
Abstract
The haptoglobin (Hp) 2-2 genotype is associated with increased risk of cardiovascular disease (CVD) in diabetes (DM). We recently proposed this increased risk arises from the tethering of redox active hemoglobin (Hb) to high density lipoprotein (HDL), thereby resulting in oxidative modification of HDL. Clinical trials have demonstrated that vitamin E (α-tocopherol) decreases while vitamin C increases CVD in Hp 2-2 DM individuals. We sought to test the hypothesis that the interaction of α-tocopherol or vitamin C on CVD in Hp 2-2 DM was due to their divergent effects on HDL oxidation and function. Vitamin C significantly increased while α-tocopherol completely blocked oxidation mediated by glycosylated Hb-Hp 2-2. Vitamin C had no benefit while α-tocopherol completely restored HDL function in Hp 2-2 DM mice. Co-administration of vitamin C mitigated the protective effects of α-tocopherol on HDL. There exists a pharmacogenomic interaction between vitamin C and α-tocopherol and the Hp 2-2 genotype on HDL function and structure. Choosing the correct antioxidant in the correct subset of patients may be critical in order to demonstrate benefit from antioxidant therapy. Antioxid. Redox Signal. 12, 209–218.
Introduction
Extracorpuscular hemoglobin (Hb), released from the red blood cell into the plasma compartment during the normal turnover of red blood cells or into the extravascular space following hemorrhage, is becoming recognized as an important mediator of disease (32). Free Hb is toxic by virtue of its ability to mediate oxidative reactions via Fenton chemistry (15). The Hp protein represents the primary defense mechanism against free Hb. Hp binds essentially irreversibly to Hb and modulates the ability of Hb to promote oxidation. The binding of Hp to Hb to form an Hp–Hb complex also accelerates the removal of free Hb via the uptake of the Hp–Hb complex by the monocyte/macrophage CD163 receptor (19).
We have demonstrated and others have confirmed that the Hp 2 protein is inferior to the Hp 1 protein in preventing the oxidation of a variety of lipid and protein substrates by Hb (5, 15). These differences in the antioxidant activity of the Hp 1 and Hp 2 proteins against Hb are dramatically exaggerated when Hb is glycosylated (1), as occurs to a greater extent in DM. Furthermore, despite the greater urgency of clearing the Hp 2–Hb complex in DM due to its increased redox activity, the clearance of the Hp 2–Hb complex by the CD163 receptor is impaired in DM, resulting in a further amplification of oxidative injury that may be mediated by Hb in Hp 2-2 DM individuals (3). We have recently demonstrated that an important target for Hp 2–Hb-mediated oxidation in DM is the high density lipoprotein (HDL) particle. Hp 2 binds to the ApoA1 protein of HDL and thereby can tether Hb to HDL. The HDL of Hp 2-2 DM individuals therefore contains redox active Hb-derived iron that may oxidatively modify the HDL of Hp 2-2 DM individuals, not only impairing the ability of Hp 2-2 DM HDL to mediate reverse cholesterol transport but also paradoxically turning Hp 2-2 DM HDL into a proatherogenic proinflammatory mediator (2, 3).
The HDL oxidative modification hypothesis linking Hp 2-2 to CVD in DM would suggest that antioxidant therapy might be beneficial to Hp 2-2 DM individuals. Indeed, in the Heart Outcomes Prevention Evaluation (HOPE) study, there was a greater than 50% reduction in the incidence of CV death and 40% reduction in the incidence of MI in Hp 2-2 DM individuals who received vitamin E (α-tocopherol) (24). These findings were confirmed prospectively in the Israel Cardiovascular Vitamin E (ICARE) study (27) where α-tocopherol administration resulted in an ∼50% reduction in the combined endpoint of MI, stroke, and CV death in Hp 2-2 DM individuals. However, potentially in conflict with the concept that antioxidant therapy may be efficacious in preventing CVD in Hp 2-2 DM, we demonstrated that in the Women's Antioxidant Vitamin Estrogen (WAVE) study, individuals with Hp 2-2 DM who received high dose vitamin C (in addition to α-tocopherol) did not receive any CV benefit and appeared to have an accelerated progression of atherosclerotic lesions (25).
In this study, we sought to reconcile the apparent divergent effects of vitamin C and α-tocopherol on CVD risk in Hp 2-2 DM by assessing the interaction between vitamin C and α-tocopherol on HDL oxidation and function using in vitro studies with purified Hp 1 and Hp 2, as well as in vivo studies with Hp transgenic mice.
Materials and Methods
Preparation of Hp and Hb
Human Hp was purified from plasma using a goat anti-Hp antibody affinity column. Hb was isolated from fresh human blood, as previously described (15). Glycosylated Hb was prepared using glycolaldehyde, as previously described (1). Briefly, fresh Hb (10 mM) was incubated with glycolaldehyde (20 mM) for 3 days at 37°C. Glycosylated Hb was then dialyzed three times against PBS at 4°C. In all experiments comparing glycosylated to nonglycosylated Hb, we used nonglycosylated Hb that had been treated the same as glycosylated Hb except for the addition of glycolaldehyde. MetHb was prepared by incubating 1 mM of Hb with 1.1 mM of potassium ferricyanide (K3Fe(CN)6) at room temperature for 30 min. The metHb was then purified on a PD-10 column. The concentration of Hp and Hb was calculated both by using the Bradford reagent and a spectrophotometer based on known extinction coefficients. The molar concentration of Hp that is reported was based on the monomer properties of that particular type of Hp because each Hp monomer (whether in the Hp 1-1 or Hp 2-2 complex) is thought to be capable of binding a single Hb dimer.
HDL purification
HDL was purified from the plasma of fasted normolipidemic healthy human volunteers by density gradient ultracentrifugation, as previously described (4).
Vitamin E
Water-miscible vitamin E (DL-α-tocopherol acetate) was obtained from Merck (Dermstadt, Germany cat #500862).
Measurement of iron-mediated oxidation
We measured the amount of oxidation mediated by elemental iron using the probe dihydrorhodamine 123 (DHR), as described previously by Cabantchick (1, 13). DHR is converted to its fluorescent form when oxidized. An assay to measure the rate of DHR oxidation has been previously described. Briefly, duplicates of 20 μl of Fe(III)/NTA complexes of different concentrations were transferred to clear-bottom 96-well plates. 180 μl of iron-free Hepes-buffered saline (HBS) containing 50 μM DHR with or without ascorbic acid and/or α-tocopherol was then added. Demonstration that the oxidation of DHR was in fact due to iron was shown by performing these incubations in the presence and absence of 50 μM of the iron chelator L1 (deferiprone). Immediately after the addition of DHR, the kinetics of fluorescence increase was followed in a BMG Galaxy Fluostar microplate reader (MTX Lab Systems, Vienna, VA) at 37°C with a 485/538 nm excitation/emission filter pair with repeated readings every 2 min up to 40 min. Results are reported as the rate of DHR oxidation (fluorescent units (FU)/min) as determined for each well.
Measurement of the amount of redox active iron associated with the Hp–Hb complex
We measured the amount of redox active iron associated with the Hp–Hb complex using a modification of the method described above. The iron-dependent component of DHR oxidation by Hp–Hb was determined by monitoring oxidation of DHR in the presence and absence of the iron chelator L1. Quadruplicates of 20 μl of different types of Hb–Hp complexes (glycosylated and nonglycosylated Hb, Hp 1, Hp 2) were transferred to 96-well plates and mixed with 180 μl of iron-free HBS containing 50 μM DHR with or without ascorbic acid (2.5 μM) or α-tocopherol (2.5 μM) and with or without 40 μM of L1. The slopes of DHR oxidation were measured using the same method and kinetics as above. The free redox active iron concentration (in μM) for a given Hp–Hb preparation was calculated from calibration curves relating the difference in slopes to the Fe concentration as previously described (1, 13).
Measurement of HDL-associated lipid peroxides
200 μg of purified HDL, 20 μM glycosylated or nonglycosylated Hb, and 10 μM hydrogen peroxide were incubated in PBS in the presence or absence of 10 μM ascorbic acid or/and 10 μM α-tocopherol overnight at 37°C. When Hp 1-1 or Hp 2-2 was added, the Hp–Hb complex was formed by incubating Hb (20 μM) with Hp 1-1 or Hp 2-2 (1 to 1 molar ratio based on the monomer concentration of Hp) for 20 min at room temperature before the Hp–Hb complex was incubated with HDL.
After overnight incubation of the mixture, lipid peroxides were determined by the addition of 1 cc of lipid peroxide reagent (0.2 M potassium hydrophosphate, 0.12 M potassium iodide, 0.15 M sodium azide, 2 g/l igepal, 0.1 g/l alkylbenzldimethyl ammonium chloride, 10 μM ammonium molybdate), as previously described (12). After a 45 min incubation in the dark, the mixture was centrifuged and the OD of the sample measured at 365 nM. The amount of lipid peroxides in nmol was determined using the extinction coefficient of lipid peroxides (2.46 × 104 M −1).
Animals and diabetes
All protocols involving animals were approved by the Animal Care and Use Committee of the Technion. The murine Hp is a type 1 Hp allele with over 95% homology to the human Hp 1 allele. Since the Hp 2 allele exists only in humans, a murine Hp 2 allele was created by molecular engineering of the murine Hp 1 allele (26). This murine Hp 2 allele was then introduced into the murine genome at the murine Hp locus by targeted homologous recombination. DM was produced by intraperitoneal injection of streptozotocin (200 mg/kg) dissolved in 50 mM citrate buffer (pH 4.5) at 8 weeks of age and antioxidant supplementation started at age 16 weeks. Glucose levels were monitored with a glucometer and HbA1c was measured using a diagnostic kit from Sigma (Rehovot, Israel). Mice were fed a standard chow diet and antioxidant supplementation consisting of vitamin C (200 mg/kg/day), α-tocopherol (600 mg/kg/day), or combination of vitamin C and α-tocopherol was provided in the drinking water for 45 days. Serum from these mice was assessed for its ability to elicit efflux of 3H-cholesterol from J774 macrophages.
Measurement of Hb binding capacity of mouse serum
The Hp concentration of Hp 1-1 and Hp 2-2 mice was measured as previously described and was similar in Hp 1-1 and Hp 2-2 mice (2, 26). The Hb binding capacity of Hp 1-1 and Hp 2-2 mouse serum was determined by adding a known amount of Hb to mouse plasma and then separating the free and bound Hb by size exclusion chromatography using a 100 kD centricon device (Millipore, Bedford, MA), as previously described (15). The maximal amount of Hb which could be bound by 1 ml of Hp 1-1 mice plasma was 18.9 nmoles of Hb dimer, and the maximal amount of Hb which could be bound by 1 ml of Hp 2-2 mouse plasma was 19.4 nmoles of Hb dimer.
Cell culture
J774 A.1 murine macrophage cells (American Type Culture Collection, Manassas, VA) were grown in DMEM supplemented with 5% FBS, 1% glutamine, 1% penicillin/streptomycin, and 1% sodium pyruvate.
Cholesterol efflux assay
The ability of serum from mice to promote cholesterol efflux from macrophages was assessed as previously described (2). Murine J774 cells were plated in 24-well plates for 48 h, washed and incubated in serum-free DMEM containing 3H-cholesterol for 1 h. After the cells were washed three times, they were incubated with 1 cc serum-free DMEM containing 10 μl serum from diabetic mice treated with placebo, vitamin C, α-tocopherol, or both vitamin C and α-tocopherol. After incubating the cells overnight at 37°C to enable cholesterol to efflux from the cells into the medium, 500 μl of the medium was taken and the amount of 3H-cholesterol was determined by liquid scintillation counting (LSC). Cells were then washed three times and lysed with 1 cc NaOH (0.2 N) and the amount of 3H-cholesterol in 500 μl of the lysate determined by LSC. The percentage of cholesterol efflux was calculated by dividing the amount of cpm in the medium by the sum of the cpm in medium and the cell after subtraction for nonspecific efflux obtained from cells incubated without serum.
Statistical analysis
Results are reported as the mean ± SEM. Differences between groups were compared using Student's t test or ANOVA with a p-value of ≤0.05 considered statistically significant.
Results
Effect of vitamin C and α-tocopherol on iron-mediated oxidation of DHR
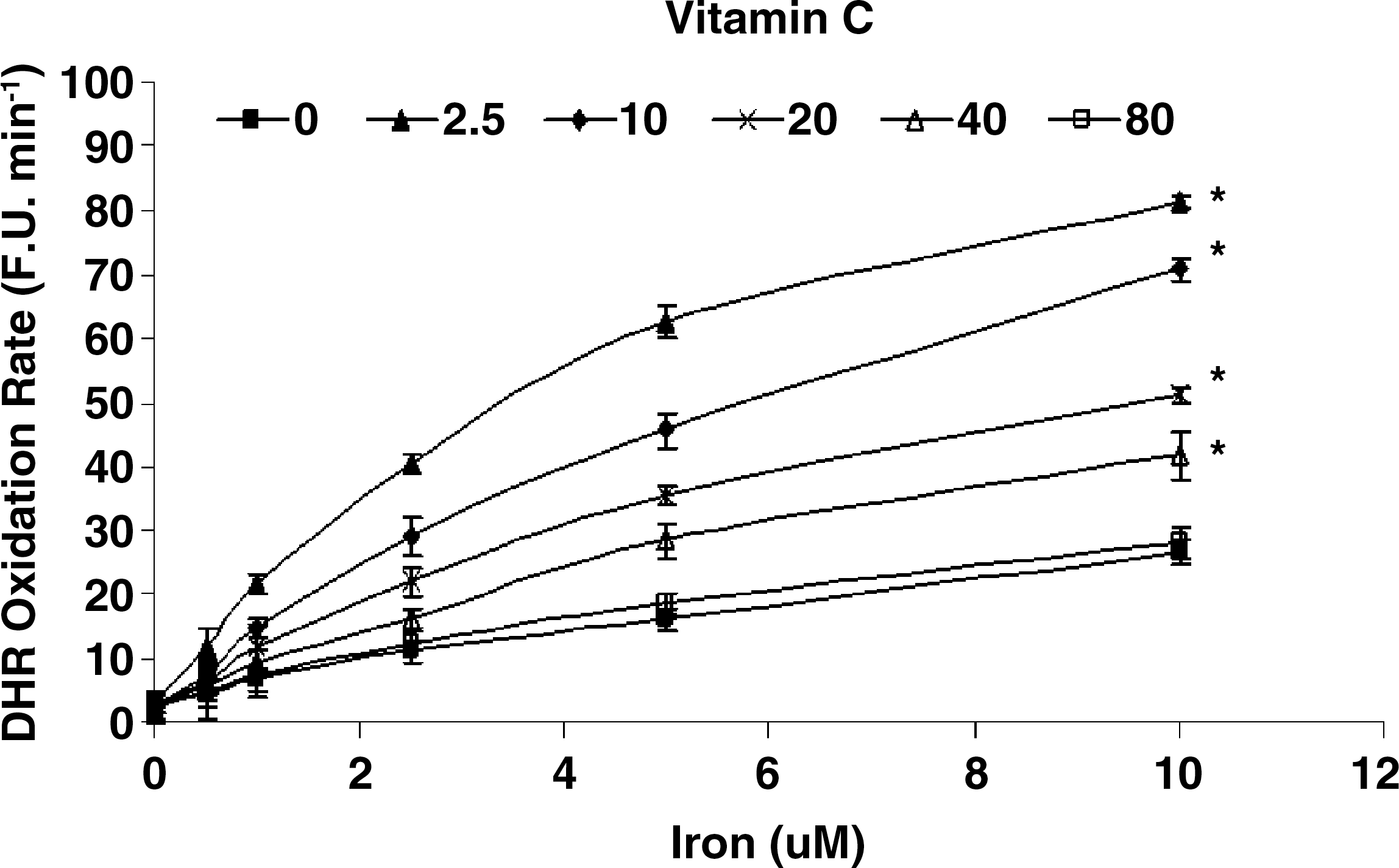
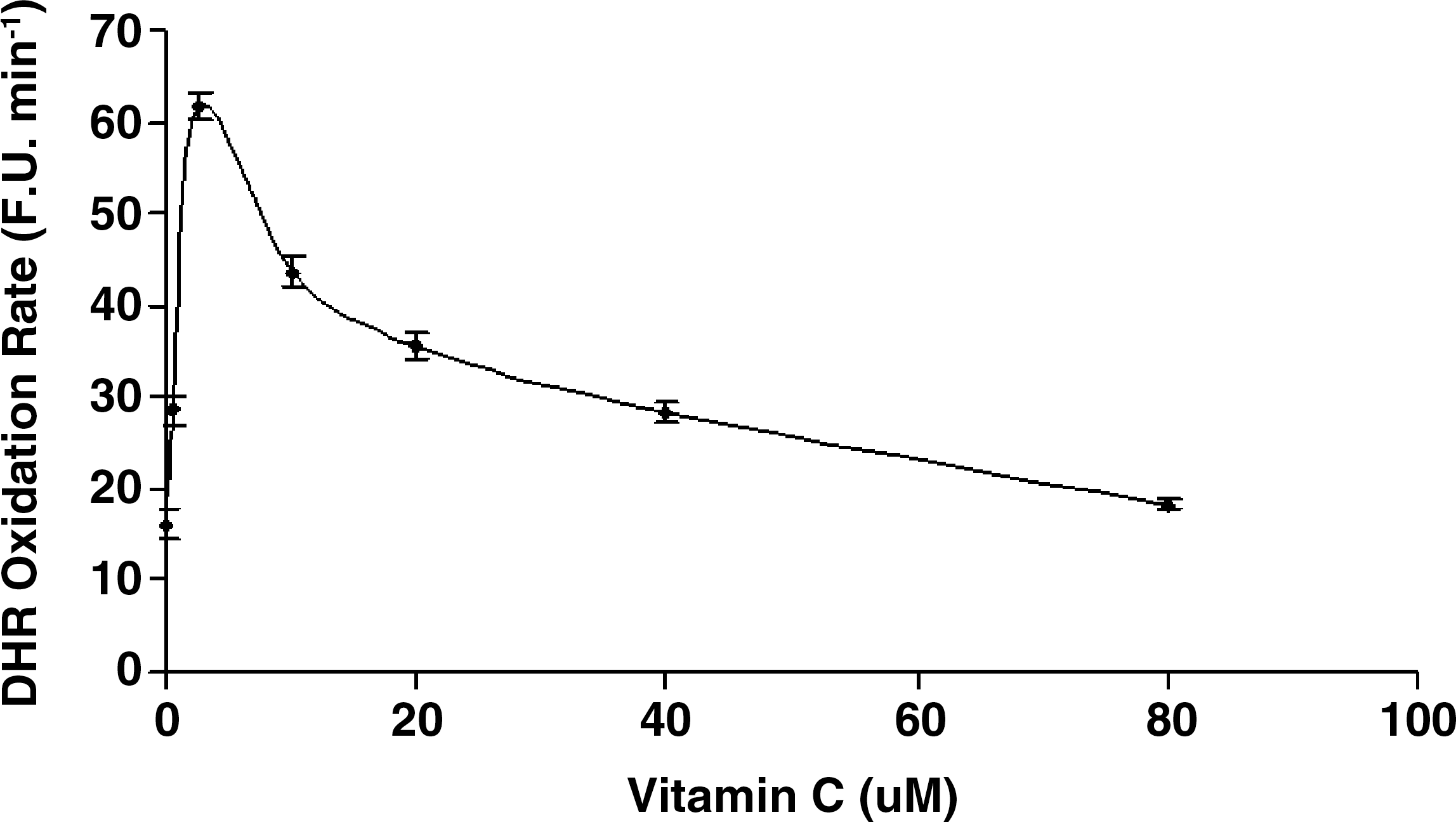
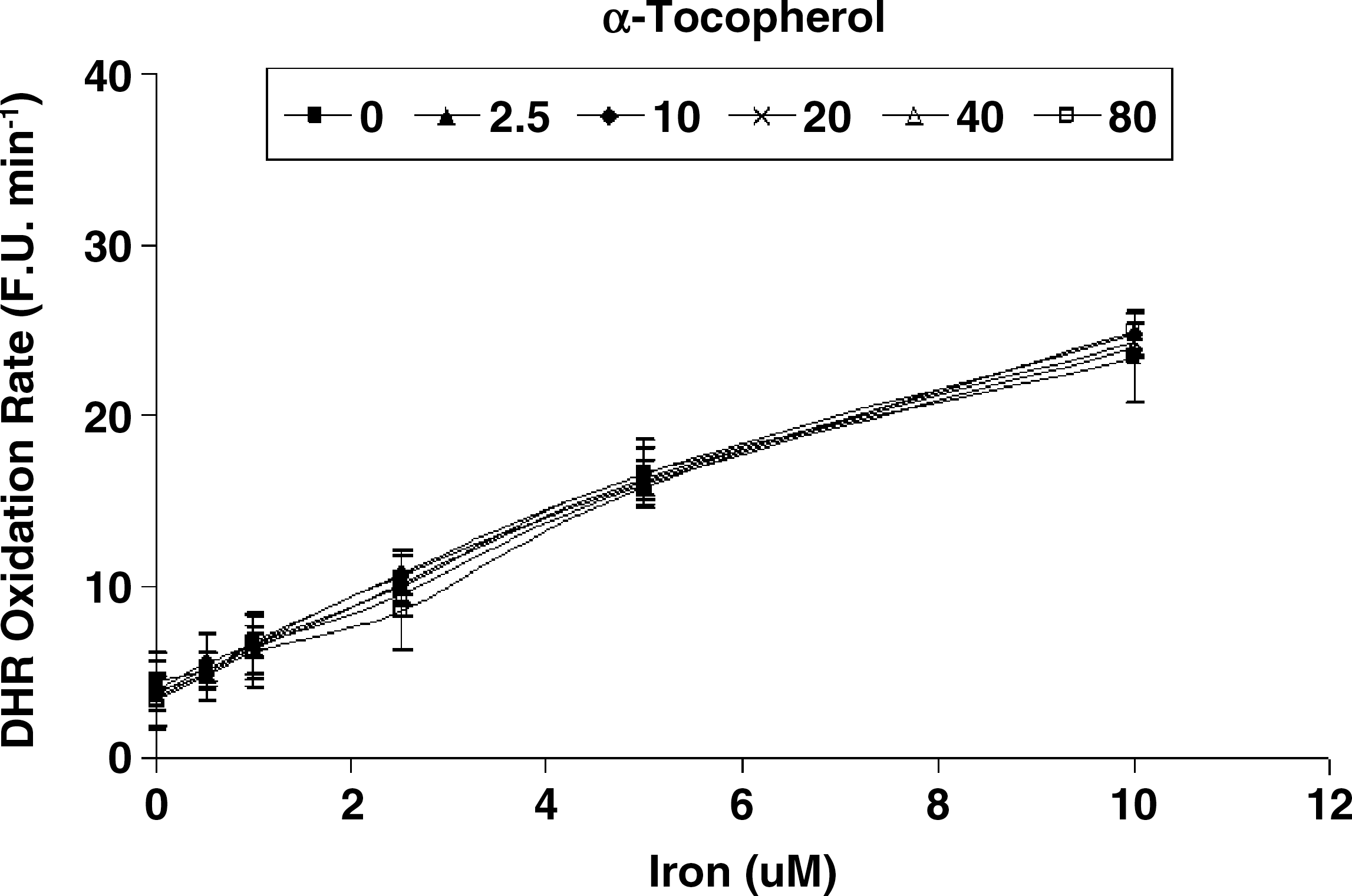
While vitamin C and α-tocopherol are traditionally thought of as preventing oxidation, it has been suggested that vitamin C might promote iron-mediated oxidation under certain conditions by promoting the redox cycling of iron (29). We assessed the rate of DHR oxidation using increasing concentrations of elemental iron in the presence or absence of vitamin C or α-tocopherol. As demonstrated in Fig. 1. we found that vitamin C at low concentrations was able to increase the rate of DHR oxidation. Regardless of iron concentration, vitamin C had a biphasic effect on DHR oxidation, reaching a maximal oxidation capacity at 2.5 μM of vitamin C. At concentrations below 40 μM, vitamin C appeared to act as a pro-oxidant, and above 40 μM vitamin C appeared to act as an antioxidant in this system (Fig. 2). α-Tocopherol, on the other hand, was not able to promote iron-mediated oxidation of DHR. As shown in Fig. 3, the rate of DHR oxidation in the presence of α-tocopherol was equal to the oxidation rate in the absence of α-tocopherol. Moreover, the combination of vitamin C and α-tocopherol had similar effect on DHR oxidation to that shown in the presence of vitamin C alone (Fig. 4), demonstrating again that α-tocopherol is not an active participant in the redox cycling driven by ascorbate. DHR is a sensitive probe that can be oxidized by different oxidants. In Fig. 5, we demonstrate that the increased oxidation of DHR by ascorbate in this system was due to iron by showing that the iron chelator L1 could completely block the increased DHR oxidation associated with ascorbate.

Redox active iron associated with Hb and Hp–Hb complexes
Free Hb is a potent oxidant due to the iron found in its heme pocket, and glycosylation of Hb results in a significant increase in the redox activity of the iron associated with Hb (termed labile redox active iron as described in Methods) (1). We sought to determine if there were differences in the amount of redox active iron associated with normal and glycosylated Hb in the presence and absence of vitamin C or α-tocopherol, and if these differences were affected by the addition of Hp 1-1 or Hp 2-2.
As previously described, we found a significant increase in redox active iron associated with glycosylated Hb as compared with normal Hb (1). Vitamin C resulted in a dramatic increase while the addition of α-tocopherol resulted in no change in the amount of the redox active iron associated with Hb, (oxidized) MetHb or glycosylated Hb (Fig. 6). α-Tocopherol significantly reduced the amount of redox active iron associated with glycosylated metHb (Fig. 6). Hp 2-2 significantly increased while Hp 1-1 significantly decreased the amount of redox active iron associated with glycosylated Hb. Furthermore, the addition of vitamin C resulted in a significant increase while the addition of α-tocopherol resulted in a significant decrease in the amount of redox active iron associated with glycosylated Hp 2-2-Hb complexes (Fig. 7).


HDL lipid peroxidation induced by Hb and Hp–Hb and the effect of vitamin C and α-tocopherol
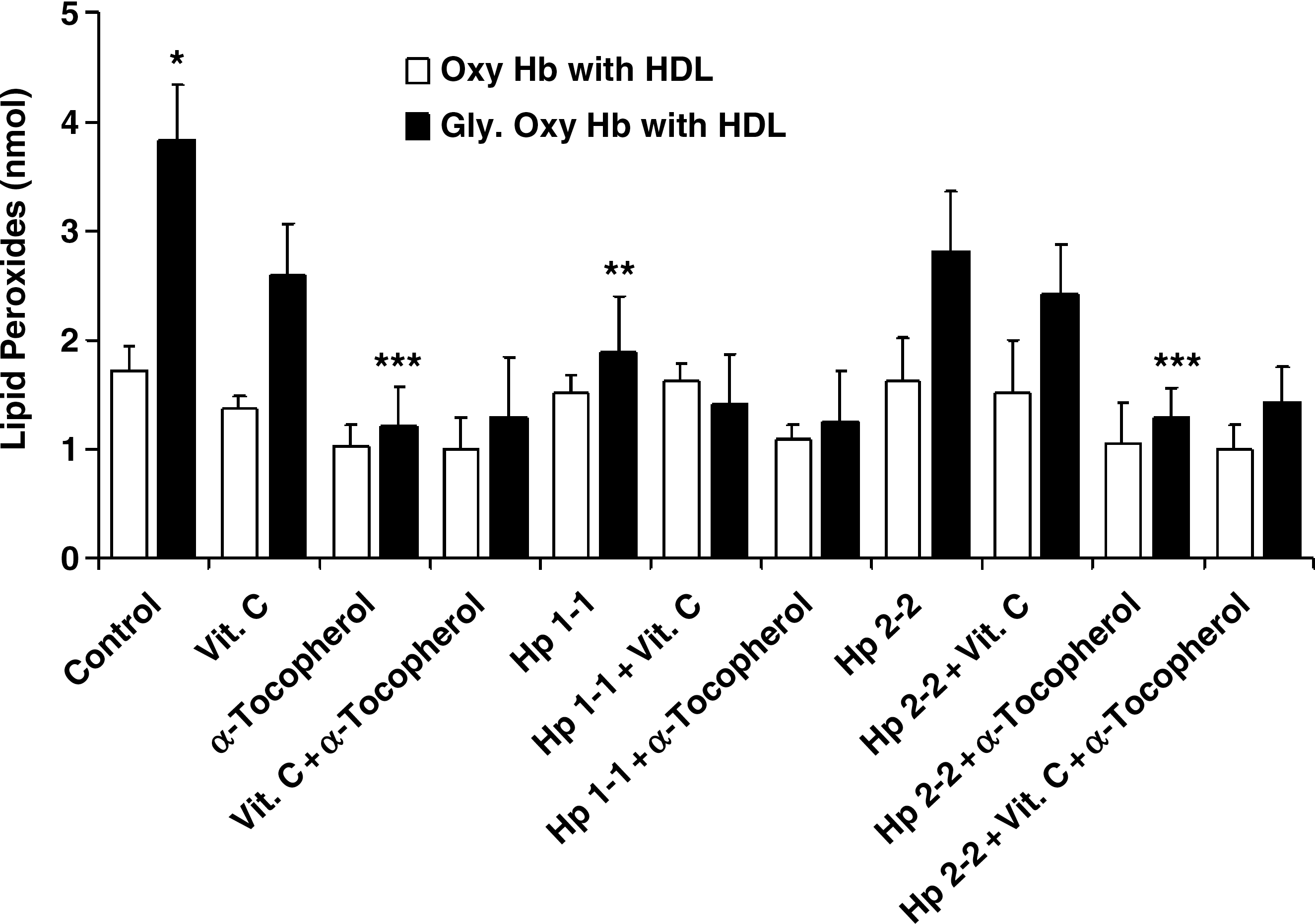
HDL is oxidatively modified in Hp 2-2 DM individuals (3). We assessed the amount of HDL lipid peroxidation after the co-incubation of HDL with Hb or Hp–Hb and whether vitamin C or α-tocopherol could interfere with this oxidative process (Fig. 8). We found that HDL lipid peroxidation was significantly increased when HDL was incubated with glycosylated Hb as compared with native Hb (1.73 ± 0.44 vs. 3.84 ± 0.98 nmol for glycosylated and native Hb, respectively; n = 4, p = 0.007). While incubation of HDL with glycosylated Hb complexed with Hp 1-1 resulted in a reduction in the amount of HDL-associated lipid peroxides (1.89 ± 1.01 vs. 3.84 ± 0.98 nmol for glycosylated Hb with and without Hp 1-1, respectively, n = 4 for each group, p = 0.032) the complexing of glycosylated Hb with Hp 2-2 did not block HDL peroxidation (2.81 ± 1.09 vs. 3.84 ± 0.98 nmol lipid peroxides for glycosylated hemoglobin with and without Hp2-2, respectively; n = 4, p = 0.211). When vitamin C was added to Hb or Hp–Hb, the amount of HDL-associated lipid peroxidation was not reduced (2.95 ± 0.94 nmol for glycosylated Hb with vitamin C vs. 2.42 ± 0.92 nmol for glycosylated Hb-Hp 2-2 complex with vitamin C vs. 3.84 ± 0.98 nmol for glycosylated Hb with no vitamin C; n = 4 for each group, p > 0.07). However, α-tocopherol significantly reduced HDL lipid peroxidation by glycosylated Hb or glycosylated Hb–Hp 2-2 (1.21 ± 0.73 nmol for glycosylated Hb with α-tocopherol vs. 1.29 ± 0.55 nmol for glycosylated Hb–Hp 2-2 complex with α-tocopherol vs. 3.84 ± 0.98 nmol for glycosylated Hb without α-tocopherol; n = 4 for each group, p < 0.01).
Cholesterol efflux from macrophages elicited by serum from Hp 1 and Hp 2 DM mice treated with vitamin C and/or α-tocopherol.
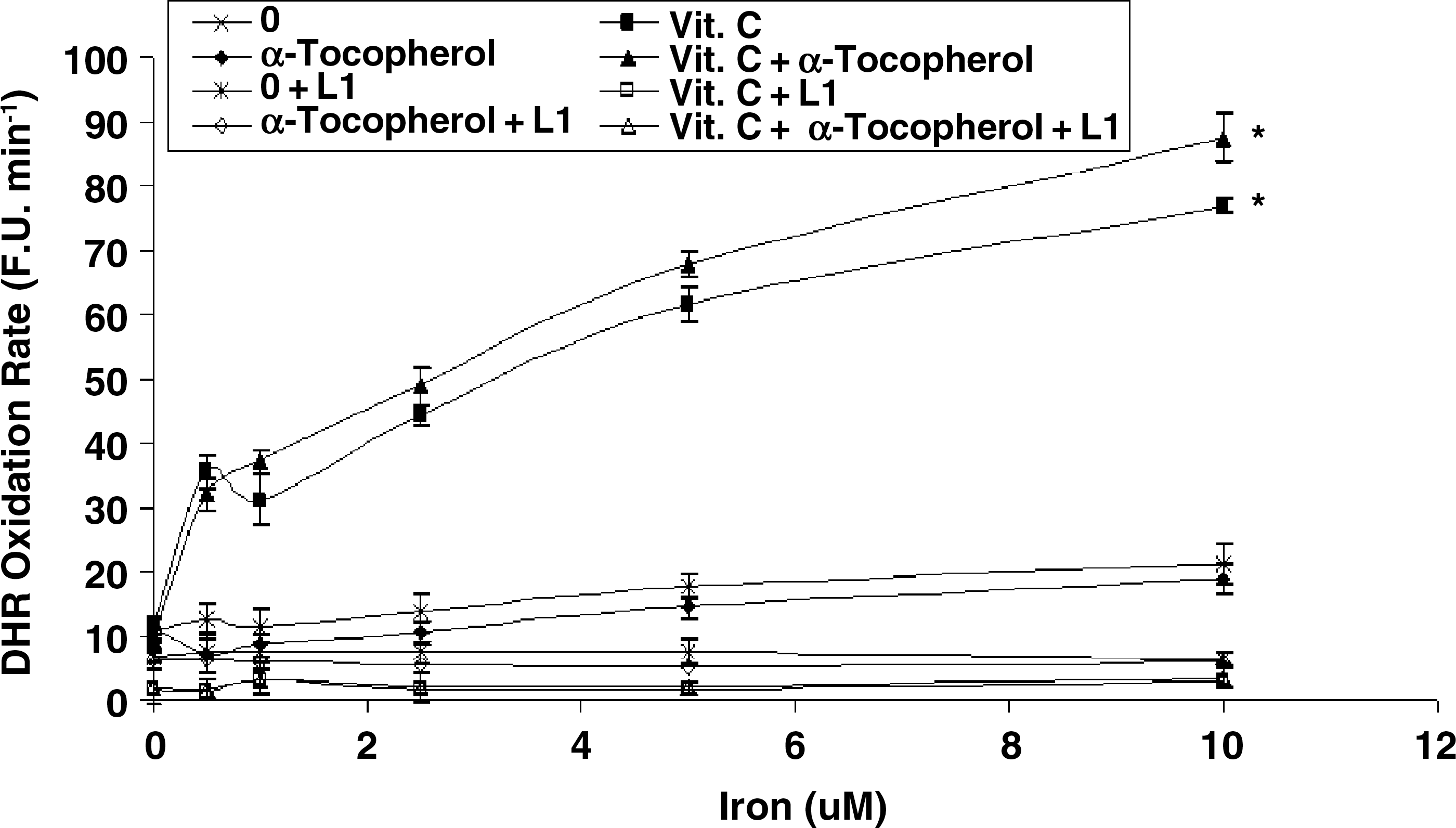
We have previously demonstrated that HDL function in Hp 2-2 DM individuals and mice (measured as the ability of serum HDL to elicit cholesterol efflux from 3H-cholesterol-labeled macrophages) is impaired (2, 3). We sought to examine whether administration of vitamin C or α-tocopherol or a combination of both to DM Hp 2-2 mice could improve HDL function. Hp 1-1 and Hp 2-2 DM mice were treated with placebo, vitamin C, α-tocopherol, or the combination of vitamin C and α-tocopherol for 45 days and the ability of the serum from these mice to elicit cholesterol efflux was determined (Fig. 9). Consistent with our previous findings, we found that cholesterol efflux from J774 cells elicited by serum from Hp 2-2 DM mice was significantly reduced as compared with efflux elicited by serum from Hp 1-1 DM mice [12.79 ± 0.54 (n = 11) vs. 8.96 ± 0.38 (n = 13) for Hp 1-1 and Hp 2-2 DM mice, respectively, p < 0.001]. Incubation of serum from Hp 2-2 DM mice treated with vitamin C had no effect on cholesterol efflux [9.3 ± 0.91 (n = 8) vs. 8.96 ± 0.38 (n = 13) for mice treated with vitamin C and placebo, respectively, p = 0.7]. However, α-tocopherol significantly improved the ability of serum from Hp 2-2 DM mice to promote cholesterol efflux [11.54 ± 0.4 (n = 15) vs. 8.96 ± 0.38 (n = 13) for mice treated with α-tocopherol and placebo, respectively, p < 0.001]. Supplementation with vitamin C blocked the protective effect of α-tocopherol on HDL function in Hp 2-2 DM mice. There were no significant differences in cholesterol efflux elicited by the serum of Hp 1-1 DM mice supplemented with vitamin C, α-tocopherol or their combination as compared to placebo.

Discussion
In this study we have demonstrated clear differences between vitamin C and α-tocopherol in terms of how they interact with the glycosylated Hb–Hp 2 complex to either promote or inhibit the generation of reactive oxygen species. We have shown that vitamin C cannot block the oxidation of HDL by glycosylated Hb–Hp 2 complexes nor can it restore HDL function to Hp 2-2 DM mice. However, α-tocopherol can block oxidation of HDL by glycosylated Hb–Hp 2-2 and can restore HDL function in Hp 2-2 DM mice. Furthermore, the addition of vitamin C to α-tocopherol appears to mitigate the protective affect of α-tocopherol on HDL function. We propose that these studies serve to reconcile the apparent divergent clinical effects of vitamin C and α-tocopherol in Hp 2-2 DM individuals.
Vitamin C is an essential water-soluble antioxidant that is capable of scavenging various types of reactive oxygen species and inhibiting lipid peroxidation. However, vitamin C can readily be demonstrated to promote oxidation in the presence of iron (14, 36). The combination of vitamin C, hydrogen peroxide, and free iron form a highly pro-oxidant mixture generating hydroxyl radicals by Fenton chemistry that can oxidize almost any type of target molecule. The key participation of vitamin C in these processes is its ability to reduce ferric to ferrous iron and thereby allow recycling of iron for another round of oxidation (29). The consumption of vitamin C by this redox cycling with iron can be readily demonstrated in human plasma supplemented with iron whereby all of the vitamin C is observed to be consumed within hours (36). Concerns about this pro-oxidant activity of vitamin C in the presence of free iron have been rebuffed due to the belief that free redox active iron does not exist in human plasma (36). Indeed, under normal physiological conditions, over 99% of all plasma iron is bound to transferrin and is redox inactive. However, DM individuals with the Hp 2-2 genotype have significant amounts of non-transferrin-bound iron (NTBI) (iron within the Hp 2-2–Hb complex) in their plasma which is capable of redox cycling with vitamin C to generate free radicals (1). This increased NTBI in Hp 2-2 DM individuals is due to both the impaired antioxidant activity of Hp 2-2 as well as the impaired clearance of Hp 2-2–Hb in DM. The demonstration in independent cohorts that vitamin C levels are lower in Hp 2-2 individuals due to a greater rate of its oxidative consumption provides further support for this paradigm (20, 28).
α-Tocopherol is the most important lipid-soluble antioxidant and has been extensively studied with respect to its ability to protect against iron-induced oxidation. In contrast to vitamin C, α-tocopherol does not promote the redox cycling of free iron (29, 36). Consequently, the incubation of iron or Hb or Hp 2-2–Hb with α-tocopherol and a protein or lipid substrate does not result in oxidation of the substrate. Furthermore, α-tocopherol does not promote Fenton chemistry and hydroxyl radical formation in the presence of hydrogen peroxide and iron. However, α-tocopherol can serve to terminate free radical chain reactions initiated by free iron (14). Therefore, the increased NTBI present in Hp 2-2 DM individuals would be expected to be associated with a decreased amount of free radical propagation in individuals receiving α-tocopherol (as opposed to vitamin C where an increased amount of free radicals would be produced).
The oxidative modification hypothesis of atherosclerosis suggests that agents which may augment the endogenous antioxidant defense system may prevent the development and progression of atherosclerosis (33). This hypothesis is particularly attractive to explain the accelerated atherosclerosis seen in individuals with DM who are known to have enhanced oxidative stress and decreased levels of antioxidants such as vitamin C and α-tocopherol (9). While observational studies appeared to support the notion that antioxidants might be protective against atherosclerosis, numerous prospective interventional clinical trials employing the antioxidant vitamins C and α-tocopherol have failed to show any clinical benefit (10, 16 –18, 22, 35, 38, 39). To the contrary, there is strong evidence that these antioxidants may in fact be harmful and paradoxically increase mortality when given as high dose supplements (8, 21, 30).
Examination of the results from HOPE, ICARE, and WAVE appears to provide two reasons why these trials of antioxidant vitamins may have failed to show benefit and potentially induce harm (34). First, the type of antioxidant used in these trials may not have been appropriate. Vitamin C and α-tocopherol do not provide equivalent protection against all types of oxidant stress. Paradoxically, vitamin C may actually promote oxidative stress in the presence of certain oxidants, as has been demonstrated here. In the clinic, proof of concept that supplementation with vitamin C and vitamin E differ in their effect on cardiovascular disease in the setting of DM was recently demonstrated in the Iowa Women's Health Study (IWHS) (21). The IWHS assessed the relationship between vitamin intake and mortality from CVD. In DM participants the relative risk for CVD mortality across quintiles of vitamin C intake were 1.0, 0.97, 1.11, 1.47, 1.84 (p for trend < 0.01). Relative risks for coronary artery disease were 1.0, 0.8, 0.99, 1.26, and 1.91 (p for trend = 0.01) and for stroke were 1.0, 0.52, 1.23, 2.22, and 2.57 (p for trend < 0.01). This association between vitamin C intake and increased cardiovascular mortality was shown to be due to vitamin C supplements (rather than increased vitamin C from food) of greater than 300 mg/d. In contrast, in the IWHS vitamin E intake was not associated with an increased incidence of CVD mortality in women with DM (21). Second, all of the antioxidant trials that failed to show benefit provided antioxidant therapy to all patients regardless of the patient's endogenous antioxidant status. Antioxidant therapy may only be beneficial to a subset of individuals under particularly high levels of oxidative stress (34). Analysis of the results of HOPE, ICARE, and WAVE suggests that there exists an interaction between certain subsets of individuals (i.e., individuals with DM and the Hp 2-2 genotype) and the supplementary antioxidant (vitamin C or α-tocopherol) on the development of cardiovascular disease. We propose that the differences in clinical outcomes observed with supplementation with α-tocopherol and vitamin C to Hp 2-2 DM individuals serves to underscore the importance of proper patient selection and proper antioxidant selection in the design of clinical trials targeting oxidant stress to reduce disease in Hp 2-2 DM individuals (6). We hope that additional pharmacogenomic trials based on this paradigm will be initiated to directly test this hypothesis.
Footnotes
Acknowledgments
This work was supported by grants from the Israel Science Foundation, the Kennedy Leigh Charitable Trust, the Juvenile Diabetes Foundation, and NIH 1R01DK085226, all to APL.
Author Disclosure Statement
Dr. Levy has served in the past as a consultant for Synvista Therapeutics which owns a patent that claims that the Hp genotype determines the risk of cardiovascular disease in individuals with diabetes.