
Abstract
In recent years, it has become clear that balanced regulation of reactive oxygen species is of critical significance for cell-fate determination as well as for stem cell development, function, and survival. Although many questions regarding intracellular redox status regulation of stem cell fate remain, we review here what is known regarding the impact of cell-fate signaling as shown with a variety of human cancer cells and more recently on cancer-initiating cells and on the regenerative capacity of skeletal muscle and hematopoietic tissue and their stem cells. We also discuss the role of altered intracellular redox status as a potential primary pathogenic mechanism in muscular dystrophy and hematopoietic pathologies. Studies discussed here illustrate how understanding altered redox regulation of stem cell behavior may contribute to the development of novel stem cell therapies. Antioxid. Redox Signal. 11, 2777–2789.
Introduction
Along similar lines, accumulation of ROS resulting from defects in ROS scavenging is believed to affect cellular aging and the senescence process, whereas the ability to withstand oxidative stress has been correlated with enhanced longevity in several species (34). As a result, an abnormal ROS accumulation has been implicated in the pathogenesis of various diseases, including cancer, atherosclerosis, diabetes mellitus, neurodegenerative disorders, and in oxidative protein damage associated with age-related sarcopenia (muscle wasting, reviewed in (29).
This review aims to present a critical account of the data vis á vis the divergent signaling by intracellular ROS and its relevance to human disease pathology, focusing in particular on tissues with high regenerative capacity, such as hematopoietic tissue, muscle tissue, and the cancer cell. Not only do we attempt to provide a succinct account of the signaling networks affected by cells under mild or excessive oxidative stress, but we also delve into the potential implications in terms of novel targets for therapeutic intervention. With regard to the latter, could one exploit the slightly elevated redox milieu of cancer cells as a therapeutic window to trigger cell death in cancer cells with compounds that kill through elevated ROS production? A recent report provides impetus to this idea by demonstrating that hyperphosphorylation of the survival kinase Akt/PKB results in massive ROS production and cell death (82), thus highlighting the pivotal involvement of ROS in divergent signaling by this kinase, better known for its prosurvival and carcinogenic activity. As activated Akt is a feature of many cancers, could one envision a scenario in which this might be exploited for the terminal extinction of the cancer cell? Alternatively, strategies to alleviate oxidative stress could be beneficial for pathologic states in which increased ROS accumulation is the underlying etiology.
Intracellular Sources and Regulation of ROS
Nox family and the mitochondrial electron-transport chain
ROS include radical species such as superoxide (O2 −), hydroxyl (OH•), hydroperoxy (HO2 •), peroxy (RO2 •), and alkoxy (RO•) radicals as well as nonradical species, such as H2O2 and singlet oxygen (1O2). The two prime suspects in the intracellular generation of ROS are the membrane-bound NADPH oxidase complex and the electron-transport chain (ETC). The NADPH oxidase complex includes the membrane-associated cytochrome b 558 complex, comprising gp91 phox and p22 phox and the cytosolic proteins p47 and p67, and the small GTPase, Rac (Rac 1 in macrophages and monocytes, and Rac2 in neutrophils) (4). This oxidase complex is activated on membrane recruitment of the cytosolic subunits and facilitates the one-electron reduction of molecular oxygen to generate superoxide radical anion (O2 −), which is an essential defense mechanism used by phagocytic cells against microbial products such as lipopolysaccharide (LPS) and cytokines (42). However, the expression of this O2 −-generating complex is not restricted to phagocytic cells, as recent evidence points to a larger family of similar oxidase enzyme complexes, termed the NOX family (NOX 1, 3, 4, 5, Duox1, and Duox 2; NOX 2 is the NADPH oxidase of phagocytic cells) (9). The nonphagocytic NOX members are implicated in a variety of cellular responses and signaling circuits involving endothelial cells, vascular smooth muscle cells, cardiomyocytes, and transformed cells.
The second major intracellular source of ROS is the mitochondrial ETC. Mitochondria use almost 90% of the consumed O2 for oxidative phosphorylation, which involves shuttling of electrons across the ETC. Because of this high flux, leakage of electrons on to O2 is unavoidable, resulting in the generation of O2 −, predominantly involving complexes I and III of the ETC, complex I being associated with O2 − production primarily on the matrix side, whereas complex III–induced O2 − appears on both sides (86). The dismutation of O2 − by superoxide dismutase generates H2O2, and these two species readily enter the Haber-Weiss reaction to form the highly reactive OH• radical. Although the accepted dogma has it that increases in mitochondrial O2 consumption do not result in increased O2 − generation, an association between increased O2 consumption and mitochondrial O2 − generation in human cancer cells overexpressing the oncoprotein Bcl-2 was recently demonstrated (19). More recently, a similar effect of increased O2 consumption and ROS generation was reported in cells on hyperphosphorylation of Akt/PKB (82).
Aside from these two major sources, the presence of the monoamine oxidase (MAO) localized to the outer mitochondrial membrane also contributes to ROS generation, in particular H2O2. Other sources include xanthine oxidase, Cyclooxygenase, and lipoxygenase, and exogenous redox-cycling compounds, such as paraquat (39).
Regulation of intracellular redox status
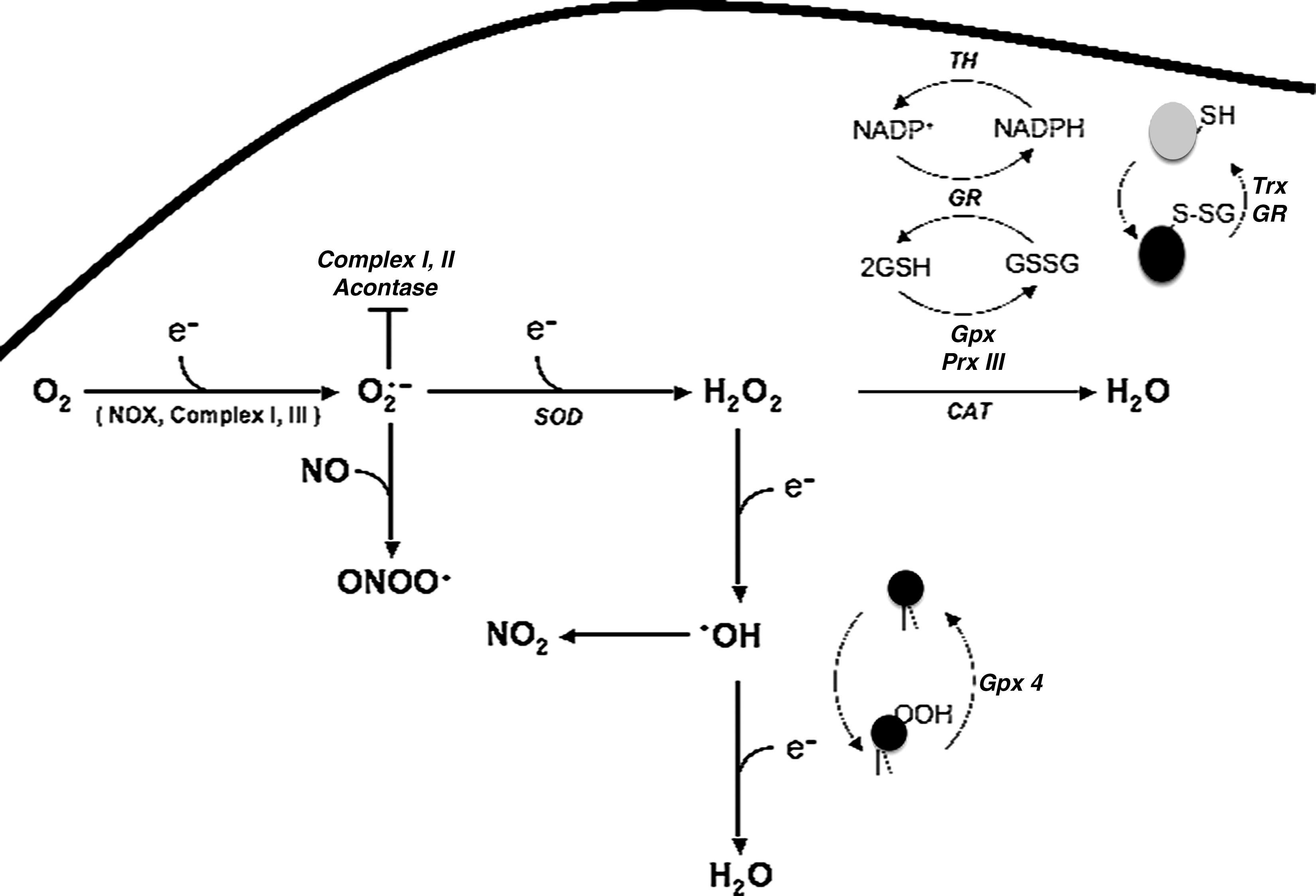
During normal homeostasis, the intracellular ROS levels are tightly regulated by an elaborate antioxidant defense comprising enzymatic and nonenzymatic systems (see Fig. 1). Outside of the mitochondria, the glutathione-based systems [glutathione S-transferase and the thioredoxin system (including peroxiredoxin)] serve as the major redox-regulatory mechanisms (86). Because of its relatively high concentration in the cell, the GSH/GSSG pool in the cells serves as the major redox system, and together with glutathione peroxidase, (Gpx) is the major H2O2-scavenging mechanism through its conversion to water (86). The presence of Mn superoxide dismutase (MnSOD) in the mitochondrial matrix coordinates with GSH to scavenge O2 − and, in doing so, limits the excessive accumulation of H2O2. A similar role is performed by the cytosolic Cu/Zn SOD for scavenging O2 −, generated outside of the mitochondria or directly released from the mitochondria. It should be pointed out that the presence of a mitochondria-specific pool of GSH has also been reported; matrix glutathione accounts for ∼15% of the total cellular glutathione (33). Gpx works much faster than peroxiredoxins; however, the relatively higher content of peroxiredoxins in the cells compensates for this, and therefore, peroxiredoxins are the most important H2O2-removing systems in eukaryotes and prokaryotes (16).
Thioredoxin is concentrated in the endoplasmic reticulum and reduces disulfide bonds of oxidatively modified proteins (71). In addition, peroxisomes contribute to the breakdown of H2O2 to H2O through the activity of the enzyme catalase; however, this system is not the most important and efficient because of the absence of catalase expression in the mitochondria, the primary source of ROS.
ROS as Regulators of Cell Fate
Excessive stimulation of the NADPH oxidase complex or a defect in the antioxidant defense arsenal results in overt and severe oxidative stress, which compromises normal cellular function and contributes to the development of pathologic states, such as neurodegeneration, diabetes mellitus, rheumatoid arthritis, atherosclerosis, cancer, and aging (29). One of the critical factors in this is the damage inflicted by ROS to the major ROS-producing organelles, the mitochondria. It is well recognized that the mitochondria are highly susceptible to ROS-induced injury because of the close proximity of the mitochondrial DNA to the ETC, as well as the lack of the protective histones, present in the genomic DNA. Furthermore, the antioxidant capacity of the mitochondria decreases with age, and the accumulation of ROS impairs mitochondrial metabolism and induces oxidative modification of proteins, resulting in abnormal activation of oncogenes or loss of function of tumor suppressors. In addition to mitochondrial DNA damage, excessive accumulation of ROS in the mitochondria could lead to the induction of the mitochondrial permeability transition (MPT), a state that allows the mitochondrial outer membrane to become leaky and facilitate the egress of apoptosis-amplification factors such as cytochrome c, AIF, and Smac/Diablo. Another strong stimulus for the induction of MPT is a decrease in the mitochondrial Ca2+ buffering capacity, as mitochondria are one of the major intracellular sources of Ca2+. This leads to mitochondrial Ca2+ overload and subsequent ROS generation, changes associated with a variety of human pathologic states, such as myocardial ischemia/reperfusion injury, neurologic diseases, and aging (29). Therefore, avoiding electron leakage through the mitochondrial ETC to restrict abnormal generation of O2 − could have tremendous implications in maintenance of normal life span, enhancing longevity, and disease prevention.
Historically, intracellular accumulation of ROS has been associated with tissue injury and cell death; however, a mild prooxidant milieu also has been linked to a variety of physiologic responses, such as signal transduction, gene transcription, and proliferation (15). Examples of these include growth-factor stimulation involving nerve growth factor (NGF), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-receptor activation, activation of cytoplasmic kinases, such as mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), AKT/PKB, and induction of NF-κB, AP-1, and HIF-1a transcriptional activity. Many of these activities are a function of a slight increase in intracellular O2 − or H2O2 (and reactive nitrogen species such as NO) that does not overwhelm the cellular antioxidant defense systems. Along these lines, a slight increase in intracellular O2 − was shown to inhibit apoptotic execution in human tumor cells in a variety of model systems, irrespective of the trigger (death receptor– or drug-induced) (1, 23, 88). Intracellular increase in O2 − also has been linked to oncogene-induced (Bcl-2, Rac, and Akt/PKB) survival or death resistance of tumor cells or both. Corroborating the prosurvival activity of an increase in intracellular O2 −, pharmacologic downregulation of intracellular O2 − with the NADPH oxidase inhibitor, diphenyleneiodonium (DPI), or on transfection with a dominant-negative form of Rac (RacN17) reverted the death-inhibitory effect of Bcl-2 in human tumor cells (20). Notably, the prooxidant effect of Bcl-2 was linked to an increase in mitochondrial O2 consumption and complex IV (cytochrome c oxidase) activity (19).
Although an effect similar to that of O2
− has also been reported with very low concentrations of H2O2, this effect could be attributed to activation of NADPH oxidase and, in turn, an increase in intracellular O2
− production. Otherwise, a tilt in the intracellular ratio of H2O2 to O2
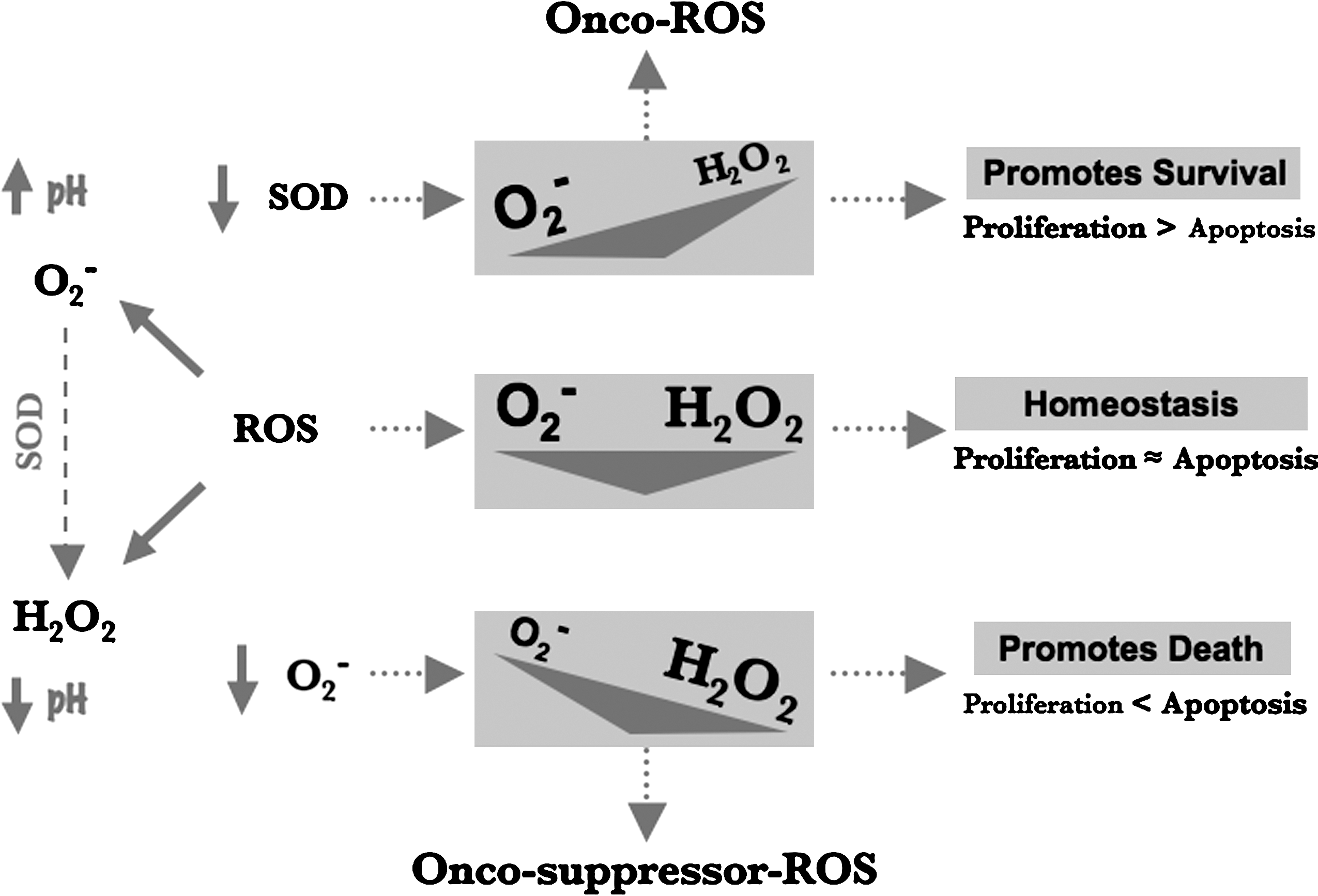
− in favor of H2O2 creates a permissive environment for apoptotic execution (21, 22). More recently, the differential activity of these two major ROS species has been linked to a distinctly opposing effect on the membrane pH regulator, Na+/H+ exchanger-1 (NHE-1), which has been associated with various phases of carcinogenesis; an increase in O2
− increases the promoter activity and expression of NHE-1, whereas a higher H2O2 concentration inhibits both its transcription and protein expression and induces cytosolic acidification (3, 59). These data suggest that O2
− could be the ROS species implicated in promoting oncogenesis, whereas a higher H2O2 concentration could function as a tumor-suppressor ROS through its potent death-sensitizing activity (Fig. 2). This ties in well with the reported tumor-suppressor effect of MnSOD, which could favorably tilt the balance in favor of H2O2 through its O2
− dismutation activity. Interestingly, upregulation of mitochondrial ROS has also been associated with p53-mediated apoptosis through induction of the ROS-inducing enzyme proline oxidase (100). Along similar lines, intracellular H2O2 generation downstream of CD95 (Apo-1/Fas) ligation was associated with downregulation of the cFLIP (
Free Radicals in Skeletal Muscle
ROS in skeletal muscles are derived from superoxide anions that give rise to hydrogen peroxide and hydroxyl radicals, whereas reactive nitrogen species (RNS) originate from nitric oxide (NO) that reacts to form peroxinitrous acid and peroxinitrite. In skeletal muscles, both ROS and RNS are produced at low levels under normal resting conditions, and many studies have demonstrated higher levels of free radicals during muscle contraction (5, 41, 54, 56, 58, 81, 83, 87). Under normal physiologic conditions, ROS are produced through the mitochondrial ETC, NADPH oxidase, and through phospholipase A2 (36, 119). Conversely, two isoforms of nitric oxide synthase (NOS), which include the neuronal isoform (nNOS) that is expressed by fast-twitch muscle fibers and is associated with the dystrophin glycoprotein complex (DGC), and the endothelial isoform of NOS (eNOS), which is associated with muscle mitochondria (54, 55), may give rise to RNS. In the face of constant free radical production, skeletal muscles are geared to adapt and respond physiologically to oxidative challenge.
In presence of ROS, the redox-sensitive transcription factors NF-κB and AP1 are activated and regulate the expression of antioxidant enzymes that include superoxide dismutases (SOD1 and SOD2), catalase (CAT), and glutathione peroxidase (Gpx). SOD1, catalase, and Gpx are present in the sarcoplasm, whereas the mitochondria contain SOD2 and Gpx (61). In addition, peroxiredoxins thioredoxin and thioredoxin reductase also are expressed in skeletal muscles (40, 72, 98). Because antioxidant enzyme expression is essential for redox homeostasis, a direct correlation exists between the oxygen-consumption rate and antioxidant enzyme expression in different types of skeletal muscles. For instance, the slow-twitch oxidative fibers (type I) have very high levels of myoglobin, an oxygen-binding protein, whereas the glycolytic fast-twitch muscle fibers (type IIa and type IIb) contain low levels of myoglobin. The presence of myoglobin in skeletal muscles renders them susceptible to oxidative stress. Correspondingly, type I fibers express higher levels of antioxidant enzymes compared with type II fibers (52). The contractile activity of muscle has been reported to cause an increase in the production of ROS (26, 83, 96), and several studies reported antioxidant responses to prolonged exercise (51, 95, 106). Interestingly, not all antioxidant enzymes are involved in this adaptive response. For instance, SOD2 and Gpx activity is increased with exercise. Conversely, SOD1 activity is not, and the effects on CAT activity are unclear (43, 60, 63, 92).
Role of Free Radicals in Muscle Stem Cells and Muscle Pathologies
Oxidative stress arising from excessive accumulation of ROS due either to increased production or to defective cellular responses that impair its removal is associated with many disease states, including aging, muscular dystrophies, muscle atrophy, and other chronic diseases that result in muscle wasting (38). Skeletal muscle regeneration is dependent on muscle stem cells, also known as satellite cells. In response to injury or myotrauma, quiescent satellite cells are activated and reenter the cell cycle to generate a pool of proliferating myoblasts that fuse to form new myofibers (25). The exact role(s) ROS plays in satellite cell activation, self-renewal, and the differentiation of muscle-precursor cells is not clearly understood. However, deregulated free radicals, leading to oxidative stress, lead to the death of proliferating myoblasts and differentiated myotubes (93, 119) and has been proposed as a key mechanism leading to degeneration of skeletal muscles in Duchenne muscular dystrophy (DMD) (92, 113). DMD is one of the most prevalent and devastating human genetic diseases caused by mutations in the dystrophin gene (45) and is characterized by degeneration and necrosis of skeletal muscles (25). Clinical symptoms are first evident in DMD patients between 3–5 years of age. As the disease progresses, patients are typically wheelchair bound by the early teens and die in the early 20 s because of progressive wasting and weakness of respiratory and locomotor muscles.
Dystrophin is a large cytoskeletal protein that associates with many membrane glycoproteins to form the dystrophin–glycoprotein (DGC) complex (12, 30) that connects the cytoskeleton to the extracellular matrix (ECM). Based on interactions of DGC with ECM and the cytoskeleton, it appears that at least one function of DGC is to maintain membrane integrity during cycles of contraction and relaxation. The absence of dystrophin disrupts these interactions, rendering the sarcolemma susceptible to damage from muscle contraction, leading to muscle cell necrosis in patients with DMD (Fig. 3). Similarly, in mdx mice that lack dystrophin and have been widely studied as a model for DMD, skeletal muscles are formed but subsequently degenerate. Given the progressive nature of the disease, it is likely that dystrophin deficiency alone is not sufficient for death of dystrophic muscles, but rather enhances the sensitivity of muscle cells to stimuli that eventually lead to muscle degeneration (73). Among the various hypotheses that have been proposed to explain the degeneration of dystrophin-deficient muscles (79, 90, 102, 117, 121), free radical–mediated cellular injury leading to muscle necrosis is supported by several lines of evidence (93, 114). These include the similarities in muscle pathology seen in conditions of vitamin E deficiency, as well as in ischemia, attributed to an increase in free radical–mediated injury, are similar to DMD (11, 91). Moreover, overt signs of oxidative injury, such as lipid and protein oxidation, are evident in dystrophic muscles (79). It is interesting to note that in DMD, a selective loss of type IIb fibers occurs (124), which may be due to their reduced antioxidant enzyme levels.
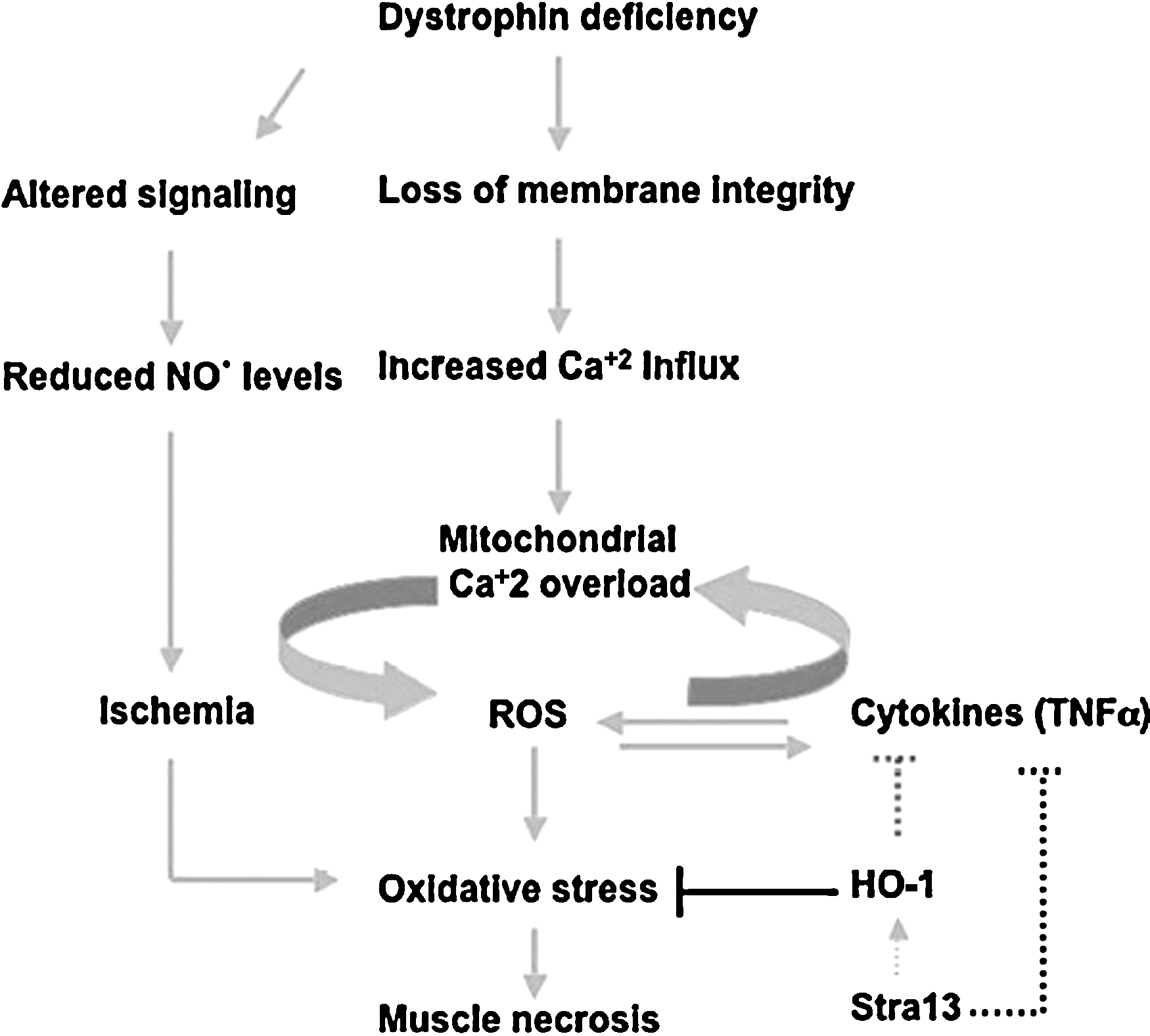
Although many such studies established a correlation of oxidative stress in dystrophic muscles, Rando and colleagues (28) were the first to demonstrate that increased levels of antioxidant enzymes are evident before muscle degeneration in the dystrophin-deficient mdx mice (28). Furthermore, they also demonstrated that dystrophin-deficient muscles exhibit increased susceptibility to oxidative damage (94). Together, these studies were a key to indicate that free radicals may play a causal role in the degeneration of muscles in DMD. One potential mechanism by which dystrophin deficiency could lead to deregulated redox homeostasis is through the association of the DGC complex with nNOS that regulates metabolism of the free radical NO•. The absence of dystrophin results in reduced nNOS activity and considerably reduced cellular NO• levels (13, 17). However, the role of NO• in dystrophy remains unclear, as the muscle pathology in mdx mice was found to be independent of nNOS perturbation (24). Moreover, NO•-mediated injury is not apparent in dystrophic mdx muscles, although hallmarks of oxidative stress are (28). Nonetheless, repeated expression of nNOS in mdx muscle was found to result in improved histopathology (125). One way of reconciling these different observations is that NO• is a highly reactive molecule that easily and readily reacts with other free radicals. The altered NO• levels in mdx mice due to reduced nNOS expression could have a major influence on other free radicals in skeletal muscle. It is likely that reexpression of nNOS in mdx muscles restores redox homeostasis, resulting in amelioration of the dystrophic phenotype. Thus, although free radical–mediated injury is evident in DMD, the role of oxidative damage as a primary cause for the degeneration of dystrophic muscles in DMD has not been clarified. In addition, the source of oxidative stress in dystrophic muscles, and the mechanisms by which it is regulated, are largely unclear. In this regard, we recently demonstrated that mice lacking the transcription factor Stra13 exhibit a dystrophic pathology in response to injury that is characterized by muscle necrosis, reduced membrane integrity, calcium deposition, and fibrosis (110, 120). Interestingly, although no structural changes or fiber-type alterations are apparent, Stra13−/− mutant muscle shows obvious signs of damage before degeneration. Hallmarks of oxidative stress, such as protein carbonylation and antioxidant enzyme levels, are altered, and the mutant muscles display an increased sensitivity to necrosis in the presence of prooxidants. Consistent with oxidative stress as a key pathogenetic mechanism in the degeneration of muscles, treatment of Stra13−/− mice with the antioxidant N-acetylcysteine prevents muscle necrosis and results in significantly reduced dystrophic pathology. Together, these strongly suggest that Stra13 regulates oxidative stress–mediated muscle degeneration (Fig. 3) (120) and underscore its role in muscular dystrophy.
Antioxidants as Therapeutic Agents
Given the association of oxidative stress in DMD, clinical trials with various antioxidants, including vitamin E, SOD, penicillamine, and selenium, have been performed (10, 32, 35, 101, 109, 122). However, the outcome of such trials has not been promising in preventing the pathologic degenerative changes in dystrophic muscles (93, 114). One simplistic interpretation of these studies is that oxidative stress does not underlie muscle necrosis in DMD. However, an important factor that may have influenced the outcome of such trials is the timing of administration of the antioxidants. Most studies were performed on DMD boys that were older than 10 years with an advanced stage of the disease. Once the degenerative changes are initiated, it may not be possible to reverse pathologic changes with antioxidants. Thus, unless the antioxidants are administered before the onset of disease symptoms, they may not be effective in preventing or slowing the degeneration of muscles. Recent studies using the antioxidant N-acetylcysteine (NAC) demonstrated improved pathology of isolated muscles in mdx mutants (126). Moreover, as mentioned earlier, administration of NAC in Stra13−/− mice had a major impact on preventing muscle necrosis (120), strongly supporting the view that antioxidants could be of considerable therapeutic value in muscular dystrophy.
Hematopoietic Stem Cells and ROS
It has become evident that uncontrolled accumulation of ROS in hematopoietic stem cells leads to abnormal hematopoiesis (18, 48, 76, 115, 129). However, the source of ROS in hematopoietic stem cells and the mechanism by which this occurs are not clear.
ATM and FoxO
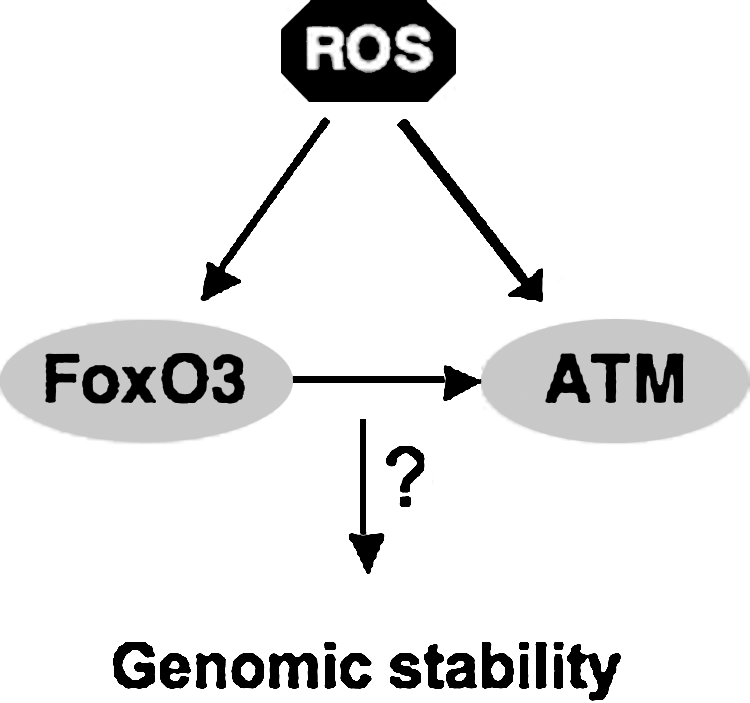
The ataxia telangiectasia mutated gene (ATM) is essential for the regulation of genomic stability. ATM serine threonine protein kinase is a critical enzyme in the regulation of stress response to DNA damage (8). Loss-of-function mutations in ATM results in a clinical phenotype characterized mainly by immunodeficiencies and predisposition to malignancies (6). Loss of ATM also results in ROS-mediated depletion of hematopoietic stem cell pool, leading to bone marrow failure in old mice (48). Accumulation of ROS in ATM-null hematopoietic stem cells leads to the activation of the retinoblastoma pathway, which blocks hematopoietic stem cell activity (48). Treatment with the antioxidant reagent N-acetyl cysteine (NAC) or with a MAPK inhibitor restores the quiescence and reconstitution capacity of ATM-null hematopoietic stem cells (49). These findings provided the first direct evidence for a role for ROS in the regulation of hematopoietic stem cell activity. Although ROS are found accumulated in several cell types and tissues, and antioxidant enzymes are deregulated in ATM-mutant cells, the mechanism of ATM regulation of ROS is not clear (8, 89).
Like ATM, FoxO transcription factors are mediators of the antioxidative stress response in hematopoietic stem cells (115). Active DAF-16 promotes dauer formation (diapause) in Caenorhabditis elegans under certain harsh environmental conditions, such as when food is scarce or the surrounding is too crowded (67, 84). The dauer is in juvenile form and reproductively immature. Dauer is a reversible condition in which the metabolic rate is highly reduced and life span is markedly enhanced. Mutations of DAF-2/Age-1/AKT1/2 signaling pathway in C. elegans (highly related to the insulin-insulin-like growth factor (IGF) receptor/PI3-kinase/AKT in mammals) leads to constitutive entry into the dauer stage and extends markedly the worm's life span (62, 65, 67, 68, 78, 84, 85). These phenotypes are abolished by the DAF-16 mutation, indicating that DAF-16 is the downstream effector of this pathway. DAF-16 regulation of longevity is mediated at least partly by its control of genes involved in stress resistance, including oxidative stress (62, 64, 77, 78, 85, 127).
Loss of three FoxOs (FoxO1, FoxO3, and FoxO4) led to defective hematopoietic stem cell numbers and activity that was associated with increased accumulation of ROS, as measured by the DCF-DA assay (115). FoxO3 appears to be the main mediator of antioxidant response in hematopoietic stem cells (76, 129). FoxO3 is the most highly expressed FoxO in the bone marrow (70) and is the principal nuclear-active FoxO in hematopoietic stem cells (129, 130). In contrast to FoxO1, which is mostly cytoplasmic, >95% of FoxO3 is in the nucleus of primitive hematopoietic cells therefore presumed transcriptionally active (129). FoxO3 modulation of oxidative stress in hematopoietic stem cells is critical for the regulation of hematopoietic stem cell activity (129). FoxO3 regulates the hematopoietic stem cell pool mainly by controlling the transcription of several antioxidant enzymes, including SOD2 and catalase (76, 129).
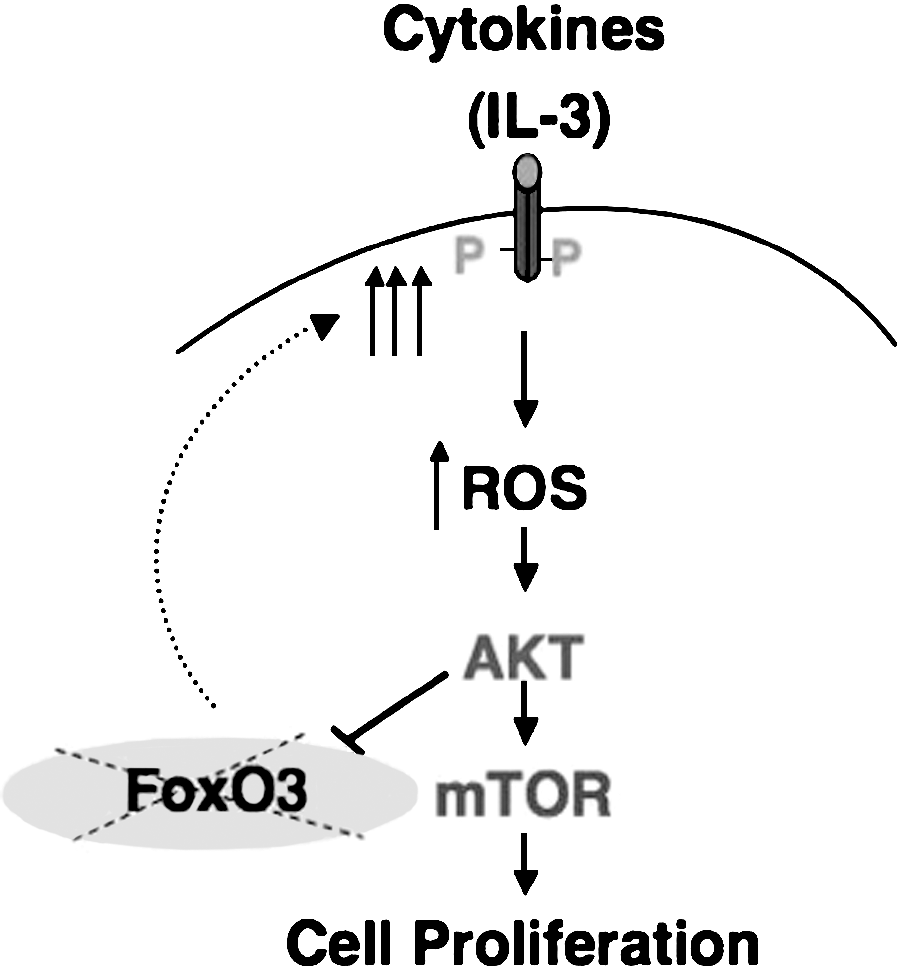
Similar to the triple deletion of FoxO1, 3, and 4 (115), loss of FoxO3 leads also to a myeloproliferative-like syndrome (129). The myeloproliferation in FoxO-null mice is accompanied with a significant portion of hematopoietic stem cell population exiting quiescence and highly cycling (115). The source of myeloproliferation in FoxO3-null mice is likely to be distinct (128, 129). FoxO3-null hematopoietic stem cells exit quiescence but show a delay in the G2/M phase of the cell cycle and are impaired in their cycling. In addition, these cells do not produce adequate numbers of hematopoietic progenitors when isolated and cultured in vitro. These findings suggest that generation of elevated numbers of hematopoietic progenitors in FoxO3-null mice is not the result of uncontrolled cycling of hematopoietic stem cells in these mice. Data from our laboratory suggest that FoxO3 modulation of ROS-mediated cytokine signaling is critical in restricting the expansion of the primitive hematopoietic progenitor cell compartment. Our data implicate oxidative stress–mediated activation of the AKT/mTOR signaling pathway, possibly as part of a feedback loop, in the increased proliferation of the FoxO3-null hematopoietic progenitor compartment (129). Interestingly, as predicted by recent studies (50), ROS accumulation is higher in FoxO3-null hematopoietic progenitors as compared with FoxO3-null stem cells. Together these data indicate that FoxO3 mediates distinct functions in hematopoietic stem cells that are mostly dormant cytokine resistant as opposed to those in hematopoietic progenitors that are highly sensitive to cytokines.
ROS are produced by growth factor, cytokine receptor, and oncoprotein signaling (46, 47, 80, 104, 111, 112, 118, 134, reviewed in 113). Stimulation of growth-factor receptors such as EGF, PDGF, and insulin signaling is associated with transient increase in ROS production. Accumulation of ROS also participates in cell proliferation by inhibition of phosphatases, such as phosphatase and tensin homology (PTEN), protein tyrosine phosphatase 1B (PTP1B), and CDC25 (74, 75, 105, 107). Unlike superoxide, hydrogen peroxide is membrane permeable, diffusible, and long lived. Hydrogen peroxide affects cellular signaling through protein modification, such as an intramolecular disulfide bridge, sulfenyl-amide bond formation, direct activation of tyrosine kinases by Cys oxidation, or by inhibition of phosphatases [reviewed in (113)]. In turn, the catalytic activity of antioxidant enzymes such as peroxiredoxins, catalase, and glutathione peroxidase is modified by signaling molecules [reviewed in (98, 99)], suggesting a dynamic balance between cellular signaling and regulation of oxidative stress.
Abnormal accumulation of ROS is known to induce apoptosis in various cell types (113). Although in hematopoietic stem cells, ROS accumulation has been associated with apoptosis (115), this has not been universal (76, 129), and the role of ROS in induction of apoptosis in hematopoietic stem cells is not clear. Nonetheless, data generated in our laboratory (128) are consistent with a model in which physiologic ROS provide a balance between hematopoietic progenitor cell proliferation and apoptosis (see Fig. 6).
Recent findings (53, 57, 103) raise the possibility that ROS may be involved in myelopoliferative diseases, such as chronic myeloid leukemia or polycythemia vera, in which the hematopoietic progenitor compartment is expanded and exhibits a high sensitivity to cytokines. This is strongly supported by our recent data demonstrating that many of myeloproliferative symptoms observed in FoxO3-null animals are highly sensitive to NAC (128). These data also demonstrate that ROS are produced by cytokine signaling in primary hematopoietic progenitor cells and participate in the regulation of their proliferation (Fig. 4). Increased generation of ROS in FoxO3-null myeloid progenitors leads to their increased proliferation through activation of the AKT/mTOR signaling pathway (Fig. 5) (128). Based on these and our previous (129) findings, we propose that FoxO3 plays distinct roles in hematopoietic stem and progenitor cells (Fig. 6). These combined observations suggest that mechanisms regulating ROS may be exploited in the treatment of hematopoietic malignancies.
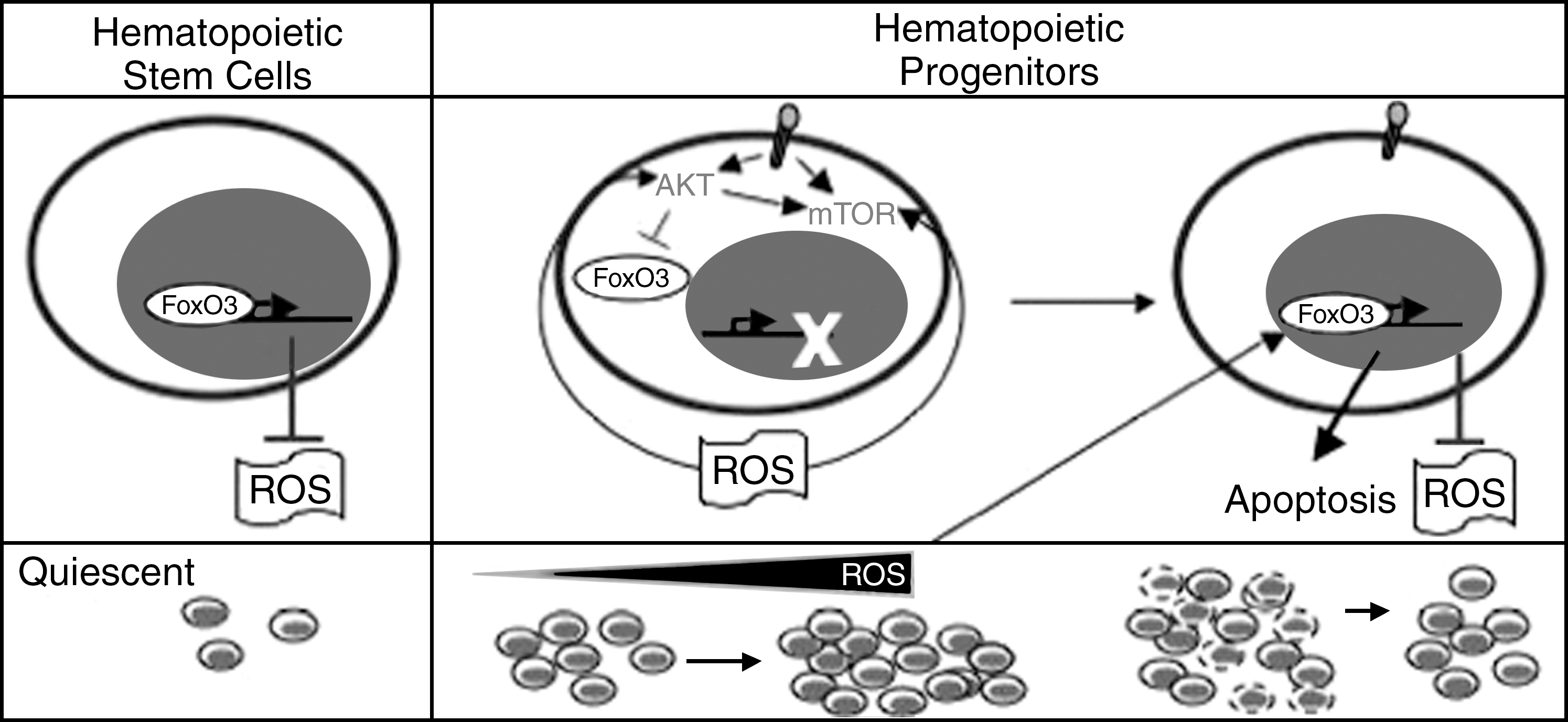
Although FoxO-null mice also displayed myeloproliferation, relative increased accumulation of ROS was limited to the hematopoietic stem cell compartment and was not observed in myeloid progenitors (115). FoxO-null hematopoietic stem cells exit quiescence and enter the cell cycle, suggesting that ROS enhance cell-cycle entry and loss of quiescence in hematopoietic stem cells (115). Taken together, these studies suggest a critical role for FoxO transcription factors in the repression of ROS and regulation of hematopoietic stem cell quiescence.
It was recently shown that deletion of tuberous sclerosis complex 1 (Tsc1) that inhibits the mammalian target of rapamycin (mTOR) has a critical effect on hematopoietic stem cell function (18). mTOR is a key regulator of cellular metabolism. Activation of mTOR in Tsc1-deleted mice leads to loss of quiescence and rapid cycling of hematopoietic stem cells (18). These abnormalities are associated with increased mitochondrial biogenesis and ROS accumulation in hematopoietic stem cells. In addition, NAC treatment of chimeric mice transplanted with Tsc1-deleted hematopoietic stem cells rescues significantly the Tsc1-deficient hematopoietic stem cell population (18), strongly suggesting that oxidative stress mediates the activation of the hematopoietic stem cell compartment in these mice. Thus, the mTOR signaling pathway is another regulator of hematopoietic stem cell quiescence.
Together these findings highlight the essential function of the regulation of oxidative stress in the maintenance of the hematopoietic stem cell pool and quiescence. In addition, they underline the critical role that ATM, FoxO (specifically FoxO3), and mTOR play in these processes. Hematopoietic stem cell deficiencies in mice that are deficient for FoxO3 or phosphatase and tensin homology (PTEN), which is a negative regulator of PI3-kinase/AKT) (131, 133), are similar. However, it is not known whether oxidative stress has any impact on PTEN-null hematopoiesis.
ROS in the Regulation of Hematopoietic Stem Cell Activity
As discussed earlier, both ATM serine threonine kinase and FoxO3 are essential for the regulation of oxidative stress in hematopoietic stem cells. Recent data suggest that FoxO3 regulates the expression of ATM (129). In addition to the regulation of ATM expression, studies demonstrate that FoxO3 directly interacts with ATM and is critical for enhanced ATM phosphorylation and activation in response to DNA damage (116). Thus, given the key functions of both ATM and FoxO3 in the regulation of oxidative stress and DNA damage, it is conceivable that FoxO3 and ATM play critical role in the regulation of oxidative stress–mediated DNA damage.
Both ATM and FoxO3 are linked to p53 through multiple pathways (7, 132). These combined findings suggest a model in which FoxO3, together with ATM and p53, provides a strong tumor-suppressor network in protecting hematopoietic stem cells from damage that may undermine their genomic stability and result in their clonal expansion. Alternatively, sustained activation of these pathways may explain the defective hematopoietic stem cell activity that is observed in aging FoxO3-null mice (76).
FoxO also interacts with Sir2/SIRT1 in response to oxidative stress. Sir2/SIRT1 is a NAD-dependent histone deacetylase that modulates the life span in various species (14, 69). It is postulated that, in response to oxidative stress, Sir2/SIRT1 interacts directly with FoxO in cultured cells and enhances its resistance to oxidative stress while limiting its ability to induce apoptosis (14). Whether this interaction occurs in hematopoietic stem cells or has any function in modulating hematopoietic stem cell activity or aging ( or a combination of these) is not known.
Understanding mechanisms of regulation of FoxO3 in hematopoietic stem cells and, in particular, its potential interactions with coactivators and co-repressors and identification of its targets in stem cells, will be critical for better elucidation of mechanisms of hematopoietic stem cell aging and malignant transformation.
FoxOs are likely to play a role downstream of the Wnt signaling pathway in hematopoietic stem cells. Wnt signaling is critical for hematopoietic stem cell self- renewal (97). FoxO association with β-catenin, the principal effector of the Wnt signaling pathway, is enhanced by oxidative stress (31) and is critical for FoxO regulation of resistance to oxidative damage (31).
Unresolved Questions
One caveat of all these studies is that they are based heavily on the use of NAC as an antioxidant. NAC is a precursor of glutathione, a powerful cellular antioxidant. NAC also is a strong chelator of heavy metals and a mucolytic. To substantiate the specificity of NAC function in these studies, it would be suitable to provide data with additional antioxidants such as catalase.
Another problem is how ROS are measured and how the source of ROS is determined. Most if not all data concerning oxidative stress in hematopoietic stem cells have thus far been derived from using the probe dichlorofluorescein diacetate and by measuring the fluorescent product, dichlorofluorescein, which it produces when it is oxidized. The major problem with this probe is that the intracellular fluorescence is based on the balance between the rate of oxidation of the probe and the rate of leakage of the probe from the cell [see (37)]. Introduction of novel methods of redox detection and measurements, such as protein-based fluorescence resonance energy transfer (FRET) sensor and inhibition of Rieske iron–sulfur protein by using short-hairpin RNAs, are likely to address some of these concerns (37).
Many questions remain regarding the function and the source of ROS in stem cells. Among these, one major question is to address the relation between the function of ROS in stem versus progenitor cells. How ROS or different levels of ROS may contribute to the hypoxic regulation of stem cells is another important topic. Future elucidations of these questions in both embryonic and adult stem cell contexts are likely to have a significant impact on our understanding of hypoxic niche and its regulation of stem cell behavior.