
Abstract
Nitric oxide (NO), plays multiple roles in the nervous system. In addition to regulating proliferation, survival and differentiation of neurons, NO is involved in synaptic activity, neural plasticity, and memory function. Nitric oxide promotes survival and differentiation of neural cells and exerts long-lasting effects through regulation of transcription factors and modulation of gene expression. Signaling by reactive nitrogen species is carried out mainly by targeted modifications of critical cysteine residues in proteins, including S-nitrosylation and S-oxidation, as well as by lipid nitration. NO and other reactive nitrogen species are also involved in neuroinflammation and neurodegeneration, such as in Alzheimer disease, amyotrophic lateral sclerosis, Parkinson disease, multiple sclerosis, Friedreich ataxia, and Huntington disease. Susceptibility to NO and peroxynitrite exposure may depend on factors such as the intracellular reduced glutathione and cellular stress resistance signaling pathways. Thus, neurons, in contrast to astrocytes, appear particularly vulnerable to the effects of nitrosative stress. This article reviews the current understanding of the cytotoxic versus cytoprotective effects of NO in the central nervous system, highlighting the Janus-faced properties of this small molecule. The significance of NO in redox signaling and modulation of the adaptive cellular stress responses and its exciting future perspectives also are discussed. Antioxid. Redox Signal. 11, 2717–2739.
Introduction
The main targets of NO under physiologic conditions are metal centered, particularly iron in iron-heme proteins. The most striking example is the formation of a nitrosyl complex with iron (II) of sGC, inducing a conformational change of the enzyme and its activation to produce cGMP and the subsequent activation of protein kinase G (PKG) and protein phosphorylation (32). Another major pathway by which NO mediates its biologic effects (cGMP-independent pathway) is the S-nitrosylation of cysteine residues in multiple cell types and tissues (72). Alternatively, NO has been shown to mediate posttranslational modification of proteins, such as ADP ribosylation (68) and nitration of tyrosine (Tyr) residues (20). These modifications may prevent normal interactions between proteins involved in presynaptic membrane-specific interactions occurring during exocytosis or in inhibition of cell death by apoptosis (171).
A role for NO as an intercellular diffusible messenger in the brain was demonstrated for the first time a few decades ago (78). Since then, both its physiologic role, mainly related to regulation of neuronal proliferation/survival/differentiation as well as to mediation of synaptic activity/neural plasticity, and its contribution to several neuropathologic states have been addressed in thousands of articles (for comprehensive reviews, see 31, 57). Of particular interest is the finding that NO, known to exert an antiproliferative action toward many cell types (33), also promotes a similar effect toward neural-derived tumor cells, as well as toward neuronal precursors (156). NO itself is a poorly reactive species; however, it is able to undergo secondary reactions to form highly oxidizing and nitrating species (viz., NO2, N2O3, and ONOO−). These secondary reactive nitrogen species (RNS) are capable of modifying a diversity of biomolecular structures in the cell. Nitric oxide is produced from
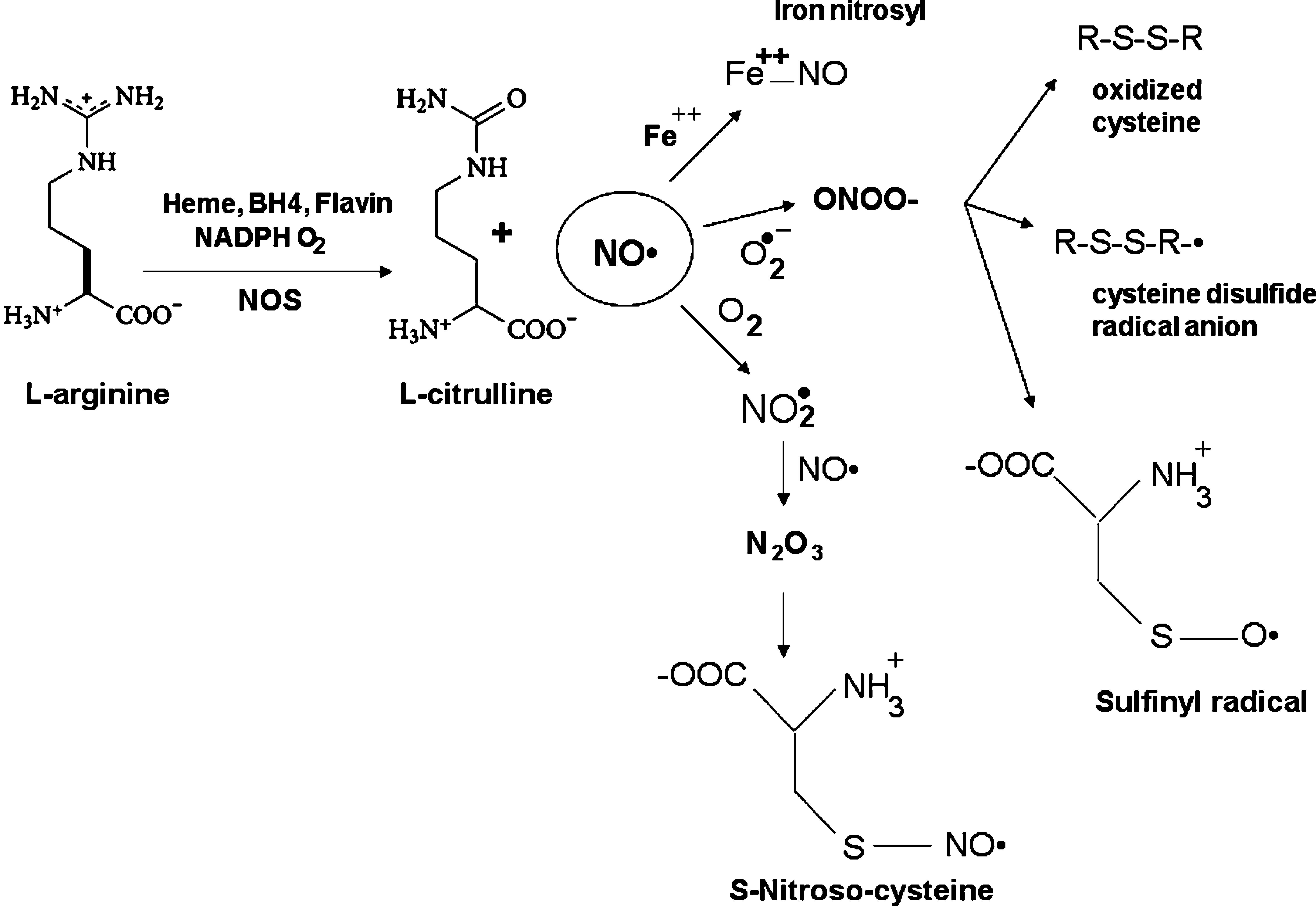

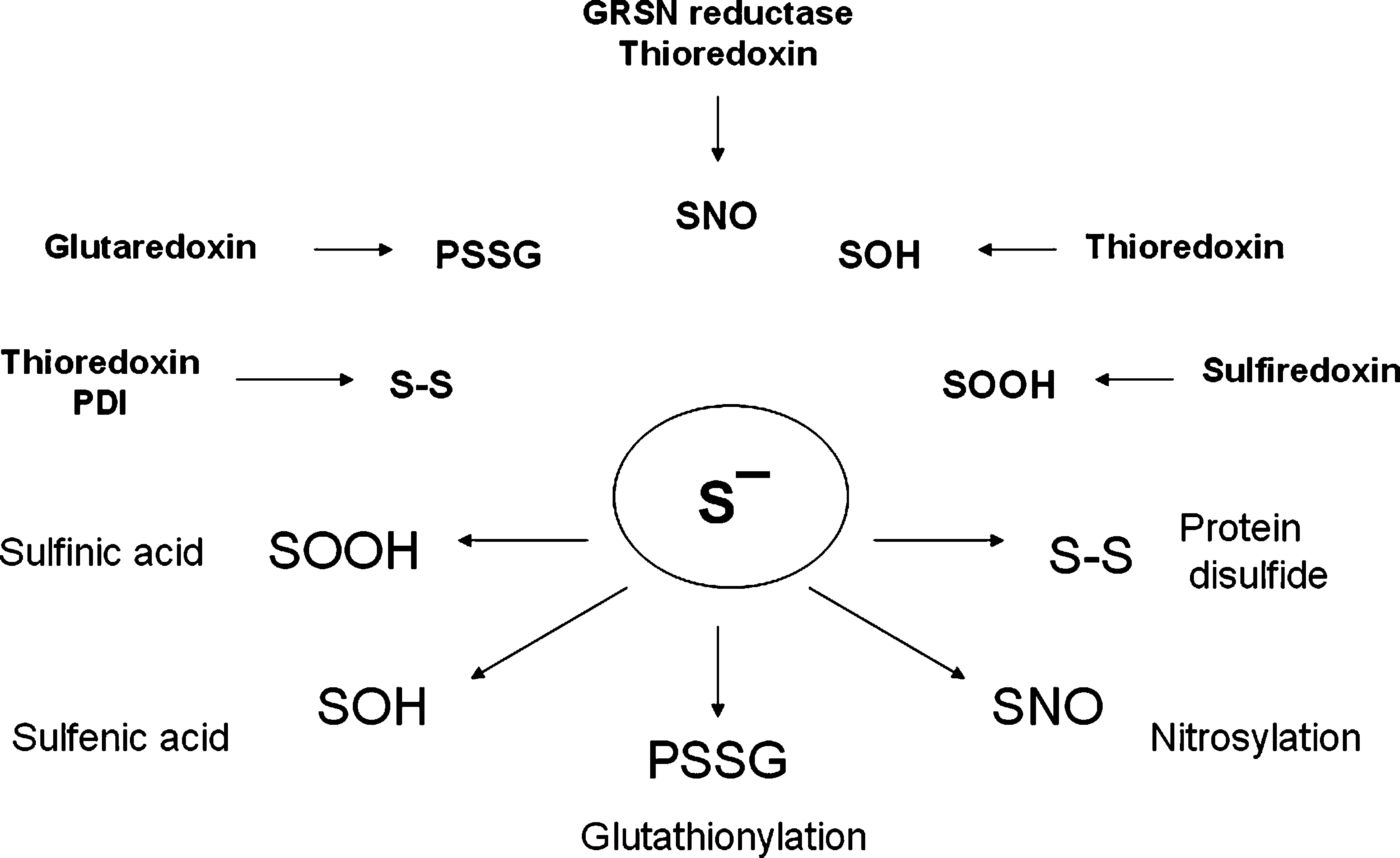
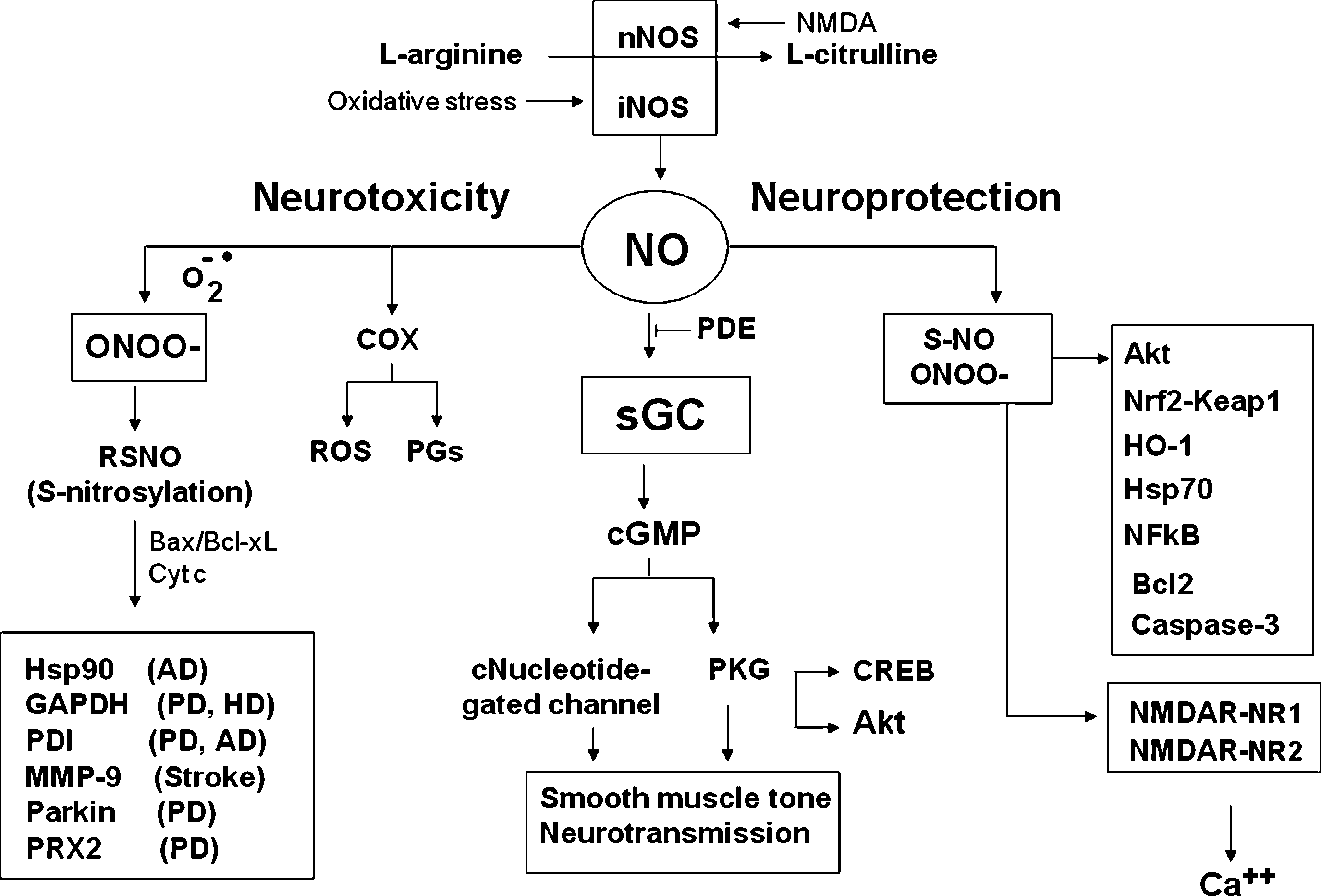
In contrast, excess NO, which can be produced by activated microglia in an uncontrolled inflammatory response (16) or by the activation of nitric oxide synthase in excitotoxicity (59), has been reported to participate in acute and chronic neurodegenerative diseases including stroke, multiple sclerosis, Parkinson disease (PD), and Alzheimer disease (AD) (22, 23, 194) (Fig. 4). RNS-induced modifications to proteins affect individual amino acids (primarily cysteine, tyrosine, and tryptophan), as well as the peptide backbone and metal cofactors of enzymes (Figs. 1 and 3). Recently, the term “nitrosative stress” has been used to indicate the cellular damage elicited by NO and its sequelae, mainly peroxynitrite and N2O3 (170). In recent years, S-nitrosylation of proteins by NO, as a cGMP-independent pathway, has been widely investigated. S-Nitrosylation is likely a prototypic redox-based signaling mechanism (99) because the S-nitrosothiols (RSNO) not only can be reduced to form thiols (Fig. 3), but can also be oxidized to form either S-glutathionylation (–SSG), cysteine sulfenic acid (–SOH), cysteine sulfinic acid (–SO2H), or cysteine sulfonic acid (–SO3H). The substantially different roles of these different types of modification are implicated in various physiologic and pathologic processes (88, 193). Numerous S-nitrosylated proteins have been identified in vivo, including serum albumin (193), hemoglobin β-subunits (81), the ryanodine-sensitive calcium-release channels (210), N-methyl-
The NO System
Chemistry of nitric oxide
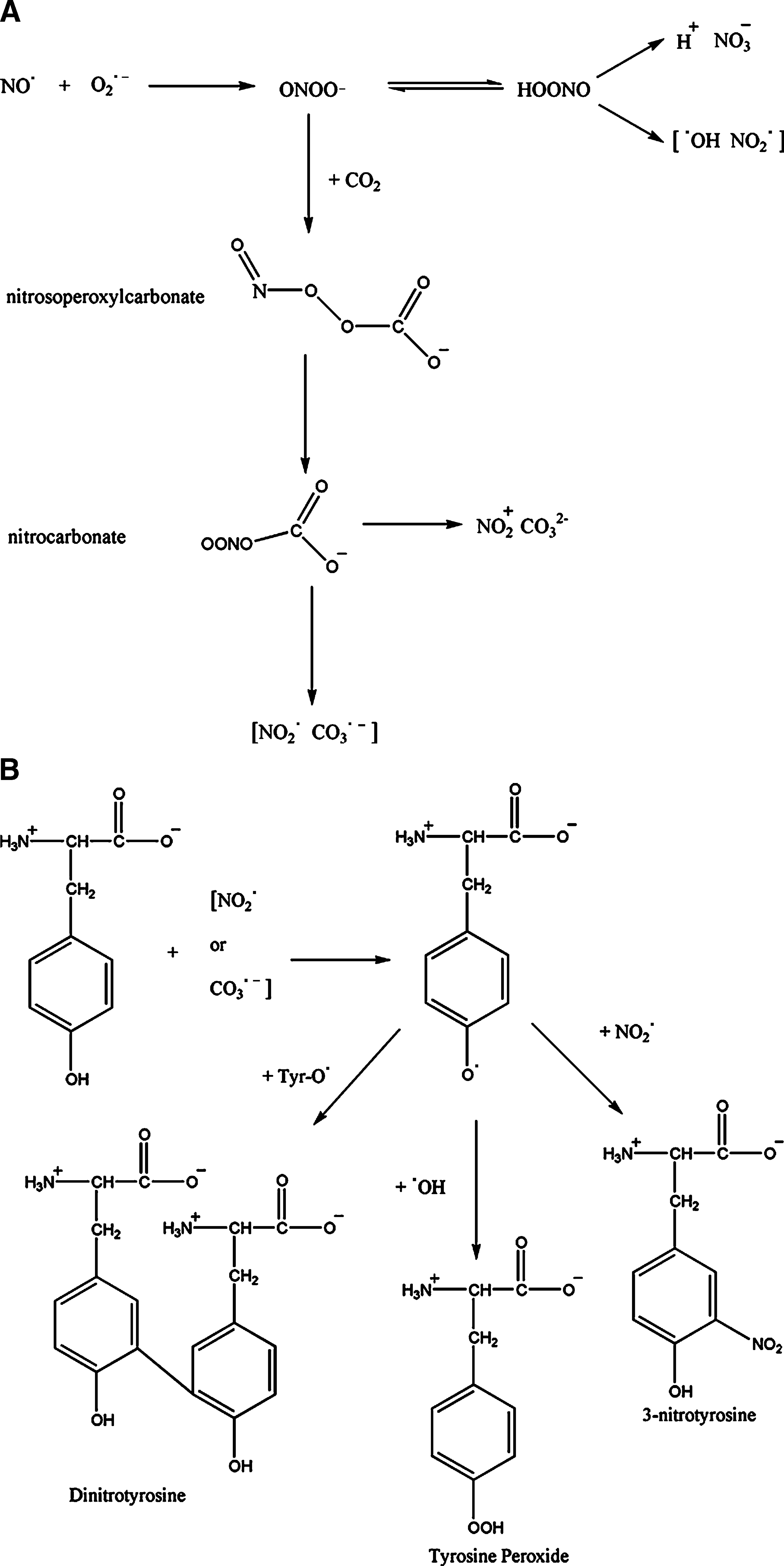
Nitric oxide (NO) is produced from the catalytic conversion of arginine to citrulline by nitric oxide synthases (NOSs) (32) (Figs. 1 and 4). Nitric oxide is neither a strong oxidant nor a strong reductant. Typically, nitric oxide does not react rapidly, with the exception of reaction with superoxide radical anion (O2 • −) (23). Nitric oxide reacts with superoxide radical anion at a diffusion-controlled rate (common among radical–radical recombination reactions) to produce peroxynitrite (ONOO−) (Fig. 5A). About 15% of superoxide produced by mitochondria goes toward the formation of peroxynitrite (the other 85% forms hydrogen peroxide) (108). Importantly, ONOO− formation in mitochondria induces cytochrome c release, an indicator of mitochondrial distress and potential inducer of cell death, and also irreversibly blocks the respiratory chain by competing with molecular oxygen (137). Two other reactions that involve NO modification of proteins, nitrosylation and nitrotyrosylination, are important to both the physiologic and pathophysiologic roles of NO. Nitrosylation is the reaction of NO with the amino acid cysteine to form nitrosothiols on interacting proteins. Nitrotyrosylination is the reaction of the amino acid tyrosine in target proteins, with ONOO−-derived •NO2 to form 3-nitrotyrosine, which has been used as a marker in neurodegeneration models (see later). Several factors influence the steady-state concentration of NO within the biologic system, including its rate of production by the various forms of NO synthase, and its rate of removal by oxidation, diffusion, NO dioxygenase, and oxyhemoglobin (60), whereas the concentration of superoxide anion is regulated by the activity of superoxide dismutase (SOD).
Being first order with respect to both reactants, the kinetics of the reaction of nitric oxide with superoxide anion depend on the activities of NOS and SOD. Cells can regulate the concentration of superoxide anion and, hence, the intracellular formation of peroxynitrite, through cytosolic CuZnSOD and mitochondrial MnSOD. The formation of extracellular peroxynitrite is controlled by secreted or extracellular SOD (EC-SOD) (23, 24), located on the cell-surface membrane:
Peroxynitrite can exist in two forms: the nucleophilic peroxynitrite anion (ONOO−), or the protonated peroxynitrous acid (HONOO) (2):
Peroxynitrite can react with carbon dioxide faster than the uncatalyzed decomposition of peroxynitrite; hence, the possibility exists that the in vivo oxidative damage attributed to peroxyinitrite may actually be mediated by the reactive intermediates from the reaction of peroxynitrite and carbon dioxide, rather than by the peroxynitrite itself (62). However, in certain instances, the presence of carbon dioxide actually inhibits the reaction of peroxynitrite with a biotarget, as is the case with the reaction of peroxynitrite with glutathione (23).
The reaction of peroxynitrite and carbon dioxide proceeds with the ONOO−/CO2 reactive pair (Fig. 5A) (122). The first intermediate in the reaction of peroxynitrite and carbon dioxide is nitrosoperoxycarbonate, which rearranges to form the second intermediate nitrocarbonate. Homolysis of nitrocarbonate is not favored but can occur to generate a nitrite radical (NO2
•) and a carbonate radical anion (CO3
•
−) (4) (189). These nitrating and oxidizing species can then react with biomolecules, forming modifications such as 3-nitrotyrosine (5) (Fig. 5B). 3-Nitrotyrosine is a covalent protein modification that has been used as a marker of nitrosative stress under a variety of cellular conditions, especially diseased versus control states (94). Dityrosine and tyrosine peroxide may also be formed from the reactions (6) and (7) (205).
Alternatively, nitrocarbonate can undergo heterolytic cleavage, resulting in the formation of NO2 + and CO3 2−. NO2 + can nitrate tyrosine residues in proteins in hydrophobic environments by electrophilic substitution (205). Figure 5A and B provide a summary of the reactions discussed earlier. Peroxynitrite can also react with sulfhydryl compounds intracellularly because of the high concentration of free thiols within the cell (99).
Reversible modifications of reactive cysteine
Proteins
Within proteins, amino acids that are targets for reversible oxidation include cysteines with a low-pK a sulfhydryl group (98), which causes unique susceptibility to oxidation compared with the typical pKa of nonreactive cysteines (170). Cysteine has been shown to undergo S-nitrosylation and oxidation on reaction with N2O3 and ONOO−, respectively (Fig. 3). The individual cysteines targeted for modification may not be located within a conserved primary amino acid sequence; however, the proximal positioning of acidic and basic residues has been proposed to accelerate S-nitrosylation. Cysteines within hydrophobic regions of proteins can be targeted for nitrosylation. Cysteine can be directly oxidized by ONOO−. Of all the amino acids, cysteine reacts the fastest with ONOO−, with a second-order rate constant of ∼3 × 103 M/s at pH 7.4. Oxidation of cysteine by ONOO− yields disulfide bonds (with neighboring cysteines) through a mechanism with a sulfenic acid (RSOH) intermediate. In addition, homolysis of ONOO− in either the presence or absence of CO2 can result in a variety of products, including disulfide radical anion (RSSR•−), sulfinyl radical (RSO•), RSNO, and R-S-NO2. Numerous classes of proteins contain free cysteine residues that are highly conserved across species, suggesting regulatory possibilities beyond structural roles and metal ion coordination (159). Cysteines, however, can be modified through alternative redox-based modifications (SNO, SOH, SSG, S-S), serving as molecular switches to integrate redox-based signals into specific functional responses. Recent studies have also shown that the sulfinic acid form of 2-Cys peroxiredoxins can be reduced back to the sulfhydryl in an ATP-dependent enzyme (sulfiredoxin; Srx)-catalyzed reaction (172), and a reversible thioesterification has been reported to modulate transcriptional activity (86). Cysteines, in addition, are uniquely targeted by alkylating species (such as cyclopentenone prostaglandins, acrolein), by hydroxynonenal, and by acylation (99).
Like NO and SNO, H2O2 also oxidizes reactive cysteines, leading to the formation of SOH, which can be further oxidized to sulfinic (SO2H) and sulfonic (SO3H) acids (185). Sulfenic (SOH) acids also are byproducts of S-nitrosylation and targets of S-glutathionylation. In particular, S-glutathionylation, the formation of a disulfide between the cysteine of glutathione and a cysteine moiety of a protein (protein mixed disulfide, or PSSG), is believed to be a protective mechanism that prevents further oxidation to sulfinic and sulfonic acids, which are not readily regenerated (133). PSSG formation is also a consequence of denitrosylation by GSNO reductase. Recently, in HEK cells transiently expressing iNOS, it has been shown that Trx forms mixed disulfides with multiple proteins in an NO-dependent manner (11), which demonstrates the capability of Trx1 to interact with SNO–caspase-3 in vivo and to denitrosylate, by a mixed-disulfide intermediate mechanism, also a broad spectrum of proteins. These finding indicates that SNO-protein denitrosylation (and thus regulation of NO-based signaling) represents an additional function of the Trx system (11). Accordingly, bilirubin (BR), an endogenous molecule with strong antioxidant properties (33, 34) was shown to act as a denitrosylating agent within a concentration range of 0.1–20 μM, which corresponds to those existing within the cell (2.5 μM) or in the plasma (20 μM), increasing the release of NO from RSNO. Mass spectrometry analysis has revealed that NO binds to BR, forming an N-nitroso derivative (BR-NO). Formation of BR-NO was confirmed also in rat fibroblasts exposed to prooxidant stimuli. This finding provides novel insights into the antioxidant potential of BR, suggesting the potential usefulness as a novel biomarker of nitrosative stress (9).
Glutathione
Reduced glutathione (GSH) is a likely target of reaction with peroxynitrite, resulting in the oxidation of GSH to GSSG, which can be recycled by glutathione reductase (99). Whereas the precise physiologic mechanism of nitrosylation is unclear, it has been hypothesized that NO reacts with oxygen to form N2O3, which reacts with glutathione to form the S-nitrosoglutathione (GSNO) group, which is an important intermediate in S-nitrosylation signal transduction. In 1997, Gow et al. (81) proposed a mechanism for the formation of S-nitrosothiols in vivo. The proposed mechanism involves the direct reaction of NO with free sulfhydryls to form a reactive intermediate that is reduced by an electron acceptor, such as molecular oxygen (O2), to produce RSNO (81). It was determined that cysteine accelerates the consumption of NO under physiologic conditions. Likewise, the consumption of O2 was determined to increase in the presence of cysteine and NO. The reaction of NO and cysteine, in the presence of CuZnSOD, produced H2O2. Taken together, these results suggest that the reaction of cysteine and NO leads to the reduction of O2 to O2 • − and the production of a nitrosothiol. Such nitrosothiols have been shown to be involved in intracellular signaling, ion channel regulation, immunologic responses, and apoptosis (23, 24).
S-Nitrosylation
S-Nitrosylation is a ubiquitous redox-related modification of cysteine thiol by nitric oxide (NO), which transduces NO bioactivity. Accumulating evidence suggests that the products of S-nitrosylation, S-nitrosothiols (SNOs), play key roles in human health and disease (72). S-Nitrosylation of specific cysteine residues has been detected in >100 proteins of all classes and, presumably, represents the major mechanism by which NO signals (92). Similar to phosphorylation by kinases, S-nitrosylation by NOSs influences protein activity, protein–protein interactions, and protein localization. Thus, S-nitrosylation has been implicated in transmitting signals downstream of all classes of receptors, including GPCR, RTK, TNF, Toll, glutaminergic, as well as transducing signals within intracellular compartments, such as in the case of modification of transcription factors in mitochondria and the nucleus (99). S-Nitrosylation exhibits a number of essential features that are prototypical of biologic signals: (a) temporal regulation on physiologic time scales (S-nitrosylation is stimulus coupled and rapid), (b) the existence of motifs within proteins that facilitate S-nitrosylation or provide specificity, or both, (c) colocalization of target proteins with a source of NO (e.g., NOS enzyme) (97), (d) reversibility, and (e) enzymatic control [e.g., GSNO reductase, thioredoxin 2 (12) controls S-nitrosothiol tone (99)]. In addition, dysregulation of protein S-nitrosylation is associated with a growing number of diseases, including asthma, heart disease, and neurodegenerative diseases (12, 146). This process, which may be mediated by NO, SNO, or several oxidants, may involve nitrosative chemistry (nitrosation refers to chemical addition of NO+), but is not restricted to reactions of NO+. The term nitrosyl describes the attachment of the NO moiety to a nucleophilic group or transition metal. S-Nitrosylation is therefore the preferred terminology in biologic systems in which “-ylation” (70) is standard nomenclature for posttranslational modifications of proteins. Given these considerations, the NO-associated posttranslational modification of cysteine thiols is here referred to as S-nitrosylation.
At high concentrations, NO is oxidized by O2 to produce N2O3, a potent nitrosating agent. Other nitrosating agents include nitrosonium ion (NO+), nitrous acidium ion (NO-OH2 +), and N2O4; however, at physiologic pH, N2O3 is the dominant nitrosating species. N2O3 is formed by a second-order reaction (k = 8.4 × 106 M/s at 37°C), which occurs in two steps: the reaction of NO with O2 to produce NO2 and the reaction of NO2 with a second molecule of NO to produce N2O3. NO2 itself is a strong oxidant (E°′ = +1.2 V); it does not accumulate during NO oxidation reactions in aqueous solutions. Although N2O3 is not a strong oxidant (E°′ = +0.7 V), it is capable of nitrosating secondary amines and reduced thiol groups (170). Oxidants used in physiologic signal transduction exhibit substrate specificity and produce reversible oxidations. Highly reactive oxidant species, such as ozone, hypochlorous acid, or hydroxyl radical, oxidize biomolecules without preference or specificity, produce oxidative modifications, such as the 3-nitrotyrosine and protein carbonyls, and do not put in motion signaling events that are also not easily reduced. These protein modifications generally lead to aberrant activation of signal-transduction cascades, thus inducing pathophysiologic effects. Conversely, physiologic oxidants, such as NO/SNO, have been implicated in reversible Cys-based modifications that underlie homeostatic control of a wide array of cellular responses.
Lipid nitration
NO has a partition coefficient in octanol/water (Poctanol) of 5.5 (2), predisposing NO to accumulate in hydrophobic regions of the cell, such as lipid bilayers and lipoproteins. Superoxide, as it may be generated by uncoupling the electron-transport chain, particularly at complex I, or by cytosolic NAD(P)H oxidase and xanthine oxidase, can be protonated to form the neutrally charged and lipid-soluble hydroperoxyl radical. Accumulation of NO and hydroxyperoxyl radicals in hydrophobic environments make these areas particularly susceptible to peroxynitrite formation and subsequent damage. Once formed, peroxynitrite undergoes heterolytic cleavage, creating hydroxyl and nitrite radicals that initiate lipid peroxidation chain reactions. Hydroxyl and nitrite radicals abstract hydrogen atoms from polyunsaturated fatty acids (PUFAs) to generate carbon-centered radicals that undergo further reactions with oxygen, reactive aldehydes, neighboring lipids, and additional radical species to generate lipid hydroperoxyradicals, Michael-addition adducts, and nitrosolipids.
Emerging evidence indicates oxidized lipids as a primary target for NO nitrating effects with formation of nitro derivatives (NO2-FA). These pluripotent signaling molecules are generated in vivo as an adaptive response to oxidative inflammatory conditions and manifest predominantly antiinflammatory signaling reactions (7). It is also important to note that directly nitrosylated lipids have been implicated in cell-signaling processes (6). Peroxynitrite has been shown to oxidize low-density lipoproteins (LDLs) (113). Binding of nitrated LDS to macrophage-scavenging receptors leads to oxidized cholesteryl ester and foam cell accumulation, and these events are believed to occur early in atherogenesis (91). NO and its derivatives can influence eicosanoid activity and product distribution by multiple mechanisms. For example, alkyl and peroxyl radical intermediates of eicosanoid synthesis react with · NO at diffusion-limited rates (7).
NO also readily reacts with critical tyrosyl radical catalytic intermediates and reduced iron centers within the synthetic enzymes, thereby modulating eicosanoid formation. For instance, lipoxygenases catalyze the specific dioxygenation of PUFAs to yield reactive lipid peroxides such as leukotrienes and HETEs (7). These oxidized species are generally proinflammatory, acting as chemotactic and chemokinetic agents. Nitric oxide reversibly inhibits LOX, concomitantly being catalytically consumed through reaction with an enzyme-bound lipid peroxyl radical intermediate. LOX catalytic turnover thus inhibits NO-dependent activation of sGC and acts as catalytic sink of NO. Under conditions in which NO is converted to the more powerful oxidant ONOO−, LOX isoforms are inhibited by tyrosine nitration at concentrations as low as 1 μM (50), although it remains to be determined whether such ONOO− concentrations occur physiologically. Protonation of NO2 − under acidic conditions produces nitrous acid (HNO2, pK a ∼3), a reaction possible in the gastric compartment (pH 1.0–5.0), phagolysosomes (pH 4.0–6.0), airways of asthmatic patients, and possibly ischemic and, hence, acidotic microenvironments.
Decomposition of HNO2 in aqueous media proceeds by a series of reactions ultimately to yield ·NO2, N2O3, and N2O4, all viable candidates for mediating nitration reactions. Although acidic nitration is not kinetically rapid, the residence time of lipids, proteins, and NO2 − in acidic tissue compartments suggests that this may be a significant pathway for nitrating reactions. Stable, linoleic acid–derived nitrated products, with levels increasing in hyperlipidemic subjects, as well as trans isomers, shorter-chain nitroalkenes (putative β-oxidation products of 18 carbon and longer precursors), nitroalkenes that have been further oxidized, and nitroalkanes presumably derived from nitroalkene precursors, have been found recently in human plasma and, more generally, in healthy human specimens (7).
NO and oxidized lipids, once thought to transduce metabolic and inflammatory information via discrete and independent pathways, thus are recognized as interdependent regulators of the immune response and metabolic homeostasis. The relation between the two pathways extends beyond co-regulation of NO and eicosanoid formation, converging, through the nitration of unsaturated fatty acids, in the production of antiinflammatory products.
Nitric Oxide Synthase Family
Responsible for NO synthesis is the NOS family of enzymes, which, in the presence of oxygen, catalyze the conversion of
NO, nitric oxide; NOS, nitric oxide synthase; RNS, reactive nitrogen species; sGC, soluble guanylyl cyclase.
Neuronal NOS is expressed in neurons of both developing and mature brain of several species, including humans. Brain areas in which nNOS is highly abundant are cerebral cortex, hippocampus (in particular, the CA1 region and dentate gyrus), hypothalamus (supraoptic and paraventricular nuclei), striatum, lateral dorsal and pedunculopontine tegmental nuclei, and cerebellum (58). In the CNS, this isoform also has been found in astrocytes and cerebral blood vessels (84). In addition to this central localization, nNOS has been found in peripheral NANC neurons, which innervate the smooth muscle in the gastrointestinal tract, as well as in penile corpora cavernosa, urethra, and prostate (32, 102). Endothelial NOS (eNOS) in the brain is expressed in cerebral endothelial cells and regulates cerebral blood flow; however, a small population of neurons in the pyramidal cells of CA1, CA2, and CA3 subfields of the hippocampus and granule cells of the dentate gyrus express eNOS (32). eNOS also has been found in rat astrocytes (84).
In the periphery, eNOS has been described in vascular/sinusoidal endothelium and in the smooth muscle of rodent or human corpora cavernosa (80). Inducible NOS levels in the CNS are low, but iNOS can be induced in astrocytes or microglial cells after inflammatory conditions, viral infections, and trauma (101). In a still-evolving and somewhat controversial concept, mitochondrial NOS (mtNOS) is proposed to be bound to mitochondrial PDZ domains of COX and to complex I (73), thus favoring a steric relation between the released NO, vectorially directed to the matrix and inner membrane, and the control of respiration (38). In pathologic conditions such as extreme hypoxia, mitochondrial NO could come either from stimulated NOS or from the reduction of nitrite by COX (37). These enzymes are bound to anchoring proteins such as caveolin in cell membrane or to heat-shock protein (Hsp) 90 or 70 chaperones (111). The interaction with these proteins may evoke different effects: (a) increased (Hsp90-NOS) or decreased (caveolin-NOS) enzyme activity, (b) modified subcellular traffic (dystrophin impedes NOS I traffic to mitochondria), and (c) participation in ubiquitination and degradation (37). Neuronal NOS and eNOS are constitutively expressed and require the formation of calcium/calmodulin complexes for their activation, whereas iNOS is the inducible form and exerts its activity in a calcium-independent manner. All three isoforms of NOS for catalytic activity need cofactors such as heme, tetrahydrobiopterin (BH4), FAD, FMN, and NADPH (31). NOS II gene expression is modulated by inflammatory mediators, like cytokines TNF-α, IFN-γ, and LPS, that activate transcription factors like NF-κB or AP-1 (38). The mtNOS is nNOS-α translocated to mitochondria with specific posttranslational modifications, such as myristoylation and phosphorylation of Ser-1412 in an Akt-dependent motif (RXRXXS/T); it is constitutively expressed and Ca2+-dependent, and it is subjected to modulation by drugs or hormones and during development (37). The activities of cytosolic NOS isoforms are able to sustain NO cytosolic concentrations large enough to reach mitochondria. The diffusion coefficient of NO in aqueous solutions is similar to that of O2, ∼4 × 10−6 cm2/s; activation of endothelium is compatible with a concentration in the arterial wall of 2 μM NO. Most NO binds to cytosolic compounds, like myoglobin (27). Therefore, mitochondrial NO coming from cytosolic NOS results in a considerably lower concentration, about 20–100 nM; however, it may increase by fivefold after induction of inducible NOS in endotoxemia (208). In this condition, both increased NO and NO-derived superoxide anions favor the formation of intramitochondrial peroxynitrite (37).
In mitochondria, NO reversibly binds to Cu2+ B center of COX and consequently inhibits electron transfer to O2 and mitochondrial O2 uptake. NO-dependent inhibition of O2 use is achieved at very low physiologic NO concentrations of 50–100 nM; in these conditions, NO inhibits by half the activity of COX (165). NO inhibits COX and increases the reduction levels of the components of the electron-transfer chain, including ubiquinol and ubisemiquinone, on the substrate side of COX and reacts directly with ubiquinol to produce nitroxyl anion (NO−) and ubisemiquinone (165). Interestingly, the cortex of human postmortem AD brains exhibits a loss of COX activity and increased levels of iNOS (108). Studies also have shown inhibition of mitochondrial respiratory complex I in isolated brain mitochondria and intact neurons by ONOO− under certain conditions. In these studies, ONOO− inhibited complex I only after either disruption of mitochondrial integrity or decreased reduced glutathione (GSH) levels (90). This suggests a potential role for GSH in neuroprotection in PD (108). Interestingly, presymptomatic PD patients are deficient in both GSH in the substantia nigra and complex I activity (31, 108).
NO and Neurotransmission
The first evidence of a role for NO as a neurotransmitter was reported by Garthwaite et al. (78), who demonstrated that stimulation of cerebellar NMDA (N-methyl-
Nitric oxide is also involved in the regulation of the release of many “classic” neurotransmitters. NO has been shown to stimulate acetylcholine release in basal forebrain and ventral striatum, not directly, but rather stimulating adjacent glutamatergic neurons (84). Basal NO concentrations have been shown to reduce, whereas high levels increase, GABA release in calcium- and sodium-dependent processes (84). Nitric oxide donors stimulated, whereas hemoglobin, an endogenous scavenger of NO, inhibited noradrenaline and glutamate release in brain areas such as hippocampus (120). In rat striatum and medial preoptic area, NO increased both dopamine and serotonin release in a cGMP-dependent way (100, 202).
Neuroprotective versus Neurotoxic Effects of NO
NO in neuroprotection
NO confers a neuroprotective effect through multiple mechanisms. This effect has been demonstrated in several experimental models (Figs. 2 and 4). Activation of soluble guanylate cyclase by NO, through reaction with its heme center, followed by increased production of cGMP and activation of protein kinases G (PKGI and PKGII), was the first identified cellular target for transduction of NO-mediated signals (93, 144). This transduction pathway is involved in the response of many different cell types to the NO signal and can affect the function of a wide array of proteins, as well as modulate the function of other cellular messengers, such as cAMP and calcium (84). As noted earlier, NO also reacts with thiol groups of amino acid residues, especially with cysteine, forming S-nitrosylated derivatives (193). Both nitration and S-nitrosylation affect the function of the target proteins, ensuring that a multiplicity of cell-specific effects stem from the same initial signaling molecule (84). The two main modalities able to elicit cellular responses to NO (i.e., through cGMP messenger or through conformational modification of protein function by direct chemical reaction) are not mutually exclusive, even if they may preferentially occur at different concentrations of NO (52).
NO and redox signaling
Signaling mechanisms adopted by regulatory proteins to control gene expression in response to increased levels of reactive oxygen and nitrogen species are common in prokaryotes. Gene activation by oxidative stress was first described in bacteria in which regulatory proteins such as OxyR and SoxR were discovered to activate the expression of antioxidant and stress-response genes (87). The activity of OxyR is turned on in response to hydrogen peroxide and S-nitrosothiols, and the first model postulated that OxyR is activated through the formation of a disulfide bond between two of its cysteine residues, C199 and C208, and is deactivated by enzymatic reduction of this disulfide bond with glutaredoxin 1 (219, 220). Recent studies suggested that modifications of a single cysteine (C199) in response to different stresses to (a) sulfenic acid, C199-SOH, by oxidative stress; (b) S-nitrosothiol, C199-SNO, by nitrosative stress; or (c) mixed disulfide with glutathione (C199-S-S-G) by disulfide stress may represent the sensor mechanism (105, 159). The expression of OxyR-dependent genes renders the bacteria more resistant to oxidant damage (167). The Escherichia coli SoxR and SoxS proteins (which principally mediate responses to superoxide) have also been implicated in responses to nitric oxide (190). SoxR activation occurs when its binuclear iron-sulfur [2Fe-2S] clusters undergo a reversible one-electron oxidation. Nitrosylation of the [2Fe-2S] clusters to form protein-bound dinitrosyl-iron-dithiol adducts has been demonstrated after treatment of intact bacteria or purified SoxR with nitric oxide (64). SoxR turns on the transcription of SoxS, which in turn activates the expression of the SoxRS regulon-encoding proteins with protective functions, including sodA (manganese-containing superoxide dismutase), zwf (glucose-6-phosphate dehydrogenase), fldA and fldB (two distinct flavodoxins), and fpr (NADPH-ferredoxin reductase).
Cyclic AMP response element–binding protein and Akt
In primary rat cerebellar granule cells cultured for 7 days, inhibition of NO synthesis resulted in a significant increase in apoptotic cell death through the activation of caspase-3. Interestingly, apoptosis secondary to NO deprivation in these cells was mimicked by the sGC inhibitor ODQ and was reversed by treatment of these neurons with NO donors or cGMP analogues (47). With this experimental system, the intracellular pathways through which NO exerts neuroprotective effects also were delineated (48), where the involvement of the kinase Akt and the transcription factor CREB in the survival pathway elicited by NO in cerebellar granule cells was clearly demonstrated (49); notably, both proteins are important as signal transducers of neurotrophin-mediated survival and protection against various neurodegenerative challenges, and this similarity contributes to the neuroprotective role of NO (32, 52) (Fig. 4). cAMP response element–binding protein (CREB) is a leucine-zipper transcription factor, which activates transcription of a large set of genes having the consensus sequence CRE in their promoters and whose functions in neural cells have been extensively studied during the past decade. This stimulus-induced transcription factor is activated through phosphorylation at Ser-133 by a large array of extracellular signals essential for neuronal function, such as neurotrophins and neurotransmitters, and regulates the expression of genes that in turn promote neuronal survival, protection from neurodegenerative insults, neurogenesis, synaptic plasticity, and memory formation and consolidation (138).
The contribution of NO to the mechanisms of synaptic plasticity, such as hippocampal LTP, and to the learning and memory has been demonstrated by several researchers and is supported by genetic or pharmacological manipulation of the NO/cGMP system (189). Although some of these effects are related to retrograde messages conveyed by diffusion of NO from the postsynaptic neurons, other effects have been linked to the activation of CREB-mediated transcription in the postsynaptic neuron (52). Recent evidence has clearly demonstrated that NO promotes neuron survival through regulation of CREB transcriptional activity. Pharmacologic dissection of the cellular pathway elicited by NO pointed to the essential role of the cGMP/PKG system and at the increased expression of the antiapoptotic gene Bcl-2 in survival promotion (49). Further refinements of this finding are supported by recent studies indicating that, instead of being constitutively bound to CRE sequences, CREB binds to the DNA consensus sequences of several genes through an NO-dependent pathway targeted to S-nitrosylation of nuclear proteins (173).
Neuroprotection through S-nitrosylation
Similar to phosphorylation by kinases, S-nitrosylation by NOSs influences protein activity, protein–protein interactions, and protein localization. S-Nitrosylation thus serves as the fundamental redox-based signal, integrating the NO signaling by guanylate cyclase. S-Nitrosylation has been implicated in transmitting receptor-mediated signals, as well as transducing, through modification of transcription factors, signals within mitochondria and the nuclear compartment (92). NO also confers neuroprotection in the NMDA-mediated neurotoxicity model, in which prolonged stimulation of the NMDA receptor causes excitotoxic cell death (32). NO protects against such excitotoxicity by S-nitrosylating the NR1 and NR2 subunits of the NMDA receptor (98, 117), which reduces the intracellular Ca2+ influx responsible for neuronal death (116).
Interestingly, prolonged nNOS stimulation, which occurs in response to sustained NMDA-receptor activation, generates superoxide radicals, which, in turn, react with NO to form peroxynitrite (143). Consequently, NO formed during excessive NMDA activation S-nitrosylates the NMDA subunits, thereby diminishing either the formation of peroxynitrite or Ca2+ influx to promote neuronal survival (Fig. 4). NO can also confer cytoprotection through the inhibition of caspase activity by S-nitrosylation of the catalytic-site cysteines (133, 118). S-Nitrosylation has been shown to reduce the activity of caspases in several cell lines, including neurons (126, 201, 203). Cortical neurons treated with several NO donors, including S-nitrosothiols, exhibited a significant reduction in staurosporin-induced caspase-3 and caspase-9 activation, probably owing to the NO-mediated S-nitrosylation of the cysteine residue in the catalytic site of these caspases In addition, NO treatment inhibited the appearance of the classic apoptotic nuclear morphology (221). Surprisingly, caspase-3 and caspase-9 inhibition by NO was not paralleled by a significant increase in neuronal cell viability, which therefore implies the occurrence of an alternative, caspase-independent, form of cell death in neurons exposed to NO, in accordance with previous findings (133).
Neuroprotection through transcriptional control of gene expression
It has been demonstrated that in mammalian cells, NO induces temporally distinct waves of gene activation. Whereas NO may affect gene transcription by controlling the methylation of CpG islands in the promoter regions of some genes, most of its activity in regulating gene expression occurs through regulation of transcription factors (178). Nuclear factor-κB (NF-κB) is an important transcription factor whose biologic activities are influenced by NO. Originally found to be active in responses of immune cells, such as inflammatory responses, by inducing the expression of cytokines and adhesion molecules, this transcription factor is particularly relevant for NO-related modulation of gene expression, as ample evidence exists for its regulation through mechanisms of nitrosylation and nitration (163, 198).
In most mammalian cells, NF-κB is constitutively expressed in the form of the p50/p65 heterodimer, and p50 S-nitrosylation at Cys-62 residue inhibits gene transcription, dependent on NF-κB binding at the DNA promoter site (164). Nitric oxide can regulate the DNA-binding and transcriptional activity of NF-κB either directly, by interacting with the factor itself, or indirectly through its endogenous inhibitor IκB proteins (51, 52). In addition, the NO-dependent decreased phosphorylation and proteasomal degradation of IκB seems to play a critical role in the cellular mechanisms leading to NF-κB activation (129, 160). A novel mechanism for NO-mediated inactivation of NF-κB transcriptional activity, targeted on p65 regulation, was recently described in cell lines (52). Exposure of cells to sodium nitroprusside resulted in rapid inactivation of NF-κB, an effect that was reverted by providing the cells with a peroxinitrite scavenger, thus suggesting the presence of a mechanism based on tyrosine nitration. Further chemical analysis through mass spectrometry revealed nitration on Tyr-66 and Tyr-152 residues of p65 (162). Centered on activation of nitrosative conformational changes of proteins, complementary cellular transduction pathways have been recently described in cortical neurons, which include brain-derived neurotrophic factor (BDNF)-induced S-nitrosylation of histone deacetylase 2 (HDAC2) and the Akt/PKB pathways (49). BDNF-induced S-nitrosylation of HDAC2 has been shown to result in histone modifications and activation of prosurvival genes, such as Fos, Vgf, and Egr1 genes (150). Similarly, the antiapoptotic kinase Akt is activated by phosphorylation, promoted by phosphatidylinositol-dependent kinase 1 (PDK1). This kinase, in turn, is activated by the effector phosphatidylinositol-3,4,5-trisphosphate (PIP3), which is the product of PI3K activity. Activation of AKT by phosphatidylinositol 3-kinase (PI3K) serves as a multifunctional regulator of apoptotic cell death, cell growth, and glucose metabolism. With respect to neuronal cell function, AKT has been shown to be required for prevention of apoptosis and promotion of cell survival through the phosphorylation of the proapoptotic Bcl-2 family protein, Bad (56), and procaspase 9 (36). Incubation of primary cortical neurons in culture with peroxinitrite rapidly triggers phosphorylation of Akt at Ser473 and protects neurons against the apoptotic death caused by etoposide. Such effects were abolished by inhibition of PI3K activity with wortmannin, or by Akt shRNA knockdown, suggesting that the PI3K/Akt pathway mediated the antiapoptotic effect of peroxinitrite (61).
Neuroprotection through overexpression of heme oxygenase and the Keap1/Nrf2/ARE pathway
Eukaryotes are equipped with sophisticated protective mechanisms against environmental challenges. Typical examples are proteins such as heme oxygenase 1 (HO-1), thioredoxin, thioredoxin reductase, superoxide dismutase, glutathione S-transferases (GST), and NAD(P)H:quinone oxidoreductase 1 (NQO1), whose gene expression is upregulated in response to a variety of chemical and physical stresses (34, 35). The induction of heme oxygenase-1 is considered an early event in the cellular response to oxidative stress and exerts a neuroprotective function (35, 40). Moreover, under prooxidant conditions, upregulation of iNOS occurs in the brain and is followed by HO-1 induction in rat astrocytes and microglia, as well as in the hippocampus (106). The upregulation of heme oxygenase-1 protein and the following increase in biliverdin, which is further reduced by biliverdin reductase into the antioxidant and antinitrosative molecule bilirubin, can be considered a secondary mechanism through which NO can exert neuroprotective effects (21, 27).
The notion that the antioxidant protein heme oxygenase could “sense” NO and thus protect against ROS and RNS insults is supported by the following findings: (a) NO and NO-related species induce HO-1 expression and increase heme oxygenase activity in human glioblastoma cells, hepatocytes, and aortic vascular cells; (b) cells pretreated with various NO-releasing molecules acquire increased resistance to H2O2-mediated cytotoxicity at the time heme oxygenase is maximally activated; and (c) bilirubin, one of the end products of heme degradation by heme oxygenase, protects against the cytotoxic effects caused by strong oxidants such as H2O2 and ONOO− (141). The concept that NO and RNS can be directly involved in the modulation of HO-1 expression in eukaryotes is based on the evidence that different NO-releasing agents can markedly increase HO-1 mRNA and protein, as well as heme oxygenase activity, in a variety of tissues, including brain cells (182). In rat glial cells, treatment with lipopolysaccharide (LPS) and interferon-γ (IFN-γ) results in a rapid increase in both iNOS expression and nitrite levels followed by enhanced expression of HO-1 protein. In the same study, the presence of NOS inhibitors suppressed both nitrite accumulation and HO-1 mRNA expression (26).
Modulation of HO-1 mRNA expression by iNOS-derived NO after stimulation with LPS has also been reported in different brain regions, particularly in the hippocampus and substantia nigra in the in vivo rat model of septic shock (183). Moreover, the early increase in iNOS protein levels observed in endothelial cells exposed to low oxygen tension seems to precede the stimulation of HO-1 expression and activity, an effect that appears to be finely regulated by redox reactions involving glutathione (140, 215). Taken together, these findings point to the central role of the NO as a signaling molecule that, by triggering expression of cytoprotective genes such as HO-1, may lead to adaptation and resistance to subsequent, eventually more severe, nitrosative and oxidative stress insults (35).
Thus, a direct interaction of NO with selective chemical sites localized in transcription factors that can be activated through nitrosative reactions could effectively contribute to the enhancement of both HO-1 gene expression and stress tolerance. Recent knowledge concerning the modulation (by altering the thiol redox state) of the activity of several transcription factors that recognize specific binding sites within the promoter and distal enhancer regions of the ho-1 gene include Fos/Jun [activator protein-1 (AP-1)], nuclear factor-κB (NF-κB), and the more recently identified Nrf proteins (8, 33). Both AP-1 and NF-κB contain cysteine residues whose interaction with oxidative or nitrosative species might be crucial for determining the binding activity to DNA (141). Data in the literature show that NO can either activate or inhibit these transcription factors, and that in many circumstances, activation depends on the reversibility of the posttranslational modification elicited by the various RNS (27, 175).
Recently, accumulating experimental evidence suggests that one of the protective functions of NO could be mediated through transcriptional activation of the Keap1/Nrf2/ARE pathway (Fig. 6), a quintessential cytoprotective pathway in eukaryotes. This pathway is composed of three major elements: (a) the sensor protein, Keap1 (Kelch-like ECH-associated protein 1) (96); (b) transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) (95, 135); and (c) specific promoter sequences ARE/EpRE (antioxidant/electrophile response element, consensus sequence 5'-A/CTGAC/GNNNGCA/G-3') (74, 148, 176, 177, 198) present in the upstream regulatory regions of a battery of >100 cytoprotective genes, among which are HO-1, thioredoxin, thioredoxin reductase, GST, and NQO1. Activation of the Keap1/Nrf2/ARE pathway leads to rapid adaptation to a variety of stress conditions and promotes cell survival (103, 110, 139).
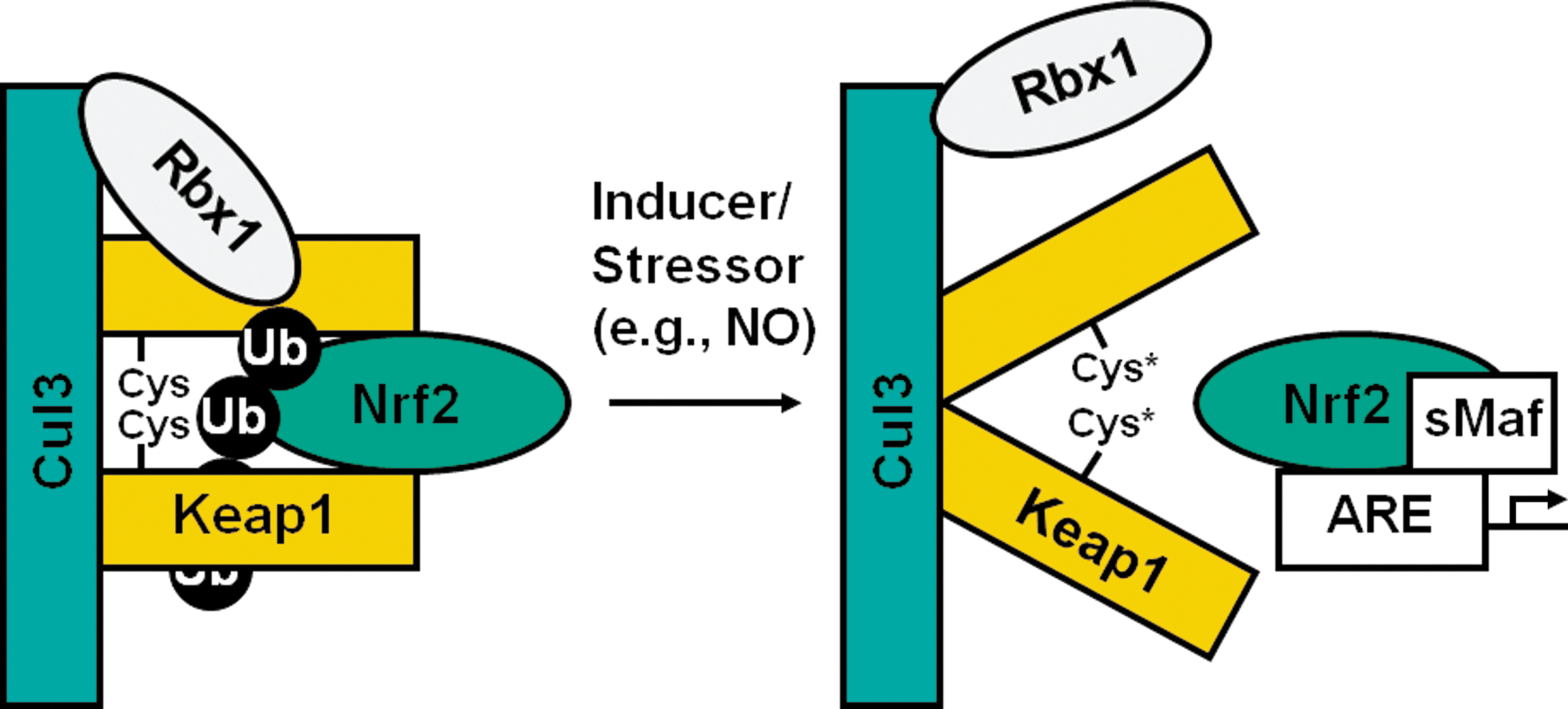
Several models have been proposed to explain how this pathway operates under both basal and stress conditions, and they were recently comprehensively reviewed by Hayes and McMahon (89). Under basal homeostatic conditions, the homodimeric sensor protein Keap1 binds one molecule of transcription factor Nrf2 and targets it for ubiquitination and subsequent proteasomal degradation (132). Keap1 is a multidomain protein equipped with several protein–protein binding sites (66). Thus, in addition to Nrf2, Keap1 binds to Cullin 3 (Cul3), and, in doing so, Keap1 presents Nrf2 for ubiquitination to the Cullin 3 (Cul3)-Ring box 1 (Rbx1) E3 ubiquitin ligase complex (54, 76, 109, 217). A critical feature of the Keap1 protein is that it contains highly reactive cysteine residues that serve as sensors for various stressors/inducers of the Keap1/Nrf2/ARE pathway (65, 66, 207). Depending on the inducer, distinct cysteines are modified, most frequently by participating in Michael addition or oxidation–reduction reactions. We and others reported that C151, C273, and C288 are of particular importance in the sensor function of Keap1 (65, 216). Inducer sensing by Keap1 ultimately leads to stabilization of the transcription factor, allowing Nrf2 to bind (in heterodimeric combinations with small Maf proteins) to the antioxidant response elements of its target genes and to activate their transcription. This transcriptional upregulation results ultimately in enhanced cytoprotection (67, 199, 200).
In human neuroblastoma cells, nitric oxide was shown to cause nuclear accumulation of Nrf2, binding to the ARE, and upregulation of Nrf2-dependent genes, and this induction counteracted NO-induced apoptosis (63). RNA interference, or transfection with a dominant negative Nrf2 construct, resulted in reduced levels of cytoprotective genes and sensitization to NO-induced apoptosis, whereas overexpression (stable transfection) of Nrf2 was protective. Treatment with NO-donating aspirin also increased the levels of several Nrf2-dependent genes in human colon (HT-29) and murine liver (Hepa1c1c7) adenocarcinoma cells, as well as in vivo in the liver and intestine of Apc Min/+ mice (77), and inhibited by about 60% the formation of intestinal tumors in this mouse model (209). Exposure of cardiac cells to nitric oxide donors in combination with hemin increased HO-1 mRNA and protein expression under both normoxic and hypoxic conditions (147). The HO-1 increase was accompanied by elevated nuclear levels of Nrf2, but this elevation was abolished by N-acetylcysteine. Similarly, exposure to nitric oxide donors increases the expression of Nrf2-dependent genes (e.g., HO-1, γ-glutamylcysteine synthetase) in vascular endothelial cells (18) and smooth muscle cells (119). Thus, in primary cultures of bovine aortic endothelial cells, nuclear levels of Nrf2 were increased after exposure to spermine NONOate, and this increase was accompanied by elevation in HO-1 expression and total glutathione levels (18). Inhibitors of ERK, p38 MAPK, as well as pretreatment with N-acetylcysteine, decreased the NO-dependent stabilization of Nrf2 and upregulation of HO-1, suggesting the importance of both phosphorylation and redox mechanism(s). In rat aortic smooth muscle cells, nitric oxide donors (spermine NONOate, S-nitroso-N-acetylpenicillamine, sodium nitroprusside, Sin-1, or Angeli's salt) increased the levels of Nrf2, the binding of Nrf2 to the promoter of HO-1, and the expression of HO-1 (119). Mutations in the ARE of HO-1 or overexpression of a dominant-negative Nrf2 construct abrogated the NO-dependent upregulation of HO-1. In this study, inhibitors of ERK or p38 MAPK had no effect on the NO-mediated nuclear accumulation of Nrf2. In contrast, pretreatment with ascorbate or N-acetylcysteine reduced the NO-mediated increase in Nrf2 levels, suggesting the involvement of redox mechanism(s).
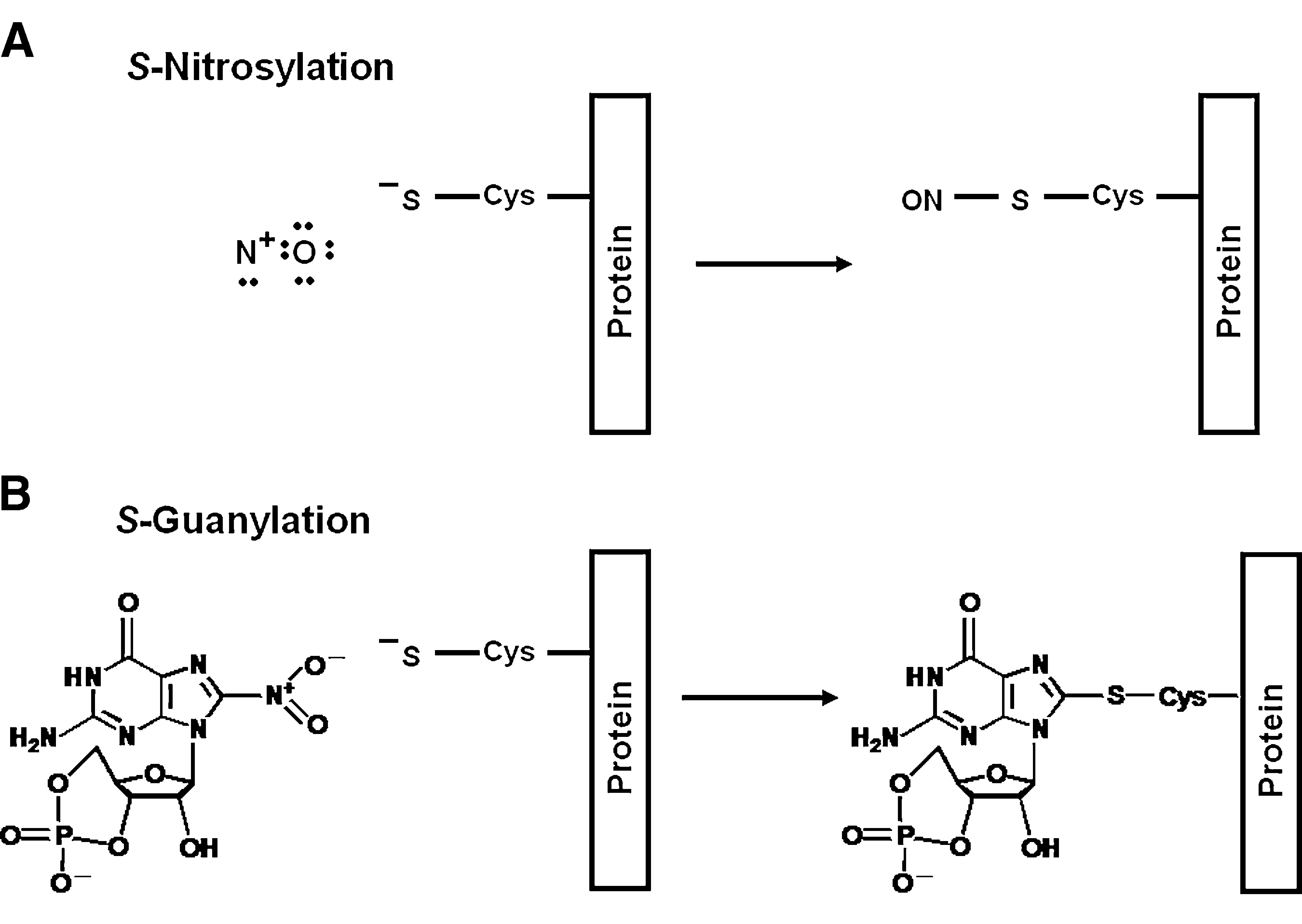
Because Keap1 is an essential regulator of Nrf2 levels in the cell and its highly reactive cysteine residues are the sensors for inducers of Nrf2-dependent genes, Keap1 is a good candidate to serve as a sensor for nitric oxide. As discussed earlier, S-nitrosylated proteins can form when a cysteine thiol reacts with nitric oxide in the presence of an electron acceptor (e.g., molecular oxygen) to form an S-NO bond (79, 180) (Fig. 1). Furthermore, S-nitrosylation is now recognized as an important mechanism for the posttranslational regulation of many proteins, thus conveying the effects of nitric oxide on signal transduction (Fig. 4) (92).
Although their exact chemical nature has not been identified, reversible cysteine modifications (that could include S-nitrosothiols, disulfide bonds, sulfenic acid derivatives) of both overexpressed and endogenous Keap1 were reported to occur when HEK293H cells were treated with spermine NONOate or S-nitrosocysteine (19). Recently, the formation of a nitrated derivative of cGMP, 8-nitroguanosine 3',5'-cyclic monophosphate (8-nitro-cGMP) was reported when RAW 264.7 cells were stimulated with LPS and IFN-γ to upregulate transcriptionally iNOS and to produce nitric oxide (181). Because of the electrophilic nature of 8-nitro-cGMP, the possibility that it might react with cellular thiols to form 8-thioalkoxy-cGMP (8-RS-cGMP) adducts was explored. Under these conditions of nitric oxide production, 8-Cys-cGMP adducts in endogenous Keap1 were identified, and the reaction was termed S-guanylation (Fig. 7). NO-dependent S-guanylation of Keap1 also was found in murine peritoneal macrophages after infection with Salmonella enterica var. Typhimurium. In vitro, recombinant Keap1 was shown to be highly susceptible to S-guanylation induced by 8-nitro-cGMP, even in the presence of excess (>3,000-fold) GSH.
Nitric Oxide in Neurodegeneration
Although NO has many important and beneficial physiologic functions, it also can play a role in neurodegenerative disease pathology (31). In these diseases, NO is produced in excess by iNOS induction owing to the proinflammatory response, which is a common feature of neurodegenerative disorders (108). Moreover, NO is much more harmful under pathologic conditions that involve the production of reactive oxygen species (ROS), such as superoxide anion and the formation of peroxynitrite (34) (Fig. 4).
Two important properties of NO that may contribute to its pathologic functions are its ability to modify proteins through nitrosylation and nitrotyrosination and its ability to react with oxygen to form RNS. Accordingly, studies indicate that nitrosylation and nitrotyrosination of many different proteins play a role in the pathology of multiple neurodegenerative diseases. The formation of nitrotyrosine, as a marker of nitrosative stress, has been documented in AD and PD (27, 29, 30, 33, 130, 195, 196).
Glutamate excitotoxicity is caused by overstimulation of synaptic glutamate receptors that results in excessive Ca2+ influx and subsequent neuronal injury. Excitotoxicity is a common event in many neurodegenerative disorders, including AD. NO can both protect and sensitize cells to excitotoxic cell death through a mitochondrial pathway. Specifically, under normal physiologic conditions, NO S-nitrosylates NMDA receptors, blocks Ca2+ influx, and promotes cell survival. In immature neurons, however, mtNOS is responsible for the majority of NOS activity. In these conditions, Ca2+ enters mitochondria and stimulates NO production by mtNOS. As a consequence, inhibition of the respiratory chain occurs, which reduces the mitochondrial membrane potential, collapses the ion gradient, and decreases entry of Ca2+ into the mitochondria. Decreased Ca2+ influx causes a decrease in mtNOS activity and NO production, promoting cell survival. Conversely, in mature neurons, when cytosolic nNOS is the primary producer of NO, Ca2+ entry through overactive NMDA channels stimulates nNOS; thus, NO can then enter the mitochondria, directly inhibiting complex IV (cytochrome c oxidase) of the respiratory chain, which leads to a block of ATP production and eventual cell death due to energetic failure. In contrast to immature neurons, in mature neurons, no feedback loop is present to protect the cell from damage, and degenerative cell death ensues.
Furthermore, NO has been shown to activate both the constitutive and inducible isoforms of cyclooxygenase, which are upregulated in brain cells under proinflammatory conditions (125). During the catalytic cycle of cyclooxygenase, the release of free radicals and the formation of prostaglandins occur, two events closely related to the development of neuroinflammation (43). Interestingly, the upregulation of inducible cyclooxygenase has been found in the brain of patients affected by AD, and it is considered a marker of the progression of dementia in AD; thus, activation of inducible cyclooxygenase can be considered an indirect way for NO to induce neurotoxicity.
Alzheimer's disease
Recently, a relation between glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the β-amyloid precursor protein (betaAPP) in relation to the pathogenesis of AD was suggested. Decreased dehydrogenase activity of cerebral glyceraldehyde-3-phosphate dehydrogenase has been described in different animal models of AD (185). In addition, recent evidence indicates that polymorphic variation within GAPDH genes is associated with an elevated risk of developing AD (55). GAPDH not only is important in ATP production, but also has a role as nitrosative stress sensor. Active-site cysteine 149 may undergo several modifications on reaction with NO (and RNS), which result in a reversible or irreversible inhibition of GAPDH enzymatic activity, depending on the severity of the prooxidant stimulus. As a consequence of this inhibition, the glucose metabolism is shifted toward the pentose phosphate shunt, which produces NADPH necessary to glutathione reductase and uncouples glucose metabolism from the production of ATP and oxidative intermediates. Glyceraldehyde 3-phosphate dehydrogenase also undergoes S-nitrosylation in neurons. In particular, S-glutathionylation after nitrosylation represents the effective inhibitory effect on GAPDH activity (197).
In addition to its well-known role in glycolysis, GAPDH contributes to nuclear signaling in apoptosis (108). S-Nitrosylation of GAPDH terminates its enzymatic activity and allows binding of GAPDH to Siah1, an E3 ubiquitin ligase. Siah1 has a nuclear localization signal and carries GAPDH to the nucleus. GAPDH stabilizes Siah1 in the nucleus and allows degradation of nuclear proteins through ubiquitination (108).
Protein-disulfide isomerase (PDI) is an endoplasmic reticulum (ER)-associated chaperone protein that prevents neurotoxicity caused by ER stress and protein misfolding (204) and can also function as an NO receptor or donor, depending on the cellular context (187). PDI is nitrosylated both in AD and PD. Both PD and AD patient postmortem brains exhibit increased levels of nitrosylated PDI as compared with those of healthy controls (204). PDI nitrosylation prevents PDI-mediated ER stress reduction and allows protein misfolding (204).
Molecular chaperone Hsp90 is a target of S-nitrosylation, with critical cysteine residue susceptibility in the region of the C-terminal domain that interacts with endothelial nitric oxide synthase (eNOS). Hsp90 is another protein associated with AD that undergoes nitrosylation (107). Hsp90 ATPase activity has a general positive effect on eNOS activity. Both, however, are inhibited by S-nitrosylation. S-Nitrosylation may therefore functionally regulate the general activities of Hsp90 and provide a feedback mechanism for limiting eNOS activation. Consistent with this, postmortem analysis of brain samples from patients with AD revealed increased levels of Hsp90. S-Nitrosylation of Hsp90 abolishes ATPase activity that is necessary for its chaperone function; thus, inactivation of Hsp90 may allow accumulation of tau and amyloid-β aggregates in the AD brain (108).
It is generally recognized that mitochondria continuously undergo two opposing processes, fission and fusion. The disruption of this dynamic equilibrium may herald cell injury or death and may contribute to developmental and neurodegenerative disorders. Nitric oxide produced in response to β-amyloid protein has been shown to trigger mitochondrial fission, synaptic loss, and neuronal damage, in part through S-nitrosylation of dynamin-related protein 1 (forming SNO-Drp1) (42). Preventing nitrosylation of Drp1 by cysteine mutation abrogated these neurotoxic events. Accordingly, SNO-Drp1 is increased in brains of AD patients and may thus contribute to the pathogenesis of neurodegeneration (42).
Parkinson's and Huntington's diseases
Studies have provided a considerable link between NO and other prevalent neurodegenerative diseases, such as PD and HD. PD, whose cardinal features include tremor, slowness of movement, stiffness, and poor balance, is attributed to a profound deficit in dopamine that follows the loss of dopaminergic neurons in the substantia nigra pars compacta and dopaminergic nerve terminals in the striatum (206). Although the mechanisms leading to PD are still uncertain, a large amount of experimental evidence implicates oxidative and nitrosative stress as crucial factors in the pathogenesis of PD (31). Considerable insights into the pathogenesis of PD have been achieved by use of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is commonly used to induce an experimental model of PD (167). Excessive free radical formation or antioxidant deficiency and the resulting oxidative stress are all mechanisms involved in MPTP neurotoxicity (167). Animal models of MPTP-induced neurotoxicity have shown that inhibition of NOS slows the progression of disease pathology (213). MPTP is a neurotoxin that inhibits complex I of the mitochondrial respiratory chain and mimics the symptoms of PD by killing substantia nigra neurons (31). Of note, 7-nitroindazole (7-NI), a selective inhibitor of nNOS, blocks MPTP-mediated decrease in striatal dopamine levels as well as neuronal death in mice (210) and baboons (70). 7-NI also protects against motor deficits and cognitive decline in the MPTP baboon model (108). In addition to the role of nNOS in MPTP-induced neurotoxicity, iNOS appears to play a role. MPTP treatment of mice causes massive gliosis, a proliferation of glial cells, and upregulation of iNOS, and iNOS-deficient mice are more resistant to MPTP (108).
In HD, another age-related neurodegenerative disorder that often gives rise to dementia, evidence of oxidative and nitrosative damage exists in the basal ganglia, as was recently reviewed (17). It was shown that matrix metalloproteinase 9 (MMP9), which causes neuronal apoptosis, is S-nitrosylated by NO that is derived from the endogenous nitrosothiol S-nitrosocysteine (32). Matrix metalloproteinases are involved in the pathogenesis of acute and chronic neurodegenerative disorders such as stroke, AD, HIV-associated dementia, and multiple sclerosis (11). A similar mechanism, proposed to occur in PD (46, 212), independently demonstrated that S-nitrosocysteine–derived NO is able to nitrosylate parkin, an E3 ubiquitin ligase. Mutation in parkin is known to cause autosomal recessive juvenile parkinsonism. Nitrosylation of cysteine residues of parkin initially increases but later decreases the E3 ubiquitin ligase activity of this protein, and thereby reduces its protective function. NO has also been shown to S-nitrosylate GAPDH, thereby reducing its activity and enabling GAPDH to bind to another E3 ubiquitin ligase, Siah1. The GAPDH–Siah1 complex then translocates into the nucleus to induce apoptosis. As a direct consequence of this finding, the identification of a new mechanism of action for selegiline, a drug already used to treat patients with PD, has emerged. At nanomolar concentrations, selegiline prevents S-nitrosylation of GAPDH, thereby blocking its interaction with Siah1 and the further induction of apoptosis. The neuroprotective effect selegiline is similar to that of TCH346, a derivative with no monoamine oxidase type B inhibitory activity (85). In addition, peroxiredoxin 2 (PRX2), a member of a family of abundant antioxidants most commonly found in neurons that reduce intracellular peroxides, becomes S-nitrosylated in a reaction with NO (108). S-Nitrosylation of PRX2 prevents its reaction with peroxides and inhibits its enzymatic activity and protective function against oxidative stress. Studies of human postmortem brains revealed an increase in S-nitrosylated PRX2 in PD patients (108).
Nitrated proteins identified in disease
Proteins are particularly susceptible to nitration at tyrosine, cysteine, methionine, tryptophan, and histidine residues and at transition metal redox centers (158). Tyrosine is the most studied nitrated amino acid and has been identified in >50 human diseases and >80 animal models (158). The first protein identified to be nitrated in vivo was mitochondrial Mn-SOD, and nitration of a single tyrosine residue (Tyr-34) completely inactivates the enzyme (123). Nitrated Mn-SOD has been observed in multiple human disease conditions, including rejected kidney allographs, the cerebrospinal fluid of amyotrophic lateral sclerosis, AD and PD patients, diabetic heart tissue, and vascular aging (211). Protein tyrosine nitration in the circulatory and central nervous systems has been implicated in multiple disease conditions (195, 196). Creatine kinase and the sarcoplasmic reticulum Ca2+ ATPase (SERCA) are inactivated by tyrosine nitration (114). Creatine kinase is the energetic regulator of cardiomyocyte contraction, and the SERCA is a major regulator of Ca2+ concentration in muscle cells. Inactivation of these proteins has major implications for the development or cause (or both) of cardiovascular disease (157). Voltage-gated K+ channels are also nitrated in coronary endothelium and potentially lead to the impaired coronary flow observed in cardiac disease (115). In addition, α-actin, desmin, and myosin heavy chain, all proteins involved in contraction, are nitrated (14).
Tyrosine nitration has been studied extensively in neurodegenerative diseases. In PD, α-synuclein nitration leads to oligomerization and subsequent Lewy body formation, the major pathologic hallmark of PD (5). Tyrosine-hydroxylase, the enzyme that catalyzes the rate-limiting step in dopamine synthesis, also is nitrated in PD (161). In amyotrophic lateral sclerosis (ALS), neurofilament L is nitrated and may be involved in the aberrant motor neuron function characteristic of ALS (53). In AD, studies have shown that 3,3'-dinitrotyrosine and 3-nitrotyrosine levels in the hippocampus, neocortex, and ventricular cerebrospinal fluid are five- to eightfold higher compared with age-matched controls (113). Redox proteomics techniques (Fig. 8) have been used to identify 10 proteins that show increased specific nitrotyrosine immunoreactivity in the brains of patients with AD. Specific proteins found to be nitrated in AD include α-enolase, triosephosphate isomerase, neuropolypeptide h3, β-actin,
Mild cognitive impairment (MCI), arguably the earliest form of AD, has several proteins that are nitrated, including α-enolase, aldolase, GAPDH, malate dehydrogenase, glucose-regulated protein (GRP) precursor, heat-shock protein 70, glutathione S-transferase 3 (GST3), multidrug-resistance protein 3 (MRP3), peroxiredoxin, dihydropyrimidase like-2 (DRP2), fascin 1, and 14-3-3 γ (196). α-Enolase was previously identified as specifically oxidized in AD brain (39). Taken together with the increased nitration of the metabolic enzymes triosephosphate isomerase, aldolase, and malate dehydrogenase, these results indicate a possible mechanism to explain the altered glucose tolerance and metabolism exhibited in AD (197). In addition to metabolism, GAPDH plays a role as an NO sensor (45). Neuropolypeptide h3, also known as phosphatidylethanolamine-binding protein (PEBP), hippocampal cholinergic neurostimulating peptide (HCNP), and raf-kinase inhibitor protein (RKIP), has a variety of functions in the brain, including upregulating the production of acetylcholine transferase in cholinergic neurons after NMDA-receptor activation (151). Acetylcholine transferase activity is decreased in AD, and cholinergic deficits are prominent in AD brain (188). Nitration of neuropolypeptide h3 may contribute to the decline in cognitive function due to lack of neurotrophic action on cholinergic neurons of the hippocampus and basal forebrain.
Carbonic anhydrase II is critical for maintenance of pH and control of carbon dioxide levels, and its activity is altered in AD (197). ATP synthase α chain is important in energy metabolism, and voltage-dependent anion channel protein 1 is involved in the mitochondrial permeability transition pore, with consequent apoptotic considerations, as well as in mitochondrial calcium ion homeostasis.
GRP precursor and Hsp70 are both chaperone proteins that aid in the proper folding of molecular targets and escort targets to the proteosome for degradation if proper folding is not achieved. MRP3 and GST3 are major detoxification enzymes that pump xenobiotics and glutathione-conjugated proteins out of the cell. The nitration of GRP precursor, Hsp70, MRP3, and GST3 proteins conceivably inhibits function and may contribute to the formation of the senile plaques observed in AD. DRP-2 is involved in axonal outgrowth and pathfinding to neighboring neurons. Fascin-1 is a marker of dendritic functionality, and the addition of fascin-1 has shown to be protective against oxidative stress (153). The nitration of DRP-2 and fascin-1 may contribute to the shortened dendritic length observed in AD (155).
In rodent models of traumatic brain injury (TBI), a condition in which sudden trauma and secondary injury cause brain damage, nitrated tyrosine residues were found on several proteins (169). These proteins include synapsin 1, γ-enolase, guanosine diphosphate dissociation inhibitor 1 (GDP1), phosphoglycerate mutase (PGM), Hsp70, and ATP synthase (169). Synapsin 1 aids in the maintenance of synaptic vesicle reserves for the release of neurotransmitters (152). Nitrated synapsin 1 correlates with the reduced neurotransmission and synaptic plasticity that patients with TBI exhibit (184). GDP1 inhibits the exchange of GDP for GTP, a process necessary for glycogen breakdown to form glucose. Glycogen levels are increased in TBI brain compared with controls, a downstream effect impaired by GDP1 activity (154). Similar to cysteine nitration by peroxynitrite, which inactivates numerous proteins involved in energy production, including GAPDH, creatine kinase, and complexes I, II, III, and V of the electron-transport chain, several of these proteins are susceptible to tyrosine nitration, as described earlier, making metabolic enzymes particularly vulnerable to oxidative modification by peroxynitrite (108).
Methionine is directly oxidized by peroxynitrite predominantly to form methionine sulfoxide, but ethylene and dimethylsulfide are formed in some instances (4). Methionine oxidation inactivates glutamine synthetase, an enzyme responsible for glutamine synthesis. Glutamine is a critical amino acid for immune system activity and is a factor necessary for macrophage-mediated phagocytosis (179). Methionine oxidation is reversible and catalyzed by methionine sulfoxide reductase (MsrA). MsrA reduces methionine sulfoxide back to methionine at the expense of NAD(P)H. MsrA uses the thioredoxin/thioredoxin reductase endogenous antioxidant system to regenerate the catalytic active site (104). Decreased methionine reductase levels have been associated with the onset of AD (192, 193). Peroxynitrite also modifies tryptophan and histidine, but the implications of the nitration of these amino acids are less clear and are the subject of current investigation (4).
Conclusions
As discussed in this review, traditional views of nitric oxide (NO) biology have been based around four key principles: (a) NO synthases [endothelial NO synthase (eNOS), neuronal NOS(nNOS), and inducible NOS (iNOS)] have unique and independent physiologic functions; (b) NO diffuses freely in biologic systems to carry out the tasks of each isoform; (c) the physiologic actions of NO are mediated primarily by cGMP, through the binding of NO to the heme group in soluble guanylate cyclase (sGC); and (d) the cytotoxic versus cytoprotective properties of NO are the consequence of the specific and respectively nonspecific redox chemistry of NO nitrosylation. Here we highlighted the Janus nature of NO. Depending on a number of factors, NO can be neuroprotective or neurotoxic. Clearly, additional molecular understanding of the roles of NO and their sequelae in cellular physiology, CNS injury, and CNS neuroprotection is needed. Newer, more powerful proteomics methods could be used to identify nitrated proteins that are not easily separated by using 2D gel electrophoretic methods. Methods of identifying low-abundance signaling proteins that are regulated or damaged by nitration will provide new insights into these Janus roles of NO. In addition, the possibility of counteracting NO/RNS neurotoxic effects by modulating the gene expression of endogenous enzymes and chaperones, which play key roles in defense against cellular injuries, by nutritional products or pharmacologic compounds, represents an innovative approach to therapeutic intervention in neurodegenerative disorders. The future of NO research in the CNS is bright, and much remains to be learned. Given the significant increase in the average age of the population in most Western and developing countries as health care improves, and the fact that aging is the single most important risk factor for age-related neurodegenerative disorders like AD, this is an exciting time to be involved in research aimed at deciphering the Janus roles of NO in the CNS.
Footnotes
Acknowledgments
Work from the authors' laboratories was supported by grants from MIUR, FIRB RBRN07BMCT, RCUK, the American Cancer Society (RSG-07-157-01-CNE), Tenovus, the Royal Society, the Anonymous Trust, and by “Fondi Ateneo” 2007 and 2008.