oxygen in presence of antimycin at a component located at center o, presumably ubisemiquinone (39).
Muller et al. (221) suggested that oxidation of ubiquinol at center o is biphasic, with delivery of the first electron to the Rieske iron–sulfur cluster, leaving a semiquinone that, in the absence of further oxidation by cytochrome bL , would interact with oxygen, forming superoxide.
Ubisemiquinone is relatively stable only when protein bound; therefore, the coenzyme Q (CoQ) pool in the lipid bilayer should be no source of superoxide. Exogenously administered CoQ has not been found to exert prooxidant effects in vivo: thus, the prooxidant species deriving from its antioxidant action (228) would not seem to be operative in in vivo supplementation.
D. Superoxide generation by complex II
The indirect evidence that often superoxide production is higher when electrons flow through complex II rather than through complex I, in both cases reaching complex III, is in line with the idea that complex II may be a source of superoxide (204).
Ishii et al. (137), in a study on Caenorhabditis elegans, produced evidence that a mutation in succinate dehydrogenase cytochrome b induces oxidative stress and aging. In E. coli, fumarate reductase (QFR), active under anaerobic conditions, is structurally analogous to complex II (succinate-CoQ reductase, SQR), active under aerobic conditions; in contrast with complex II, however, E. coli QFR has no b heme. Significantly, E. coli QFR is a potent source of H2O2, whereas SQR is not. The source of electrons to oxygen is fully reduced FAD. The difference between SQR and QFR has been ascribed to the electron-attracting capacity of cytochrome b, due to its high redox potential (132): thus, in the absence of cytochrome b, the electrons would be held preferentially on the flavin, favoring leak to oxygen.
Damage to the tetranuclear FeS cluster of complex II has been suggested to enhance its capacity to produce superoxide, presumably by electron leak from FAD, as shown in brain mitochondria from rotenone-treated rats (239). For this purpose, release of oxaloacetate inhibition by simultaneous pyruvate, glutamate, and malate oxidation enhanced superoxide generation, suggesting that oxaloacetate inhibition may physiologically protect against excessive ROS generation (241).
Direct demonstration of superoxide production by complex II was obtained by Zhang et al. (339) in purified succinate-CoQ reductase and succinate dehydrogenase; the enzymes were found to generate superoxide by autooxidation of flavin; reconstitution of complex II with the bc 1 complex to yield an active succinate-cytochrome c reductase inhibited superoxide formation.
E. Further sites of ROS generation in the respiratory chain
A high rate of ROS production was detected in insect flight muscle mitochondria (26) and in brown adipose tissue mitochondria when glycerol-3-phosphate was used as the respiratory substrate. This suggested that mitochondrial glycerophosphate dehydrogenase (mtGPDH) could be the source of ROS. Drahota et al. (65) demonstrated that mtGPDH in presence of antimycin is a powerful source of hydrogen peroxide, which was strongly stimulated by addition of the one-electron acceptor ferricyanide. The ferricyanide-induced H2O2 production is a specific feature of mtGPDH: it is completely inhibited by mtGPDH inhibitors, and is negligible with NADH or succinate as substrates. It is reasonable that ferricyanide takes up one electron from the enzyme, whereas the second one is used to reduce oxygen. In a study of Drosophila mitochondria (213), ROS production by mtGPDH and the relative contributions of complex I by reverse electron transfer, of center o of complex III, and of mtGPDH, were assessed by use of specific inhibitors, demonstrating that mtGPDH is the major source of superoxide in that system.
In a study on the topology of ROS production by the respiratory chain, St. Pierre et al. (296) observed a copious production of H2O2 by rat muscle and heart, but not liver, mitochondria when oxidizing palmitoyl carnitine. Because the rate was only slightly enhanced by exogenous superoxide dismutase, it was suggested that superoxide production occurred at the matrix site, therefore, it was generated by a site different from center o of complex III. The authors considered it likely that the flavoproteins ETF and ETF dehydrogenase, involved in fatty acid oxidation, were the sites for generation of superoxide.
Dihydroorotate dehydrogenase was found to be involved in the production of superoxide in liver mitochondria (89) and in malarial parasite cells (161). It is worth noting that the major domain of the enzyme, carrying the carboxyl terminal, protrudes into the intermembrane space, so that it is likely that superoxide is released in this space. Recently, an additional source of ROS in mitochondria (directly in the form of hydrogen peroxide) was demonstrated in the p66Shc protein (208). P66Shc is a splice variant of p46Shc/p52Shc, two cytoplasmic proteins involved in signal transduction from tyrosine kinases to Ras; p66Shc has the same modular structure of the former proteins but contains a unique N-terminal region and is not involved in Ras regulation. Its function has been discovered to be the regulation of ROS metabolism and apoptosis (207); expression of the protein is required for mitochondrial depolarization and release of cytochrome c after a variety of proapoptotic signals. P66Shc-/- cells are resistant to apoptosis. P66Shc deletion in mice decreases the incidence of aging-associated diseases (268) and prolongs the life span of animals (207). A fraction of p66Shc has a mitochondrial localization in the intermembrane space, where it is bound in an inactive form in a high-molecular-weight complex including the TIM/TOM protein import system (102); proapoptotic signals dissociate p66Shc from the complex and activate it to a form that induces the permeability transition by opening a high-conductance channel in the inner membrane, the permeability transition pore involved in the events leading to apoptosis. This effect is due to the intrinsic property of p66Shc to act as a redox protein, accepting electrons from cytochrome c and directly producing hydrogen peroxide (102). Because the reaction equilibrium of cytochrome c oxidation by p66Shc is low (K eq = 0.1), the reaction is thermodynamically favored when the level of reduced cytochrome c is high (102). Therefore, H2O2 production by this mechanism should be enhanced when cytochrome c oxidase is inhibited. Low oxygen tensions promote the activation of hypoxia-inducible factor (HIF) by a still-controversial mechanism (308) that triggers a series of metabolic changes, among which is the alteration of cytochrome oxidase subunits and activity. In this condition, the activation of the p66Shc pathway may in part explain the paradoxic enhancement of ROS production during hypoxia (112). Another factor leading to decrease of cytochrome oxidase activity is nitric oxide, NO•, which inhibits the enzyme competitively with oxygen (275).
F. Modulation of mitochondrial superoxide production
Most of superoxide is generated at the matrix side of the inner membrane, as appears from the observation that superoxide is detected in submitochondrial particles (SMPs) that are inside-out with respect to mitochondria. A study with suitable spin traps, however, demonstrated the formation of superoxide radical in mitoplasts, indicating that a significant aliquot of this species also is released at the outer face of the inner membrane (296). It is likely that complex I releases superoxide in the matrix, whereas complex III and glycerol phosphate dehydrogenase release it both in the matrix and in the intermembrane space (212).
Are free radicals produced by mitochondria physiologically released to the cytosol? Staniek and Nohl (293) applied a noninvasive detecting system for hydrogen peroxide and found that isolated intact rat heart mitochondria do not produce detectable H2O2, unless when succinate is used in the presence of antimycin. Korshunov et al. (154) also found no hydrogen peroxide formation by intact rat heart mitochondria, unless they were pretreated in such a way as to deplete them of endogenous antioxidants. It may be inferred that, under normal conditions, ROS are not exported out of mitochondria. Overwhelming evidence indicates that ROS production detected in different cells under pathologic conditions has a mitochondrial origin. The superoxide anion released at the intermembrane space may be exported to the cytoplasm through an anion channel related to VDAC (115). Mitochondrial superoxide production is enhanced in state 4 and when the rate of electron transfer is reduced (291). The rationale is in a more reduced state of the respiratory carriers capable of donating electrons to oxygen. For this purpose, uncoupling and release of excessive membrane proton potential may protect mitochondria from damage due to excessive free radical production. In rat hepatocytes, the futile cycle of proton pumping and proton leak may be responsible for 20 to 25% of respiration (30); in perfused rat muscle, the value is even greater. Uncoupling may be obtained by activating proton leak through uncoupling proteins (39). In such a way, a tissue may dissipate a conspicuous part of the energy conserved by its mitochondria; however, it keeps the mitochondrial respiratory chain under more-oxidized conditions, preventing the formation of damaging free radicals. The superoxide production by the respiratory-chain complexes may be under physiologic control; this is particularly evident for complex I. Events leading to a decrease of the rate of electron flow in the complex also lead to overproduction of superoxide. Physiologic states that inhibit complex I activity, such as subunit phosphorylation, may modify its superoxide-generating capacity (254). It is, therefore, tempting to speculate that endocrine alterations may affect the capacity of superoxide formation by hyper- or hypophosphorylation of the complex.
Increasing evidence suggests that pathologic states in which complex I activity is impaired also lead to superoxide overproduction; in cell lines from patients with complex I deficiency, an inverse relation was found between superoxide production and residual enzyme activity (79). This observation also is relevant to more-common pathologies, such as Parkinson disease; in a rotenone model of the disease, an increased superoxide production was ascribed to complex I but also substantial appearance of superoxide generation by complex II (239).
Mitochondria can respond to elevated ROS concentrations, originating from either endogenous or exogenous sources, by increasing their own ROS generation (ROS-induced ROS release) (29, 342). This phenomenon, having high relevance to the effects of ischemia/reperfusion injury, can occur in two modes. The first one involves ROS-induced opening of the mitochondrial permeability transition pore followed by a wave of ROS production by a hitherto not clearly understood mechanism; the second mode involves ROS-induced opening of the inner membrane anion channel, also followed by a short wave of ROS production. The secondary ROS production in both modes originates from the respiratory chain. The secondary release of ROS may be transmitted to neighboring mitochondria, thus establishing a wave of damage extension in the myocardium.
IV. Biogenesis and Assembly of Respiratory Complexes
The biogenesis of respiratory complexes is an intricate process that requires the synthesis of different subunits, their assembly, the incorporation of various types of metal or organic cofactors, and the anchoring of the complex to the inner mitochondrial membrane.
Yet little is known regarding the kinetics of mitochondrial complex assembly during embryonic and fetal life. It has been found that the fetal respiratory-chain complexes are fully assembled and functional at early stages of development in several human organs, although their absolute activity values are lower than those observed in postnatal tissues (210). However, at birth, a spectacular activation of nuclear and mitochondrial gene expression occurs, consistent with the adaptation to life at higher partial oxygen pressures, and is related to the switch from fetal mostly anaerobic glycolysis to neonatal oxidative phosphorylation (242).
The OXPHOS system is unique, as it takes contributions from two physically separated genomes, the nuclear genome (nDNA), and the mitochondrial genome (mtDNA). Steroid and thyroid hormones have been identified as major regulators of the mitochondrial energy capacity (283, 328) which coordinately upregulate nuclear and mitochondrial genes participating in the de novo biosynthesis of some OXPHOS subunits. Hormonal regulation of genes could also rapidly adapt mitochondria to the increased energy needs of the cell, if the redox enzyme components are constitutively available. The molecular mechanism by which the hormones coordinate transcription in nDNA and mtDNA is still under consideration (Fig. 10): the presence of common regulatory sequences for steroid—and other—hormone receptors in nuclear OXPHOS genes and in genes encoding mitochondrial transcription factors, as well as in mitochondria (252, 328), offers the possibility of coordinating OXPHOS gene transcription by primary, direct action of the steroid receptors on both the nuclear and mitochondrial OXPHOS genes, or by parallel induction of the mitochondrial transcription factors affecting secondarily mitochondrial gene transcription, or both.

Nisoli and colleagues (236) recently showed that long-term exposure to low concentrations of NO induces mitochondrial biogenesis in various mammalian cells as well as in animal tissues. This process is mediated by cGMP, resulting from NO-dependent activation of soluble guanylate cyclase, and involves increased expression of peroxisome proliferator–activated receptor γ coactivator 1α (PGC-1α), nuclear respiratory factor 1 (NRF-1), and mitochondrial transcription factor A (Tfam). Similarly, it is accompanied by increased mtDNA content and expression of both COXIV and cytochrome c proteins, as detected with immunoblot analysis. Nisoli and colleagues (237) also demonstrated that the NO/cGMP-dependent mitochondrial biogenesis is associated with enhanced respiration coupled with generation of ATP, thus yielding functionally active mitochondria, in terms of respiratory function and metabolic activity. It was further established that endogenous NO plays a critical role in mitochondrial biogenesis in vivo in animal models (215).
The mitochondrial respiratory complexes contain a number of subunits much larger than their bacterial counterparts; besides the involvement in functional control of the enzymes, the additional protein subunits that are not essential to catalytic activity must play a role in the assembly and stability of the enzyme architecture. Further complexity derives from the observation that individual respiratory complexes agglomerate into gigantic supercomplexes (cf. section V). The understanding of the spatial and temporal dissection of their assembly pathway still represents a major challenge.
Many studies on the yeast Saccharomyces cerevisiae have disclosed the basic mechanisms underlying the processes of assembly of complex II, III, IV, and V and the role of many assembly factors, some of which have orthologues in humans. In the case of complex I, which is lacking in S. cerevisiae, other organisms, such as Neurospora crassa and Yarrowa lipolytica, or mammals like Bos taurus, have served as models for the study of the structure and assembly of this enzyme. Furthermore, the study of mitochondrial disorders in human patients in whom enzyme assembly is compromised by defects in either structural subunits or chaperones has been helping to unravel the mechanistic aspects of respiratory chain biogenesis in mammalian systems (83).
A. The complex I assembly model
All eukaryotic complex Is have a dual genetic origin: seven to no subunits (ND subunits) are encoded in the mitochondrial genome, whereas the remaining subunits are nuclear gene products (cf. section I.A.1). It has been suggested that the high degree of conservation of certain modules of the complex between various organisms may be reflected by similarities in the assembly system. Here we give only a brief survey of the current understanding of complex I biogenesis, referring the reader to a comprehensive review by Vogel et al. (322) for a more-detailed summary on this topic.
Through the years, the assembly process of complex I has been investigated, either indirectly in mutants or directly by using conditional assembly systems, in bacteria, in algae, and in fungi, as well as in higher plants and in mammals (259, 282, 284, 322).
From the analysis of the assembly models proposed for these organisms, a consensus model has been extracted (Fig. 11A) in which an early subcomplex of nuclear DNA-encoded subunits is expanded to form a peripheral arm intermediate that is then anchored to the mitochondrial inner membrane by joining with the membrane arm. Further extension with additional membrane and peripheral subunits leads to the formation of the holoenzyme (322). Particularly in humans, a generalized model scheme based on subunit analysis of the subassembly intermediates in patients with different genetic defects (167) suggests that a 100/200-kDa subassembly of the peripheral arm, containing at least subunits NDUFS2 and NDUFS3, and a membrane-arm scaffold, consisting of at least ND1, come together to form an early 400- to 500-kDa intermediate before completion of each arm of the complex (Fig. 11B).
The involvement of dedicated factors for complex I assembly is documented by some proteins bound to the large assembly intermediates of the complex, but not to the mature enzyme, whose encoding genes are essential for complex I assembly into the active form. Such a role was demonstrated for the two non-subunit proteins, CIA84 and CIA30, in N. crassa (163) and for the homologue chaperone NDUFAF1 in human cell lines (67). Other identified assembly factors in humans are Ecsit, a signaling molecule that interacts with CIA30/NDUFAF1 (322); the apoptosis-inducing factor AIF (314); the chaperone B17.2L/NDUFA12L that, unlike CIA30/NDUFAF1, seems to play a role in the late assembly stages of complex I (229); B14.7, a subunit homologous to translocases that are included in the machinery for importing proteins into mitochondria; and the mitochondrial protein C6ORF66, whose functional significance in complex I biogenesis remains to be investigated, although homozygous mutation in C6ORF66 has been already associated with severe complex I deficiency (271). Based on our understanding of the assembly process of photosynthetic complexes, it is expected that putative complex I assembly factors might function in the delivery of the multiple FeS clusters to their subunits or in the insertion/stability of hydrophobic subunits. Bych and colleagues (36) recently characterized a novel mitochondrial protein called Ind1, belonging to the Mrp-like P loop NTPases, that can bind a [4Fe–4S] cluster that is readily transferred to an acceptor Fe–S protein. Deletion of the IND1 gene in Yarrowia lipolytica results in a very specific defect on respiratory complex I, which is decreased by 80% in amount and activity, whereas the activities of other mitochondrial Fe–S enzymes, including aconitase and succinate dehydrogenase, are not affected. This suggests that Ind1 specifically facilitates the effective assembly of Fe–S cofactors and subunits of complex I.
The fact that complex I is associated with complex III and complex IV into supercomplexes (cf. section V.B) implies that additional levels of control exist for the assembly of this multimeric enzyme. Interestingly, Lazarou and colleagues (167) demonstrated that the chaperone B17.2L associates with an 830-kDa subassembly intermediate of complex I and with a combination of this intermediate with a complex III dimer before complex I assembly is completed (Fig. 11). This supports the hypothesis that supercomplex assembly occurs in conjunction with the formation of the individual redox complexes and correlates with experimental evidence (2, 103) showing that critical amounts of complexes III and IV are required for supercomplexes to form and to provide stability to complex I integrity (cf. section VI.A.2.).
B. The assembly of complex II
In the eukaryotic organisms, with only a few exceptions, all the genes for complex II are single and nuclear encoded (85). The complex also contains FAD, Fe-S centers, and UQ as prosthetic groups (cf. section I.B.1).
Most of the information on complex II biogenesis is available from yeast and other organisms (172), although the investigation of complex II mutations in humans (269) has also provided a contribution to the elucidation of the assembly process for this flavoprotein. Studies in E. coli mutants that are unable to synthesize porphyrins and hemes, or that lack the ability to insert iron into the porphyrin ring, have demonstrated a requirement for heme biosynthesis in the posttranslational assembly process and insertion of a functional SQR into the membrane (227). A possible mechanism for the molecular assembly has been proposed (Fig. 12) in which heme b is bound by axial ligands of SdhD followed by SdhC (i.e., His-71 and His-84, respectively), thus bridging the two subunits in the membrane.

A general consensus indicates that the covalent linkage of FAD to the succinate dehydrogenase flavoprotein (SdhA, Fp) is a self-catalytic process, not mediated by another enzyme, that occurs within the mitochondrial matrix once the imported protein has folded into the proper scaffold, sufficiently to recognize the flavin cofactor (40). FAD attachment is stimulated by, but not dependent on, the presence of the iron–sulfur subunit (SdhB, Ip), and a mitochondrial chaperone (hsp60) has been shown to assist in the flavinylation process in S. cerevisiae mitochondria (265).
Then the soluble catalytic portion binds to the membrane by docking onto SdhC when intact cytochrome b556 containing heme is formed.
Dibrov et al. (63) also identified a 64-kDa membrane-spanning protein (Tcm62p), with 17.3% sequence identity to hsp60 in yeast, which forms a complex with at least three of the Sdh subunits and behaves as a chaperone specifically required for complex II biogenesis.
C. The complex III assembly process
Although mammalian complex III contains an additional subunit (subunit 9) compared with S. cerevisiae, the structural similarity between the yeast and mammalian enzymes has made the former a useful paradigm with which to understand the assembly process of the latter (cf. section I.A.2). Cytochrome b is the only bc1 subunit encoded by mtDNA, whereas all the other nine to ten subunits are of nuclear origin. These latter are synthesized in the cytosol and posttranslationally imported into mitochondria. The model formulated in recent years for the bc1 assembly has been very recently updated on the basis of new data obtained with the two-dimensional analysis of the mitochondrial membranes from yeast-deletion strains. The new tentative model proposed by Zara and colleagues (336) partially overlaps with the previous one in several aspects, but depicts additional steps introducing novel subcomplexes of bc1 as putative intermediates in the biogenesis of the enzyme. In this model (Fig. 11C), the central core, composed of cytochrome b associated with the small supernumerary subunits Qcr7p and Qcr8p, is stabilized in the inner mitochondrial membrane on interaction with core-protein-1 and core-protein-2, but, in addition, the ability of cytochrome c1 to interact with each of the two core proteins emerges as an early step. Zara et al. (336) suggested that this novel interaction, never proposed in the past, may be the molecular basis for the high stability of these three proteins in all the mutant yeast strains tested. Once the central core and the two core proteins plus cytochrome c1 assemble together to form a 500-kDa precomplex, the binding of Qcr6p occurs, and the precomplex is then able to bind Qcr9p, Qcr10p, and RIP1. Incidentally, these observations seem to indicate that dimerization of the complex occurs before full assembly of each monomer. In yeast as in humans, a protein chaperone (i.e., Bcs1p and BCS1L, respectively) intervenes in the final steps by promoting the incorporation of the Rieske Fe-S protein into the nascent complex III dimer (84, 336).
Overall, most details about the mechanism of assembly of the bc 1 complex are still largely unknown, including the precise sequence in which the subcomplexes associate, the addition of the prosthetic groups to the redox subunits, and also, in light of the recent structural models of the complex III–containing supercomplexes in various organisms (c.f. section V.B), the gluing together with other respiratory complexes.
D. The assembly line of complex IV
The assembly pathway of cytochrome c oxidase (Fig. 11D) is characterized by the sequential incorporation of COX subunits around a seed formed by COX1, one of the three mtDNA-encoded subunits (cf. section I.A.3). Analysis of assembly intermediates in defective yeast mutants, as well as in conditioned or deficient human cells, has given insights into the overview of the process and has provided information about nonstructural ancillary factors whose functions, required for all steps of the process and significantly conserved from yeast to humans, have been reviewed elsewhere (125). Indeed, COX biogenesis has received significant attention over the last decade because of its medical relevance (reviewed in refs. 10, 83). Rare disease-related mutations have been described in the mtDNA-encoded subunits, all of them affecting the assembly/stability of complex IV (272), whereas mutations in nuclear-encoded structural subunits were sought but never found in COX-deficient patients, thus raising the conjecture that they are incompatible with extrauterine life. All mendelian disorders with a COX deficiency have been assigned to mutations in COX-specific chaperones (i.e., SURF1, SCO1, and SCO2; COX10 and COX15; LRPPRC) that are involved in the biogenesis but are not part of the mature enzyme (246). To our knowledge, the only exception is a pathogenic mutation of the COX6B1 subunit in two cases of mitochondrial encephalomyopathy and isolated COX deficiency (200).
In this section, we briefly summarize the critical steps of COX biogenesis, while introducing the reader to contributions from other authors for further details on this sophisticated biologic process.
The first step of COX assembly (Fig. 11D) is the insertion of COX1 into the inner mitochondrial membrane (assembly intermediate S1), followed by subunits COX4 and COX5A (assembly intermediate S2). Heme a insertion is likely to occur just after formation of S1 or during the formation of S2 and is accompanied by the insertion of CuB and heme a3 . Maturation of the CuA center is necessary for the addition of COX2 to the S2 intermediate. Then a cascade-like series of events, initiated by the insertion of COX3, the last of the mitochondrially encoded subunits, produces the formation of the monomeric holocomplex (S4) by the incorporation of small subunits that associate with the surface of the complex core. A number of ancillary gene products were identified that are required for incorporation of the heme and copper cofactors into the protein subunits and for maintaining the nascent intermediates in an assembly-competent state (10, 206, 295). Moreover, it should be noted that COX1, COX2, and COX3 are synthesized on mitochondrial ribosomes, and their membrane insertion involves an homooligomeric complex (Oxa1) that is present in the inner mitochondrial membrane, whereas the remaining COX subunits are encoded by the nuclear genome and, on synthesis on cytoplasmic polysomes found in the vicinity of the mitochondrion, they are imported into the mitochondrial matrix through the TOM/TIM machinery (198).
It is generally believed that dimerization of cytochrome oxidase occurs in vivo on completion of the monomeric holocomplex; evidence suggests that the contact between monomers is mediated by COX1 and by some of the nuclear-encoded subunits, COX6A, COX6B, and COX5B (171, 222). Conversely, it is currently not clearly understood which subunits are physically localized at the interface of complex IV within the supramolecular assemblies containing other OXPHOS enzymes. Although ongoing single-particle EM analysis of multiple forms of supercomplexes from bovine, plant, and yeast mitochondria is addressing this important issue, the resolution of the 3D maps is still too low to position complex IV precisely (cf. section V.B). Interestingly, Mick and colleagues (206) showed that when S. cerevisiae mitochondria are solubilized in the mild detergent digitonin, which leaves supercomplexes intact, a significant fraction of Shy1, the yeast assembly factor homologue of human SURF1, interacts with complex III through fully and partially assembled forms of complex IV that have already passed the stage of COX4 incorporation.
V. Structural Organization of the Respiratory Chain
The systematic resolution and reconstitution of four respiratory complexes from mitochondria was accomplished by Hatefi (118), leading Green (107) to postulate that the overall respiratory activity is the result of both intracomplex electron transfer in solid state between redox components having fixed steric relations and, in addition, of intercomplex electron transfer ensured by rapid diffusion of the mobile components acting as co-substrates [i.e., CoQ and cytochrome c (cyt. c)]. This proposal was substantially confirmed over the following years, leading to the postulation by Hackenbrock et al. (114) of the Random Diffusion Model of Electron Transfer. The organization of the respiratory chain has represented a major research subject in the 1970s through the 1980s, culminating with acceptance of the Random Collision Model by the majority of investigators in the field. The fluid mosaic model of membrane structure (290) had already predicted random distribution of integral membrane proteins as a two-dimensional oriented solution in the viscous phospholipid bilayer with high diffusional freedom in the plane of the membrane.
Evidence in favor of the random diffusion model stems from three major kinds of observations: The integral proteins of the inner membrane are randomly distributed in the bilayer, and phospholipid dilution of the mitochondrial membrane proteins slows electron transfer. Electron transfer in the CoQ and cytochrome c region obeys pool behaviour, according to the Kröger-Klingenberg equation developed for CoQ. Electron transfer follows saturation kinetics with respect to CoQ and cytochrome c concentrations.
A critical discussion of the interpretation of these experimental observations can be found in previous reviews (181 –183).
A. Evidence for supercomplex organization
It is worth remembering that the original view derived from the spectrophotometric pioneering studies of Chance and Williams (42) depicted the respiratory chain as a solid-state assembly of flavins and cytochromes in a protein matrix.
Circumstantial evidence against a random distribution of respiratory complexes comes from the early investigations reporting isolation of complex I/complex III (131) and complex II/compIex III units (335), indicating that such units may be preferentially associated in the native membrane.
Nevertheless, not many researchers paid attention to a potential supramolecular organization of the respiratory chain, and available data on the presence of stable supercomplexes of complex III and IV isolated from some bacteria (20, 138, 292) as well as circumstantial evidence produced by Ozawa et al. (235) and by Hochman et al. (131) of the existence of OXPHOS aggregates was usually overlooked by subsequent studies.
Much more recently, new evidence of multicomplex units in yeast and mammalian mitochondria was obtained by introducing a quantitative approach: a mild one-step separation protocol for the isolation of membrane protein complexes, BN-PAGE (53, 278, 281). In particular, BN-PAGE in digitonin-solubilized mitochondria of Saccharomyces cerevisiae, which possesses no complex I, revealed two bands with apparent masses of ∼750 and 1,000 kDa containing the subunits of complexes III and IV, as assigned after two dimensional SDS-PAGE followed by N-terminal protein sequencing (280). The smaller supercomplex (III2IV1) consisted of a complex III dimer and a complex IV monomer while the larger supercomplex (III2IV2) represented a complex III dimer associated with two complex IV monomers. The minimal effect of increasing the digitonin/protein ratio for solubilization of mitochondria seemed to indicate a stable rather than a dynamic association of proteins.
Similar interactions of supercomplexes were investigated in bovine heart mitochondria: complex I to III interactions were apparent from the presence of ∼17% of total complex I in the form of a I1III2 supercomplex that was found further assembled into two major supercomplexes (respirasomes) comprising different copy numbers of complex IV (I1III2IV1 and I1III2IV2 contain 54% and 9% of total complex I, respectively).
Only 14 to 16% of total complex I was found in free form in the presence of digitonin (281), so it seems likely that all complex I is bound to complex III in physiologic conditions (i.e., in the absence of detergents), whereas approximately one third of total complex III remains unbound because of stoichiometric excess. The fraction of complex IV in free form represents more than 85% of total cytochrome oxidase of mitochondria. So far, some putative associations of complex II with other complexes of the OXPHOS system under the conditions of BN-PAGE were described only in mouse liver samples (3). Interestingly enough, complex II in older studies (cf. 113, 335) seemed to be the most attractive candidate for physiologic association with complex III (cf. section VI.A.1 for discussion).
BN-PAGE has become a popular experimental strategy for the structural analysis of the protein-complex composition of the respiratory chain in different systems. Based on this procedure, the existence of respirasome-like supercomplexes also was reported for bacteria (186, 298), fungi (159), and higher plant mitochondria (75, 157), as well as for human mitochondria (279).
In addition to BN-PAGE, the technique of colorless-native-PAGE (CN-Page) has been used for detection of supercomplexes (156): this technique has actually proven to be superior in allowing higher yields of supercomplexes to be detected. The reason is in the limited use of the Coomassie dye, the presence of which may induce the dissociation of the weaker supercomplexes (158, 199, 331).
A major I1III2IV1 supercomplex also was found in the wild type of the filamentous fungus Podospora anserina, although long-lived mutants deficient in complex IV and overexpressing the alternative oxidase AOX had a different arrangement, including a I2III2 supercomplex that was revealed to be a major one by a gentle colorless-native PAGE (159). Interestingly, in those mutants, the I2III2 supercomplex would release electrons from complex I directly to the CoQ pool and then to AOX, whereas complex III would be kinetically inactive; presumably its sole function would be in stabilization of complex I (159) (cf. section VI.A.2). To date, few studies addressed the problem of possible protein-protein–specific interactions of AOX with respiratory chain complexes or supercomplexes. Navet et al. (224) showed an apparent co-migration of AOX and dimeric complex III (located at around 480 kDa in BN-PAGE) in both a free-living soil amoeba (Acanthamoeba castellanii) and a higher plant fruit (Lycopersicon esculentum), and he suggested that a kinetic interplay between the cytochrome pathway and the alternative oxidase pathway may exist, based on the physical interaction of those two protein enzymes. Conversely, in most cases, an exclusive interaction of AOX with OXPHOS complexes is not proved (74, 159); therefore, it is commonly assumed that the alternative oxidase is present in the unbound form and that cyanide-insensitive respiration occurs through CoQ pool behavior.
The elution of respiratory supercomplex assemblies after electrophoresis is mandatory for further biochemical analysis; techniques for preparative electrophoresis of mitochondrial complexes and supercomplexes have been described in detail by Seelert and Krause (286).
B. Molecular structure of supercomplexes
A distinct structure was observed for all supercomplexes that were investigated, supporting the idea of highly ordered associations of the respiratory supercomplexes and discarding most doubts on artificial interactions. Moreover, from the limited data available, it appears that such interactions may be species or kingdom specific (324). Three-dimensional models of the I1III2 supercomplex isolated from plant (66, 248) and mammalian mitochondria (Fig. 13) were generated by comparison of the 2D projection map of the supercomplex, as revealed with electron microscopy analysis (EM) and single-particle image processing, with known EM and x-ray structures of complex I and complex III. In Arabidopsis (66), the specific orientation observed for the two respiratory-chain complexes indicates an interaction within the plane of the membrane, whereas the matrix-exposed protein domains are in one another's vicinity but probably do not (strongly) interact. Positions and orientations of all the individual complexes were determined in more detail in a bovine supercomplex consisting of complex I, dimeric complex III and complex IV (I1III2IV1); Schäfer and colleagues demonstrated that complex III and IV are both associated with the membrane arm of complex I and are in contact with each other, and they showed that the concave face of complex IV, which is the dimer interface in the x-ray structure, is the contact surface with the rest of the supercomplex (277). This is in contrast to the proposed interaction in yeast (119) and in potato (34), in which the complex IV monomer is assumed to be attached by its convex side to complex III2. On the basis of the structural information gained from the 3D map (277), the putative mobile electron carrier (ubiquinone or cytochrome c) binding site of each complex is facing the corresponding binding site of the succeeding complex in the respiratory chain, supporting the notion of a more-efficient electron transfer through the supercomplex due to the short diffusion distances of substrates. Interestingly, analysis of the characteristic features of the III2IV2 supercomplex in S. cerevisiae revealed additional EM densities on the intermembrane-space–exposed side of complex III in the supercomplex, which could be interpreted as bound cytochrome c molecules (119).
It also has been proposed (34, 297) that the OXPHOS complexes may assemble into higher types of organization, forming row-like megacomplexes composed by supercomplexes as building blocks, which seem to be important for the morphology of the inner mitochondrial membrane (cf. section VI.C).
Systematic chemical studies aimed directly to determine the contacting subunits and the protein–protein interaction sites of associated complexes are needed to gain substantial progress in the molecular structure of supercomplexes. Early cross-linking investigations of bovine heart mitochondria showed that it was not possible to purify complex IV independent of complex III and to separate cytochrome c from complex IV (304). No study of this kind has ever been performed, dealing with complex I and its putative partners. Moreover, a number of recent observations highlighted a new level of complexity in the higher-organization state of yeast and plant supercomplexes due to the presence of further interaction partners, such as the TIM23 protein translocase and its associated import motor (i.e., the PAM machinery), as well as the ADP/ATP carrier, some COX assembly proteins, and the carbonic anhydrase subunits that co-assemble with the OXPHOS supercomplexes. Co-precipitation of cytochrome oxidase with lactate transporter(s) and mitochondrial lactate dehydrogenase was demonstrated in muscle (116) and neurons (117) as part of the intracellular lactate shuttle.
C. Kinetic evidence of supercomplex organization
The characterization of the supercomplexes by biochemical functional analysis is still poor, despite the fact that biochemical characterization is required for ascertaining a functional role of the supramolecular associations. Most evidence available has been obtained by indirect observations of deviations from “pool” behavior of electron transfer (182, 183) and from studies directly aimed to prove substrate channeling by metabolic flux-control analysis of electron transfer in mitochondrial membranes, whereas study of the kinetic properties of isolated supercomplexes is still in its infancy.
In their original article on the CoQ pool function, Kröger and Klingenberg (160) already noticed that 10 to 20% of CoQ in submitochondrial particles is not reduced by any substrate. In a recent publication, Benard et al. (17), with flux-control analysis in state 3 (phosphorylating) conditions with succinate as substrate and by measurement of the reduction levels of CoQ and cytochrome c, described the existence of three different pools of CoQ (and cytochrome c) in rat liver and muscle mitochondria: one used during steady-state respiration, another mobilizable [i.e., as a reserve used in case of a perturbation to maintain the energy fluxes at normal values (e.g., due to inhibition of the respiratory complexes or in case of mitochondrial diseases)], and a third one that is not mobilizable and is unable to participate in succinate-dependent respiration. The reserve of CoQ was ∼8% in muscle and 23% in liver, whereas the unusable CoQ was ∼79% in muscle and 21% in liver. These results are compatible with CoQ compartmentation, although similar results with NADH oxidation were not provided.
Boumans et al. (28) conducted a careful study in Saccharomyces cerevisiae mitochondria; they investigated the pool behavior of CoQ in succinate oxidation by inhibition of complex III with antimycin titration; the pool behavior of cytochrome c also was investigated with antimycin titration of complex III by using NADH oxidation through the alternative dehydrogenases (because S. cerevisiae does not have complex I) that feed electrons to complex III at a much higher rate so that complex III becomes part of Vred for cytochrome c. It was shown that neither ubiquinone nor cytochrome c exhibits pool behavior, as determined from the linearity of the inhibitor-titration experiments, implying that the respiratory chain in yeast is one functional and physical unit of the respiratory complexes involved. This study anticipated the exploitation of metabolic flux-control analysis to detect substrate channeling in the respiratory chain (section V.C.1).
Conversely, addition of high phosphate or trichloroacetate, acting as chaotropic agents, restored pool behavior for both electron carriers. The authors concluded that the respiratory chain of yeast is organized as a supramolecular unit, but ascribed their findings to a special feature of the respiratory chain in yeast as different from that of higher eukaryotes, in which CoQ pool behavior is normally found in electron transfer between complex II and complex III. It is noteworthy, however, that the original study of Cruciat et al. (53) in yeast did not find complex II as part of a supercomplex, because its subunits did not co-fractionate with the III–IV supercomplex. No other studies dealing with complex II in relation to supercomplexes in S. cerevisiae are available.
1. Flux-control analysis of the respiratory complexes
As explained in section II.A, metabolic flux-control analysis allows a quantitative measurement of the control exerted on a composite pathway by its individual enzymatic steps. It is assumed by the theory of this analysis that the individual steps consist of separate enzymes joined by the diffusion of common intermediates. In any such system, the control would be differently exerted by one or more steps in the pathway, but the sum of the control coefficients of all steps would approach 1 and not overcome unity.
The situation would be different, however, in enzymatic supercomplexes in which the interactions between active sites are fixed and substrates and intermediates are channeled from one defined site to another one without leaving the protein environment; in such a supercomplex, the metabolic pathway would behave as a single enzyme unit, and inhibition of any one of the enzyme components would elicit the same flux control. In particular, in a system in which the respiratory chain is totally dissociated from other components of the OXPHOS apparatus (i.e., ATP synthase, membrane potential, and carriers), such as open nonphosphorylating submitochondrial particles, the existence of a supercomplex would elicit a flux-control coefficient near unity at any of the respiratory complexes, and the sum of all apparent coefficients would be greater than 1 (148). The two alternatives (random diffusion or channeling) in terms of flux-control analysis are depicted for the mitochondrial respiratory chain in the scheme in Fig. 14.
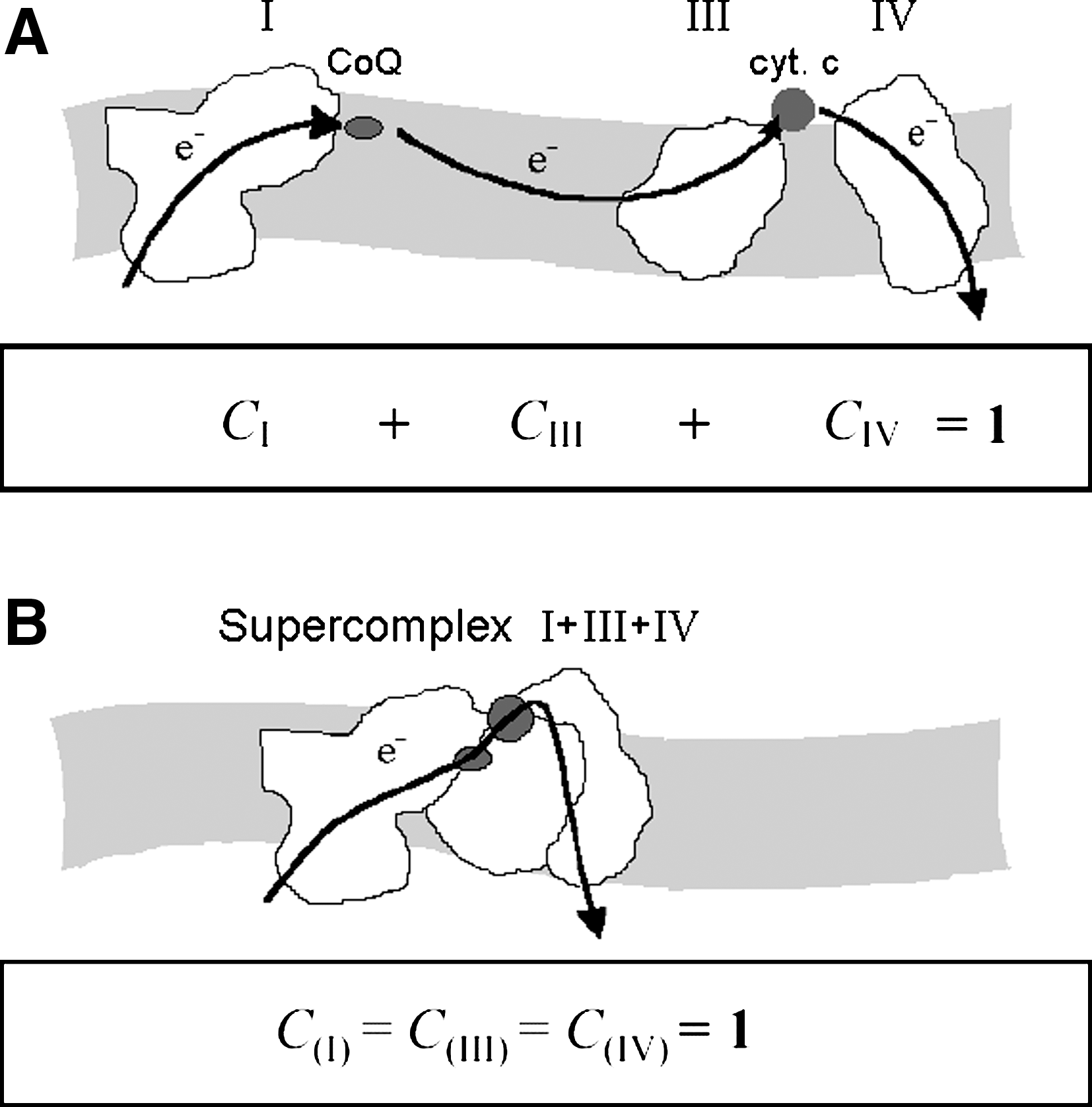
The use of flux-control coefficients for the assessment of pool behavior, although based on different theoretic grounds, has a practical exploitation very similar to that of inhibitor titrations: the presence of a lag in the inhibition of the integrated activity, as an indication that the inhibited step is not rate limiting, should be common to both types of analysis.
We addressed the problem in mammalian and in plant mitochondria. The flux-control coefficients of the respiratory complexes (I, II, III, IV) were investigated by using bovine heart mitochondria and submitochondrial particles devoid of substrate permeability barriers and by performing titrations with specific inhibitors of each complex (23). Both complexes I and III were found to be highly rate controlling over NADH oxidation (CI = 1.06; CIII = 0.99), with the strong kinetic evidence suggesting the existence of a functionally relevant association between the two complexes. On the contrary, complex IV appears to be randomly distributed (CIV = 0.26), although it is possible that if any stable interaction with complex IV exists in mammalian mitochondria, it escaped detection, most likely because of a pronounced abundance of molecules in nonassembled form. Moreover, complex II is fully rate limiting for succinate oxidation (CII = 0.88; CIII = 0.34; CIV = 0.20), clearly indicating the absence of substrate channeling toward complexes III and IV (23).
In permeabilized mitochondria from freshly harvested potato tubers, in which no activity of the so-called alternative oxidase, AOX, is present at the level of ubiquinone (5), inhibitor-titration experiments indicate that complexes III and IV are involved in the formation of a supercomplex assembly comprising complex I (98), whereas the alternative dehydrogenases, as well as the molecules of complex II, are considered to be independent structures within the inner mitochondrial membrane (Genova et al., unpublished data).
The electrophoretic demonstration of the existence of respiratory supercomplexes requires the use of digitonin that alters the membrane and destroys the permeability barriers; Conversely, our initial demonstration with flux-control analysis that a complex I to III aggregate exists in respiring mitochondrial membrane fragments (23) was done under nonphosphorylating conditions in a system lacking possible rate-limiting steps external to the respiratory chain per se (dehydrogenases, substrate carriers, membrane potential, ATP synthase, adenine nucleotide carrier).
Flux-control analysis in intact mitochondria under phosphorylating or uncoupled conditions usually exhibits low flux-control coefficients for respiratory complexes in mitochondria isolated from various tissues (cf. 54, 136, 219, 319) because the control is distributed among other components. We obtained recent evidence of the existence of a complex I to III aggregate in mitochondria under phosphorylating conditions (state 3) by exploiting flux-control analysis in freshly prepared liver mitochondria from old rats (98), in which, being respiration-rate limiting over other accessory activities because of aging, the old rats showed high control by both complex I and complex III. No information about the presence of supramolecular assemblies can be inferred based on the results obtained in state 4 (controlled respiration) because, in this condition, the proton leak across the inner membrane becomes the rate-limiting step, and therefore, a very low metabolic control over respiration is exerted by the enzyme complexes regardless of their assembly status.
2. Electron-transfer activity of isolated supercomplexes
Very recently, Acin-Perez et al. (3), in a detailed study of mouse liver mitochondria, confirmed the presence of different forms of supercomplexes after solubilization in different detergents and BN-PAGE; different from previous studies, some supercomplexes also contained complex II and ATP synthase (complex V). One particular subfraction (band 3) contained all complexes I, II, III, and IV and, in addition, cytochrome c and CoQ9. The band was excised from the gel, and respiration was measured with a Clark electrode, showing full respiratory activity from either NADH or succinate, which was sensitive to the specific respiratory inhibitors of all involved complexes. The simultaneous inclusion in the oxygen electrode chamber of the individual respiratory complexes did not allow an integrated respiration. Therefore, the oxygen consumption shown by the supercomplex bands not only is the consequence of the presence of all the respiratory complexes and “mobile” carriers needed, but also reflects the proper arrangement into a functional structure.
This finding directly confirms that supercomplex organization is compatible with electron transfer; however, it does not discard the idea that electron transfer is possible in the membrane in the absence of supercomplex organization in accordance with the random-collision model. Acin-Perez et al. (3) confirmed that most or all complex I is indeed in the form of a supercomplex, but most of complexes II, III, and IV appear to be free as isolated complexes. The authors propose a “plasticity model” in which both types of organization are possible and functional, depending on the different mitochondrial systems and on the particular physiological states: for example, a supercomplex I to III devoid of complex IV may still be useful under brief hypoxic conditions to generate a local membrane potential and ATP synthesis, in absence of cytochrome oxidase activity but in presence of excess cytochrome c.
The plasticity model well suits the information obtained by us with flux-control analysis suggesting that electron transfer between complex I and complex III is effected only by CoQ channelling, whereas that between complexes II and III and complexes III and IV seems to occur mostly by the pools of CoQ and cytochrome c, respectively (at least in beef heart and rat liver mitochondria).
A special case may be represented by electron transfer between complexes III and IV: the adherence to pool behavior shown in beef heart mitochondria by flux-control analysis may be dictated by the high percentage of individual free complexes in excess with respect to the III to IV supercomplex, so that overall flux-control analysis detects only pool behavior; thus, substrate (cytochrome c) channeling is present but negligible. In addition to that, however, the behavior in that region may depend on the strength of binding of cytochrome c to the supercomplex; unpublished studies from our laboratory suggest that in potato tuber mitochondria, where channeling occurs between complex III and complex IV, cytochrome c is tightly bound and detected in the III to IV supercomplex with BN-PAGE, whereas in beef heart and rat liver mitochondria (where no channeling is detected between complex III and cytochrome oxidase), cytochrome c is not found in the supercomplex, unless forced by a high concentration of exogenous cytochrome c.
VI. Implications of Supercomplex Organization
The (partial) organization of the respiratory chain in the form of stable associations between complexes is strongly changing our understanding of mitochondrial respiration. The integrationist approach to describe biologic systems originated from the understanding that they can no more be described as random collections of molecules governed by the physical and chemical laws of diffusion and casual interactions (317). This new understanding of the organization of mitochondrial respiration has deep novel implications, both in the direction of investigating the structural and functional properties that are consequent to this organization, and in elucidating the physical, chemical, and metabolic reasons at the basis of this association.
A. Structural and functional consequences of supercomplex organization
1. Kinetic advantage: substrate channeling and metabolic consequences
The physiological implications of the specific interaction between complex I and complex III to form a supercomplex are not yet fully understood. It was speculated that they include enhancing of electron flow by direct channeling of ubiquinone. We now examine this question in more detail.
The key question concerns the compatibility/incompatibility of the stoichiometric channelling of CoQ between complex I and complex III with the existence of pool behavior by the bulk of CoQ molecules free in the bilayer. First, the problem appears to be confined to the interaction between complex I and complex III, because no clear demonstration exists that complex II is part of a supercomplex. Complex II kinetically follows pool behavior in mitochondrial membranes, in reconstituted systems, and in the double-inhibitor titration experiments in intact mitochondria (cf. 182), in accordance with the lack of supercomplexes found with both BN-PAGE (281) and MCA (23). Nevertheless, the isolation of discrete units having succinate-cytochrome c reductase activity (335), the biophysical studies by Gwak et al. (113), and the antimycin-titration experiments in yeast (28) appear in strong contrast to this indication. The only possible explanation so far is in the existence of very loose but specific contacts between complex II and complex III, not giving rise to any kind of channelling, even in phosphorylating mitochondria. Significantly, in the recent article by Acin-Perez et al. (3), minute amounts of functional supercomplexes containing complex II and endowed with succinate oxidase activity were detected.
a. Mechanism of channeling: electron tunneling or microdiffusion?
The fundamental design of electron-transfer proteins is two catalytic sites connected by redox chains; the two catalytic sites and the connecting chain may be entirely within a single protein or belong to different protein subunits. In the respiratory chain of mitochondria, the redox complexes are composed by several subunits containing a trail of cofactors needed to allow a sufficiently short distance for electron transfer to occur. Intraprotein electron transfer is typically limited by tunneling through the insulating protein medium between the edges of the interacting centers: electron tunneling in protein is reasonably well described by a simple exponential decay with distance, so that the maximal distances allowing physiologic electron transfer should not exceed 13 to 14 Å (220).
Interprotein electron tunneling follows the same exponential rate dependence on distance as does intraprotein electron transfer; however, small-scale constrained diffusive motions are sometimes necessary to bring redox centers within the 14-Å tunneling limit: electron-transfer rates reflect diffusional motion of domains of the proteins after a protein–protein complex has been formed. For this purpose, electron transfer between QA and QB in photosystem II may include, besides tunneling, a movement of QB (in this case, retarding rate compared with that expected by theory) (96).
In the case of supercomplexes formed by apposition of individual complexes connected by potentially mobile cofactors, what is the mechanism of electron transfer?
Ideally, we should have a detailed knowledge of the molecular structure of the interacting sites, and this knowledge is still in an infancy stage. Obviously, we may have the extremes from close docking of the active sites with real interprotein tunneling, up to relatively long distances that may be covered either by important conformation changes or by restricted diffusion (microdiffusion) of the mobile components within the space between the two active sites. All of these alternatives have in common obligate channeling between two fixed sites, so that even the last situation, microdiffusion, would be quite distinguishable from pool behavior where the interaction of the mobile component may stochastically occur with a great number of possible sites reached by random diffusion. However, the previously described kinetic analysis cannot distinguish among different possible mechanisms of channeling.
In the interaction between complex I and complex III within a supercomplex, if the sites are connected by CoQ microdiffusion, it is possible that it takes place within a lipid milieu, although we cannot exclude that the sites are put together by movement of CoQ on the protein or by movement of the protein itself. If lipid is involved, then indirect indications may be obtained from studies on the temperature dependence of mitochondrial membrane–bound enzymes. In the late 1970s and early 1980s, many studies dealt with breaks and discontinuities in Arrhenius plots of membrane-bound enzymes, taken to reflect abrupt changes in activation energies in coincidence with a lipid-phase transition in the membrane bilayer (for reviews, see 184). The change in activation energy was generally ascribed to hindrance, exerted by lipid crystallization, of conformational changes required for activity (173). Heron et al. (122) studied the temperature dependence of NADH-cytochrome c reductase activity reconstituted by interaction of isolated complexes I and III with CoQ10 and dimyristoyl lecithin, which has a transition temperature at ∼24°C. At a 2:1 CIII/CI ratio, a break occurred at 20 to 24°C in NADH-cytochrome c reductase activity with activation energies (EA) around 60 to 72 kJ/mol above the transition and 100 kJ/mol below the transition. The changes in EA were interpreted not to be due to a temperature-dependent conformational change, because they were not present in either isolated complex I or complex III activities, and therefore were taken to represent a change from Q-pool behavior above the transition to stoichiometric association due to lipid-phase separation below the transition; according to Heron et al. (122) the increased EA in the case of stoichiometric association may “reflect changes in mobility and/or concentration of ubiquinone-10 in the interface between the complexes.”
Studies of this kind in natural membranes as a function of CoQ pool behavior and supercomplex association as in succinate and NADH oxidation, respectively, might be useful to throw light on the problem.
b. Metabolic consequences
A recent study of human neutrophil mitochondria (316) clearly points out the consequences of lack of supercomplex organization. In neutrophils, energy is provided mainly by a high glycolytic rate; mitochondria are present, but their respiration with NAD-linked substrates is rather low and is not apparently used for ATP synthesis. It was shown that respiratory supercomplexes are present in the myeloid cell line HL-60, but during differentiation to neutrophil-like cells, HL-60 loses supercomplex organization with a consequent lack of respiration. Interestingly, a high mitochondrial membrane potential is produced by complex III during glycerol phosphate oxidation through the glycerol phosphate shuttle (316). Because the delivery of electrons to complex IV is hampered in neutrophils, the operation of the shuttle may be maintained by delivery of electrons to oxygen with formation of superoxide (90). The observation that the delivery of electrons from glycerol phosphate dehydrogenase to complex III takes place in the absence of supercomplex organization is in line with the notion that glycerol phosphate dehydrogenase operates in mitochondria through the CoQ pool (258). Although not discussed, the work of Van Raam et al. (316) indicates a similar behavior for succinate, because this substrate generates a membrane potential only slightly lower than that of glycerol phosphate: it is well known and amply discussed previously that complex II does not form supercomplexes.
Similarly, clear indications were obtained in another recent study (266) of canine cardiac mitochondria in heart failure induced experimentally by microembolization. Although the activity of individual complexes I, III, and IV was normal, respiration with NAD-linked substrates in state 3 or after treatment with uncoupler was severely affected; BN-PAGE showed a similar severe reduction of supramolecular organization with particular decrease of the major I–III2–IV supercomplex. Clearly, the OXPHOS defect is to be ascribed to the supramolecular assembly rather than to the individual components of the respiratory chain; the reason for such diminished assembly was not discussed, but it is tempting to speculate that enhanced ROS production due to ischemia and reperfusion in the microembolized vessels modifies the membrane environment as a consequence of lipid peroxidation, thus disrupting supercomplex assembly (see section VI.B.2.). Interestingly, individual complexes, including free complex I, were not modified or disassembled in this condition (see next section).
2. Stability and assembly of individual complexes
The first chromatographic isolation of a complete respirasome (I1III4IV4) from digitonin-solubilized membranes of Paracoccus denitrificans indicated that complex I is stabilized by assembly into the NADH oxidase supercomplex, because attempts to isolate complex I from mutant strains lacking complexes III or IV led to the complete dissociation of complex I under the conditions of BN-PAGE. Reduced stability of complex I in those mutant strains was also apparent from an almost complete loss of NADH-ubiquinone oxidoreductase activity when the same protocol as that for parental strain was applied (298).
Analysis of the state of supercomplexes in human patients with an isolated deficiency of single complexes (279) and in cultured-cell models harboring cytochrome b mutations (2, 56) also provided evidence that the formation of respirasomes is essential for the assembly/stability of complex I. Genetic alterations leading to a loss of complex III prevented respirasome formation and led to secondary loss of complex I; therefore, primary complex III–assembly deficiencies were seen as complex III/I defects. Conversely, complex III stability was not influenced by the absence of complex I.
D'Aurelio et al. (56) studied mtDNA complementation in human cells by fusing two cell lines, one containing a homoplasmic mutation in the COXI subunit of complex IV, and the other, a distinct homoplasmic mutation in cytochrome b of complex III. With cell fusion, respiration was recovered in hybrids cells, indicating that mitochondria fuse and exchange genetic and protein materials. The recovery of mitochondrial respiration correlated with the presence of supercomplexes containing complexes I, III, and IV; critical amounts of complexes III or IV are therefore required for supercomplexes to form and provide mitochondrial functional complementation. From these findings, supercomplex assembly emerged as a necessary step for respiration, its defect setting the threshold for respiratory impairment in mtDNA mutant cells. Several other studies in mutants and in patients having specific defects in a single respiratory complex have suggested that complex III and, to a lesser extent, complex IV are involved in the assembly and stabilisation of complex I in mammals (62, 203). This is not the case in fungi, because a P. anserina mutant lacking both complexes III and IV possesses a normal complex I, presumably as a consequence of special fungal features such as the presence of AOX and a dimerized complex I (193).
Conversely, mutations of complex I had controversial effects, because in some studies, they did not affect the amounts of other complexes, whereas in others, they significantly reduced the amounts of complexes III and IV (105, 279, 311). The reason for this discrepancy is not known, but might be related to the specificity of the mutation affecting subunits of complex I involved in the contacts with the other complexes.
3. Possible role of supercomplex organization in limiting ROS formation
Although no direct study is available on the effect of supramolecular organization on ROS production by the respiratory chain, indirect circumstantial evidence suggests that supercomplex assembly may limit the extent of superoxide generation by the respiratory chain. Zhang et al. (339) showed that reconstitution of succinate CoQ reductase with the bc 1 complex to yield an active succinate cytochrome c reductase inhibited superoxide formation. Although the relevance of a II to III supercomplex in vivo is questionable in light of that amply discussed in this review, the principle that a tighter organization of the respiratory enzymes may hide autooxidizable prosthetic groups hindering their reaction with oxygen may have a wider application.
Similarly, it was discussed in section III.B that two potential sites for oxygen reduction exist in complex I, represented by FMN and iron–sulfur cluster N2; controversial results from different laboratories working either on isolated complex I (72, 95) or on mitochondrial membranes (79, 180) would generally indicate that N2 as a source of ROS would be predominant in membrane particles, whereas FMN might become available after complex I isolation. A reasonable hypothesis is that FMN becomes exposed to oxygen only when complex I is dissociated from complex III. Although the molecular structure of the individual complexes does not allow us to envisage a close apposition of the matrix arm of complex I, where FMN is localized, with either complex III or IV (66, 248, 277), the actual shape of the I1III2IV1 supercomplex from bovine heart (277) suggests a slightly different conformation of complex I in the supercomplex, with a smaller angle of the matrix arm, with the membrane arm showing a greater bending toward the membrane (and presumably complex III), in line with the notion that complex I may undergo important conformational changes (253). Moreover, the observed destabilization of complex I in the absence of supercomplex (section VI.A.2) may render the 51-kDa subunit containing the FMN more “loose,” allowing it to interact with oxygen. The elevated ROS production observed in P. anserina respiring on AOX, in which the major form of complex I is a I2III2 supercomplex rather than the usual I1III2 supercomplex (159), is in line with this reasoning, because it is likely that the complex I dimer may undergo a looser interaction than a complex I monomer with the complex III dimer.
On the basis of their studies on rat brain mitochondria oxidizing different substrates, Panov and collaborators (240) suggested that the supercomplex organization of complex I within the chain prevents excessive superoxide production on oxidation of NAD-linked substrates because the efficient channeling helps to maintain the chain in the oxidized state, whereas on succinate oxidation, the backward electron flow keeps the centers more reduced in complex I, favoring production of superoxide; it is interesting to note that complex II does not form a respirasome.
Additional circumstantial evidence on the role of supercomplex organization may come from the observation that high mitochondrial membrane potential elicits ROS generation, whereas uncoupling strongly reduces ROS production (176); although likely explanations have been given for these observations, they are compatible with the suggestion by Piccoli et al. (251) that high membrane potential may dissociate the supercomplexes into the individual units (described in section VI.B.3).
B. Determinants of supercomplex organization
1. Protein concentration
At low protein concentrations in lipid bilayers, proteins are usually randomly dispersed, whereas at high concentrations, they tend to aggregate. Integral membrane proteins become surrounded by a lipid domain of boundary lipids; the possibility that the wetting lipid layer is shared by two or more proteins also gives rise to aggregation phenomena that are at a longer-range scale than those involved in direct protein–protein interactions (168).
In the mitochondrial inner membrane, integral proteins of the OXPHOS system are densely packed so that the average distance between complexes may be calculated to be a few nanometers (174); in addition, the presence of different types of lipids would enhance wetting and capillary condensation, as well as the possibility of phase separations. Thus, the presence of protein aggregates of the transmembrane respiratory complexes would not be at all unexpected on theoretic grounds. The immobilization of the proteins would be also favored by the presence of outer membrane–inner membrane contacts, by the narrow tubular connections of the cristae (197), and by the high viscosity of the matrix, which would exert a slowing effect on the diffusion of proteins spanning the inner membrane.
In addition, direct binding of matrix enzymes with respiratory chain complexes may occur, as proposed for complex I with several NAD-linked matrix dehydrogenases, including pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes, malate dehydrogenase, and β-hydroxyacyl-CoA dehydrogenase (233, 300).
Early experiments reported by Heron et al. (123) provided evidence that purified complex I and complex III, when mixed as concentrated solutions in detergent and then co-dialyzed, combine reversibly in a 1:1 molar ratio to form a complex I to III unit (NADH-cytochrome c oxidoreductase) that contains equimolar FMN and cytochrome c1 (clearly at difference with the supercomplexes found by BN-PAGE, where complex III is present as a dimer, cf. section V.A) and 2 to 3 moles of ubiquinone-10 per mole of protein unit. Activation-energy measurements for NADH-cytochrome c oxidoreductase activity showed that oxidoreduction of endogenous ubiquinone-10 proceeds somewhat differently from the oxidation and the reduction of exogenous quinones, supporting the idea that the mobility of ubiquinone-10 in the complex I to III unit is highly restricted and suggesting that CoQ10 is effectively trapped between the component complexes in an environment that may be partly protein and partly derived from the lipid annuli of those complexes. However, CoQ-pool behavior could be restored, and complex I and complex III could be made to operate independent of each other by increasing the concentrations of phospholipid and ubiquinone (approximately a twofold and a sixfold increase, respectively) in the concentrated mixture (123). Inclusion of phospholipids in the reconstituted system may have a number of effects on the physical state of the system. Heron and co-workers (123) proposed that, when phospholipids in excess of those needed to form an annulus are absent, relative mobility is lost, and complexes are frozen in their complex I to III assembly, favoring a stable orientation of the site of reduction of ubiquinone with respect to the site of oxidation. Heron et al. (123) also reported that endogenous ubiquinone-10 leaks out of the complex I to III unit when extra phospholipids are present, causing a decrease in activity that could be alleviated by adding more ubiquinone. It is likely that the function of the large amount of ubiquinone in the natural membrane may be, therefore, to maintain the ubiquinone-10 content in the supercomplex unit when it is formed (cf. section VII.A).
An analogous system, obtained by fusing a crude mitochondrial fraction (R4B) (262) enriched in complex I and complex III with different amounts of phospholipids and CoQ10 (179), was used in our laboratory to discriminate whether the reconstituted protein fraction behaves as individual enzymes (CoQ-pool behavior) or as assembled supercomplexes, depending on the experimental distances between the intramembrane particles. The comparison of the experimentally determined NADH-cytochrome c reductase activity with the values expected by theoretic calculation applying the pool equation showed overlapping results at phospholipid dilutions (wt/wt) from 1:10 on (i.e., for distances >50 nm), whereas at shorter distances between complex I and complex III, resembling the mean nearest-neighbor distance between respiratory complexes in mitochondria, pool behavior was no longer effective (22, 179). In the two experimental models, kinetic testing according to the metabolic flux-control analysis (Table 6) validated the hypothesis of a random organization and of a functional association between complex I and complex III, respectively (98)
AAPH = 2,2′-azobis-(2-amidinopropane) dihydrochloride
Fraction R4B from bovine heart mitochondria was fused with phospholipids (PL, Asolectin) and coenzyme Q10 by cholate dilution (179).
2. Lipid composition
The possibility that a protein exhibits preference for a given lipid species so that a lipid annulus wets the protein surface may be another source of protein aggregation (168); changing the nature of the lipids would dramatically change their matching conditions to the proteins. Changes in membrane protein aggregation induced by lipid peroxidation may well be expected on theoretic grounds as a consequence of profound biophysical changes in the lipid bilayer. All purified preparations of mitochondrial electron-transfer complexes are isolated as lipoprotein complexes, the extent of associated lipid depending on the particular method used for isolation, and the phospholipid composition reflecting that for the mitochondrial inner membrane. Predominant phospholipids present include cardiolipin, phosphatidyl choline, phosphatidyl ethanolamine, and lesser amounts of neutral lipids and phosphatidyl inositol.
Two roles of phospholipids have been distinguished: (a) a dispersive solubilization effect that can be duplicated by appropriate detergents; and (b) a catalytic effect that can be specifically fulfilled only by tightly bound cardiolipin, more likely buried within the protein complexes (93, 163, 168, 264). Two additional possible roles may have to be met, particularly in the cases of complex I and complex III. These roles might be to provide a sufficiently lipophilic environment for the interaction of the lipophilic electron carrier, ubiquinone, and to participate in linking together components of the respiratory chain.
The absolute requirement of cardiolipin (CL) for the activity of cytochrome oxidase, complex I, and complex III, as well as for that of several mitochondrial carriers, suggests that this phospholipid plays a crucial role in the coupled electron-transfer process (93), but recent results seem also to indicate that cardiolipin stabilizes respiratory-chain supercomplexes as well as the individual complexes. The availability of a CL-lacking yeast mutant (Δcrd1 null) provided the opportunity to demonstrate that mitochondrial membranes still contained the III2–IV2 supercomplex, but that it was significantly less stable than supercomplexes in the parental strain. Other phospholipids that are increased in the mutant, including phosphatidyl-ethanolamine and phosphatidyl-glycerol, could not substitute for cardiolipin and could not prevent dissociation of supercomplexes, showing 90% of the individual homodimers of complex III and IV not organized into supercomplex under BN-PAGE conditions (250, 340).
The putative direct protein–protein interaction of cytochrome oxidase and complex III in yeast (250) is proposed also to involve two phospholipid molecules (i.e., cardiolipin and phosphatidyl-ethanolamine), tightly bound in a cavity of the membrane-imbedded domain of complex III (165), suggesting that they can provide a flexible linkage between the interacting subunits of complexes III and IV. It also was found that, in the absence of cardiolipin, most supramolecular associations, including those involving the adenine nucleotide carrier, are destabilized or undetectable (47).
In a different study, the stability and assembly of complex IV was found to be reduced in yeast cells lacking Taz1 (31), the hortologue of human Tafazzin, an acyl transferase involved in the synthesis of mature tetralinoleyl cardiolipin; mutations of Tafazzin in humans result in Barth syndrome, a cardioskeletal myopathy with neutropenia, characterized by respiratory-chain dysfunction. It was later found (203) that the cardiolipin defect in Barth syndrome results in destabilization of the supercomplexes by weakening the interactions between respiratory complexes. Tafazzin is found in the inner membrane in a complex including ATP synthase and the adenine nucleotide carrier; the absence of this complex due to Taz1 mutations also induces altered cristae morphology (46).
It is well documented that exposure of mitochondria to reactive oxygen species (ROS) can affect the respiratory activity through oxidative damage of cardiolipin (249). We have demonstrated with flux-control analysis that the maintenance of a I–III supercomplex after reconstitution of a protein fraction enriched with complex I and III (R4B) into phospholipid vesicles at high protein-to-lipid ratios (see earlier) is abolished if lipid peroxidation is induced by 2,2′-azobis-(2-amidinopropane) dihydrochloride (AAPH) before reconstitution (22). Evidently, the distortion of the lipid bilayer induced by peroxidation and the alteration of the tightly bound phospholipids determine dissociation of the supercomplex originally present in the lipid-poor preparation (Table 6).
In addition to lipid peroxidation, changes in membrane shape and lipid composition, as caused by modification of lipid biosynthesis enzymes, can be putative actors in the regulation of the oxidative phosphorylation system through supercomplexes stabilization. In their recent review (94), Furt and Moreau described the role of different lipids in mitochondrial morphodynamics and also illustrated how the lipid-induced membrane curvature can affect the activity of various protein partners by conditioning their recruitment and segregation into microdomains. The challenge in the future will be to unravel the detailed mechanisms.
3. Functional state
The impact of the mitochondrial transmembrane potential (ΔμH+) on the flux control exerted by cytochrome c oxidase on the respiratory activity in intact cells and isolated mitochondria was evaluated by Piccoli et al. (251). The results indicate that, under conditions mimicking the mitochondrial state 4 respiration, the control strength of the oxidase is decreased in respect to endogenous state 3 respiration. Although the interpretation of the results in such a complex system is very difficult, the authors suggest that such a change in control strength might be featured in terms of equilibrium between a random organization of cytochrome oxidase with respect to other complexes (at high membrane potential, state 4) and a respirasome organization (at lower membrane potential, state 3). Because the respiratory rate is high in state 3 conditions, the supercomplex organization would produce an extra advantage by increasing the rate with channeling. Although the driving forces leading to the assembly/disassembly of the supercomplexes have been not yet defined, it is not inconceivable that, given the membrane-integrated nature of the single complexes, electrostatic/hydrophobic interactions may enter into play in response to ΔμH+. No direct demonstration of the association state of respiratory complexes as a function of the physiological states of the OXPHOS system is available.
The regulatory role of complex I and complex IV phosphorylation has been treated in section II.B. No direct study is available investigating the supramolecular organization of the respiratory chain as a function of phosphorylation/dephosphorylation events, but it is tempting to speculate that the increase of activity of complex I and the decrease of ROS generation elicited by phosphorylation of the NDUFS4 subunit may be the result in part of enhanced stability of the I to III supercomplex.
In cytochrome oxidase, one may speculate on the role of phosphorylation of subunit COXI on tyrosine-304. This amino acid is located in the intermembrane-space domain of subunit I, on a helix that is in contact with the other catalytic subunit (COXII), and facing the interface region of the two monomers. The enhancement of the monomer–monomer interaction might be a plausible effect of phosphorylation (170). Interestingly, the concave face of complex IV, which is the dimer interface in the x-ray structure, is also the contact surface of the monomer with its protein partners when it is assembled in the I1III2IV1 bovine supercomplex (277).
C. Why has the supercomplex organization been overlooked?
One of the major points in favor of the Random Collision Model of electron transfer was the axiom that “The integral proteins of the inner membrane are randomly distributed in the bilayer and phospholipid dilution of the mitochondrial membrane proteins slows down electron transfer” (114).
Freeze-fracture electron microscopy of giant mitochondria (megamitochondria obtained by cuprazone treatment) showed that the intramembrane protein particles are randomly distributed in the inner membrane (114). It is possible that weak hydrophobic forces keeping together the respirasomes are broken by the freeze-fracture technique. Close inspection of the fractured faces in the electron micrographs (130), however, shows long-range random distribution of the intramembrane particles, but in the short range, it reveals apposition of small clusters, hinting at possible associations of the integral proteins, which were not considered by the authors. In a subsequent study with the rapid-freeze deep-etch technique of the inner membranes of Paramecium, it was not possible to obtain fracture faces in the tubular cristae (6); however, the study revealed projections of 9-nm diameter, identified as F1-ATPase, located entirely out of the cristae toward the matrix. These particles were arranged in a nonrandom tightly ordered pattern, contrasting the idea of a random distribution of membrane components; moreover, other regularly arranged projections to the matrix, 13 nm wide and 13 to 22 nm long, were seen aligned at regular intervals and were identified as complex I, possibly in a dimeric state. A better interpretation based on the recent knowledge of supercomplex organization should identify these particles as I1III2IV1–2 supercomplexes (see section V.A). This study revealed a highly ordered state of the proteins in the mitochondrial cristae, different from previous interpretations of freeze-fracture EM studies. Single-particle electron microscopy confirms that supercomplexes are arranged in strings of larger structures or megacomplexes (34): the individual units of these megacomplexes in potato mitochondria appear to be composed of I2III2IV2 units, different from other types of supercomplexes. In a recent study, Thomas et al. (309) also found regular rows of ATP synthase dimers in yeast mitochondrial membranes. A tomographic analysis of beef heart mitochondrial membranes showed the structure of ribbons of ATP synthase in great detail (297).
The decrease of respiratory activities in mitochondria fused with phospholipids (114) and the restoration of activity by incorporating excess CoQ can be easily explained by dilution of the CoQ pool together with the proteins when the phospholipids-to-protein ratio increases: such a condition closely corresponds to point (b) later in this section VI.C. Thus, we may easily explain these results with dissociation of CoQ from the I to III supercomplex after membrane dilution with phospholipids. In other words, the CoQ concentration in the lipids decreases with dilution of the membrane, and consequently, dissociation of bound CoQ is favored: therefore, its concentration in the supercomplex decreases, leaving empty a progressively greater number of sites where electron transfer becomes interrupted. Reconstitution of the membrane with excess CoQ would restore electron transfer by shifting the equilibrium toward bound CoQ in the supercomplex.
Furthermore, at high phospholipids-to-protein ratio, dissociation of supercomplexes also occurs, as shown by our studies measuring the rates of electron transfer and by flux-control analysis by fusing a crude mitochondrial fraction (R4B) enriched in complex I and complex III (262) with different amounts of phospholipids and CoQ10 (22, 98, 179) (section VI.B.1 and Table 6).
Most evidence on the random-diffusion model concerns the CoQ region, whereas fewer studies are available on cytochrome c. Conversely, evidence in favor of channeling in the cytochrome c region is much less solid, and the state of the art was discussed in the previous sections. We therefore restrict our considerations to electron transfer between complex I and complex III.
We can summarize the available evidence accumulated against channeling in three major points: (a) electron transfer in the CoQ region obeys pool behaviour, according to the Kröger-Klingenberg equation; (b) electron transfer in the CoQ region follows saturation kinetics with respect to CoQ; and (c) phospholipid dilution of the mitochondrial membrane proteins slows electron transfer. Point (c) was addressed in the previous section VI.B. A detailed analysis of this issue was described in a previous publication (182).
An additional comment to this critical analysis: the functional relevance of supercomplex organization in electron transfer may vary with the physiological conditions in the intact cells: clearly under physiologic conditions, the respiration rate is under control of energy-consuming processes (respiratory control). Under prevalent state 4 conditions (high ATP/ADP ratio), the electrochemical proton gradient is largely the rate-limiting step, so that operation of the respiratory chain as tunneling or diffusion may be of minor importance for the flux control. Conversely, the situation may be dramatically different under conditions of high energy demand (state 3) or under uncoupling conditions. The latter may be more important in vivo than originally thought: it has been suggested that muscle and liver mitochondria may dissipate 30 to 50% of the electrochemical potential as heat, through the action of the uncoupling proteins (cf. section VI.B.3).
VII. Role of the Coenzyme Q and Cytochrome c Pools
A great deal of data in the literature, reviewed in the previous sections, indicates that most certainly, a mobile pool of CoQ exists in the inner mitochondrial membrane, and that this pool coexists with protein-bound CoQ. Is this pool just a reservoir of an excess of CoQ molecules without a specific function, or is the pool necessary for functioning of the respiratory chain or for additional functions or both?
A. Dissociation equilibrium of bound components
All available evidence, already reviewed, points out that complex I is almost totally associated in a supercomplex with complex III, with electron channelling of bound CoQ in the boundary between the two complexes. However, this does not exclude that free CoQ in the pool is also necessary for proper channeling between the complexes. The bound intercomplex quinone that allows electron flow directly from complex I to complex III may well be in dissociation equilibrium with the CoQ pool, so that its amount, at steady state, would be dictated by the size of the pool: this equilibrium would explain the saturation kinetics for total ubiquinone exhibited by the integrated activity of complex I and complex III and the decrease of respiratory activities in mitochondria fused with phospholipids with subsequent dilution of the CoQ pool (114). To be in agreement with the experimental observation obtained with metabolic flux analysis, this proposition must, however, require that the dissociation-rate constant of bound CoQ be considerably slower than the rates of intercomplex electron transfer via the same bound quinone molecules (Fig. 15). To explain the high apparent K m found for CoQ in NADH oxidase activity, the rate constant for CoQ binding to complex I must be slow (99).
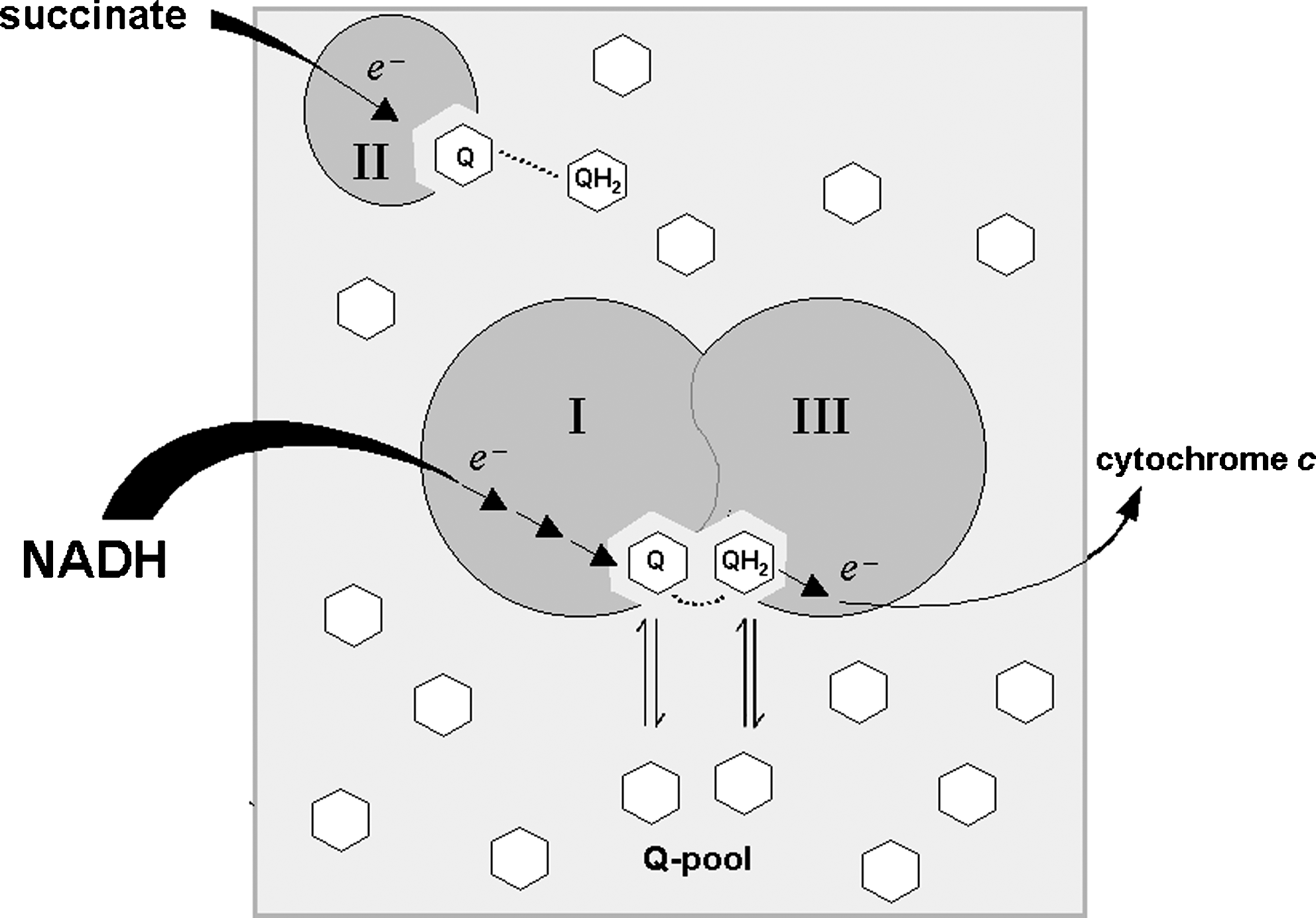
In this way, free CoQ behaves as a reservoir for binding to the I to III supercomplex; in addition, free CoQ may be a reservoir for other functions believed to require CoQ binding to specific proteins, such as uncoupling proteins (69) and the permeability transition pore (8, 326), and it also represents the main antioxidant species in the inner mitochondrial membrane (70).
A different question is the following: can electron transfer between complex I and complex III occur through the CoQ pool in absence of supercomplex organization? The question is much less simple than may be envisaged; analysis of the literature does not offer clear-cut examples of electron flow between complexes I and III in mitochondrial membranes, certainly mediated by the CoQ pool. The studies reported in section VI.A.1.B. appear to indicate that electron transfer in the absence of supercomplex organization is lost even if activity of the individual complexes is normal. However, additional data, such as the CoQ concentration in the membrane, were not provided. Some reconstitution studies, however, show that electron transfer is possible in both modes: the association of complex I with complex III (123) allows both channeling (electron transfer stoichiometric with the percentage amount of complex III associated with complex I) and CoQ pool behavior (hyperbolic relation). In our reconstitution studies reported in section VI.B.1, we showed that NADH-cytochrome c reductase activity followed the pool equation at phospholipid dilutions (wt/wt) from 1:10 on, whereas at lower dilutions, pool behavior was not effective.
B. Electron transfer between individual complexes not involved in supercomplex organization
Certainly the CoQ pool is required for electron transfer from complex II to complex III: as mentioned in the previous sections, complex II kinetically follows pool behavior in reconstitution experiments and in intact mitochondria, in complete accordance with the lack of supercomplexes found with both BN-PAGE and flux-control analysis (23). The existence of small amounts of supercomplexes containing complex II, recently described by Acin-Perez et al. (3), does not contradict the knowledge that most of succinate oxidation takes place by the CoQ pool between complexes II and III. An exception is the study of Boumans et al. (28) in yeast.
Furthermore, other enzymes, such as glycerol-3-phosphate dehydrogenase, ETF dehydrogenase, dihydroorotate dehydrogenase, that are likely to be in minor amounts and strongly rate limiting in electron transfer, are probably integrated by interaction through the CoQ pool. The only direct study addressing this problem (258) demonstrated that in brown fat mitochondria, the inhibition curve of glycerol phosphate-cytochrome c reductase is sigmoidal in the presence of myxothiazol and antimycin, suggesting the presence of a homogeneous CoQ pool between glycerol phosphate dehydrogenase and complex III.
A detailed model based on CoQ pool behavior also was proposed by Siedow and Moore (288) to explain the kinetic characteristic of alternative oxidase. In addition, reverse electron transfer from succinate to NAD+, involving sequential interaction of complexes II and I by means of CoQ, must take place by collisional interactions in the CoQ pool, because no aggregation was demonstrated between complexes I and II. The hyperbolic relation experimentally found by Gutman (111) between the rate of reverse electron transfer and succinate oxidase is in complete accordance with the pool equation. This observation poses a particularly puzzling question (181): if all or most complex I units are associated with complex III, and the interaction of CoQ in the pool with the quinone-binding site in common between the two enzymes is necessarily slow, then how can CoQH2 reduced by complex II interact from the pool with the CoQ site in complex I at a rate compatible with the steady-state kinetics of reverse electron transfer? The intriguing idea that complex I may possess two different quinone-binding sites for direct and for reverse electron transfer, respectively, is compatible with the proposal (109) that two different routes exist for forward and reverse electron transfer within complex I (Fig. 16). These two sites might become alternatively accessible, depending on the magnitude of the membrane potential. It must be noted that the ATP-driven reverse electron transfer from succinate to NAD+ occurs in the presence of a high mitochondrial transmembrane proton-motive force that, according to Piccoli et al. (251), might be the physiologic signal and, at the same time, the trigger causing the structural reorganization of the enzymatic complexes of the mitochondrial OXPHOS system. The model hypothesis depicted by Piccoli et al. from data on cytochrome oxidase might be extended to other enzymes of the respiratory chain, suggesting also that the I to III supercomplex would dissociate its constituting complexes under high ΔμH+ conditions, and this would no longer limit the access to the CoQ binding site in complex I.

VIII. Mitochondrial Complexes and Supercomplexes in Pathology
An overwhelming body of evidence accumulated in the last decades has demonstrated that mitochondria have a central role in the etiology and pathogenesis of most major chronic diseases and in aging itself (cf. 183 for a list of references). The involvement of mitochondria in disease, which has generated the term “mitochondrial medicine” (64), has been largely ascribed to their central role in production of ROS and to the damaging effect of ROS on these organelles. In particular, damage to mitochondrial DNA (mtDNA) would induce alterations of the polypeptides encoded by mtDNA in the respiratory complexes, with consequent decrease of electron-transfer activity, leading to further production of ROS, and thus establishing a vicious circle of oxidative stress and energetic decline. This decrease in mitochondrial energetic capacity is considered to be the cause of aging and age-related degenerative diseases (177), although the picture is further complicated by the complex interplay and cross-talk with nuclear DNA and the rest of the cell (177, 270).
In this scenario, it is easy to foresee the deep implications of supercomplex organization as a missing link between oxidative stress and energy failure (181). It is tempting to speculate that under conditions of oxidative stress, a dissociation of complex I to III aggregates occurs, with loss of facilitated electron channeling and resumption of the less-efficient pool behaviour of the free ubiquinone molecules. In this regard, the study on loss of supercomplex organization and of efficient electron transfer in heart failure (266) is a clear indication of the deleterious consequence of lack of supramolecular organization.
As predicted by Lenaz and Genova (181), dissociation of supercomplexes might have further deleterious consequences, such as disassembly of complex I and III subunits and loss of electron transfer or proton translocation or both; the consequent alteration of electron transfer may elicit further induction of ROS generation (181). The observation that complex III alterations prevent proper assembly of complex I has therefore deep pathologic implications beyond the field of genetic mitochondrial cytopathies. The different susceptibilities of different types of cells and tissues to ROS damage may depend, among other factors, on the extent and tightness of supercomplex organization of their respiratory chains that depend, conversely, on phospholipids content and composition of the mitochondrial membranes. These changes may have deep metabolic consequences, as depicted in the scheme in Fig. 17. An initial enhanced ROS generation for different possible reasons and originating in different districts of the cell besides mitochondria (cf. 176) would induce supercomplex disorganization, eventually leading to possible decrease of complex I assembly; both the lack of efficient electron channeling and the loss of complex I would decrease NAD-linked respiration and ATP synthesis; the defect in OXPHOS might then lead to compensatory enhancement of glycolysis to overcome the energetic deficiency. These considerations are highly relevant not only to cancer cell biology, but also may apply to ischemic diseases, neurodegeneration, and all conditions in which an oxidative stress originally takes place (176).

Mitochondrial DNA mutations have been consistently found in cancerous cells; they have been found to be associated with enhanced ROS production, and ROS act both as mutagens and as cellular mitogens (153); thus, the involvement of mtDNA mutations in cancer may well be of pathogenic importance.
We have shown that a cell line of a malignant thyroid oncocytoma, characterized by abnormal mitochondrial proliferation (44), contains a mutation of mitochondrial DNA preventing expression of subunit ND1 (27). These cells exhibit a dramatic decline of ATP synthesis supported by NAD-dependent substrates, whereas in the mitochondria isolated from these cells, the complex I activity is strongly depressed (27). Accordingly, the cell line produces much greater amounts of ROS compared with a line derived from a nononcocytic thyroid tumor (294). We also found (98) with BN-PAGE of mitochondrial proteins from the oncocytic cell line, a complete absence of high-molecular-weight aggregates containing either complex I or complex IV, which are instead present in a control cell line from a nononcocytic thyroid tumor. If the absence of supercomplexes comprising complex I is in line with its disassembly due to lack of ND1, the absence of aggregated complex IV must be a secondary phenomenon, possibly due to the alteration of the lipid environment by the excessive ROS production. In another study (185, Sgarbi et al. unpublished data), we reported that downregulation of the respiratory chain in Ras-transformed fibroblasts is operated through strong decrease of complex I activity and content, probably due to lack of correct assembly of the subunits, as a consequence of altered supercomplex organization, as demonstrated by loss of the highest molecular weight I1III2IV1–2 supercomplex. Clearly, the role of derangement of supercomplex organization in pathologic states is a new area of research deserving extensive investigation.
IX. Conclusions
The evolution of our understanding of mitochondrial respiration reflects the evolution of all biologic research. In the 1950s, when the structure of biomembranes was dominated by the rigid Danielli model, and membranes were accessible by microscopy more than by biochemistry, the impenetrable structure of the mitochondrial inner membrane allowed Chance (42) to envisage the respiratory chain as a sequence of redox-active prosthetic groups embedded in a rigid protein matrix. Subsequently, membranes became a matter of massive enzymologic research by David Green in Wisconsin, and Hatefi (118) in his laboratory was able to dissect the respiratory chain into four major enzymatic complexes that were purified and partly characterized; at the same time, new models of membrane structure appeared that had to consider these new revolutionary developments, and culminated in the Singer-Nicolson fluid mosaic model (290). Most research in the subsequent decades followed the reductionistic approach of gaining deeper and deeper details on the structure of each complex, and very little attention was paid to the possibility of functional supramolecular associations between complexes. The discovery of mitochondrial supercomplexes coincided with the increasing attention to a different, holistic approach in all areas of biology. It is not by chance that the investigators that discovered mitochondrial respiratory complexes had the key for supercomplex associations, but they were unaware of it: the individual complexes were isolated from intermediate steps in the purification procedure that were enriched in what we would now call supercomplexes (118). We are now at the edge of exciting new developments in mitochondrial bioenergetics, rising from these new concepts and projecting to important areas of physiology and pathology.
Footnotes
Acknowledgments
The list of references is by all means incomplete for editorial reasons. We apologize to the authors not quoted in this review.
Abbreviations Used
Reviewing Editors: Srinivas Bharath, Enrique Cadenas, Gino Cortopassi, Sergey Dikalov, Maria N. Gadaleta, Ana Navarro, Sergio Papa, Rodrigue Rossignol, and Raj S. Sohal