
Abstract
We review here current evidence on the role of reactive oxygen species (ROS) and of the intracellular redox state in governing crucial steps of the metastatic process, from cell detachment from the primary tumor to final colonization of the distant site. In particular, we discuss the redox-dependent aspects of cell glycolytic metabolism (Warburg effect), of cell juggling between different motility styles (epithelial-to-mesenchymal and mesenchymal-to-amoeboid transition), of cell resistance to anoikis and of cell interaction with the stromal components of the metastatic niche. Central to this overview is the concept that metastasis can be viewed as an integrated “escape program” triggered by redox changes and instrumental at avoiding oxidative stress within the primary tumor. In this novel perspective, metabolic, motility, and prosurvival choices of the cell along the entire metastatic process can be interpreted as exploiting redox-signaling cascades to monitor oxidative/reductive environmental cues and escape oxidative damage. We also propose that this theoretic framework be applied to “normal” evasion/invasion programs such as in inflammation and development. Furthermore, we suggest that the intimate connection between metastasis, inflammation, and stem cells results, at least in part, by the sharing of a common redox-dependent strategy for infiltration, survival, dissemination, and patterning. Antioxid. Redox Signal. 11, 2791–2806.
Introduction
Recent advances in elucidating the role of ROS as signaling intermediates downstream from activated growth-factor receptors and adhesion molecules have provided a biochemical framework for understanding how intracellular redox changes can affect cellular functions relevant to the metastatic processes, like proliferation (104, 131), motility/adhesion (23), and survival. In particular, ligand-induced ROS generation through membrane-bound NADPH oxidases can directly regulate the enzymatic activities of tyrosine phosphatases and tyrosine kinases through the controlled and fully reversible oxidation of their critical cysteine residues (19, 118, 143). This finding has contributed to the recognition of ROS as signaling molecules, thus allowing the inclusion of redox changes observed in many models of malignancy in the domain of oncogene-driven signal-transduction derangement. However, although this perspective opens novel and interesting scenarios for the understanding and, we hope, the management of cancer metastasis, it should not obscure the notion that actively proliferating cells are extremely prone to oxygen toxicity (58, 117, 132), and that resistance to indiscriminate oxidation should be legitimately included among the “hallmarks of cancer” (55). This is particularly true for malignant cells bound to a metastatic fate.
Prolonged or even intermittent hypoxia, innate as well as adaptive immunity, and inflammation are responsible for extensive cell death within a primary tumor site. It is now clear that oxygen radicals and oxidative stress significantly contribute to environmental selection against tumor cells. ROS are generated in mitochondria of cells exposed to low oxygen (4), a phenomenon further amplified by cyclic reoxygenation (80). In addition, ROS are released within the tumor and at the tumor–host interface by infiltrating immune and inflammatory cells. Finally, radiotherapy and anticancer drugs kill cancer cells largely by generating ROS. Hence, malignant cells capable of leaving the primary tumor site and entering the metastatic route are likely to have developed a remarkable resistance to oxidative insults. Interestingly, hypoxia, inflammation, and resistance to chemoradiation therapy have all been mechanistically linked to an increased risk of tumor dissemination (30, 36, 51, 92). Therefore, the link between ROS and metastasis proves unexpectedly complex, in that environmental pressure to resist and avoid oxidative stress and the capacity to activate and harness redox-dependent signaling cascades for proliferation, migration, and survival must be balanced for malignant cells to disseminate to a distant site (99).
The aim of this review is to summarize recent insights into the role of ROS and redox reactions in multiple steps of the metastatic process, from cell adaptation to hypoxia and acidosis, to cell invasiveness and anchorage independence, to cell homing at distant sites and interplay with local inflammatory cues. Central to this overview is the concept of metastasis as an integrated “escape program” triggered by redox changes and instrumental in avoiding oxidative stress within the primary tumor. In this novel perspective, cell preferences for aerobic glycolysis switch (Warburg effect), increased cell motility, and enhanced survival represent different aspects of a global “antioxidant response,” which also explains why these processes so extensively involve oxygen species as signaling intermediates.
Cell Adaptation at the Primary Tumor Site: “Glycolytic Switch” as an Antioxidant Strategy
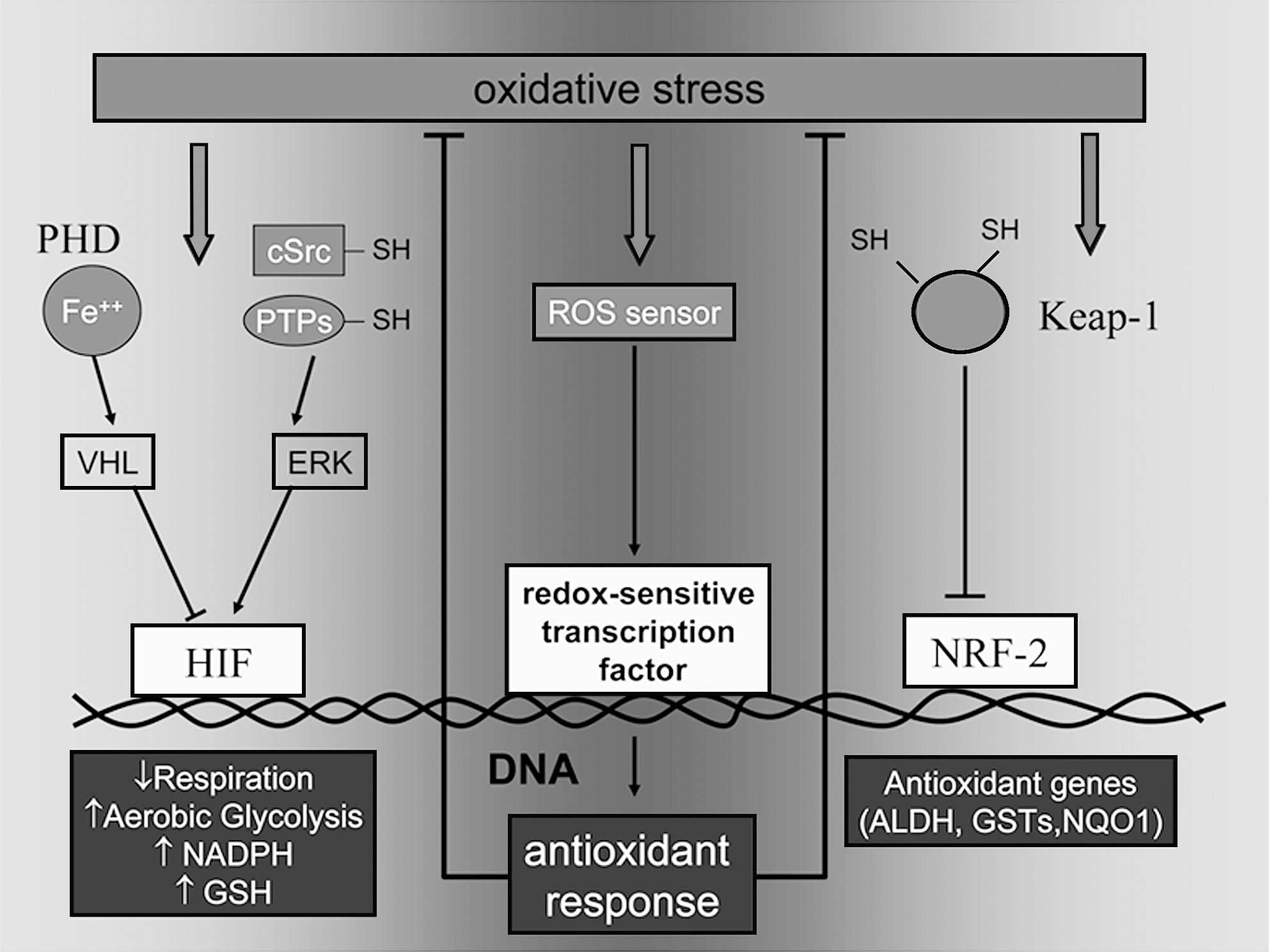
Reduced oxygen tension and reiterated hypoxia/reoxygenation cycles are normally faced by cancer cells within a rapidly growing tumor mass (29, 60). Adaptation of cells to low oxygen, in the current view, underlies the Warburg effect (i.e., metabolic switch to glycolysis even in the presence of an adequate oxygen supply (61). Metabolic reprogramming toward glycolytic ATP generation involves several molecular players and biochemical mechanisms, including primary mitochondrial dysfunction and mtDNA mutations (62), oncogenic transformation (126), loss of tumor-suppressor genes such as Trp53 (5, 93) and von Hippel–Lindau (VHL) (123), and, primarily, hypoxic signaling through the hypoxia-induced transcription factor 1 (HIF-1) (112). HIF-1 is a heterodimer composed of constitutive, stable β subunits and one of the unstable α (alpha1, alpha2, and alpha3) subunits, which undergo rapid proteasomal degradation because of sequential actions of oxygen-dependent prolyl hydroxylases (PHDs) and the VHL ubiquitin ligase (reviewed in ref. 65). HIF-1 stabilization in low oxygen leads to transcriptional activation of several glycolysis-related genes, including the glucose transporter GLUT-1, hexokinase, and lactate dehydrogenase (LDH) (112). Additionally, HIF-1 indirectly inhibits mitochondrial respiration through at least two distinct mechanisms: (a) upregulation of the pyruvate dehydrogenase kinase (PDK-1), an enzyme capable of phosphorylating and inactivating mitochondrial pyruvate dehydrogenase, thereby diverting the metabolic fate of pyruvate toward cytosolic reduction to lactate (72); and (b) altered composition and function of the cytochrome oxidase (COX) complex, through transcriptional activation of the subunit COX-IV-2 and protease-mediated degradation of the subunit COX-IV-1 (42). These coordinated changes are important to hypoxic adaptation of the cell in that they translate into increased ATP generation, reduced formation of mitochondrial hydrogen peroxide, and enhanced survival of poorly oxygenated cells. Finally, by transactivation of the carbonic anhydrase IX (CA-IX) gene, HIF-1 prevents intracellular acidification and allows the transfer of acidic equivalents generated by excess production of lactate and CO2 (112) to the extracellular milieu, an event in turn favoring matrix degradation and cell invasion (119). The role of HIF-1 in metabolic cell reprogramming, coupled with the finding that HIF-1 expression levels positively correlate with cancer invasiveness and poor prognosis in experimental and clinical settings (7), suggests that HIF-1 may be critical in linking tumor hypoxia, glycolytic energy metabolism, and cancer metastasis. This idea also is corroborated by recent breakthrough findings from microarray studies, revealing that the expression of lysyl-oxidase (LOX), a matrix-modifying enzyme whose expression in breast cancer cells is associated with EMT and lung metastasis, is also strongly induced by HIF-1 (35).
It is becoming increasingly clear that ROS play a primary role in HIF-1 activation during tumor progression (113). Hydrogen peroxide appears to be essential for HIF-1 accumulation under mild (1% O2) hypoxia, but is dispensable for cell response to anoxia (54). This reactive intermediate is generated by the mitochondrial electron-transport chain (ETC), complex III, and promotes HIF-1 accumulation, at least in part, by interfering with Fe2+-dependent activity of PHDs (45). Chemical inhibitors of ETC reportedly inhibit hypoxic induction of HIF-1 (133), whereas mitochondrial DNA mutations associated with increased generation of ROS promote normoxic accumulation of the factor and, in parallel, metastatic growth (62). ROS also mediate oncogene-driven accumulation of HIF-1 in normoxia, a phenomenon crucial for the establishment of the Warburg effect. In particular, Nox-1, a component of the NADPH oxidase complex, can trigger the angiogenic switch in prostate cancer cells (2), and Rac-1, another component of the same enzyme complex, may be necessary for full HIF-1 activation by low oxygen (59). Additionally, H-Ras stabilizes the hypoxic factor through the generation of the superoxide anion in normal osteoblasts (145) and conceivably also in cancer cells. In these examples, direct PHD inactivation by ROS, as well as redox-dependent activation of protein phosphorylation cascades, may both contribute to HIF-1 stabilization (10).
Oncogene-dependent activation of HIF-1 in normoxia suggests that glycolysis in cancer cells may represent a molecular strategy for malignant progression rather than a simple adaptation to lack of oxygen or mitochondrial failure. With the trade-off of reduced ATP generation, aerobic glycolysis provides substrates for biosynthetic pathways, including nucleotide, fatty acid, and protein synthesis, necessary to sustain uncontrolled cell proliferation (27). Additionally, glucose binding and phosphorylation by hexokinase, even in the absence of further catabolic steps, promotes enzyme association with mitochondria and is necessary for the antiapoptotic signaling of the Akt/PKB serine-threonine kinase (50). More important, glucose metabolism through the pentose shunt, reportedly favored by HIF (45 –52), increases the pool of NADPH and, through glutathione reductase, that of the major cellular antioxidant buffer, GSH, as well. NADPH is also necessary for NADPH oxidase activity, and, by extension, for redox signaling downstream of RTKs and cytokine receptors. NADPH oxidase activity may also benefit, especially under low-oxygen conditions, from increased availability of O2 shunted from inhibited mitochondria. Additionally, acidification of the extracellular milieu by lactate and carbonic acid may promote matrix degradation and cell invasion (119). Finally, loss of mitochondrial respiration reduces the mitochondrial oxidative burden and decreases the probability of apoptosis initiation by the organelle (28).
Several lines of evidence support the relevance of glycolytic metabolism to malignant cell behavior. First, inhibition of lactate dehydrogenase in aggressive cancer cells restores mitochondrial respiration and, in parallel, reduces proliferation and tumorigenicity in vivo (37). Second, M2, the embryonic variant of pyruvate kinase, which is catalytically inefficient and allows upstream accumulation of glycolytic intermediates, is preferentially expressed in malignant cells (24). Third, metabolic accumulation of succinate and fumarate in the cytosol, due to genetic mitochondrial defects, stabilizes HIF-1 through PDH inhibition (74), and, in human gliomas and other cancer cell lines, the accumulation of HIF-1α protein under aerobic conditions requires the metabolism of glucose to pyruvate (86). Finally, a glycolytic switch is induced in cancer cells by loss of the p53 tumor-suppressor function, one of the most frequent molecular signatures of malignancies. Molecular details of the latter phenomenon have been recently clarified. Loss of p53 leads to reduced expression of two p53 target genes, synthesis of cytochrome oxidase-2 (SCO 2) (93) and Tp53-induced glycolysis and apoptosis regulator (TIGAR) (5). Consequently, glycolysis is promoted by preventing the assembly of mitochondrial DNA-encoded COX II subunit (MTCO2 gene) into the COX complex and by increasing the concentration of fructose 2-6 bis-phosphate, a potent activator of glycolysis and inhibitor of gluconeogenesis, respectively. Additionally, functional p53 may inhibit glycolysis by restraining the activity of the proinflammatory factor NF-κB (69). Interestingly, an increased resistance to oxidative stress represents a major consequence of p53 inactivation in cancer cells (103), in parallel with the metabolic shift to glycolysis.
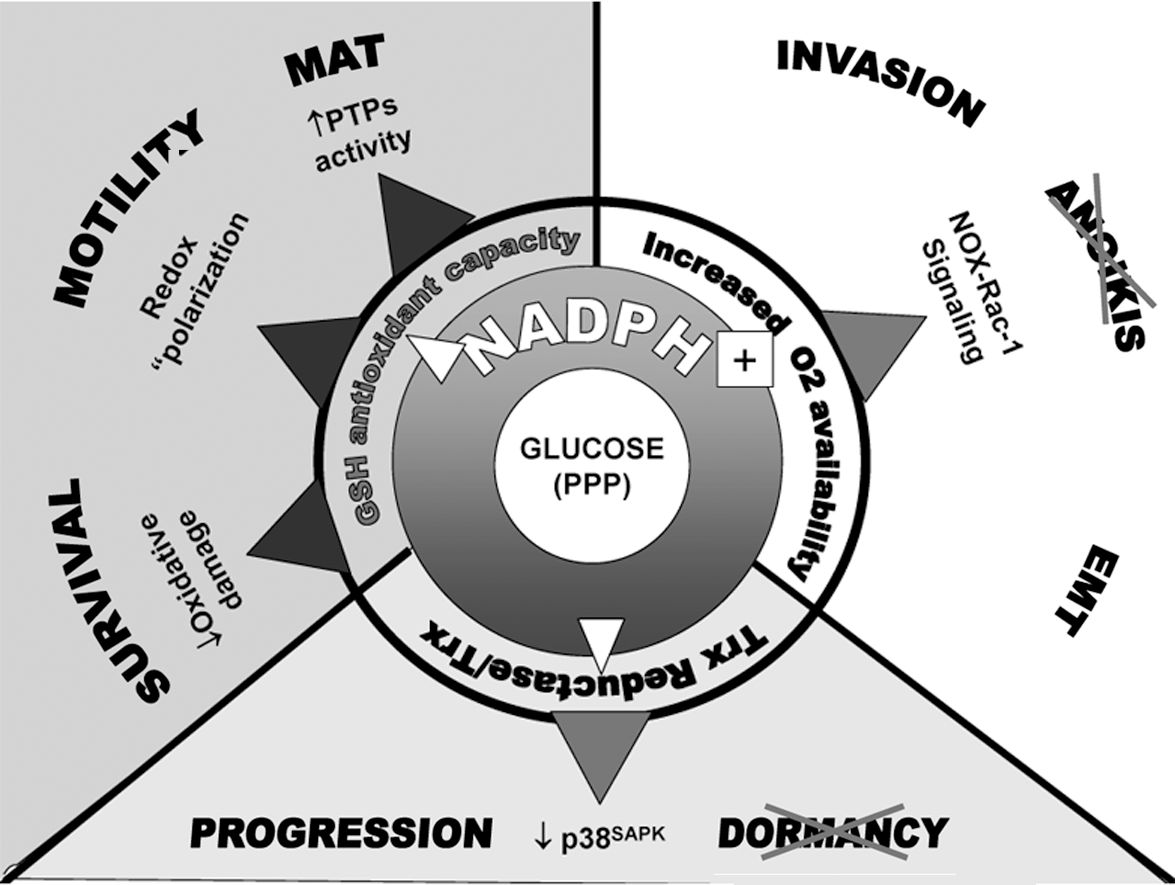
Although these considerations underline the many facets of the long-sought link between glycolysis and cell malignancy, they also support the idea that glycolytic switch may primarily represent a cancer cell strategy to resist oxidative stress (11). Potential antioxidant mechanisms activated by glycolysis include GSH repletion through the pentose phosphate pathway and NADPH (44, 52) and a decrease in ROS generated by mitochondrial respiration. Accordingly, glucose deprivation leads to an increase of intracellular ROS and to glutathione oxidation in several tumor cell lines, in a fashion largely targetable by ETC inhibitors (128; Pani et al., unpublished results). Additionally, pyruvate may contribute to lower oxidative stress by directly scavenging H2O2. Because HIF-1, the major determinant of the glycolytic switch in malignant cells, is highly sensitive to oxidative stress (42, 54, 113), it is intriguing to represent the ROS–HIF-1 pathway, which is triggered by hypoxia, oncogene activation, or mitochondrial dysfunction, as part of a typical antioxidant circuit (83). In an example of such an antioxidant switch, the Keap-1–Nrf2 (nuclear respiratory factor 2) cascade, the “sensor” Keap-1 dissociates from NRF-2 in response to oxidative stress, allowing its translocation to the nucleus and the efficient transactivation of multiple antioxidant genes harboring an antioxidant response element (ARE) (83). In a parallel fashion, HIF activation in response to oxidative inhibition of PHDs or tyrosine phosphatases (“redox sensors”) translates into a complex genetic program, which culminates in the Warburg effect as an integrated antioxidant strategy (Fig. 1). Although growth-promoting functions of moderate levels of ROS clearly contribute to cancer development and metastasis (130, 137, 149), excess ROS can inhibit cancer cell growth and induce senescence, oxidative damage, and cell death. Antioxidant protection may be particularly critical to the fate of metastatic cells when they lose contact with neighboring cells and undergo EMT (104), as well as for survival in the bloodstream and during colonization of inflammation-primed metastatic sites (see later). Thus, enhanced glycolysis, which can reduce the ROS, might be a crucial mechanism for fine-tuning ROS levels to allow enhanced tumor cell growth and dissemination, while protecting them from ROS-induced senescence and apoptosis (6).
The Engagement of the Motile Phenotype: Escaping from a Hostile Environment
One main feature of a successful cancer is its ability to activate an epigenetic program of motility, allowing cancer cells to escape an unfavorable primary site and invade a secondary and more favorable site (17, 64). Reasons to escape from the original site may include a limited supply of oxygen or nutrients or both, unfavorable extracellular matrix (ECM) proteins, as well as the interplay with the stromal counterpart of the tumor microenvironment (96, 100). It is noteworthy that these cues all induce oxidative stress and are the same as those that specifically engage the transcriptional program of transdifferentiation toward a motile tumor cell. Hence, in addition to the genetic changes that tumor cells have undergone during multistep tumorigenesis, their ability to invade and metastasize may be enhanced by microenvironmental cues, including hypoxia/ischemia or stromal cell interaction. It has been clear for a long time that tumor cells do not proliferate and progress in isolation. Rather, they are dependent on support from their surroundings, which include ECM and various supporting or stromal cells. The microenvironment of the cancer cell is a strong determinant of whether it acquires the potential to survive and metastasize (32, 91, 100). Thus, during primary tumor formation, the cells in a carcinoma engage a multifaceted collection of mesenchymal cells, jointly forming the tumor-associated stroma. As tumor progression proceeds, the stromal cells create a “reactive stroma” that releases a variety of signals that induce changes in carcinoma phenotype. The stroma consists of cancer-associated fibroblasts (CAFs), converted into myofibroblasts by not yet completely identified cancer cell–released factors, endothelial cells, and pericytes that form blood and lymph vessels, as well as inflammatory cells attracted by cancer chemokines (32, 91, 100). All these host cells engage in continuous molecular cross-talks with the cancer cells, secreting large amounts of factors/cytokines to influence survival and invasion. CAFs are known to play a key role within the tumor milieu, as they undergo a process called mesenchymal–mesenchymal transition (MMT) or fibroblast activation in response to factors released from epithelial cells, macrophages, or cancer cells themselves (66). CAFs are in turn able to modify the behavior of cancer cells through release of soluble factors such as transforming growth factor β1 (TGF-β1), hepatocyte growth factor (HGF), or fibroblast growth factor-2, or the composition and the amount of ECM (155). Recently it was reported that TGF-β1 can induce a redox-dependent MMT in skin fibroblasts, involving lipid hydroperoxidation and expression of MMPs, as well as secretion of HGF, vascular endothelial growth factor (VEGF), and interleukin-6 (16). Tumor-associated host cells are themselves invasive, and some of them arrive at the site of metastasis in advance of the cancer cells, thereby protecting cancer cells when circulating and facilitating their invasion (43, 120).
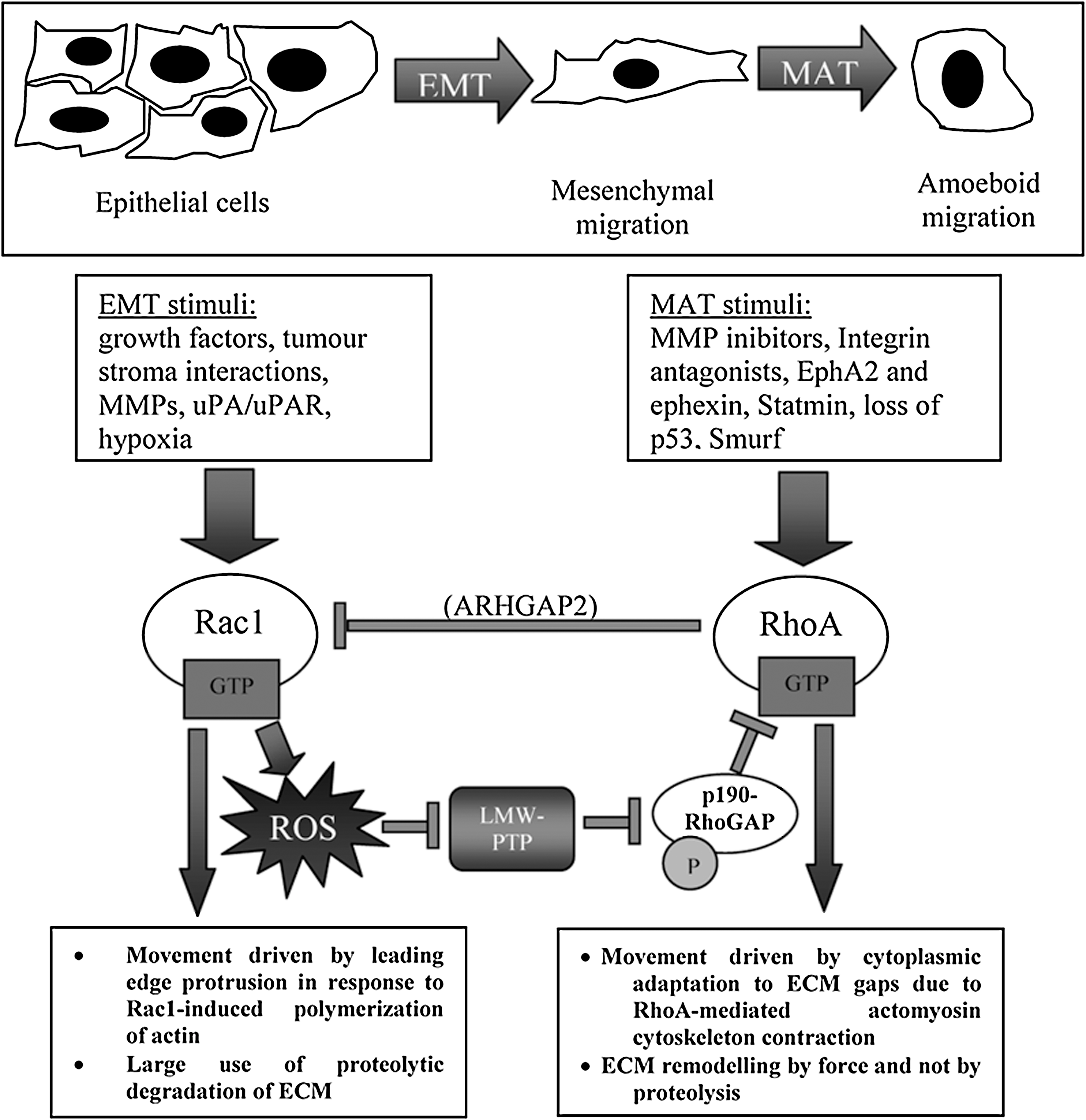
Several studies describe the nature of the signals that are released by the tumor microenvironment and serve to induce EMT or the mesenchymal-to-amoeboid transition (MAT). The latter transition involves profound changes in cell phenotype that cause immotile epithelial cells to acquire traits such as motility, invasiveness, resistance to apoptosis, and the ability to adapt to environmental changes and continue to invade successfully (41). These signals serve to induce expression of a series of transcription factors, which, in turn, are capable of inducing the plasticity of cell motility. In the EMT process, the cells lose their epithelial characteristics, including their polarity and specialized cell–cell contacts, and acquire a mesenchymal migratory behavior, allowing them to move away from their original site to remote locations. EMT illustrates the differentiation plasticity during development, but is commonly exploited by cancer cells to invade and metastasize (111, 153). Mesenchymal motility is characterized by elongated and polarized cell morphology. It depends on ECM degradation by the moving cells, which, through production of matrix metalloproteinases (MMPs), generates a “path.” This also requires activation of Rac1 at the leading edge of the cell and to inhibition of RhoA GTPase (Table 1) (41, 122).
Although the involvement of ROS as procarcinogenic agents has been strongly supported by the past literature (20, 21, 49, 149), their role in the acquisition of an invasive and metastasizing phenotype is still not clear. ROS have been implicated in the activation of EMT in several cancer models. It is well established that NADPH oxidase is responsible for the generation of intracellular ROS induced by several growth factors, thereby triggering cell proliferation, migration, cytoskeletal remodeling, angiogenesis, and regulation of genes associated with tumor metastasis (49, 142). Recently, a differentially spliced form of Rac1, Rac1b, was found to be involved in MMP3-induced EMT of mammary epithelial cells, and the consequent burst in oxidants due to Rac1b activation has been correlated with carcinogenesis through DNA damage (115). Consistent with this, Rac1-mediated redox signaling has been reported to be vital for melanoma aggressiveness driven by expression and activation of Met, a known regulator of EMT (38). Furthermore, ROS play a key role in driving EMT in response to both TGF-β1 and aldosterone in renal models (38, 156). In addition, hypoxia has been correlated with EMT and the consequent acquisition of an invasive behavior (15). Of note, hypoxia has been causally linked to an increase in intracellular/mitochondrial ROS, thereby stressing that hypoxic conditions and ROS can act synergistically in inducing metastatic outcomes (75).
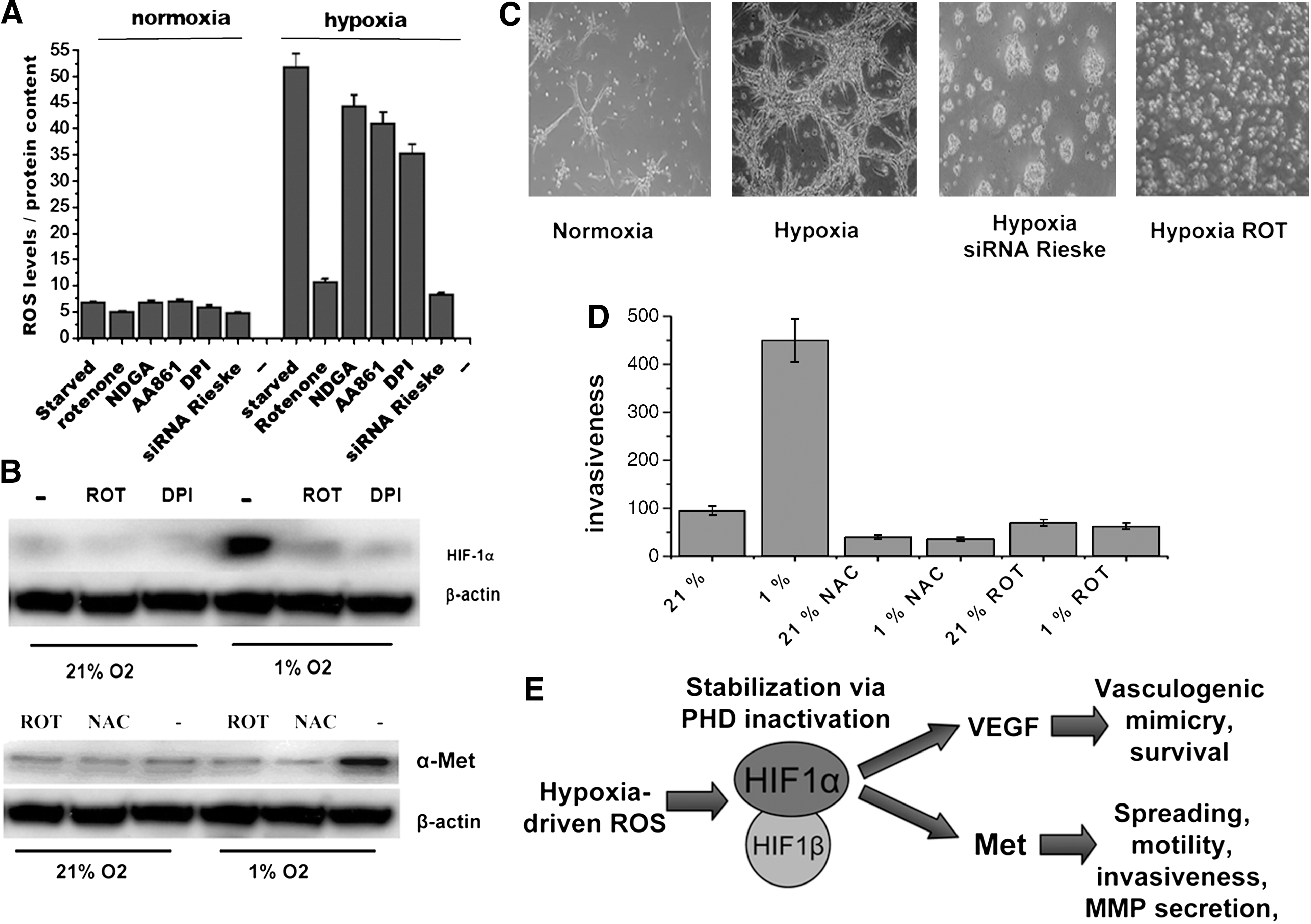
In agreement with a role for ROS in hypoxia-mediated EMT, in several epithelial cancers, specific inhibition of mitochondrial generation of ROS revealed that early EMT-related events induced by hypoxia are redox regulated by the activation of Snail and Twist transcription factors, whereas the motility and invasiveness responses are sustained mainly by HIF-1α and VEGF-dependent mechanisms (15). Furthermore, in keeping with a prosurvival and promigratory role of ROS in cancer cells, we have evidence that in human aggressive melanoma, both HIF1-α stabilization and VEGF secretion are under the control of mitochondrial ROS release, guiding a cMet-dependent motility response and a VEGF-dependent survival of cancer cells (Fig. 2). Hypoxia has also been linked with the ability of melanoma cells to organize into perfusable vascular-like networks, a process called vasculogenic mimicry for metastatic dissemination (57). Our preliminary data indicate that ROS and HIF-1α stabilization are strongly involved in hypoxia-mediated vasculogenic mimicry, thereby confirming ROS as central players in the different strategies adopted by cancer cells to escape from unfavorable sites. In addition, hypoxia-generated mitochondrial ROS in hepatoma, neuroblastoma, and colon carcinoma cells, contribute to the survival of cells during hypoxia. These mitochondrial ROS play a dual role. First, they enhance HIF-1α stabilization through inhibition of PHD and, second, they activate, in a redox- dependent manner, the Src kinase leading to NF-κB activation through IκB tyrosine phosphorylation (84).
Moreover, Weinberg (90, 111) recently reported that breast cancer cells undergoing EMT acquire many of the properties of cancer stem cells (CSCs). These self-renewing tumor-seeding cells found in several solid tumors are the subpopulation endowed with particular type of plasticity in motility, after undergoing the EMT transcriptional program. The “stemness” of a cancer population is a mechanism to survive in hostile microenvironment once the primitive cancer has grown. CSCs have often been reported as quiescent cells, still in G0 cell phase of the cell cycle, which, on receiving activating stimuli, can regenerate the original cancer in a different site, thereby facilitating survival of neoplastic cells (71). The link between cancer stemness and EMT, therefore, establishes the motility prompt as a prosurvival response, a strategy used to escape the primitive site and disseminate motile cancer cells. Unfortunately, the real contribution of redox circuitries to CSCs is far from being elucidated. Indeed, whereas EMT has been correlated with enhanced ROS production, the quiescent G0 phenotype of both hematopoietic stem cells and epithelial cancer stem cells has been associated with low ROS levels (30, 63). Although further studies are necessary for a better understanding of how ROS may affect cancer cell stemness, this evidence points to interesting although speculative scenarios. For instance, exposure to moderately increased levels of ROS, as elicited by surrounding hypoxia/inflammation, may “alert” stem cells to resume proliferation and, through redox-sensitive components of the motility apparatus, migrate out of their no-longer safe niches, in an effort to (a) “repair” and (b) reproduce elsewhere what is perceived as a threatened cancer tissue.
Another differentiation plasticity observed in cancer cells is MAT (i.e., the shift from a mesenchymal to an amoeboid motility). The latter is a primitive form of cell migration that allows cells to glide through, rather than degrade, ECM barriers through weakened cell–ECM attachments (41). In contrast to mesenchymal motility, cells moving through an amoeboid mode show inhibition of Rac1 and strong activation of RhoA (41, 106, 122). Depending on the context of invasion, adaptation responses can cause MAT, thereby generating sustained dissemination of single cells. MAT has been reported for several cancer cells on inhibition of integrin or MMP function, or expression of Rho-modulating agents such as EphA2 or Snurf (106, 121, 122). Of note, aggressive melanoma cells are able to shift ad hoc between mesenchymal and amoeboid motility, after different microenvironmental prompts. In response to proinflammatory cytokines, they undergo EMT, whereas after reexpression of the embryonic EphA2 receptor, which strongly activates the small GTPase RhoA, they undergo MAT (106). Although the identification of molecular players of this plasticity in aggressive cancers is still in its infancy, the two GTPases Rho and Rac have been found to act as sensors for the shift in motility programs (122, 151). Indeed, EMT is characterized by activation of Rac1 and inhibition of Rho, whereas MAT has always been associated with the opposite behavior of the two GTPases. Both GTPase have been reported to be key regulators of redox signaling. Rac1 is a widely known upstream modulator of several regulated intracellular sources of ROS (39), including NADPH oxidase and LOXes, and it is thus conceivable that its activation in EMT has been correlated with the EMT transcriptional program. In addition, Rac1 activation has been linked to a redox-dependent inhibition of Rho, through regulation of its negative regulator p190RhoGAP (98). Therefore, the same redox-mediated pathways give rise to activation of the EMT program and, conversely, to repression of MAT, through inhibition of its central player, RhoA.
In contrast to EMT, MAT motility program appears to be mediated by activation of RhoA and inhibition of Rac1, thereby suggesting that it may be associated with a decrease in cell redox potential. In keeping with this hypothesis, EphA2 tyrosine kinase receptor signaling, reported to activate a Rho-mediated MAT in aggressive melanoma and metastatic prostate carcinoma through inhibition of Rac1, is able to cause a decrease in intracellular ROS (13). This leads to activation of the redox sensitive low-M r protein tyrosine phosphatase and to inactivation of its substrate p190RhoGAP. These events cause enhancement of Rho activation and the achievement of an amoeboid motility style (Fig. 3). Therefore, the cellular redox potential may be the master switch allowing a moving/escaping cell to adapt to epigenetic and microenvironmental cues shifting ad hoc between different motility strategies and allowing cancer cells to survive and metastasize.
The Intermediate Step: Surviving Inadequate ECM Adhesion
In normal tissues, adhesion to appropriate ECM proteins is essential for cell survival, whereas loss of contact from the ECM induces a death process that has been termed anoikis (127). Tumor cells that acquire malignant potential have developed mechanisms to overcome this loss of ECM support and to survive outside their normal environment in the absence of adhesion. This process is an important step within the metastatic progression of epithelial cancers, because resistance to anoikis allows cells to survive within the circulatory and lymphatic systems and to spread to distant sites away from the primary tumor.
Similar to several other kinds of apoptosis, the initiation and execution of anoikis occurs after activation of different pathways, the triggering of cell-surface death receptors (the extrinsic pathway) or the perturbation of mitochondria (the intrinsic pathway) (22, 48). To date, anoikis resistance of several metastatic cancer cells has been reported and, in some instances, a key role is played by constitutive oxidative stress, a common feature of several cancers cells. Redox regulation of anoikis resistance has been reported exclusively for the intrinsic pathway. This intrinsic process is initiated by mitochondrial permeabilization and results in the release of cytochrome c into the cytoplasm and activation of the BH3-only family of proapoptotic proteins (9). Bid and Bim are activated after detachment of cells from ECM and rapidly promote Bax/Bak oligomerization (22, 134). The apoptosis sensitizers (Bad, Bik, Bmf, Noxa, Puma, and Hrk) also contribute to cell death by inhibiting Bcl-2, thus allowing the Bax/Bak oligomer to form (77).
Evidence indicates that the BH3-only proteins play an essential role in anoikis of different cell types. In particular, after integrin engagement, both phosphoinositide-3-OH kinase (PI3K)/Akt and extracellular signal-regulated kinase (ERK)-mediated phosphorylation of Bim lead to its ubiquitin-dependent degradation (78, 114). Conversely, on cell detachment, the inhibition of PI3K/Akt and ERK signals strongly increases accumulation of Bim, which interacts with Bcl-2, neutralizing the latter's prosurvival function (135).
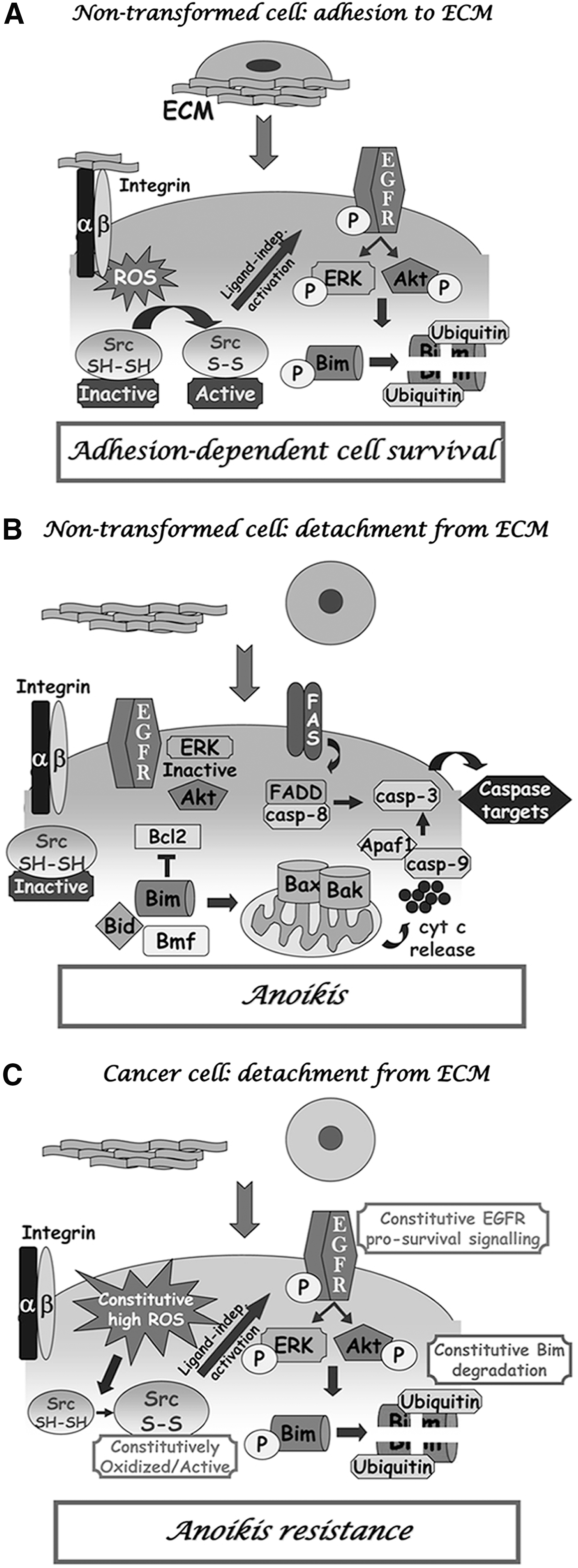
First, a redox-based strategy for allowing cancer cells to evade anoikis and enhance their metastatic potential has been proposed, thereby focusing on the crucial role of ROS as key signaling molecules eliciting prosurvival signals. Indeed, recent results by our laboratory highlighted a fundamental role of hydrogen peroxide, produced on integrin engagement in a Rac-1–dependent manner, in ensuring protection of nontransformed epithelial cells from anoikis through the oxidation of the tyrosine kinase Src (46). In particular, Src undergoes a redox-mediated activation and phosphorylates/activates EGF receptor (EFGR) in a ligand-independent manner. This event sustains an ERK- and PI3K/Akt-mediated phosphorylation of the proapoptotic protein Bim, leading to its degradation and therefore protecting cells from anoikis (46, 79) (Fig. 4A). In the absence of ECM contact, or when the redox regulation of Src is impeded through antioxidant treatments, Bim accumulates and leads cells to apoptosis (46) (Fig. 4B). Recently, it also was reported that a specific protection from anoikis has been carried out through a redox control of the tyrosine phosphatase PTEN. PTEN oxidation is associated with its enzymatic inactivation and the subsequent phosphorylation/activation of Akt, leading to a prosurvival signal (89). Data from our group further supported the key role of ROS as prosurvival molecules, demonstrating a constitutive oxidant production in metastatic prostate carcinoma cells. The redox imbalance releases amplified and deregulated signals, normally triggered by integrin engagement in a transient fashion, thus leading to sustained anchorage-related signals even in absence of ECM contact. Owing to the oxidative stress, Src is constitutively maintained in its oxidized/activated state and leads to a sustained ligand-independent activation of EGFR-related prosurvival signals (i.e., ERK and PI3K/Akt. This results in the degradation of the proapoptotic protein Bim, thereby promoting cell survival even in the absence of proper adhesion, with obvious implications for the metastatic dissemination of the original tumor (47) (Fig. 4C). Interestingly, interfering with Src redox regulation, through antioxidant intervention or genetic manipulation, restores the apoptotic phenotype in response to lack of adhesion. Conversely, a prooxidant milieu is able partially to confer anoikis resistance to nontransformed prostate epithelial cells, suggesting that exposure to exogenous oxidative stress, as well as increased expression or activation of EGFR or Src kinase, may affect anoikis sensitivity of primary prostate tumor cells, thereby serving as one of the factors affecting their metastatic dissemination (47).
The involvement of both Src and EGFR activity in protection from anoikis is not a novel theme in cancer biology. Indeed, the key role of EGFR-dependent protection from anoikis has been reported in lung fibroblasts, mammary epithelial cells, and epidermal keratinocytes (78). In addition, Src activation has been causally linked to transient protection from apoptosis during initial detachment of epithelial cells from the basal membrane (85). Furthermore, in human lung adenocarcinoma cells, Src activation compensates for the loss of cell-survival signals caused by disruption of cell–ECM interactions and contributes to anoikis resistance (147). Giannoni et al. (47) contributed to this prosurvival circuitry with the novel proposal that the specific role of Src activity during protection from anoikis is mainly redox-based and that ROS are central players of the sustained activation of the kinase (47).
In keeping with a central role of oxidants as modulators of cancer cell resistance to anoikis, it has been recently demonstrated that melanoma cells derived from immortalized nontumorigenic, melanocytes subjected to rounds of anchorage blockade (102) exhibit a significant increase in oxidative/nitrosative stress (14). This metabolic imbalance may have a causal role in modulation of DNA methylation, by inducing a significant increase in DNA methyltransferases activity, and may directly contribute to the acquisition of an anoikis-resistant phenotype through an epigenetic mechanism. Interestingly, antioxidant treatments prevent the increase in DNA methylation observed during melanocyte transformation, thus interfering with the ability to overcome anoikis (14).
Besides the proven role of ROS, several reports implicate the oxidants or oxidant-sensitive targets in anoikis resistance of cancer cells. For example, the strategy of acquiring constitutive activity of the prosurvival pathways to circumvent anoikis is often exploited by cancer cells (22). Nesbit et al. (97) described the acquisition of an autocrine loop for basic fibroblast growth factor loop as one of the most important steps during melanoma progression (97). Other growth factors, such as HGF, interleukin-8, and platelet-derived growth factor-AA, have been shown to act in an autocrine manner to aid proliferation, survival, and migration of melanoma cells (81). In addition, the neurotrophic tyrosine kinase receptor TrkB, overexpressed in several highly aggressive human tumors, was recently described as a prosurvival factor. In particular, TrkB, frequently upregulated in anoikis-resistant cells, acts as a potent suppressor of anoikis by activating the PI3K/Akt pathway (154). Although not proven, a role of ROS in anoikis resistance due to HGF, PDGF, or TrksB autocrine loops is highly likely because of the reported involvement of hydrogen peroxide downstream of these GFs.
Furthermore, in neoplastic cells, the constitutive activation of Src, Akt, and Erk1/2 pathways, which inhibit Mcl-1 degradation, likely accounts for the ability of cancer cells to evade anoikis (148). These data highlight the importance of the antiapoptotic member Mcl-1 in the maintenance of anoikis resistance (148). In adherent cells, Mcl-1 inhibits Bim, thereby preventing the activation of the apoptotic machinery. Anoikis initiation is dependent on the ubiquitination of Mcl-1 by Mcl-1 ubiquitin-E3 ligase (Mule), culminating in Mcl-1 degradation. Hence, the balance of proapoptotic and antiapoptotic factors, mainly regulated at a posttranslational level, determines the susceptibility of a given cell to anoikis.
EMT is another strategy for cancer cells, not only to become highly invasive and aggressive, but also to acquire anoikis resistance and to survive successfully in inappropriate locations. The activation of several mesenchymal genes, such as Snail, Twist, HGF-R/cMet, and NF-κB, is critical for EMT success and also overcomes the apoptotic program by constitutively activating specific prosurvival mechanisms (136). In particular, Snail, a zinc-finger transcriptional factor, confers apoptosis resistance by activating survival genes such as the PI3K/Akt cascade and by inhibiting caspase-3 activation (3). Twist, a helix–loop–helix transcriptional factor, can modulate the apoptotic machinery by increasing the Bcl-2/Bax ratio (129). Furthermore, several lines of evidence point to the essential role of NF-κB, constitutively activated in several human cancers, in conferring anoikis resistance to tumor cells, through activation of the PI3K/Akt signaling cascade (76). HGF/Met, another essential player of EMT, has been correlated to the ability of the tumor cell to escape anoikis. On EMT, the de novo–expressed Met receptor induces anoikis resistance in uterine endometrial cancer cells by upregulation of cyclooxygenase-2 expression, as well as in head and neck squamous cell carcinoma cells by activation of ERK and PI3K/Akt signaling (67). Of note, beside eicosanoids, cyclooxygenase-2 is a mixed-function oxidase producing large amount of ROS, already involved in tumor progression (107, 146). Again, a concurrent role of ROS in these kinds of insensitivity to anoikis is possible, particularly on the basis of the key role played by ROS during EMT and Met oncogenic activation (see earlier).
The Colonization and Final Step: Following the Inflammatory Route
Although the idea of an intimate connection between cancer and inflammation dates back to Virchow's pioneering observations, recent advances in the understanding of the molecular basis of metastasis have revealed a surprising correlation between cancer dissemination and the mechanisms underlying leukocyte- and bone marrow–derived cells (BMDCs) recruitment to target tissues in the context of the inflammatory process. Accordingly, genetic programs and signatures decoded by microarray profiling of metastatic malignancies constantly include a large fraction of inflammation-related genes, no matter whether “metastasis initiation,” “metastasis progression,” or “metastasis virulence” genes are specifically considered (18). From an even broader perspective, a similar degree of molecular overlap can be found between metastatic cancer dissemination and embryo growth and morphogenesis, and, more specifically, between metastatic and stem cells (see earlier). Interestingly enough, analogies among metastatic/cancer cells, inflammatory cells, and stem cells go beyond the shared properties of motility and “invasiveness” (8), and extend also to metabolic (56) and survival capacity (88), and, as is most relevant to this article, to resistance to oxidative stress (30).
The successful establishment of a cancer metastatic colony requires, even more critically than for primary tumor growth, a complex network of interactions with local microenvironment components, including ECM, stromal and inflammatory cells, and blood vessels. These interactions are crucial to the organ-specific localization of tumor cells, and to the expansion of the colonizing cells (1, 18). Mounting evidence indicates that oxidative signals may represent a nodal component of this network.
In striking similarity to the inflammatory processes, chemokines appear to be involved in the metastatic homing and proliferation of cancer cells. One of the best examples is provided by the role of the stromal cell–derived factor-1/CXCR4 axis in breast cancer metastasis to bone marrow (18). CXCR4 is a G protein–coupled receptor that can transactivate the EGFR in ovarian cancer cells and the TCR in T lymphocytes through an Src-dependent mechanism (108, 144). RTK transactivation and signaling propagation by CXCR4 and other chemokine receptors may involve hydrogen peroxide as a signaling intermediate (40, 140). Although not fully investigated, this possibility is particularly attractive, in view of the pleiotropic effect of the SDF-1/CXCR4 axis on both the cancer and the stromal component of the metastatic niche (12). Interestingly, CXCR4 is a transcriptional target of HIF-1, further supporting the multifaceted role of the latter redox-sensitive factor in different phases of tumor spreading. Along similar lines, perturbation of respiratory chain in breast cancer cells induces the expression of the CXCL14 chemokine in an ROS-dependent fashion, in association with increased proliferation and motility, and with enhanced malignancy in vitro and in vivo (110). Thus, chemokine signaling may represent another component of the “escape” program implemented by metastatic cells to avoid oxidative stress and colonize more favorable distant sites.
Angiogenesis is crucial in metastasis development (55). Again, involvement of ROS in angiogenesis is well established. ROS act at multiple levels, from induction of angiogenic factors to intracellular signaling downstream of their receptors on endothelial cells (25, 94, 141). Additionally, recruitment of endothelial cell precursors (ECPs) derived from bone marrow during neovasculogenesis requires the NOX-2–based NADPH oxidase, and NOX-2–deficient precursors display reduced mobilization from the bone marrow after hindlimb ischemia and migration in vitro in response to SDF-1 (140). This evidence further supports the role of ROS as signaling intermediates in the chemokine system, and, by extension, in the complex network of chemokine-dependent cell interactions taking place at the metastatic site.
Although angiogenesis is conceivably required at an advanced stage of metastasis to support growth of the ectopic tumor mass, the early development of metastatic cancer colonies is critically dependent on the recruitment of BMDCs of myeloid origin to arrange the so-called “premetastatic niches” (68). Factors produced by these cells, such as tumor necrosis factor-α (TNF-α) and MMPs, promote subsequent invasion and proliferation of cancer cells and probably also prevent their immune recognition and destruction.
Indirect evidence suggests that oxidant-mediated signaling may actively participate in this early stage of metastasis development. LOX, an amino-oxidase secreted by hypoxic cancer cells, and whose expression correlates with poor prognosis of breast or head-and-neck cancers (35), promotes BMDC recruitment to niches by enzymatically cross-linking basement membrane components collagen IV and elastin (34). Cross-linked matrix provides optimal mechanical supports to BMDCs and promotes their adhesion and secretion of MMPs, which in turn facilitate cancer cell invasion. LOX is an H2O2-producing enzyme, and previous studies have already shown that this ROS contributes to the chemotactic and chemokinetic activity of LOX (87). Additionally, LOX has been shown to facilitate integrin signaling through the focal adhesion kinase (FAK) and to promote invasiveness of breast cancer cells, in a fashion dependent on the intracellular release of hydrogen peroxide (109). Although the importance of H2O2 in BMDC recruitment/activation by LOX has not been directly addressed, this possibility cannot be excluded. For instance, MMPs are under the transcriptional control of redox-sensitive factors such as NF-κB (70), and a role for LOX-generated peroxide in their release by BMDC appears conceivable. At an upstream level, H2O2 may also promote FAK-dependent adhesion of myeloid cells to basement-membrane components of the niche (23). Additionally, MMPs are known to activate the Rac-1–NOX–ROS signaling cascade on the cancer cell side, favoring invasiveness and genomic instability (115). Thus, LOX, independent of its prooxidant capacity, may indirectly activate prometastatic redox signaling in cancer cells by inducing MMP release from macrophages. Intriguingly, another recent work by Karin and co-workers (73) convincingly showed that versican produced by cancer cells can activate myeloid cells through Toll-like receptors (TLR) 2 and 6 to secrete TNF-α and IL-6 (73); in turn, versican and TNF-α are both necessary for Lewis lung carcinoma cells (LLCs) experimental metastases to lung, liver, and adrenal glands. TLRs activation leads to generation of ROS (105), and TLR2 couples with NADPH oxidase (152). Thus, taken together, these findings outline a scenario whereby NADPH-oxidase–generated ROS support bidirectional signaling between cancerous and myeloid cells at the metastatic site.
Based on the recruitment and activation of myeloid cells, the local release of proinflammatory cytokines, and the active generation of hydrogen peroxide by cancer cells and during matrix remodeling by LOX, one could reasonably consider the metastatic niche to be a strongly oxidized milieu. Moreover, oxidants may well operate, in this context, as transcellular, paracrine signals, central to a complex network of interactions involving many different cellular players. This is reminiscent of the role of nitric oxide in neuronal networks (101). Although fascinating, this possibility is difficult to reconcile with the idea of metastasis as an “escape program” from oxidative stress. In a darwinistic perspective, however, one could conclude that only cells capable of maintaining a low level of intracellular ROS, coupled with a pronounced migratory behavior, can successfully spread, survive, and proliferate at distant sites; interestingly, this capacity perfectly matches that of both normal and malignant stem cells (30).
In keeping with this notion is accumulating evidence on the molecular events underlying the phenomenon of metastasis “dormancy.” The mechanisms that determine whether a tumor cell that has disseminated to a secondary site will resume growth immediately, die, or enter a state of dormancy are poorly understood. However, activation of stress-signaling cascades, initiated by p38SAPK and the endoplasmic reticulum stress-triggered kinase, a key component of the “unfolded protein response” (150), seem to play a pivotal role in the establishment of this dormant state, characterized by growth arrest in the G0/G1 phase of the cell cycle and resistance to apoptosis and chemotherapy (1). While oxidative stress has been mechanistically linked to endoplasmic reticulum stress (138), p38 is known as an effector of redox signaling, its upstream activation by the apoptosis signal–regulating kinase-1 (ASK-1) being restrained by reduced thioredoxin and relieved by thioredoxin oxidation (139). Accordingly, mitochondrial ROS, as generated under hypoxia, activate p38SAPK (33). Although metastasis dormancy may under some circumstances (as for instance, during chemotherapy) represent an adaptive strategy allowing resistance and subsequent tumor recurrence, this response is generally viewed as a mechanism of metastasis suppression, to be avoided by malignant cells for successful dissemination to ensue. This is in keeping with the evidence that p38 mediates oxidant sensing in cancer cells, and that p38-signaling inactivation is necessary for successful malignant progression (31). Because the thioredoxin redox state is regulated by NADPH as a cofactor of the thioredoxin reductase (TR) (82) and endoplasmic reticulum stress induced by glucose and nutrient deprivation concomitant with an increase of intracellular ROS (150), both stress cascades linked to metastasis dormancy appear to be redox sensitive and subject to metabolic control. This implies that an elevated reductive potential, as supported by aerobic glycolysis (see earlier), may be crucial for disseminated cancer cells to avoid quiescence dictated by stress signaling, fully to accomplish their metastatic task (Fig. 5).
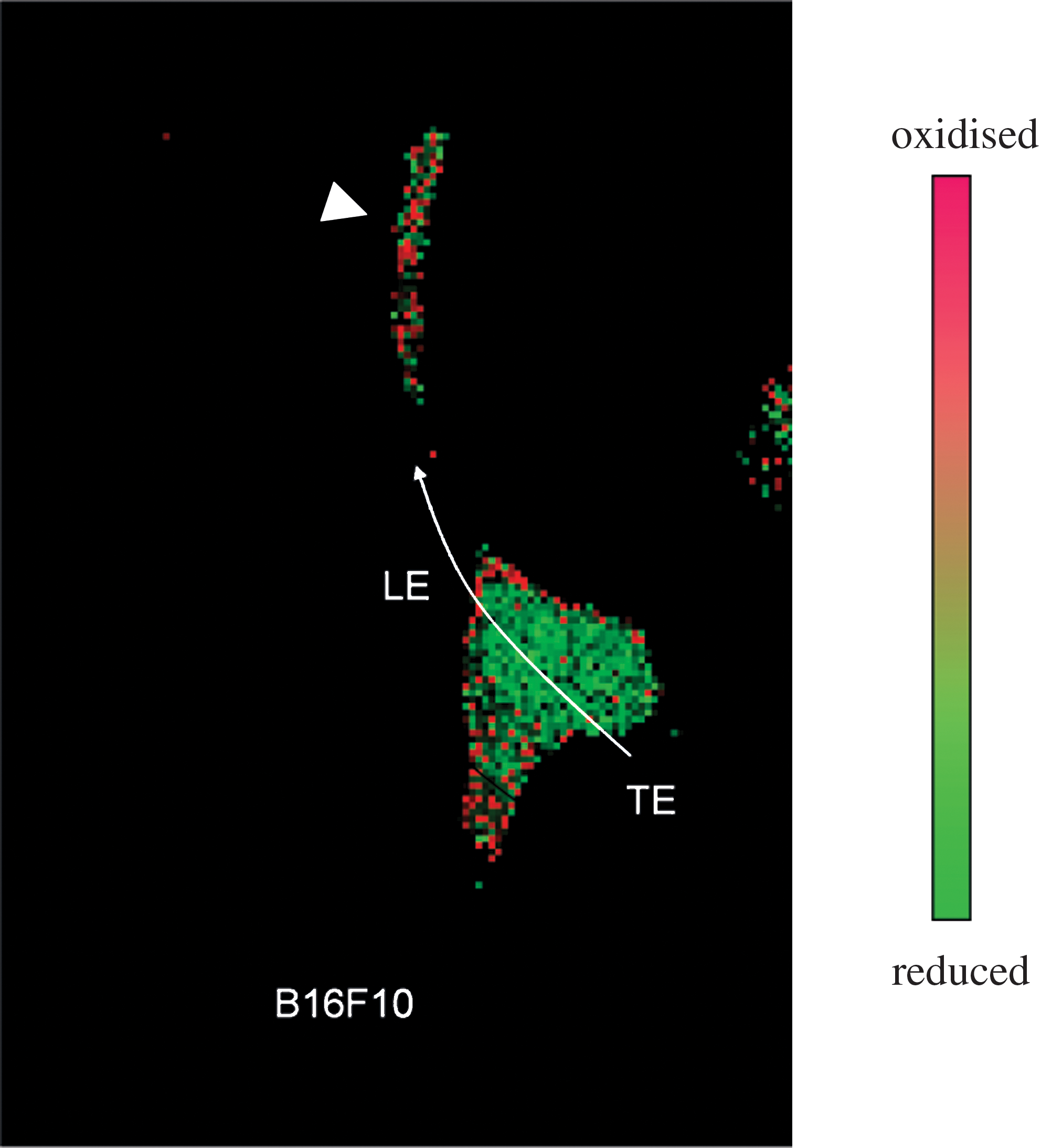
The need for an elevated intracellular redox potential is not in contradiction with prometastatic signaling events sustained by ROS and necessary for cell adhesion, migration/invasion, and anchorage-independent survival. Because these signals are generated mainly by NADPH oxidase or other membrane-bound enzymes, they are likely to be confined in the subplasmalemmal region of the cell. High-resolution imaging of redox signaling through oxidation-sensitive fluorescent proteins supports this view (53, 95). In general terms, indiscriminate oxidation is expected to interfere with redox signaling by increasing the background noise and by masking subtle, highly localized redox changes such as those triggered by physiological cues. More specifically, a reducing “background” may be necessary to maintain the redox “polarization” during cytoskeleton dynamics.
By taking advantage of a redox-sensitive mutant of the yellow fluorescent protein (rxYFP), we have observed that, in a migrating cancer cells, oxidation is confined to the leading edge of the cell body, whereas reduction prevails at the trailing edge (Pani et al., unpublished data) (Fig. 6). This asymmetric distribution may allow integrin-dependent adhesion and cytoskeleton rearrangement at the front of the cell, and cell detachment at the rear through increased phosphatase activity (116, 125). Thus, in this model, the capacity to maintain redox polarization with adequate reductive buffers is as critical as ROS generation to sustain cell migration/invasiveness (see model in Fig. 5).
What Metastasis Is Teaching Us: The “Cancer Lesson”
Cancer cells (like or even more than normal cells) are subjected to the toxic and growth-inhibitory action of reactive oxygen species, and oxidative damage and death underlie tumor response to many current anticancer therapies. We here reviewed recent literature in support of the idea that resistance to oxidative stress, coupled with active motility, represents two fundamental prerequisites of malignant cells undergoing a successful metastatic fate. In other words, we suggest that metastasis is a cancer cell strategy to escape oxidative stress. This theoretic framework integrates metabolic features of cancer cells, exemplified by the Warburg effect, with accumulating information on the role of prosurvival or inflammatory-like signals and cytoskeleton dynamics in the accomplishment of metastatic growth. Most of these signals are generated by NADPH-dependent membrane oxidases and therefore connected to the glucose-derived NADPH. NADPH also maintains the GSH redox buffers and restrains stress signaling through thioredoxin and thioredoxin reductase, thereby favoring cell expansion over dormancy (Fig. 5). The genetic program triggered by HIF-1, and by extension, by hypoxia, fulfils these prerequisites by coupling metabolic shift to glycolysis with increased expression of effectors like c-Met, CXCR4, and LOX, whose mitogenic and proinvasive activity largely involves oxidative-reductive reactions at the cell membrane.
How should we reconcile defense from oxidative stress with prometastatic signaling by ROS? We propose that (a) recently discovered redox-signaling pathways governing cell proliferation, motility/invasion, and survival in response to specific receptor ligands (i.e., growth factors, integrins) originally evolved as detectors of harmful oxidative stress coming from outside (inflammation, cell necrosis) or inside (mitochondrial dysfunction, nutrient deprivation, hypoxia) the cells; (b) activation of these pathways implements a defense response whereby cells increase antioxidant capacity by glycolysis, escape danger by increased motility, and repair cell loss by active proliferation; and (c) carcinogenesis usurps and deregulates this defensive strategy, leading to metastasis (Fig. 7).

The “escape program” we have tried to outline is conceivably not restricted to malignant cells, but can be implemented also by “normal” cells equipped with the appropriate antioxidant and motile capacity. Why are not all malignant cells metastatic, and some “normal” cells (embryonal and inflammatory) definitely are? The few cells with the capacity of resisting and exploiting ROS for dissemination at a distance are probably what we also refer to as “(normal) stem cells” or “cancer stem cells,” and to understand the molecular details of their peculiarity is both intellectually challenging and therapeutically critical. We may then turn the conventional logic (from physiology to pathology) around and use cancer metastasis as a paradigm for how redox-based strategies for cell evasion/invasion operate in “normal” invasive processes like inflammation and development. Reports on the crucial role of HIF-1 in leukocyte function and in inflammation (26), and of an “antioxidant phenotype” in “normal” stem cells maintenance (30) clearly point to this direction (124).
Footnotes
Acknowledgments
We are indebted to Dr. Giuseppe Maulucci (Catholic University, Rome) for providing unpublished results displayed in ![]() , to Prof. George van Rossum and Dr. Yongjian Wu for critically reading the article, and to members of the respective laboratories for helpful comments and suggestions. G.P.'s laboratory is funded in part by the EC-FP6-project DiMi, LSHB-CT-2005-512146.
, to Prof. George van Rossum and Dr. Yongjian Wu for critically reading the article, and to members of the respective laboratories for helpful comments and suggestions. G.P.'s laboratory is funded in part by the EC-FP6-project DiMi, LSHB-CT-2005-512146.