
Abstract
The hypoxia-inducible factor (HIF) family of transcription factors is responsible for coordinating the cellular response to low oxygen levels in animals. By regulating the expression of a large array of target genes during hypoxia, these proteins also direct adaptive changes in the hematopoietic, cardiovascular, and respiratory systems. They also play roles in pathological processes, including tumorogenesis. In recent years, several oxygenases have been identified as key molecular oxygen sensors within the HIF system. The HIF hydroxylases regulate the stability and transcriptional activity of the HIF-α subunit by catalyzing hydroxylation of specific proline and asparaginyl residues, respectively. They require oxygen and 2-oxoglutarate (2OG) as co-substrates, and depend upon non-heme ferrous iron (Fe(II)) as a cofactor. This article summarizes current understanding of the biochemistry of the HIF hydroxylases, identifies targets for their pharmacological manipulation, and discusses their potential in the therapeutic manipulation of the HIF system. Antioxid. Redox Signal. 12, 481–501.
Introduction
Substantial progress has been made in our understanding of the molecular mechanisms by which the levels and activity of HIF are regulated by oxygen. In particular, the modulation of hydroxylase enzymes that act as oxygen sensors for the HIF system holds significant therapeutic promise (75, 157, 175). This article summarizes the current understanding of the biochemistry of the HIF hydroxylases, identifies targets for their pharmacological manipulation, and discusses recent progress and potential in the field.
The Prolyl Hydroxylase—Hypoxia-Inducible Factor Axis
The non-heme Fe(II)-dependent oxygenases and structurally related oxidases are a ubiquitous superfamily of enzymes that catalyze a wide range of oxidation reactions. The largest subfamily of these oxygenases uses the citric acid cycle intermediate 2-oxoglutarate (2OG, or α-ketoglutarate) as a co-substrate, and share conserved double-stranded β-helix core (DSBH) fold and Fe(II) binding motifs (35, 39, 171). In plants and microorganisms, the 2OG-dependent oxygenases catalyze an extremely diverse range of oxidative reactions (58). In animals, their known activity is currently limited to hydroxylation (or N-methyl demethylation via hydroxylation), but sequence analyses in humans suggest the existence of more than 60 Fe(II)- and 2OG-dependent oxygenases, many of which have no assigned physiological function (117). In this review, we will focus on the four enzymes identified as direct regulators of the HIF transcriptional pathway (170).
Hypoxia-inducible factor
HIF was first identified as a hypoxia-induced nuclear factor that binds to response element enhancer regions associated with the erythropoietin (EPO) gene (178). HIF was subsequently shown to be an α/β-heterodimeric protein, both subunits of which belong to the basic helix-loop-helix PAS protein family. The α-subunit exists in humans as three isoforms, HIF-1α, HIF-2α, and HIF-3α (with splice variants). Of these, HIF-1α is the most extensively studied. In contrast to HIF-1α, which is ubiquitously expressed, HIF-2α has restricted tissue expression, but both HIF-1α and HIF-2α are positive regulators of hypoxia-inducible gene expression (176). HIF-3α seems to suppress hypoxia-inducible gene expression in the human kidney, and may therefore act as a negative regulator (71, 120).
Both subunits of HIF are constitutively expressed. Under most circumstances, the HIF-β subunit is present in excess relative to the HIF-α subunit, the abundance of which is regulated directly by oxygen availability (202). Under most normoxic conditions, HIF-1α is rapidly degraded. The key oxygen-dependent step in this degradation pathway was identified in 2001 as the C-4 hydroxylation of specific proline residues (Pro402 and Pro564, in the case of human HIF-1α) within the N- and C-terminal oxygen-dependent degradation domains of HIF-1α. Hydroxylation at these sites enables binding of the von Hippel–Lindau tumor suppression protein [pVHL, (124)] to HIF-α, which in turn targets HIF-1α for E3-ligase-mediated ubiquitination and proteasomal degradation (24, 54, 85, 86, 124) (Fig. 1). Prolyl hydroxylation is inhibited under hypoxic conditions, wherein HIF-1α accumulates and dimerizes with HIF-1β. In association with the co-activator complex p300/CBP, the HIF-1 dimer binds to the hypoxia response elements and regulates the expression of target genes.

Prolyl hydroxylase domain (PHD) enzymes
The enzymes responsible for HIF-1α prolyl hydroxylation, termed prolyl hydroxylase domain (PHD) or EGLN proteins, are members of the Fe(II)- and 2OG-dependent oxygenase superfamily (24, 54, 85, 86, 124). The three human PHD isoforms (PHD1, PHD2, and PHD3) share homology in their C-terminal catalytic domains, but differ in their N-terminal regions (82) and functionally in terms of their expression, cellular localization, tissue distribution, and catalytic activities (e.g., 4, 130) (Table 1). Like the collagen prolyl hydroxylases (CPH), which were the first 2OG-dependent oxygenase family to be identified (83), the PHDs require oxygen and 2OG as co-substrates and depend upon Fe(II) and, probably, ascorbate as co-factors. It is their requirement for oxygen, with Km values slightly higher than tissue oxygen concentrations during normoxia, that is proposed to enable the HIF–PHDs to function as effective cellular oxygen sensors (52, 77, 128) (Table 2). Nuclear magnetic resonance and labeling studies have shown that the PHDs catalyze hydroxylation at the trans-4-position of the proline ring, with retention of stereochemistry (116, 128). Crystallographic studies of PHD2 in complex with an inhibitor have verified the presence of the predicted DSBH fold, 2OG binding site and iron binding residues (125) (Fig. 2). Kinetic and mechanistic studies imply substrate binding with movement of a loop region surrounding the active site (57). Interestingly, PHD2 appears to have unusually tight binding for Fe(II) and 2OG (127). It also appears to be the most ubiquitously expressed of the PHDs; genetic studies have also defined a nonredundant role for PHD2 in the regulation of HIF-1α during normoxia (17).
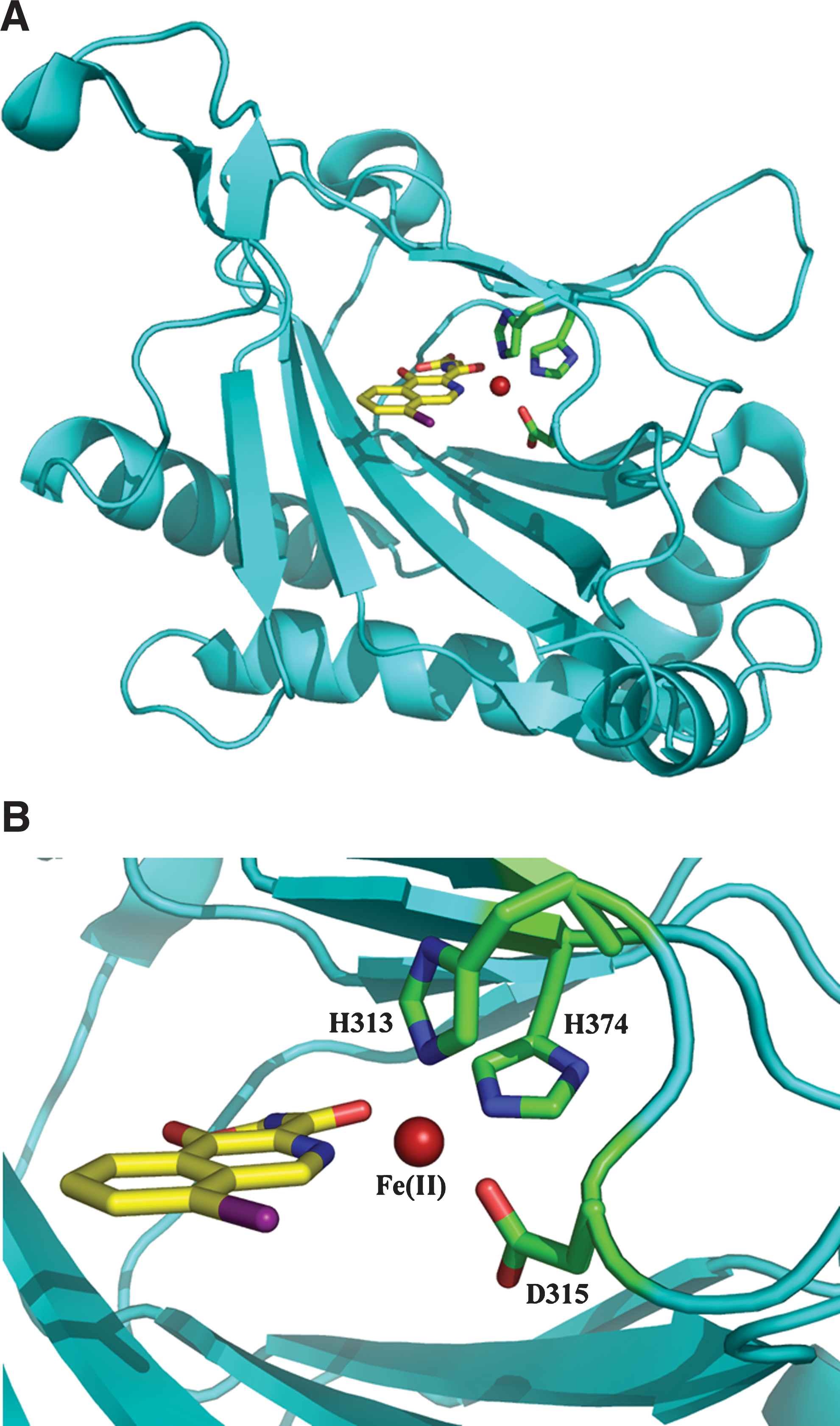
Modified from ref. 181. Pro402 and Pro564 denote the prolyl residues of HIF-1α that are hydroxylated by the active PHDs (4, 24), and these columns refer to the ability of each hydroxylase to modify these residues in HIF-1α. Asn803 and Asn851 denote the asparaginyl residues of human HIF-1α and HIF-2α, respectively, that are hydroxylated by FIH (24, 74, 109, 119). For more detail on cellular and tissue distribution of the PHDs and FIH, see refs. 130, 181, 187, and 210.
Reported apparent Km values (μM) for recombinant human collagen prolyl hydroxylases (CPH1-3, references 77, 107, and 135), HIF prolyl hydroxylases (HIF-PHD1-3, references 52, 77, 78, 102, and 127) and the HIF asparaginyl hydroxylase (FIH, references 52, 101) for co-substrates oxygen and 2-oxoglutarate and the co-factors iron (Fe(II)) and ascorbate. The ranges reflect variability in the literature, likely in part related to methodological differences, and/or the dependence of apparent Km values for HIF hydroxylases on the length of HIF1α/HIF2α substrate and nature of the oxygen dependent degradation domain (ODDD, N-terminal versus C-terminal). The Km values for oxygen are slightly higher for all the HIF hydroxylases than typical tissue oxygen concentrations during normoxia, indicating that these enzymes are well-suited to their role as cellular oxygen sensors.
N.A., value not available in the literature.
There is evidence for selective regulation of PHD isoforms by particular HIF-α isoforms. PHD2 is specifically induced by HIF-1α, whereas PHD3 is responsive to both HIF-1α and HIF-2α (5). This induction of PHD expression may serve as a feedback loop to limit HIF-mediated responses during hypoxia and/or increase the rate of HIF degradation following reoxygenation (4, 44).
Recently, a novel Fe(II)- and 2OG-dependent prolyl hydroxylase has been suggested as an additional member of the HIF-PHD family. This enzyme is widely expressed and is capable of HIF-1α hydroxylation both in vitro and in cultured cells, and of influencing cellular HIF-α protein levels (103, 145). However, unlike other PHD enzymes it contains a transmembrane domain and its catalytic region is located within the endoplasmic reticulum (145). The significance of this enzyme in the regulation of the HIF pathway remains unclear (103, 135, 145).
Factor inhibiting HIF (FIH)
In addition to regulation of HIF-α stability via prolyl hydroxylation, the transcriptional activities of HIF-1α and HIF-2α are regulated by hydroxylation of an asparagine residue in their C-terminal transactivational domains (CADs). Hydroxylation at this residue (Asn803 and Asn851 in human HIF-1α and HIF-2α, respectively) reduces the interaction between HIF and the cysteine-histidine (CH-1) domain of the p300/CBP co-activator complex that is necessary for activation of target genes (110, 119). The asparagine hydroxylase responsible for catalysing this reaction is Factor Inhibiting HIF (FIH) (24, 74, 109, 119) (Fig. 3).
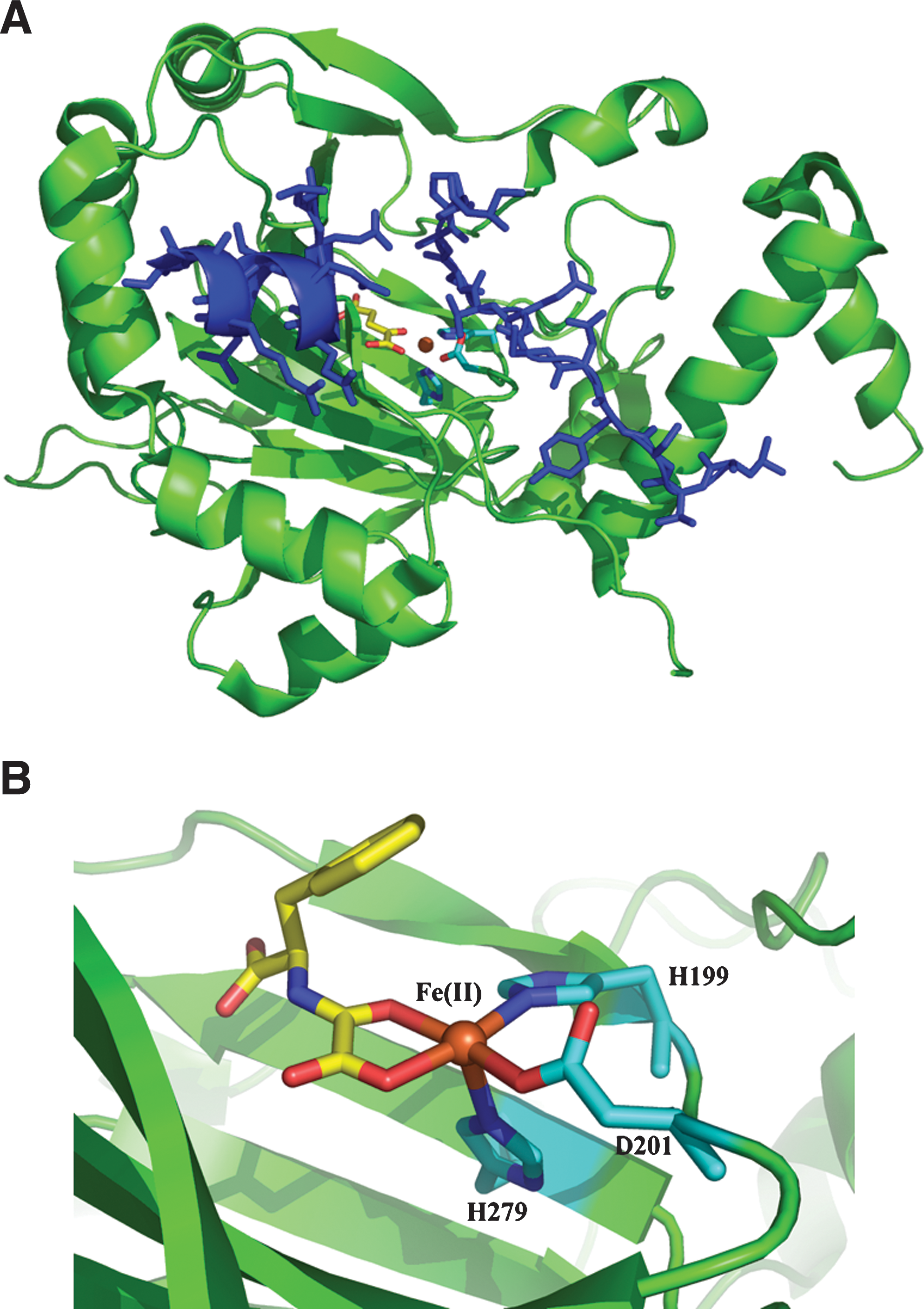
Like the PHDs, FIH is an Fe(II)- and 2OG-dependent oxygenase. Unlike the PHDs, which appear to form a discrete subfamily of human oxygenases, FIH is a member of an extended family that includes the ‘JmjC’ histone demethylases (34). There are significant structural and functional differences between PHDs and FIH, including differences in their affinities for co-factors and co-substrates. FIH, unlike the PHDs, is dimeric and does not have strong dependence on ascorbate for HIF hydroxylation, at least in vitro (Table 2). Furthermore, as well as acting on HIF-1α, FIH also catalyzes hydroxylation of conserved aparagine-residues in the ubiquitous ankyrin repeat domains (36, 37, 95). Understanding differences in the roles of FIH and the PHDs is likely to be important for the selective therapeutic manipulation of HIF system.
Insights from genetically engineered animals
Insights into the roles of the PHDs have been gained from studies of genetically engineered mice. Germ-line disruption of PHD2 (PHD2-/-) results in embryonic lethality due to severe placental and cardiac defects, but PHD1-/- and PHD3-/- mice survive to adulthood (193). Interesting abnormalities have been demonstrated in both strains. PHD1-/- mice have a remarkable tolerance to ischemia and a substantially reduced exercise tolerance (7). These observations may be linked at a biochemical level by the upregulation of pyruvate dehydrogenase kinase isoforms that restrict the normal entry of glycolytic intermediates into the citric acid cycle. The resultant shift towards anaerobic metabolism reduces the capacity for oxidative muscle performance, but also reduces the degree of oxidative stress experienced during hypoxia (7). A role for PHD3 in the regulation of apoptosis during sympathoadrenal development has also been described. In the PHD3-/- mouse there was abnormal sympathetic innervation of target organs, reduced adrenal medullary secretory capacity, and reduced systemic blood pressure (19, 112). Consistent with previous reports of isoform specificity in the PHD-HIF axis (4), PHD3 appears to regulate the sympathoadrenal axis in association with HIF-2α (19).
Despite the lethality of germ-line inactivation of PHD2 (193), an important role for PHD2 in the cardiovascular system is suggested by studies of conditional PHD2 inactivation in adult mice. Takeda et al., for example, observed widespread upregulation of angiogenesis following conditional PHD2 knock-out in ∼6 week-old mice, an effect that was associated with elevated serum levels of vascular endothelial growth factor (VEGF), but no increase in tissue VEGF mRNA levels (192). The latter is surprising, given the assumed paracrine nature of hypoxia-induced angiogenesis and the established role of HIF in the regulation of VEGF expression. Somatic PHD2 inactivation has also been shown to increase murine erythropoietin and red cell production (132, 191), and a role in erythropoiesis is supported by reports of heterozygous PHD2 mutation as a cause of human familial polycythemia (1, 108, 148, 150).
Inhibition of HIF Hydroxylases
The catalytic mechanism of the 2OG oxygenases can be divided into two half reactions (Fig. 4): the generation of a reactive oxidizing species, and its subsequent utilization for substrate hydroxylation (reviewed in refs. 34, 181). In the resting state, Fe(II) at the active site is coordinated by three residues from the enzyme (normally two histidine residues and one asparatate or glutamate residue) and by two–three water molecules. The binding of Fe(II) to the HXD/E … H motif of 2OG oxygenases is labile compared to the heme oxygenases. Indeed, the Fe(II) bound at the PHD2 active site may be substituted by other transition metal ions (129), providing a possible explanation for the pathophysiological effect of cobalt poisoning (see below). Crystallographic and kinetic analyses suggest that 2OG and substrate bind sequentially to the active site. Substrate binding is followed by binding of molecular oxygen, which is proposed to replace the remaining water molecule from the iron centre. Oxidative decarboxylation of 2OG leads to the production of carbon dioxide, succinate, and the highly reactive Fe(IV) = O intermediate that is responsible for HIF-α hydroxylation (Fig. 4).
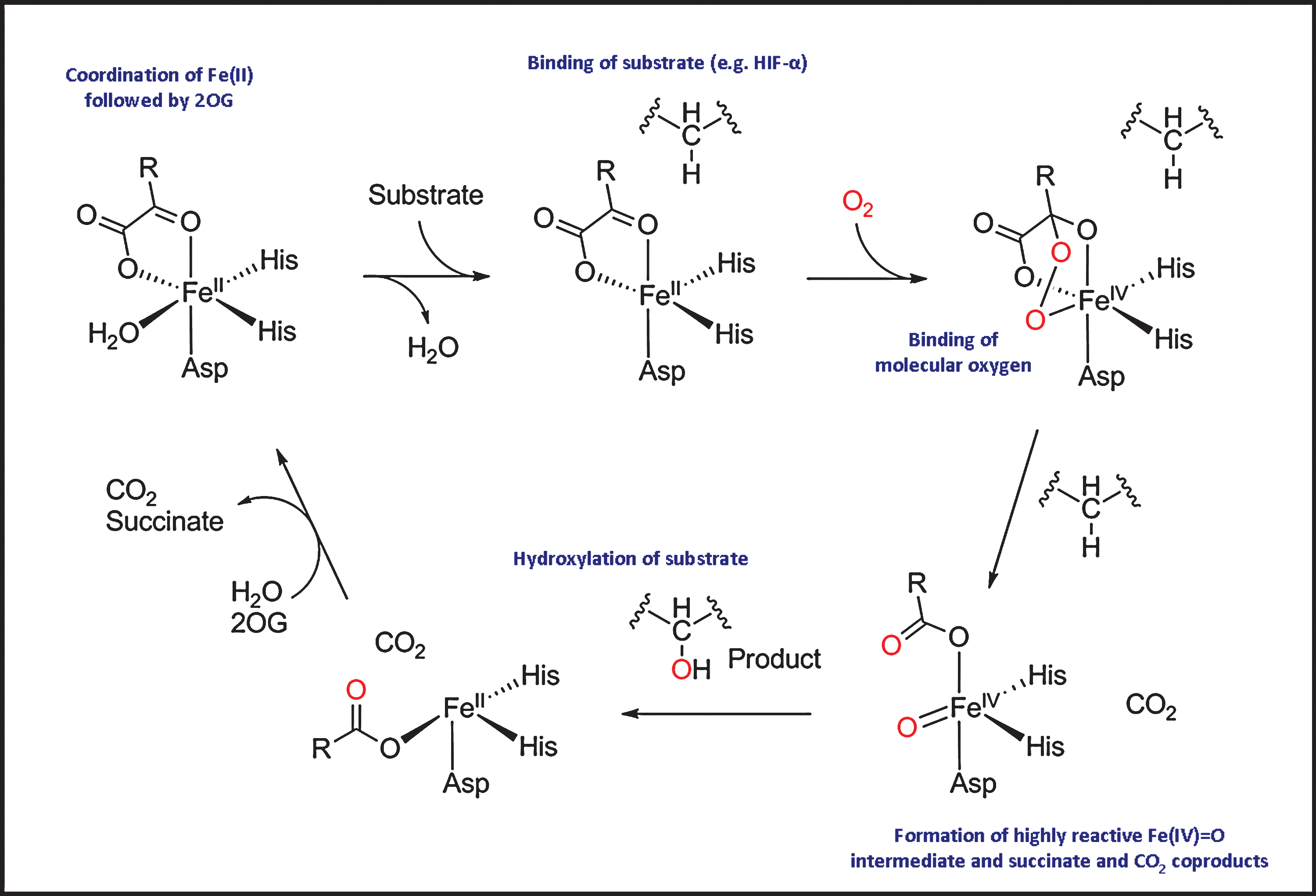
Because this general mechanism is common to most, if not all, 2OG oxygenases, selectivity is a major challenge in the development of HIF hydroxylase inhibitors. To date, compounds identified as HIF hydroxylase inhibitors (likely) act via one or more of the following four mechanisms: (a) reduction of iron availability (e.g., by chelation of iron either in solution and/or at the active site); (b) competition with iron for enzyme binding; (c) competition with 2OG for enzyme binding; (d) competition with the HIF-α substrate (Fig. 5). Here we briefly exemplify compounds from different classes of HIF hydroxylase inhibitors.
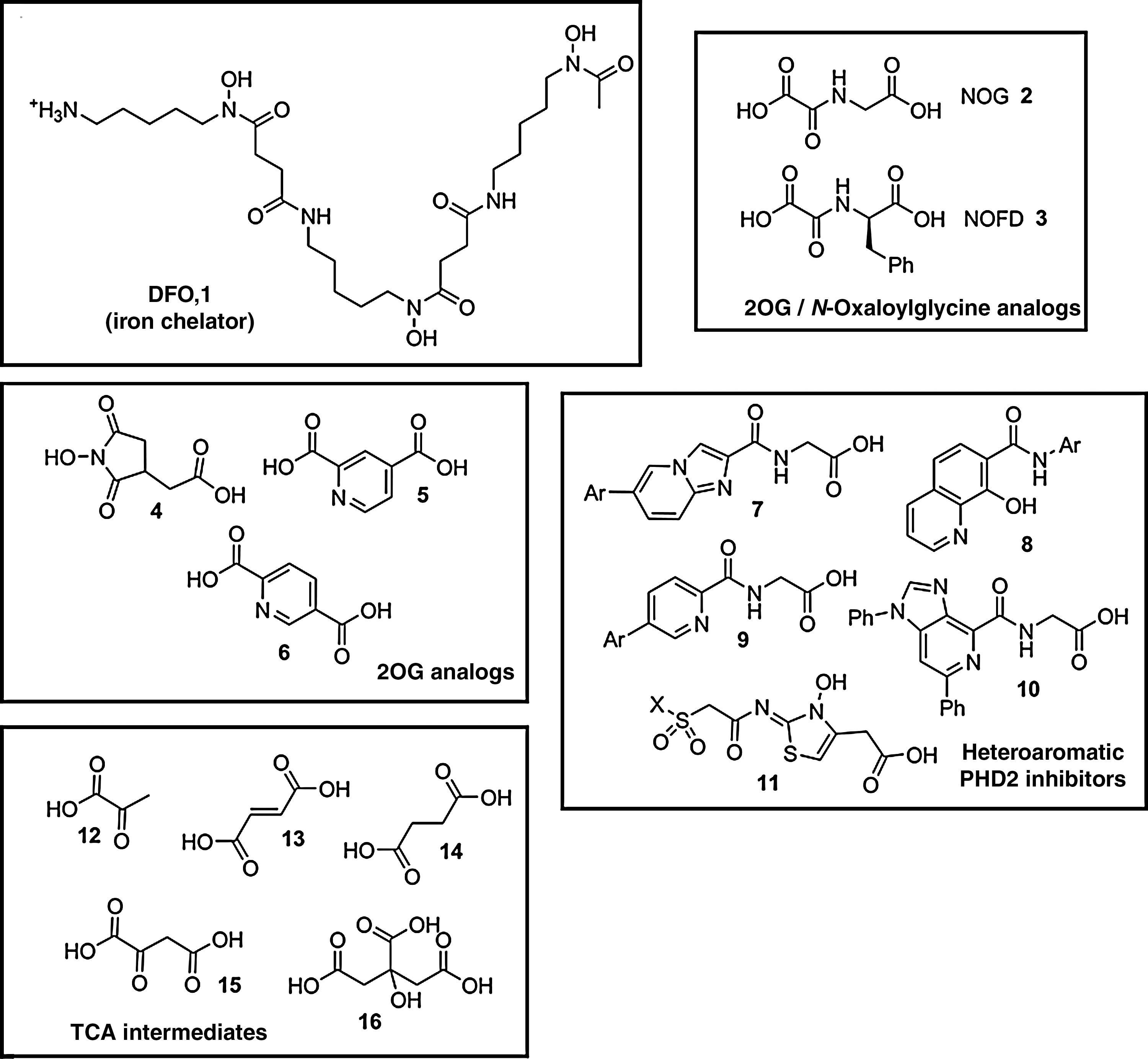
Iron chelators and competitors
Historically, cobalt [Co(II)] poisoning has been associated with stimulation of erythropoiesis (65). Indeed, Co(II) has been used in the treatment of anemia (211). The discovery of the PHD–HIF axis raised the possibility that the hypoxia-mimetic effects of cobalt and other transition metals were mediated via this pathway (54). In cell culture, Co(II), nickel [Ni(II)], and manganese [Mn(II)] are all reported to increase EPO mRNA (65), probably via HIF-α stabilization. Biochemical studies suggest competition for the metal ion binding site in the PHDs (46), but direct HIF-α-binding or depletion of cellular ascorbate are possible alternative or additional mechanisms (168, 215).
Pioneering studies also demonstrated that iron chelators [e.g., desferrioxamine (DFO, Fig. 5.1)] induce HIF-α stabilization and activate HIF transcriptional activity in cell culture (203). It is now well established that a variety of iron chelators inhibit both PHD and FIH (54, 78), very likely by decreasing the availability of Fe(II) in solution, and, possibly in some cases, by binding at the enzyme active site. DFO has been used clinically for many years in the treatment of iron-overload and aluminium-toxicity (47, 199), and has been used to examine the PHD–HIF axis in human physiology (11, 158, 183).
Kinetic studies using recombinant PHD proteins must be interpreted with a degree of caution, given the possibility of methodological differences between studies and the dependence of catalytic activity on factors including the length of HIF-α substrate and the nature of the oxygen-dependent degradation domain (e.g., N-terminal versus C-terminal). However, such experiments indicate the apparent Km values for Fe(II) binding to each PHD isoform (PHD1–3) to be ≤0.1 μM, in contrast to a higher value (∼0.5 μM) for FIH (Table 2). DFO, Ni(II), Co(II), and Mn(II) all therefore exhibit greater inhibition of FIH, compared with the PHDs, at least with isolated proteins (78). Compared with other 2OG oxygenases, PHD2 has an unusually high affinity for both Fe(II) and some other transition metal ions (127, 129). Unexpectedly, while Fe(II) binds at a single high affinity metal binding site in the catalytic domain of PHD2, some other transition metal ions can bind to the PHD2 catalytic domain at a ‘second’ metal binding site (129).
Analogues of 2OG and related compounds
Crystallographic studies reveal that, in all studied 2OG oxygenases, the 2OG binds to Fe(II) in the active site via its 1-carboxylate and 2-oxo groups. The 2OG also binds to side-chains of conserved arginine/lysine residues in the hydroxylase enzymes and to at least one active site alcohol serine/threonine/tyrosine residue via its 5-carboxylate group (35). In contrast to the binding of the 2OG to Fe(II), there are significant variations in the 2OG binding pocket. The requirement for 2OG, coupled to this variation in the 2OG binding pocket, has led the use of its analogues as HIF hydroxylase inhibitors.
N-Oxaloylglycine derivatives
N-Oxaloylglycine (NOG, Fig. 5.2) is a 2OG analogue in which the C-3 methylene group is substituted for an NH, thus making the 2-carbonyl group less susceptible to nucleophilic oxygen attack and preventing activity as a co-substrate. Originally explored as a CPH inhibitor (43), NOG is now recognized as an inhibitor of many, but not necessarily all, 2OG oxygenases, as well as other enzymes (75). Dimethyl-oxalylglycine (DMOG) is a diester derivative of NOG that penetrates cells and induces HIF-1α in cell-based assays (86).
NOG has a broad spectrum of inhibitory activity, but its derivatives hold promise as more selective inhibitors of FIH. K i values of PHD1-3 and FIH for NOG have been reported as 8–50 and 2 μM, respectively (101). Based on crystallographic data indicating that modification of the NOG side-chain may enhance FIH inhibition, N-oxalyl amino acids were synthesised and screened with recombinant FIH and PHD2 (126). For PHD2, inhibitory activity was reduced in every case, compared with NOG. For FIH, D-amino acid derivatives showed enhanced inhibitory activity. The most potent reported FIH inhibitor, N-oxalyl-D-phenylalanine (NOFD, Fig. 5.3) has also recently been shown to inhibit the histone demethylase JMJD2E, a 2OG oxygenase that is structurally more similar to FIH than PHD2 (162).
Other 2OG analogues
Inhibition of CPH by the naturally occurring cyclic hydroxymate dealanylalahopcin (76) inspired the synthesis and screening of analogues of this natural product against the PHDs (169). Although hydroxyamic acids can act as iron chelators, dealanylalahopcin analogues appear to inhibit PHDs mainly as 2OG competitors. This is supported by the greater inhibitory activity of an analogue (Fig. 5.4) with a side-chain similar in length to that of 2OG (169). Other cyclic 2OG analogues explored as inhibitors of PHDs and FIH include pyridine-2,4-dicarboxylic acid (Fig. 5.5), which was reported to be an efficient inhibitor of the PHDs and FIH, but also to inhibit other 2OG oxygenases including CPH and JMJD2E (101, 162). Its isomer, pyridine-2,5-dicarboxylic acid (Fig. 5.6), had lower activity against PHDs, FIH, and JMJD2E, but is more potent against CPH (101, 162). Just as modification of the side-chain of NOG enhanced selectivity against FIH, modifying pyridine-2,4-dicarboxylic acid or pyridine-2,5-dicarboxylic acid may be starting points for the development of selective FIH inhibitors.
Potent PHD2 inhibitors
The importance of PHD2 in human oxygen sensing has focused efforts on the identification of highly potent inhibitors of this isoform suitable for therapeutic application. A combination of crystallographic analysis and structure-based design has, for example, been employed to identify inhibitors based on analogues of an imidazo[1,2-a]pyridine scaffold with a glycine side-chain [(125, 207), Fig. 5.7] These PHD2 inhibitors induced cellular VEGF by stabilizing HIF-1α, and similar results have been demonstrated by the same group using a series of 8-hydroxyquinolines [(208), Fig. 5.8] or 5-substituted pyridine derivatives [(206), Fig. 5.9] Structure-based design has also identified substituted aza-benzimidazoles (Fig. 5.10) as PHD2 inhibitors, and provided insights into the structural requirements for tight binding and potent PHD2 inhibition (60). Hydroxy-thiazoles (Fig. 5.11) are also potent inhibitors of PHD2, with structure–activity relationships studies demonstrating particular importance of the carboxylic acid side-chain and N-hydroxy functionality (196). To date, it has not been reported whether these inhibitors are selective for PHD2 over PHD1 or PHD3, but clearly this is of interest with respect to their development as therapeutic agents.
Citric acid cycle intermediates and other inhibitors
Citric acid cycle intermediates, including pyruvate (Fig. 5.12), have been investigated as HIF hydroxylase inhibitors (73, 84, 102, 173). Among them, fumarate (Fig. 5.13) and succinate (Fig. 5.14) were identified as in vitro inhibitors of PHD1-3 with K i values of 50–80 μM for fumarate and 350–460 μM for succinate. Oxaloacetate (Fig. 5.15) showed modest inhibition of PHD1–3 (K i 400–1,000 μM) and FIH (K i 400 μM), whereas citrate (Fig. 5.16) was more selective for FIH (K i 110 μM). The concentration of these intermediates may reach sufficiently high levels to inhibit the HIF hydroxylases in some cells, in particular in tumors where mutations can lead to elevated levels of succinate and fumarate (152).
Recently, aspirin metabolites were also identified as potential inhibitors of HIF hydroxylases. Mass spectrometric binding studies and in vitro inhibition data revealed significant PHD and FIH inhibition, and some metabolites induced HIF-1α accumulation and HIF target gene expression in cell-based studies (114), though the mechanism by which this occurs is uncertain. Traces of some of these metabolites were identified in human urine following oral aspirin administration, raising the possibility that activation of the HIF pathway could contribute to the biological actions of aspirin.
Therapeutic Applications of PHD Inhibition
PHD inhibition has emerged as a potential therapeutic strategy in numerous pathophysiological settings, including myocardial ischemia, cerebrovascular disease, and anemia. Progress in these areas has to date been limited mainly to preclinical models of disease, with little translation into the clinical domain and a scarcity of published data from human studies. Nevertheless, we review some of the promising advances below, and in Table 3 provide an overview of selected PHD inhibitors that have been used to manipulate the oxygen sensing system in vivo, or have clear implications in a clinical setting.
FG-2216, FG-3019, FG-4592, and FG4539 have undergone investigation in clinical trials (see
MCAO, middle cerebral artery occlusion.
Myocardial ischemia
A link between prolyl hydroxylation and myocardial ischemia dates back to 1975, when Judd and Wexler reported that changes in myocardial collagen deposition and fibrosis following experimental myocardial infarction (MI) were associated with increased CPH activity in the rat (90). In 2001, Nwogu and colleagues administered the oral 2OG oxygenase inhibitor FG0041 to adults rats for 4 weeks following experimental MI (142). Left ventricular function was significantly enhanced, an effect attributed by the authors to reduced extracellular matrix collagen deposition, but one which could now be attributed to inhibition of the HIF hydroxylases (151) or, possibly, other 2OG-dependent oxygenases.
Ischemic preconditioning (IPC) is a process by which repeated short periods of cardiac ischemia and reperfusion protect against subsequent ischemia (134). There is now considerable evidence for involvement of the HIF system in this process, particularly in the so-called ‘late phase’, which develops a number of hours after the ischemic insult, lasts for several days, and classically involves the transcriptional activation of HIF target genes including heme-oxygenase-1 (HO-1), cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase [iNOS, (20)]. The beneficial effects of cardiac preconditioning are lost in HIF-1α+/- mice (27), or in mice treated with small interfering RNA (siRNA) against HIF-1α (50). In contrast, the effects of IPC can be mimicked in normal mice by administration of cobalt chloride, DMOG or siRNA specifically targeting PHD2 (50, 212). This effect was not seen after inhibition of either PHD1 or PHD3, implicating the PHD2-HIF-1α axis in this process (50). In similar experiments, PHD2 inhibition appeared to induce substantial HO-1 and iNOS expression both in cell culture and in adult mice, an effect that was associated with an attenuated acute inflammatory response to myocardial ischemia (138, 139, 144).
Although these findings support an important role for the PHD–HIF pathway in IPC, direct evidence remains difficult to obtain. The expression of key mediators such as HO-1, COX-1, and iNOS is under the control of several other signaling pathways that are activated (or inhibited) by ischemia. The nuclear factor-κB (NF-κB) transcription factors, for example, are well-established as important mediators of cardiac IPC (194, 213). In addition to functional interaction between the HIF and NF-κB signalling pathways at the level of gene expression, there appears to be direct molecular interaction and interdependence (36, 194). This complexity suggests considerable potential for diverse and unexpected effects of in vivo PHD inhibition.
Despite these mechanistic uncertainties, several indirect observations enhance the therapeutic potential for PHD-HIF manipulation in ischemic cardiac disease. First, sustained activation of the HIF pathway raises the possibility of post-event intervention. In mice subjected to myocardial infarction, increased levels of PHD2 and PHD3 persisted in peri-infarction zones for at least 7 days, and co-localized with increased levels of HIF-α chains and the HIF-regulated gene products Glut-1 and HO-1 (210). Second, increased levels of PHD3, with an associated decrease in HIF-1α, have been reported in the aging mouse and human heart (161), suggesting that the pathway may be particularly amenable to therapeutic manipulation in the elderly population. Similar findings have been reported in the rat brain (140). Finally, involvement of HIF-1α in human cardiac disease is supported by recent studies associating polymorphisms in the HIF-1α gene both with the formation of coronary collaterals and with the nature of initial presentation in patients with coronary artery disease (79, 159). Larger association studies are required to confirm and reconcile the findings of these preliminary studies. Resar et al. (159) reported in a group of 100 patients with coronary artery disease that the presence of the single nucleotide polymorphism at HIF-1α residue 582 (C to T) was negatively associated with the formation of coronary collaterals, and speculated that this polymorphism may reduce cardiac HIF-1 activity. Hlatky et al. (79) later reported that the same polymorphism did indeed reduce HIF-1 activity in cell culture. In a group of ∼1,350 patients in this study, its presence was a predictor of stable angina, rather than myocardial infarction, as a first presentation of coronary disease.
Cerebral hypoxia and ischemia
Over 1000 experimental pharmacological interventions have failed to prevent the progression of infarction due to cerebral ischemia (143) and only one, thrombolysis, has been shown clearly to be effective in the setting of acute stroke (68). Traditionally, treatments have been directed against one specific pathophysiological target, for example, N-methyl D-aspartate (NDMA) receptor antagonism, calcium antagonism, or free radical scavenging. More recently, research has focused on multi-modal interventions with pleiotropic effects (154). Hypothermia and IPC are probably the most effective experimental strategies (80, 136), with the former now approved for patients after cardiac arrest and in children with hypoxic-ischemic encephalopathy (15, 179). They offer protection by both inhibiting deleterious cellular pathways and upregulating antiapoptotic and neuroprotective genes, including members of the Akt/protein kinase B pathway (216)
As in the heart, there is evidence that the HIF pathway is activated by IPC in the brain (14, 63, 88). Prior to the discovery of the HIF hydroxylases, the ability of iron chelators and cobalt chloride to reduce cell loss in models of focal and global ischemia was attributed to reduced oxidative stress (10, 146, 147, 188). Subsequent studies suggest inhibition of the HIF hydroxylases as the underlying mechanism of these protective effects, and PHD inhibitors have been reported to mimic the neuroprotective effects of preconditioning both in vitro and in vivo (70, 182). Siddiq et al., for example, synthesized a peptide that binds all three PHD isoforms, stabilized HIF-1α, activated HIF target genes, and protected embryonic rat cortical neurons from oxidative stress (182). Other novel compounds, for example, TM6008 and TM6089, believed by the authors to be HIF–PHD specific, exhibited neuroprotective properties after global cerebral ischemia in the gerbil. Unusually, these compounds did not increase VEGF levels (137). This hypoxically-regulated angiogenic factor has previously been described as neuroprotective (190), but in the brain appears to mediate both neuroprotection and potentially deleterious changes in blood-brain barrier permeability, a dual role which may have implications for the therapeutic use of PHD inhibition in stroke (96, 111). Other specific HIF targets that are showing promise as neuroprotectants include insulin-like growth factor-1 (IGF-1, (105)) and erythropoietin (EPO). Indeed, the latter provides one of the few examples of clinical data on the therapeutic potential of HIF target genes, with several small clinical trials suggesting a beneficial effect of EPO on neurological outcome after cerebral ischemia (51, 69, 200). It will be interesting to see whether these exciting preliminary results can be replicated or improved upon in the future using PHD inhibitors.
Significantly, given the scarcity of post-event treatment options in acute stroke, the role of PHD inhibition after the ictus remains controversial (12, 31, 70, 182). In a mouse model of focal ischemia, post-ischemia PHD inhibition offered less protection than pre-ischemic treatments (12). In rat pups, Chen et al. found that early (i.e., immediately after 10 min of severe cerebral hypoxia and ischemia) activation of HIF-1α with DMOG worsened brain edema and blood-brain barrier disruption, compared with hypoxia/ischemia alone (31). This effect was associated with increased levels of VEGF. In this context it is of interest that in a rat model of focal cerebral ischemia, PHD inhibition by pretreatment with DMOG might not only be cytoprotective, but also improve cerebral blood flow by upregulating VEGF and endothelial nitric oxide synthase (unpublished data, Nagel et al.). The role of HIF in this effect is supported by the observation that DFO given during temporary cardiac arrest in the rat appears to enhance cerebral perfusion and improve neurologic deficit score in the first 20 min following resuscitation (113). Effects on cerebral blood flow have also been suggested in studies of other neuroprotective candidates, and can complicate interpretation of experimental results (56).
The effects of PHD inhibition in the brain may depend not only on timing of the stimulus, but also on cell type. In cell culture studies, selective loss of HIF-1α in astrocytes provides protection against hypoxia, whereas loss of HIF-1α in neuronal cells increases susceptibility to hypoxia-induced damage (201). This may relate to cell-specific differences in HIF-regulated gene expression. A perceived advantage of PHD inhibition over the administration of specific HIF target proteins, such as VEGF, EPO, or IGF-1, is the activation of a wide array of protective genes, with potential for synergistic beneficial effects. However, this also raises the potential for the activation of deleterious cellular pathways. BNIP3, for example, is a member of the BH3-only family of proteins that is known to be upregulated by HIF activation during hypoxia (23), and the expression of which has been linked to hypoxia-induced cell death in both the heart and brain (32). This balance of beneficial and deleterious effects of HIF activation in brain or other tissues may be very difficult to predict from cellular studies or animal models, and translation of promising experimental results into clinical advances seems likely to require a greater emphasis on in vivo human studies in the future (155, 157).
The efficacy of any systemic neuroprotectant will depend upon its ability to access the central nervous system. In contrast to most PHD inhibitors, the known oral antiviral agent tilorone has recently been identified as a potent HIF-activating agent that readily crosses the blood-brain barrier in rats. The mechanism of action of tilorone remains unknown (156). It is thought to be independent of HIF hydroxylase inhibition (though the metabolism of tilorone to iron chelating agents cannot be ruled out entirely).
Anemia and kidney disease
Commercial interest associated with the use of recombinant human EPO, in combination with emerging knowledge of the mechanisms regulating renal EPO expression (64, 189) has stimulated interest in the use of PHD inhibitors for the treatment of anaemia.
In the mouse, oral or intravenous PHD inhibition inhibits renal HIF hydroxylase activity and increases EPO production (81, 166). In male rhesus macaques, the oral PHD inhibitor FG-2216 induced significant and reversible EPO induction (81). FG-2216 was well-tolerated over several months, increasing erythropoiesis and preventing anemia induced by weekly phlebotomy. In July 2007, the biotechnology company FibroGen completed a phase-2 clinical trial of FG-2216 in patients with renal anemia, and is currently recruiting for a similar trial of the compound FG-4592 in patients with anemia and chronic kidney disease (see
Intravenous administration of DFO increases EPO production in healthy volunteers (11, 158, 183) and excessive erythropoiesis has been reported in families with HIF-activating mutations (3, 61, 108, 149, 150). In combination with data from genetically engineered animals (see section on Insights from genetically engineered animals), these observations have identified HIF-2α and PHD-2 as the likely isoforms regulating EPO production in humans (149, 150). Such tissue-specificity may guide the development of system-specific PHD inhibitors. In the context of anemia, it is interesting that excessive erythrocytosis in the mouse results both from somatic PHD2 inactivation and from germ-line PHD1/PHD3 double inactivation. This suggests a degree of redundancy in the system, although PHD1/PHD3 deficiency appears to stimulate erythrocytosis mainly through a hepatic pathway, whereas PHD2 deficiency has its predominant effect via renal EPO production (191).
PHD inhibitors also show promise in renal ischemic disease. Ischemia in the tubulointerstitium is thought to play a pivotal role in the pathophysiology of acute renal failure and the progression of chronic kidney disease (92). In a rat model of acute ischemic injury, pre- and post-ischemia treatment with cobalt chloride reduced tubolointerstitial damage and decreased serum creatinine levels. Cobalt reduced macrophage infiltration and increased the renal expression of several cytoprotective HIF target-gene products, including EPO, VEGF, and HO-1 (122). The PHD inhibitor FG-4487 mimics the renoprotective effects of hypoxic preconditioning in mice, reducing tissue injury and apoptosis (16), and the compound FG-4497 protects against distal tubular injury in isolated perfused rat kidneys (164). Furthermore, HIF hydroxylases may also be involved in mediating the cardioprotective effects of remote renal preconditioning ((93), see section on HIF hydroxylation substrates and co-factors).
Gastrointestinal disease
Recent evidence suggests a role for HIF-1α in mucosal barrier function in the gut (195). Both DMOG and the specific PHD inhibitor FG-4497 are reportedly protective in mouse models of inflammatory bowel disease (42, 160). In the case of DMOG, this protection was associated with reduced apoptosis of colonic epithelial cells (42). Colitis is more severe in mice with conditional intestinal epithelial cell knockout of HIF-1α (94), and in rats the iron chelator quercetin attenuated experimentally-induced colitis via activation of HIF-1 and VEGF. This effect was abolished by iron supplementation (87).
CPH inhibitors have previously been used in the treatment of experimental liver fibrosis (18, 123, 167). The small molecule S4682, for example, reduced hepatic collagen accumulation and decreased prevalence of ascites in a rat model of liver injury (18). As 2OG analogues, such inhibitors are also likely to have inhibited the HIF hydroxylases, and a growing body of evidence implicates VEGF in the pathology of hepatic injury and repair (22, 38, 165).
Other applications for HIF hydroxylase inhibition
PHD inhibition may be a therapeutic target in several other clinical settings, such as retinal diseases characterized by inadequate vascularization, including early retinopathy of prematurity and retinitis pigmentosa (reviewed in ref. 8). PHD inhibitors may also mimic the protective effects of retinal ischemic preconditioning. In the mouse, pre-treatment with DFO attenuated ischemia-induced retinal injury. This effect was associated with increased HIF-1α levels and upregulation of adrenomedullin, a HIF-dependent gene product previously identified as neuroprotective in the retina (198, 217). Induction of the HIF pathway during ischemic preconditioning may also be of value in transplantation medicine. A recent small randomized controlled trial in 60 liver transplant donors demonstrated that a 10-min period of liver ischemia in the donor significantly improved post-transplantation liver function (2). This effect was associated with increased hepatic HIF-1α levels, although no evidence was presented for induction of HIF target genes.
Finally, there has been interest in the potential role for HIF-mediated gene expression in the pathogenesis of placental disease. Placental vascularization is abnormal in HIF-1α-/- or HIF-2α-/- mouse embryos (40), and HIF-1-mediated upregulation of the soluble VEGF receptor sFlt-1 has been implicated in the etiology of pre-eclampsia (141). In contrast, however, it has been suggested by other authors that HIF activation may in fact protect against pre-eclampsia (67). This possibility is supported by the intriguing report of a higher incidence of the disease in nonanemic women taking iron supplements, which could theoretically promote HIF-α breakdown by PHD-activation (185, 218). In common with all the potential therapeutic areas discussed above, considerable further work will be needed before the potential positive and negative effects of PHD manipulation in this context can be appropriately balanced.
Human models of long-term HIF upregulation
As noted above, there are few published reports of detailed studies on the pharmacological effects of PHD inhibition and/or HIF upregulation in humans. However, important (and possibly cautionary) lessons may be learnt from patients with rare monogeneic disease involving the PHD-HIF pathway. Chuvash polycythemia, first described in the Russian Chuvash population, is an autosomal recessive disorder characterized by excessive erythrocytosis, pulmonary hypertension, and premature mortality due to cerebral vascular events and peripheral thrombosis (26, 66, 184). Chuvash polycythemia is caused by a specific 598C>T mutation in the VHL gene, which reduces the affinity of the VHL protein for hydroxylated HIF-1α, inhibits HIF-1α degradation, and results in excessive activation of target genes including EPO, VEGF, and the vasoconstrictor endothelin-1 (3). Patients with Chuvash polycythemia appear not to be at risk of developing classical von Hippel-Lindau disease, in which congenital inactivation of one VHL allele predisposes to the development of vascular tumours such as hemangioblastomas and renal cell carcinoma (91). Nevertheless, these two clinical syndromes, both of which are associated with excessive activation of the HIF pathway (3, 91), highlight the requirement for long-term studies to investigate the safety of iatrogenic PHD inhibition in humans, and reinforce the likely importance of tissue- and/or PHD isoform-specific interventions.
Activation of HIF Hydroxylases
Agents that specifically increase the hydroxylation activity of PHDs are relatively unexplored (Table 4). Such agents have potential therapeutic value for conditions in which HIF-dependent gene expression contributes to disease pathology. PHD activators identified to date fall into two categories: they are either components of the HIF hydroxylation reaction, or specific small molecule activators of the PHD enzymes.
HIF hydroxylation substrates and co-factors
In cultured human cells, supraphysiological supplementation of both iron and ascorbate enhances PHD activity and potentiates the degradation of HIF (98, 99). Ascorbate is proposed to promote the availability of Fe(II) to the active site of the enzyme (99), most likely by maintaining iron in the active reduced Fe(II) state. Among all the agents that have been shown to influence PHD activity, iron and ascorbate uniquely share the advantage of having been in routine clinical use for decades. They are readily available, safe, and inexpensive, and so deserve special attention as potential therapeutic modulators of HIF hydroxylation.
As discussed above, 2OG is a rate-limiting co-substrate for the PHDs under some circumstances (86, 121). In normoxic cultures of human cells, increasing intracellular levels of 2OG stimulates PHD activity and markedly reduces basal expression of HIF-1α protein (118), raising the possibility that 2OG itself could be used to accelerate HIF degradation therapeutically. This potential has yet to be explored in man, but 2OG has been employed as a PHD activator in animal studies. In a rat model of remote ischemic preconditioning, PHD inhibition and renal preconditioning both had cardioprotective effects that were abolished by prior intraperitoneal administration of 2OG (93).
Small molecule activators
In 2001, it was reported that hypoxia raises the intracellular level of the lipid second messenger phosphatidic acid, primarily through the action of diacylglycerol kinase (6). Pharmacological inhibition of diacylglycerol kinase using the specific inhibitor R59949 was further shown to impair the hypoxia-induced accumulation of HIF-1α (6). It has since been proposed that R59949 inhibits the accumulation of HIF-1α and HIF-2α by increasing PHD activity in cells (197). The significance of this link between lipid second messengers and PHD–HIF oxygen sensing is not yet understood, nor is the mechanism of action for R59949. Suggested activating mechanisms include increasing the affinity of PHDs for oxygen, regulating intracellular pools of ascorbate or iron, altering levels of reactive oxygen species (ROS), and inhibition of unidentified negative regulators of PHDs (197).
Targeted screening of a molecular library recently identified the compound KRH102053 as a novel small molecule activator of PHD2 (33). This compound increased PHD2 activity by 26% in vitro, and decreased levels of HIF-1α and selected HIF target gene products in a variety of cell types. This study focused on PHD2, considered to be the most important PHD isoform for the control of HIF-1α in normoxia (4). The authors hypothesized that PHD2 could be regulated via redox-related pathways, and screened a library of more than 600 chemicals with a benzopyran moiety and antioxidant properties. Several PHD2 activating compounds were identified, of which KRH102053 was the most effective. Further work is required, not least because the in vitro activation was relatively small, and the mechanism of action unknown.
The immunosuppressive drug cyclosporin-A, used after organ transplantation and in the treatment of severe autoimmune disease, has also been shown to abrogate hypoxic stabilisation of HIF-1α and HIF-1α-mediated cellular responses (45, 104, 115). This effect, which may contribute to the nephrotoxicity associated with the drug, has been attributed by some authors to a direct increase in PHD activity (45). Others studies, however, suggest a PHD-independent mechanism of action (104, 115), and further experiments will be required to resolve this issue.
Therapeutic Applications for PHD Activation
Research into therapeutic PHD activation is much less advanced than for PHD inhibition, yet the potential for therapeutic benefit is clear, particularly in the field of oncology. Other important applications could include pulmonary hypertension and proliferative retinopathies. The identification of clinically useful small molecule activators may possibly benefit from the development of structural analogues of R59949 and KRH102053 (33, 197). In the meantime, there have been interesting results from studies of ascorbate and iron in cancer and pulmonary hypertension, respectively.
Cancer therapy
HIF-1α expression and activity is increased in many human cancers as a result of intratumoral hypoxia (64, 175). HIF influences all major aspects of cancer biology, promoting cell survival in the hypoxic microenvironment by increasing the expression of proteins that regulate metabolic adaptation, resistance to apoptosis, angiogenesis, and invasion and metastasis (174). Therapeutic inhibition of HIF in solid malignancies is a target of intense investigation, and many anticancer agents under development or in clinical use have some activity as HIF inhibitors (175). The potential for inhibition of HIF through activation of the PHDs has not been specifically exploited, although the history (albeit controversial) of ascorbate as a cancer treatment provides an intriguing link.
Interest in the use of ascorbate in cancer treatment was stimulated in the 1970s when the Nobel Laureate Linus Pauling, together with British cancer surgeon Ewan Cameron, published retrospective data suggesting possible benefit of ascorbate in patients with cancer (28, 29). Subsequent double-blind placebo-controlled trials failed to replicate these findings (41, 133), and while ascorbate treatment has remained popular with alternative medicine practitioners, it has not since featured in mainstream oncology practice. However, there has recently been increasing interest in ascorbate as a cancer therapy, with in vitro and in vivo work and some limited human data suggesting that very high dose ascorbate (equivalent to intravenous human doses of up to 100 g/day) may have positive effects in some tumours (59). These results have been ascribed largely to redox-mediated effects (30), but the precise mechanism of action of ascorbate remains unknown. Further work will be needed to investigate whether activation of the HIF hydroxylases could contribute, but this possibility is supported by the recent finding of an antitumorigenic effect of ascorbate in mouse tumour models, which was dependent upon PHD2 activation and HIF-1α downregulation (62). Nevertheless, although it is possible that ascorbate therapy will find a place in combination with other cytotoxic agents, the ‘evidence that vitamin C could help human cancer patients is still thin’ (21).
Pulmonary hypertension
Through recent work in animals (25, 180, 214), including humans (26, 61, 183, 184, 186), it has become apparent that HIF plays an important role in regulating the development of hypoxia-induced pulmonary hypertension. This results from hypoxic pulmonary vasoconstriction and vascular remodeling and is a major cause of morbidity and mortality among patients with lung disease and at high altitude (13). Inhibition of HIF through increasing PHD activity (or by other means) is a promising new therapeutic avenue in pulmonary hypertension. Interestingly, cyclosporin-A attenuates the development of hypoxia-induced pulmonary hypertension in rats and mice, although this effect may well be mediated by HIF- and/or PHD-independent cellular pathways (48, 104, 106, 115).
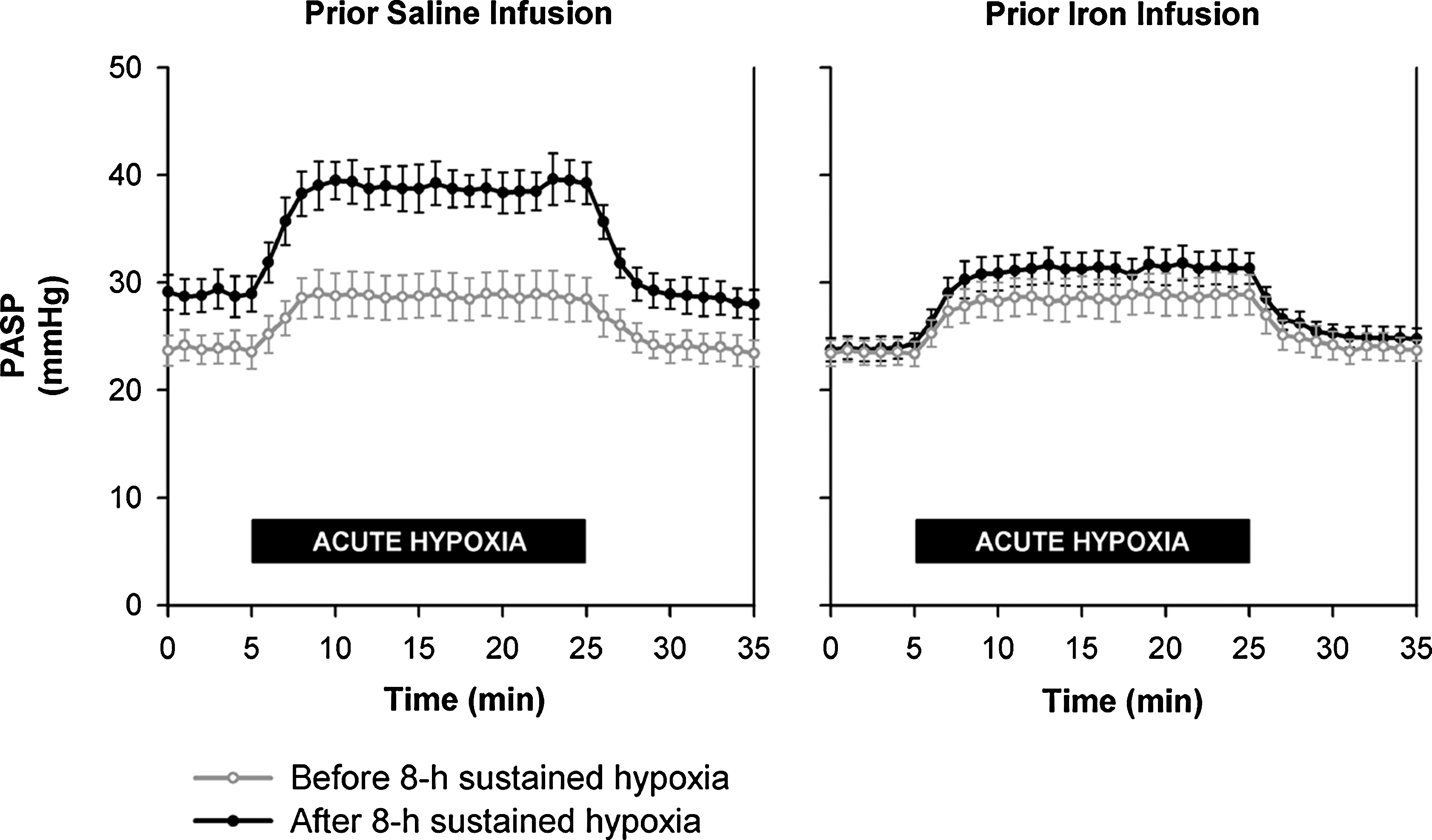
Particularly interesting is the emerging body of work investigating the effects of iron on the pulmonary vasculature in humans (89). Chelation of iron with DFO mildly elevates pulmonary arterial pressure (11), and a recent study established the existence of a substantial interaction between iron, hypoxia, and the pulmonary circulation (183). In a series of 8-h experiments in healthy volunteers, intravenous infusions of iron blunted pulmonary vasoconstrictive responses to hypoxia and chelation of iron with DFO enhanced the constrictor response (183) (Fig. 6). These studies were predicated on the known requirement for iron of HIF hydroxylation, and the established involvement of HIF in pulmonary physiology, but changes in other oxygen-sensitive pathways cannot be excluded. Such an effect could arise through the interaction of HIF hydroxylases with other pathways, through HIF-PHD-independent redox-mediated events, or through an effect of iron on previously unidentified iron dependent oxygenases or other factors. Nevertheless, these results may herald a therapeutic potential for PHD activation in hypoxic pulmonary hypertensive disease (89).
Vasoproliferative retinopathies
Vasoproliferative retinopathies include major causes of visual impairment and blindness such as proliferative diabetic retinopathy and retinopathy of prematurity. These are characterized by retinal hypoxia and neovascularization in which HIF plays a mediating or contributory role (8). Much attention has been focused on the potential for treating these conditions by inhibiting HIF activity, and early results obtained from studies in animal models have been promising (97, 172). Vasoproliferative retinopathies cause substantial morbidity, and the development of PHD-activators that are safe and effective in treating these diseases would be very welcome.
Conclusions
A growing body of experimental and preclinical data suggests that inhibition of the HIF hydroxylases may be of benefit in some of the leading diseases in the developed world, including ischemic heart disease, cerebrovascular disease, and anemia. Similarly, enhancing PHD (and maybe FIH) activity holds promise in cancer, pulmonary vascular disease, and retinal disease. The proposed benefit in all these conditions is usually attributed to the influence of these enzymes on the HIF system, and there is now good evidence for specific roles for HIF-α and/or PHD isoforms in particular physiological and pathological processes. As our understanding of tissue and temporal specificity with the HIF system develops further, so too will the potential for specific therapeutic intervention. Novel in vivo imaging techniques, for example, the use of noninvasive bioluminescent reporter assays for enzyme activity (166), may provide new insights in this area by allowing downstream physiological effects to be more accurately attributed to changes in the activity of specific enzymes or signaling pathways. In addition, recent data suggesting non-HIF targets for the HIF hydroxylases, particularly FIH (37, 209), may be very important both for targeting therapy and understanding or avoiding non-therapeutic effects.
Despite the undoubted potential in this field, promising preclinical developments have yet to translate into important clinical advances. Ongoing clinical trials investigating the use of PHD inhibitors in renal anemia appear encouraging, but peer-reviewed results are not yet available. Further clinical and in vivo studies will be necessary to clarify the effects of human HIF hydroxylase manipulation, and in particular to move towards an answer to important questions about the specific therapeutic niche of any such intervention. In ischemic vascular disease, for example, does PHD inhibition have a role in the acute setting, or should its promise be considered mainly in primary and/or secondary prevention? In the setting of chronic disease such as hypoxia-induced pulmonary hypertension or cancer, could simple correction of iron deficiency influence disease progression by altering PHD activity? Do well-established therapeutic agents such as aspirin (114) have as yet unknown effects on the HIF hydroxylases? Mirroring the importance of the HIF pathway in human health and disease, the possible therapeutic applications for HIF hydroxylase manipulation are myriad. The translation of this preclinical potential into clinical advances represents a considerable, but very exciting, challenge.
Footnotes
Acknowledgments
The authors wish to thank the Biotechnology and Biological Sciences Research Council and the European Union (JM, CJS), the Newton-Abraham Fund (JM), the Wellcome Trust (NPT, JM, TGS, CJS), the Medical Research Council (SN, AMB), and the Deutsche Forschungsgemeinschaft (SN) for financial support.
Author Disclosure Statement
CJS is a cofounder of ReOx, a company that aims to exploit research about the hypoxic response for therapeutic benefit.