
Abstract
The mechanisms underlying the effect of the renin-angiotensin-aldosterone system (RAAS) inhibition on endothelial dysfunction in type 2 diabetes are incompletely understood. This study explored a causal relationship between RAAS activation and oxidative stress involved in diabetes-associated endothelial dysfunction. Daily oral administration of valsartan or enalapril at 10 mg/kg/day to db/db mice for 6 weeks reversed the blunted acetylcholine-induced endothelium-dependent dilatations, suppressed the upregulated expression of angiotensin II type 1 receptor (AT1R) and NAD(P)H oxidase subunits (p22phox and p47phox), and reduced reactive oxygen species (ROS) production. Acute exposure to AT1R blocker losartan restored the impaired endothelium-dependent dilatations in aortas of db/db mice and also in renal arteries of diabetic patients (fasting plasma glucose level ≥7.0 mmol/l). Similar observations were also made with apocynin, diphenyliodonium, or tempol treatment in db/db mouse aortas. DHE fluorescence revealed an overproduction of ROS in db/db aortas which was sensitive to inhibition by losartan or ROS scavengers. Losartan also prevented the impairment of endothelium-dependent dilatations under hyperglycemic conditions that were accompanied by high ROS production. The present study has identified an initiative role of AT1R activation in mediating endothelial dysfunction of arteries from db/db mice and diabetic patients. Antioxid. Redox Signal. 13, 757–768.
Introduction
Elevated ROS production, which is manifest in hypertension, diabetes, and atherosclerosis, is also one of the major initiators for endothelial dysfunction (8, 41) by direct inactivation of endothelium-derived NO. It is thus of great importance to define and explore oxidative mechanisms involved in endothelial dysfunction in type 2 diabetes (19). Sources of endogenous ROS that cause endothelial dysfunction include NAD(P)H oxidases (7) and endothelial nitric oxide synthase (eNOS) uncoupling (31).
The role of the renin-angiotensin-aldosterone system (RAAS) had been best defined in hypertension due to the wide application of RAAS blockers for lowering blood pressure. Of importance, existing evidence suggests a significant role of a local RAAS in the vascular wall as a key negative regulator of endothelial function in diabetes as well. Chronic angiotensin converting enzyme (ACE) inhibition improves endothelial function and cardiovascular outcomes in type 2 diabetic patients (14, 30, 32, 47). Apart from ACE inhibitors, angiotensin receptor blockers (ARBs) are also effective in improving cardiac function and reducing arterial stiffness in diabetic patients (4, 11, 16, 35, 44). Local and circulating angiotensin II (Ang II) is an important mediator of both metabolic and vascular dysfunction in diabetes (6). Animal studies also provided evidences for RAAS blockers in diabetes. ARB improve vascular function in type I diabetic rat (2, 36). ARB may ameliorate diabetic vasculopathy and nephropathy through prevention of eNOS uncoupling (31, 34, 45). Ang II binds to both Ang II type 1 (AT1R) and type 2 receptor (AT2R) (43). Most known detrimental effects of Ang II in vasculature are attributed to AT1R which is linked to NAD(P)H oxidase activation and ROS production (21). Hyperglycemia also upregulates the AT1R in vascular smooth muscle cells (37). However, the functional implications and the precise intracellular mechanisms by which AT1R activation and subsequent oxidative stress in diabetes that in turn impairs vasodilatation are not thoroughly understood.
In the present study, we examine the hypotheses that the upregulation of AT1R together with oxidative stress plays a critical role in the induction and maintenance of endothelial dysfunction in aortas of type 2 diabetic db/db mice and in renal arteries from type 2 diabetic patients.
Materials and Methods
Animal model
All animal experiments were performed on type 2 diabetic mice (C57BL/KSJ) lacking the gene encoding for leptin receptor (db/db) and heterozygote (db/m+ ) control which were supplied by Chinese University of Hong Kong (CUHK) Laboratory Animal Service Center after an approval was obtained from the Animal Experimentation Ethics Committee, CUHK. Mice were kept in a temperature-controlled holding room (22°–24°C) with a 12-h light/dark cycle, and fed a standard diet and water ad libitum. At the age of 12 weeks, adult male db/db mice were treated for 6 weeks with valsartan or enalapril at 10 mg/kg body weight/day or vehicle via oral gavage. Plasma glucose levels were determined using a blood glucose meter (Ascenia ELITE® XL, Bayer, IN). Systolic blood pressure was measured by a tail-cuff method.
Human renal arteries
Human renal arteries were obtained during surgery after informed consent from kidney cancer patients, aged between 56 and 82 years old, undergoing nephrectomy. One artery was obtained from each patient. The group of diabetic patients had a fasting plasma glucose level ≥7.0 mmol/l (126 mg/dl) or 2-h plasma glucose ≥11.1 mmol/L (200 mg/dl).
Plasma lipid profile and insulin in mice
Plasma levels of total cholesterol and triglyceride were determined using enzymatic methods (Stanbio, Boerne, TX) and plasma insulin level was assayed by enzyme immunoassay (Mercodia, Uppsala, Sweden).
Isometric force measurement
After mice were sacrificed by CO2 inhalation, the thoracic aortas were rapidly removed and placed in oxygenated ice-cold Krebs–Henseleit solution. Changes in isometric tension of vessels were recorded in a Multi Myograph System (Danish Myo Technology, Aarhus, Denmark) as previously described (24), and changes in isometric tension were recorded. The ring was stretched to an optimal baseline tension of 3 mN and then allowed to equilibrate for 60 min before the start of the experiment. Each ring was first contracted by 60 mmol/L KCl and rinsed in Krebs solution, and after wash out, phenylephrine (1 μmol/L) was used to produce a steady contraction and relaxed by cumulative additions of acetylcholine (ACh) (10−8 to 10−5 mol/L) in control or in the presence of 3 μmol/L losartan (ARB), 100 μmol/L apocynin [NAD(P)H oxidases inhibitor], or 100 μmol/L tempol [superoxide dismutase (SOD) mimetic]. These inhibitors had no effect on acetylcholine-induced relaxations in aortas from nondiabetic db/m+ mice (data not shown). Endothelium-independent relaxations to sodium nitroprusside (SNP) (10−9 to 10−6 mol/L) were studied in rings without endothelium. Each experiment was performed on rings prepared from different mice.
Each human renal artery was cut into 2–3 ring segments (2–3 mm in length) and each set of experiments were performed on rings from different human samples. Rings were suspended in organ baths as described previously (26). Each ring was initially stretched to an optimal tension of 25 mN and then allowed to equilibrate for 90 min before the start of the experiment.
Detection of intracellular ROS by dihydroethidium fluorescence
The amount of intracellular ROS production was determined using dihydroethidium (DHE) (Molecular Probes, Eugene, OR), which binds to DNA when oxidized to emit fluorescence (33). Aortic rings from db/m+ and db/db mice were obtained as described above and treated with or without ACh. To investigate the inhibitory effects of the RAAS inhibitor on ROS production, aortas were exposed for 30 min to one of the inhibitors including losartan, apocynin, or tempol before the addition of ACh, as to mimic the conditions in the functional study. To verify the contribution of ROS production from endothelium, the endothelial layer was removed by rolling the luminal surface with the tip of a pair of fine forceps. To examine the role of extracellular calcium ions on the generation of ROS, calcium-free Krebs solution was prepared to incubate the aortic rings for 30 min before the addition of ACh. Frozen sections of the aortic ring were cut in 10-μm thickness using cryostat and incubated for 10 min at 37°C in Krebs solution containing 5 μmol/L DHE. Fluorescent intensity was measured by confocal microscope (FV1000, Olympus, Tokyo, Japan) at excitation/emission of 488/605 nm to visualize the signal. The images were analyzed by the Fluoview software (Olympus).
Immunohistochemical staining of Ang II
Aortic rings were fixed in 4% paraformaldehyde at 4°C overnight, dehydrated, processed, and embedded in paraffin. Cross sections at 5 μm were cut on microtome (Leica Microsystems, Wetzlar, Germany). After rehydrated to water, sections were microwave boiled in 0.01 mol/L citrate buffer (pH 6.0) for 10 min for antigen retrieval, then incubated for 15 min with 3% H2O2 at room temperature to block endogenous peroxidase activity. After washed with phosphate buffer saline (PBS), sections were blocked in 5% normal goat or donkey serum according to the host species (Jackson Immunoresearch, West Grove, PA) for 1 h at room temperature. Primary antibody (anti-Ang II, 1:500, Peninsula laboratory, Belmont, CA, and anti-eNOS, 1:200, Santa Cruz, CA) diluted in normal serum were incubated overnight at 4°C. The slides were washed with PBS three times (5 min each). Biotin-SP conjugated goat anti-rabbit secondary antibodies (1:500, Jackson Immunoresearch) diluted in PBS were added and incubated for 1 h at room temperature. Slides were washed with PBS three times (5 min each) and incubated for 30 min with streptavidin-HRP conjugate (1:500, Zymed laboratory, San Francisco, CA) at room temperature, and washed. Positive staining was developed as brown precipitate by 3,3’-diamonobenzidine tetrachloride (DAB) chromogen substrate (Vector laboratory, Burlingame, CA). Slides were rinsed with water and counterstained with hematoxylin. Pictures were taken under Leica DMRBE microscope with a SPOT-RT digital camera and SPOT Advanced software (Diagnostic Instruments, Sertling Heights, MI) and intensities of signals were analyzed by ImageJ (National Institute of Health, Bethedsa, MD).
Western blot analysis
Protein samples prepared from aorta homogenates were electrophoresed through a 10% SDS-poly-acrylamide gel, transferred onto an immobilon-P polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA). Nonspecific binding sites were blocked with 5% nonfat milk or 1% BSA in 0.05% Tween-20 PBS. The blots were incubated overnight at 4°C with the primary antibodies: monoclonal anti-AT1R, polyclonal anti-AT2R (1:1000, Abcam, Cambridge, UK); monoclonal anti-nitrotyrosine (1:2000, Abcam), polyclonal anti-phosphor-eNOS Ser1177 (1:1000, Upstate Biotechnology, Lake Placid, NY); polyclonal anti-ACE, anti-eNOS, anti-p22phox and anti-p47phox (1:1000, Santa Cruz); monoclonal anti-phosphor-p38 MAPK (Thr180/Tyr182), polyclonal anti-p38 MAPK, monoclonal anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204), monoclonal anti-p44/42 MAPK (Cell Signaling, Beverly, MA), followed by HRP-conjugated secondary antibody (DakoCytomation, Carpinteria, CA). Monoclonal anti-β-actin (1:5000, Abcam) was used as a housekeeping protein. Densitometry was performed using a documentation program (Flurochem, Alpha Innotech Corp., San Leandro, CA).
Organ culture of mouse arterial rings in high glucose medium
High glucose (30 mmol/L) and mannitol (osmotic control) solutions were prepared in Dulbeco's Modified Eagle's Media (DMEM, Gibco, Gaithersberg, MD) culture media supplemented with 10% fetal bovine serum (FBS, Gibco), plus 100 IU/ml penicillin and 100 μg/ml streptomycin. Mouse thoracic aortic rings (2 mm in length) were then incubated in four groups, including 5 mmol/L glucose alone (NG), 5 mmol/L glucose plus 25 mmol/L mannitol (M), 30 mmol/L glucose (HG), 30 mmol/L glucose plus 3 μmol/L losartan (HG + losartan) for 36 h in an incubator kept at 37°C. After the incubation period, the segments were transferred to fresh Krebs solution, mounted in a myograph, and changes in arterial tone were recorded.
Drugs and solutions
Acetylcholine, NG-nitro-L-arginine methyl ester (L-NAME), phenylephrine, angiotensin II, sodium nitroprusside (SNP), diphenyliodonium, and tempol were purchased from Sigma-Aldrich Chemical (St Louis, MO). Apocynin was from Calbiochem (San Diego, CA). Losartan was purchased from Cayman (Ann Arbor, MI). Besides losartan, apocynin and diphenyliodonium were dissolved in DMSO (Sigma-Aldrich), all other drugs were dissolved in double-distilled water. Krebs solution contained (mmol/L): 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1.2 KH2PO4, and 11 D-glucose. A Ca2+-free solution was identical to Krebs solution with exclusion of Ca2+ and addition of 2 mmol/L EGTA.
Statistical analysis
Results were means ± SEM from different mice or human subjects. Concentration-response curves were analyzed by nonlinear regression curve fitting using GraphPad Prism software (Version 4.0, San Diego, CA) to approximate Emax as the maximal response and pIC50 as the negative logarithm of the drug concentration that produced 50% of Emax. These values are summarized in Supplemental Table 1 (see
Results
Basic metabolic parameters
Body weight of db/db mice increased gradually from 4 to 16 weeks when compared with age-matched db/m+
lean control mice (Supplemental Fig. 1A; see
Results are means ± SEM of measurements from 6–8 different mice. *p < 0.05 relative to db/m + group; # p < 0.05 relative to db/db group.
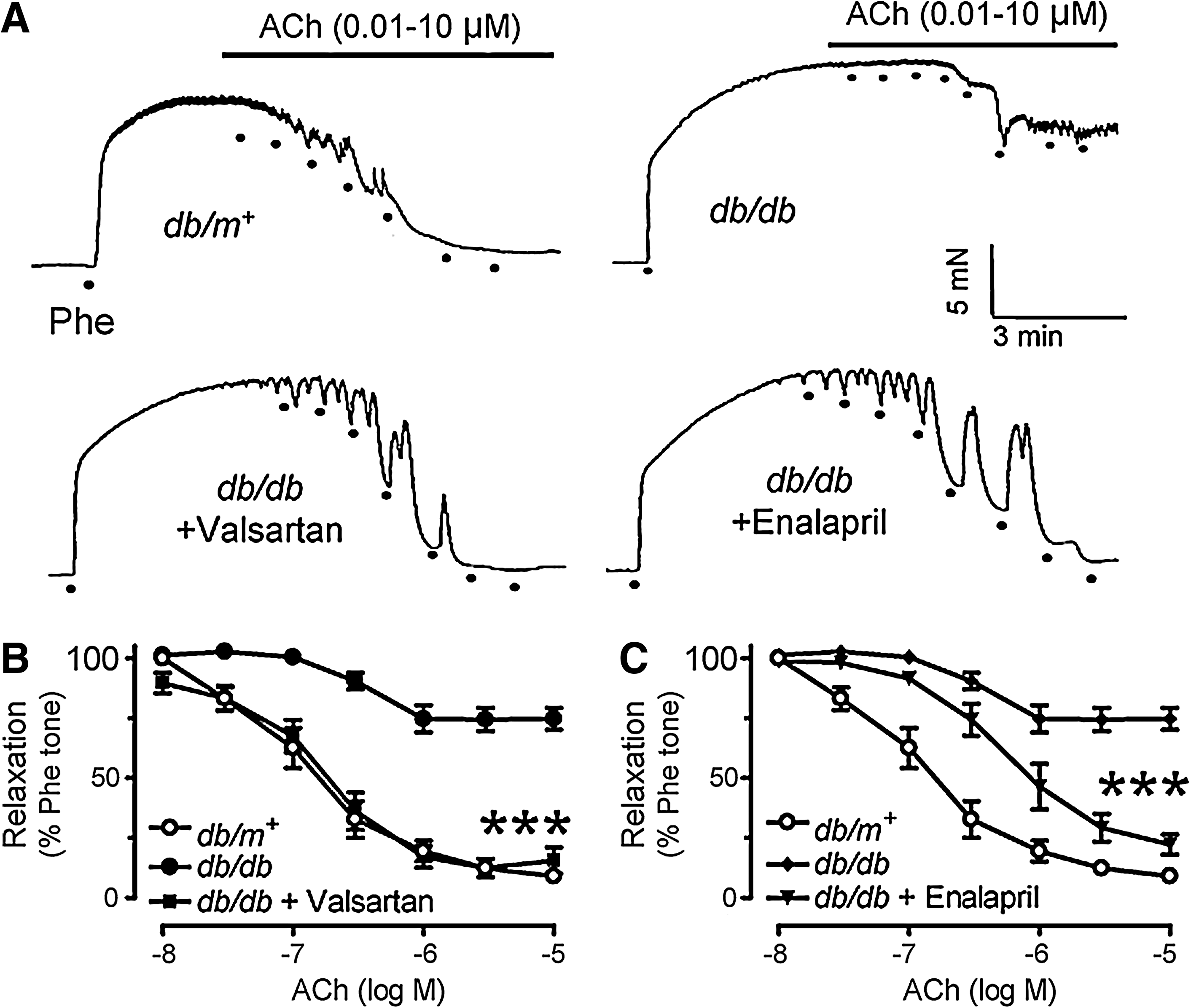
Improved endothelium-dependent dilatations in db/db mouse aortas by RAAS blockade
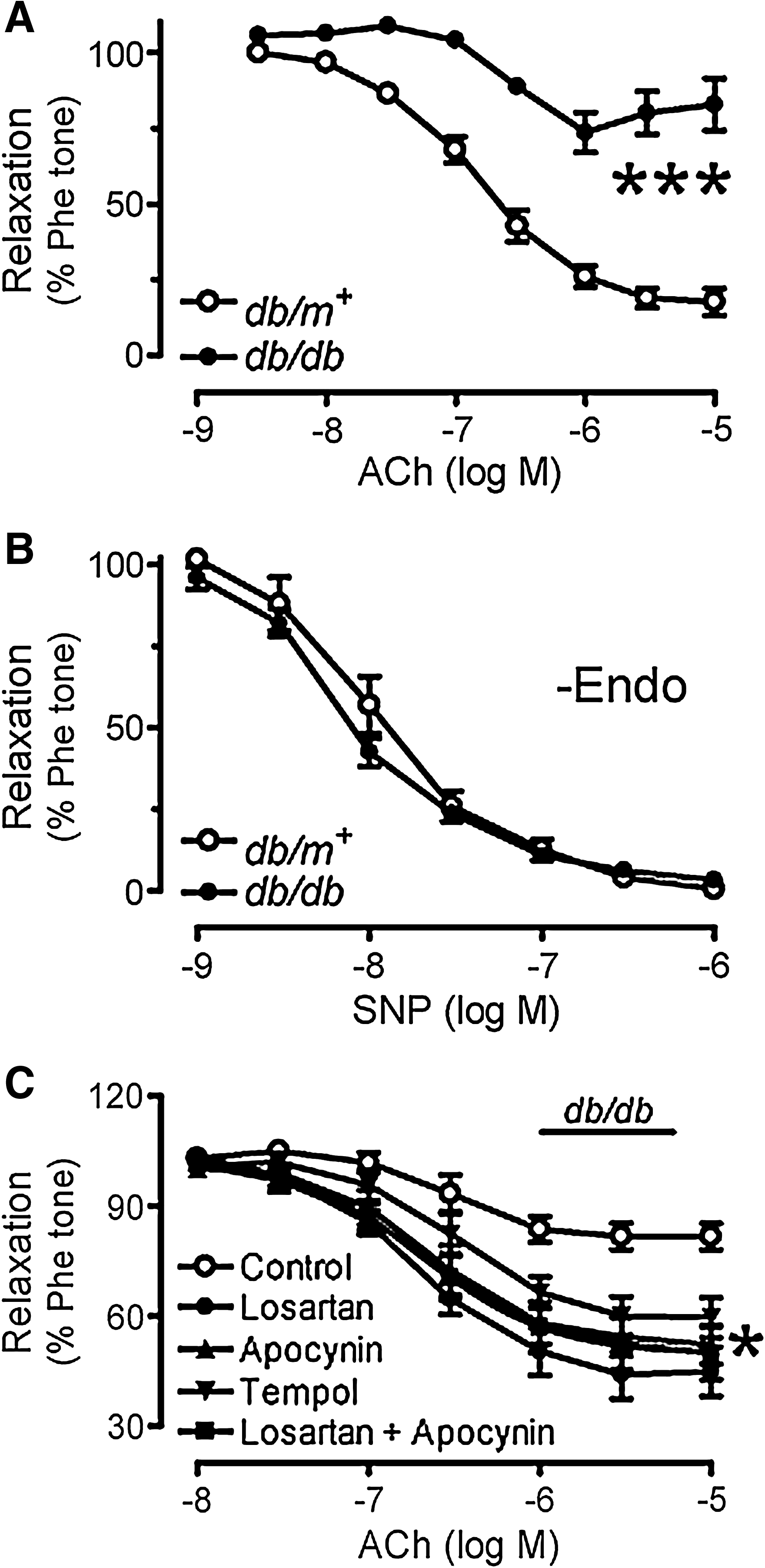
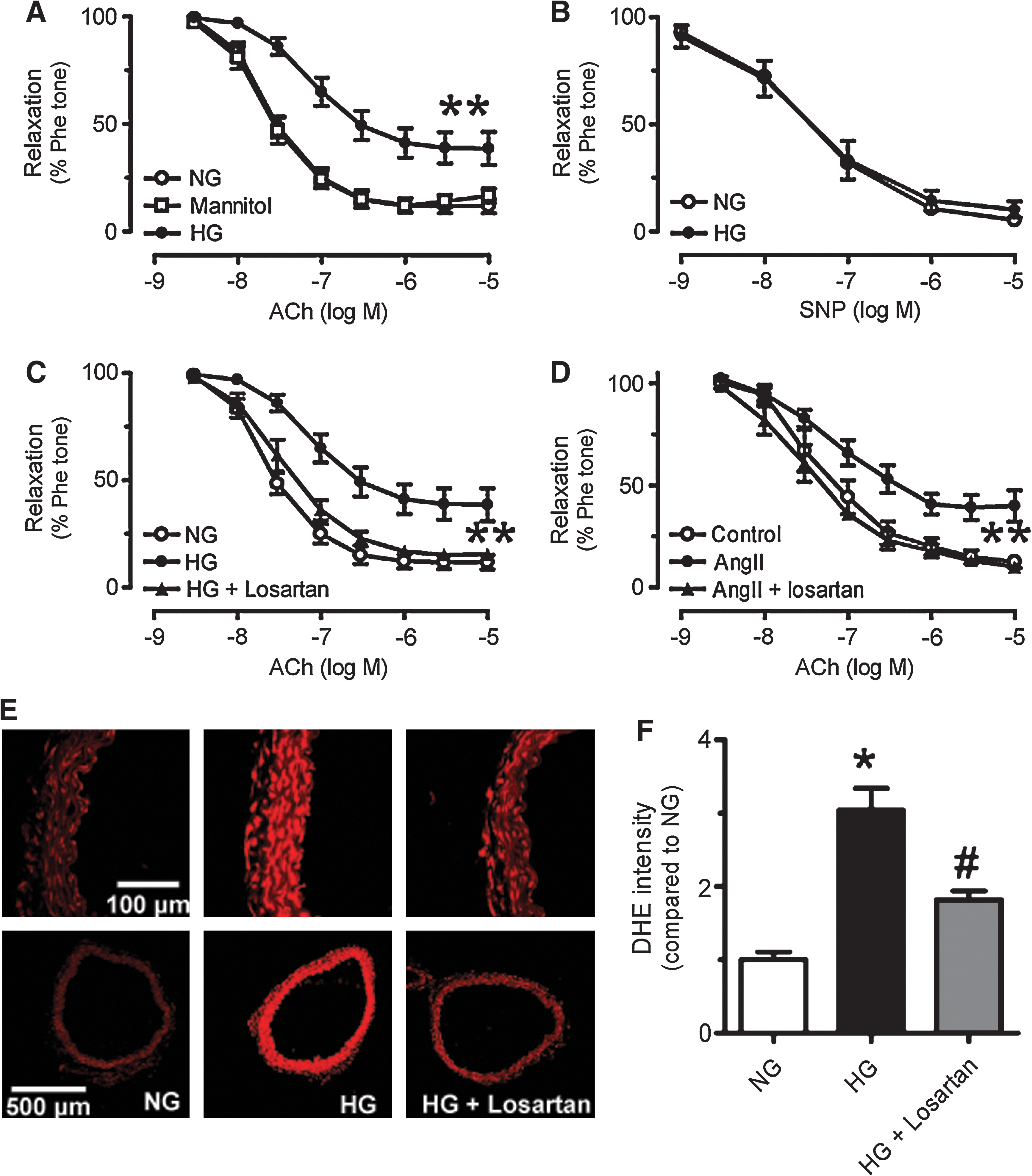
Six-week chronic treatment with valsartan or enalapril significantly improved endothelium-dependent dilatations in db/db mouse aortas as shown in representative tracings (Figs. 1A–1C). ACh-induced endothelium-dependent dilatations were impaired in db/db mouse aortas as compared with those of nondiabetic db/m+ mice (Figs. 1A and 2A), whilst sodium nitroprusside (SNP)-induced endothelium-independent dilatations were comparable between the two groups (Fig. 2B). AT1R blockade by losartan (3 μmol/L, 30-min incubation) (Fig. 2C) and inhibition of NAD(P)H oxidases by apocynin (100 μmol/L, Fig. 2C) improved ACh-induced vasodilatations, whilst combination of losartan and apocynin (Fig. 2C) did not cause further improvement (Supplemental Table 1). SOD mimetic tempol (100 μmol/L, Fig. 2C) also enhanced the blunted dilatations to ACh in db/db mouse aortas.
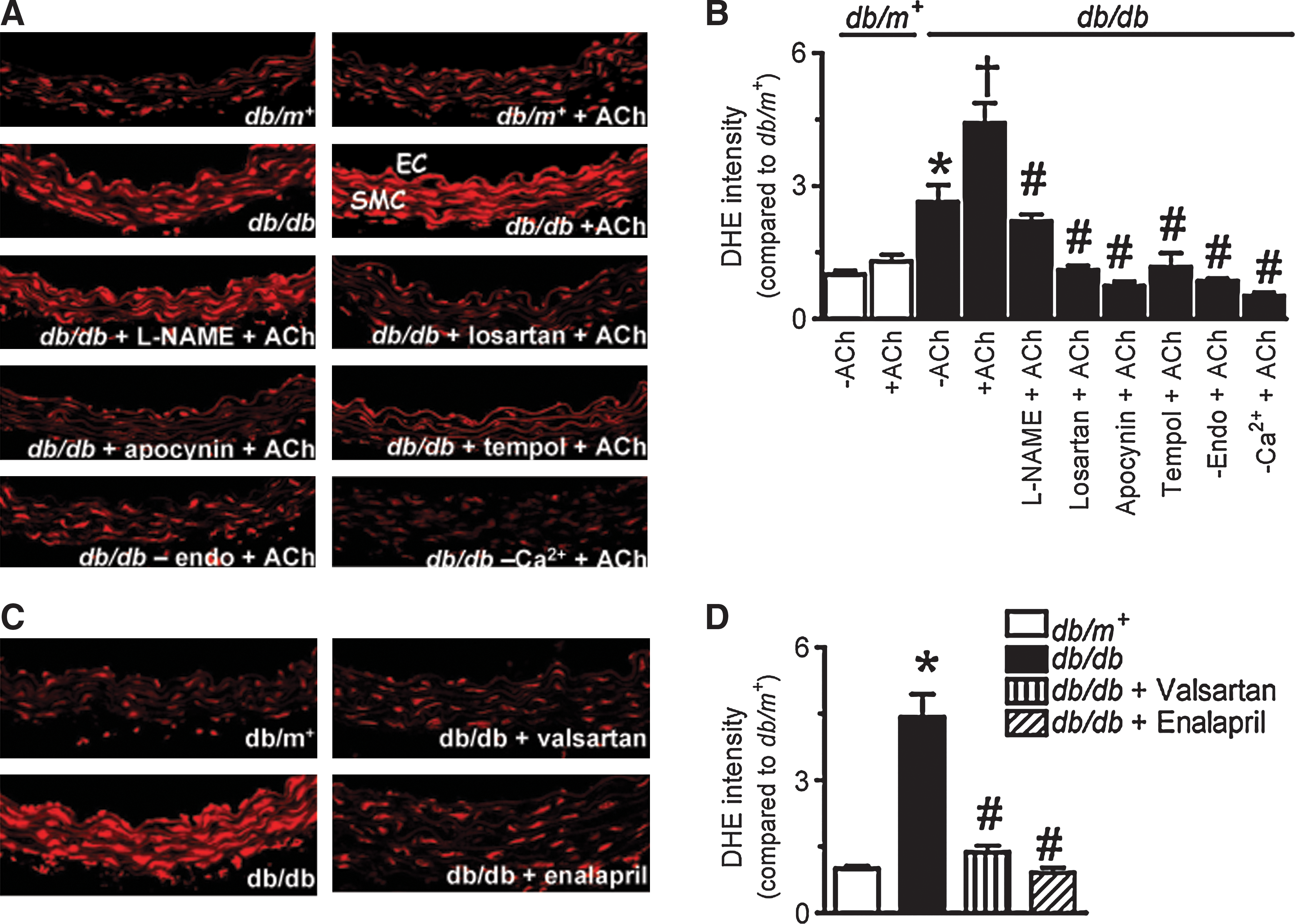
Augmented ROS production in db/db mouse aortas mediated by AT1R
The basal level of ROS reflected by the intensity of dihydroethidium (DHE) fluorescence was much higher in the wall of db/db mouse aortas (Fig. 3). The ROS level markedly increased in response to ACh (10 μmol/L), but to a greater extent in db/db mouse aortas (Figs. 3A and 3B). Acute exposure of db/db mouse aortas to L-NAME (100 μmol/L) attenuated ACh-stimulated rises in ROS. The increased ROS generation was eliminated by 30-min treatment with losartan (3 μmol/L), apocynin (100 μmol/L), or tempol (100 μmol/L) (Figs. 3A and 3B). Furthermore, the ACh-stimulated ROS increase was greatly diminished in the absence of extracellular Ca2+ ions or in aortas without endothelium (Figs. 3A and 3B). Increased ROS production in db/db mouse aortas was also abolished by chronic valsartan or enalapril treatment (Figs. 3C and 3D).
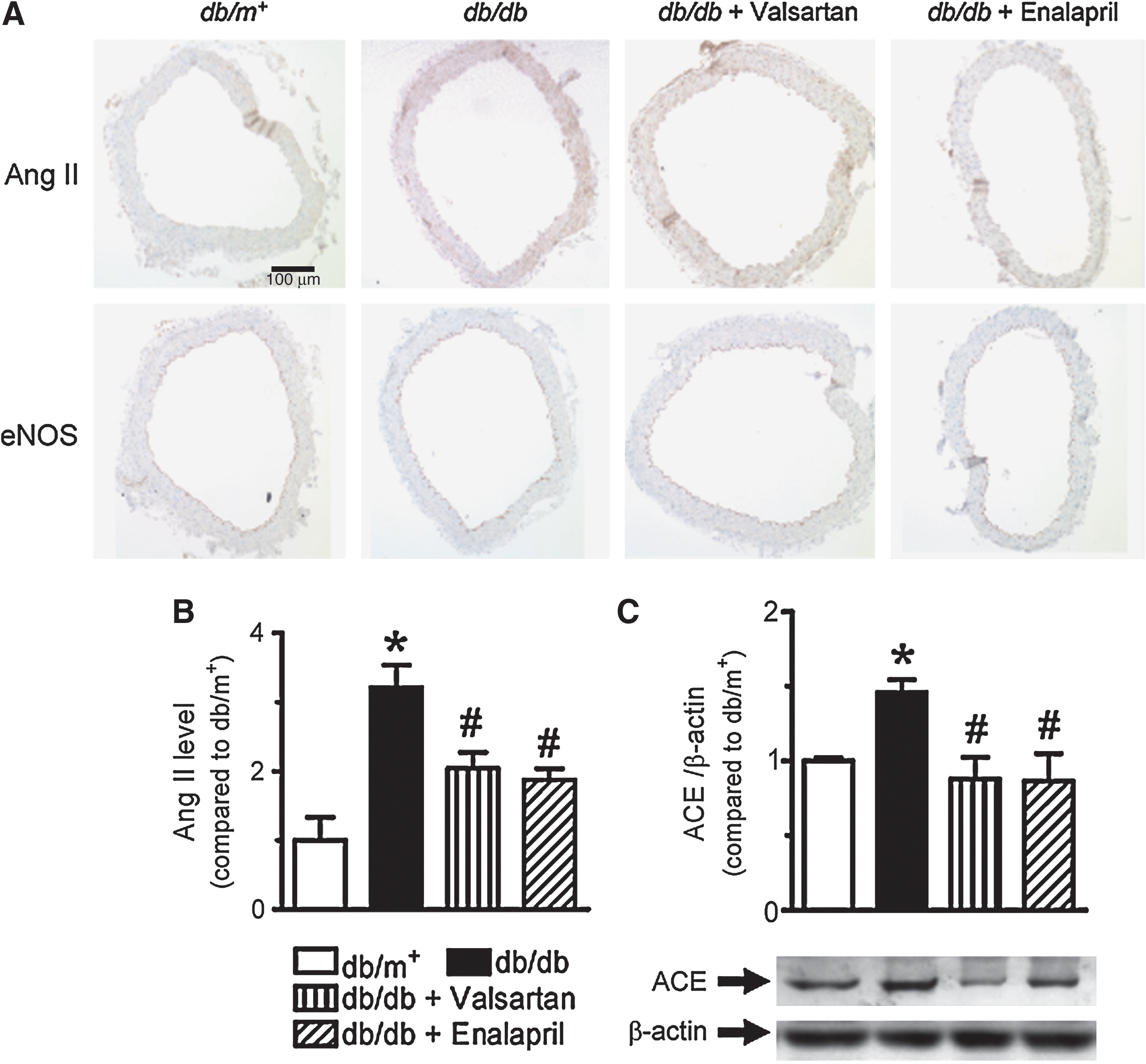
Effects of RAAS blockade on local production of Ang II in the vascular wall
Increased Ang II staining was observed in the vascular wall of aortas from db/db mice compared with db/m+ control (Figs. 4A and 4B), accompanied by ACE upregulation (Fig. 4C). Chronic RAAS blockade normalized the ACE expression and tissue Ang II levels (Figs. 4A–4C).
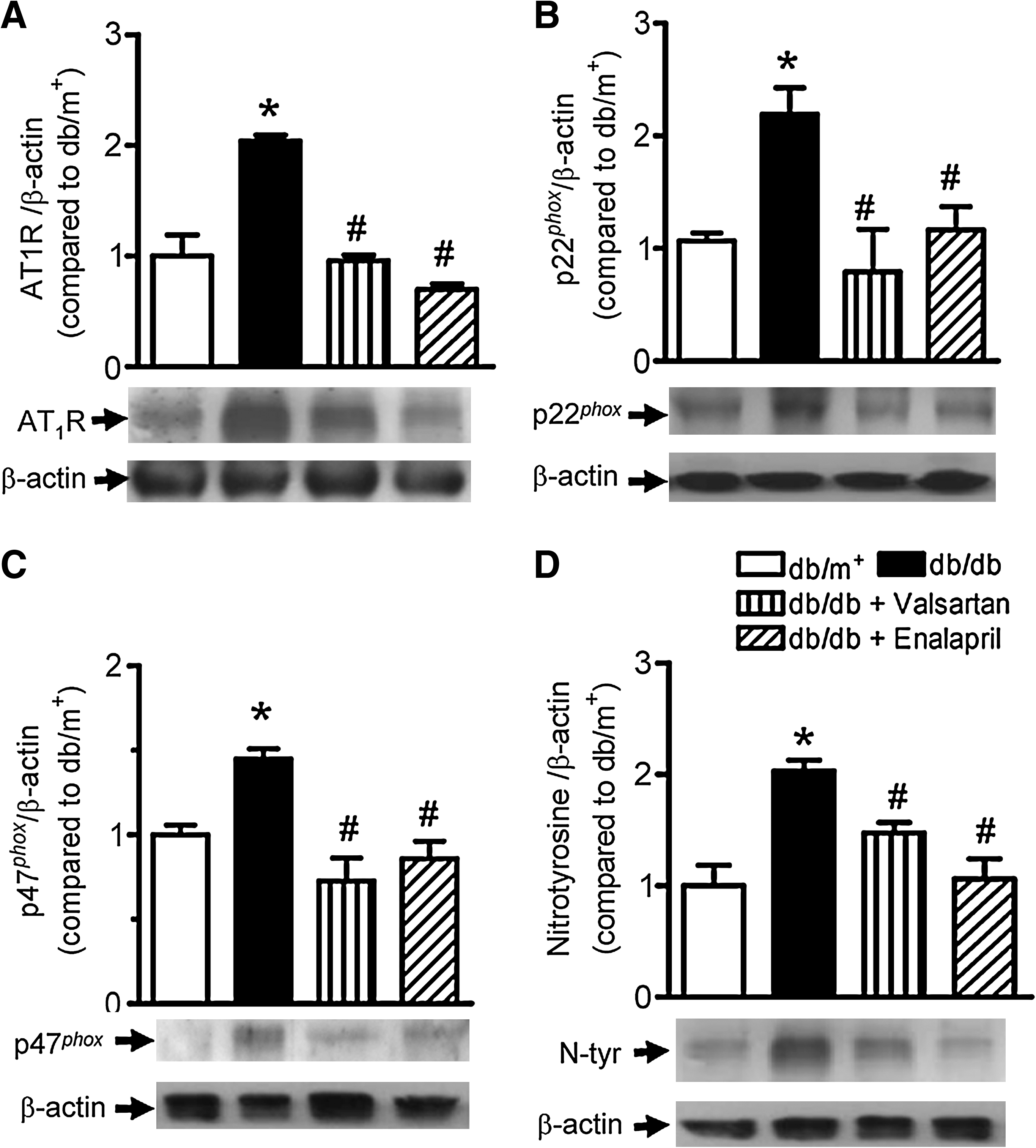
Western blot analysis of AT1R, AT2R, p22phox, p47phox, nitrotyrosine, eNOS, and p-eNOS
Immunoblotting showed that a significantly increased expression of AT1R in db/db mouse aortas was normalized by valsartan or enalapril treatment (Fig. 5A) while AT2R expression remained unaffected (Supplemental Fig. 3B; see
Impaired endothelium-dependent relaxations in renal arteries from diabetic patients rescued by AT1R blockade
Renal arteries obtained from diabetic patients relaxed significantly less in response to ACh than those from nondiabetic subjects (Figs. 6A and 6B). Acute exposure to losartan (3 μmol/L) for 30 min markedly enhanced the ACh-induced relaxations in diabetic human renal arteries (Fig. 6C) without affecting relaxations in nondiabetic human renal arteries (Fig. 6D). Renal arteries from diabetic patients have significantly higher AT1R expression as compared with those from nondiabetic control (Fig. 6E, Supplemental Fig. 5; see

High glucose-induced endothelial dysfunction mediated by AT1R
Chronic exposure (36 h) of nondiabetic mouse aortas to high glucose (30 mmol/L), but not to mannitol resulted in impaired ACh-induced dilatations (Fig. 7A), whilst SNP-induced endothelium-independent relaxations were unaffected (Fig. 7B). The presence of losartan (3 μmol/L) prevented the impairment of ACh-induced dilatations in high glucose-treated aortic rings (Fig. 7C). Likewise, losartan inhibited high glucose-stimulated increase in ROS production in the aortic wall (Fig. 7E). Losartan also restored ACh-induced dilatations which were impaired by 12-h incubation with Ang II (100 nmol/L) in nondiabetic mouse aortas (Fig. 7D).
Discussion
Our results clearly show a key role for AT1R-mediated ROS overproduction in the diminished NO bioavailability which accounts for the impairment of ACh-induced endothelium-dependent dilatations in db/db mouse aortas. Chronic administration of valsartan (ARB) or enalapril (ACE inhibitor) to 12-week old diabetic db/db mice prevents impaired endothelium-dependent dilatations, which correlates with marked downregulation of AT1R expression and reduction in ROS production. Further supporting evidence comes from our demonstration that acute exposure to inhibitors of RAAS-oxidative stress axis (losartan, apocynin, or tempol) improves endothelium-dependent dilatations in db/db mouse aortas and inhibits the ACh-stimulated ROS production. Importantly, losartan can also reverse the impaired endothelium-dependent relaxations in renal arteries from patients with diabetes. To further substantiate these findings, we also demonstrate that losartan is able to reverse the impaired dilatation that is induced by 36-h exposure of nondiabetic mouse aortas to high glucose (30 mmol/L); implicating that hyperglycaemia-induced increase in ROS generation requires AT1R activation. Taken together, the results of the present investigation support and further define the critical role of AT1R as the therapeutic target for alleviation of endothelial dysfunction and associated vascular events in diabetes.
The effect of RAAS blockade has been tested in various animal models of diabetes related vascular dysfunction. ACE inhibitors such as perindopril, zofenopril, and enalapril can prevent atherosclerosis progression in diabetic apoE-deficient mice (10, 25) by decreasing Ang II and increasing bradykinin. ACE inhibitors also restore vascular reactivity in type I diabetic mice (5). Likewise, ARBs such as candesartan, irbesartan, and valsartan also showed effectiveness in attenuating diabetes-associated atherosclerosis, retinopathy, and nephropathy through inhibiting advanced glycation, oxidative stress, and inflammatory cytokines (9, 10, 49). However, little information is available concerning the functional benefit of RAAS blockade in blood vessels of db/db mice. Previous clinical studies showed that AT1R blockade by losartan could improve endothelial dilator function in patients with type 1 and type 2 diabetes (12, 13). However, whether this protective effect is mediated through blood pressure-lowering effects or other specific mechanisms is not clear. Flammer et al. reported that losartan significantly improved endothelial function in type 2 diabetic patients with hypertension, which might be attributed to the antioxidative effect of ARB and was independent of its blood pressure-lowering action, as serum 8-isoprostane (a marker of oxidative stress) was significantly lower in losartan group, regardless of blood pressure changes (17). These results show the importance of antioxidative aspect of RAAS blockade that may contribute to the vasoprotection. While the correction of hypertension by ACE inhibitors or ARBs may partly explain the observed improvement of endothelial function in db/db mice, in the present study, we intend to investigate whether AT1R blockers could reverse the reduced vasodilatation in diabetic mice and diabetic patients through direct actions on the vascular wall.
The observation of impaired endothelium-dependent dilatations in db/db mouse aortas is consistent with recently reported results (29, 50). We conclude that AT1R mediates the impaired vasodilatation in diabetes based on the following observations. First, acute exposure of diabetic mouse aortas to ARB significantly enhances ACh-induced dilatations. Acute treatment with apocynin or tempol enhances the ACh-induced dilatations to a similar extent. In addition, a combined treatment with losartan and apocynin does not produce additive effects, implicating that Ang II signaling involves sequential steps, initial stimulation of AT1R followed by activation of NAD(P)H oxidases instead of independent actions. As apocynin was found to act as an antioxidant at concentrations higher than 300 μmol/L (1, 20), we used 100 μmol/L of apocynin in the present study. We have also demonstrated that the enhanced ROS generation in mouse aortas upon angiotensin II stimulation detected by DHE fluorescence dye was prevented by both the NADPH oxidase inhibitors while apocynin had no effect on hydrogen peroxide-stimulated ROS production (Supplemental Fig. 6; see
Chronic oral treatment with valsartan or enalapril markedly improves endothelium-dependent dilatations of db/db mouse aortas. It is postulated that ROS derived from AT1R-mediated NAD(P)H oxidases lowers the bioavailability of NO by either directly scavenging NO or by reducing the biosynthesis of NO catalyzed by endothelial nitric oxide synthase (eNOS). Our immunoblotting results clearly show that significant upregulations of AT1R and NAD(P)H oxidase subunits (p22phox and p47phox) in db/db mouse aortas can be normalized by chronic treatment with valsartan or enalapril, suggesting that RAAS blockade suppresses the stimulatory effect of Ang II on the expression and activity of NAD(P)H oxidases. NAD(P)H oxidase is the major source of ROS generation stimulated by Ang II, which is composed by membrane-bound gp91phox homolog (NOX1 in vascular smooth muscle cells and NOX2 in endothelial cells), catalytic subunit p22phox, and regulatory subunits such as p47phox, p40phox, p67phox, and Rac1 (3, 28). In addition, the activation of p38 and extracellular signal-regulated kinase (ERK) 1/2 mitogen-activated protein kinase (MAPK) in db/db mouse aortas was also inhibited by RAAS blockade (Supplemental Fig. 6). ROS stimulate the activation of MAPK pathways which further promote the expression of proinflammatory cytokines in endothelial cells (40), and ARBs can ameliorate diabetic glomerulopathy by suppressing MAPK activation (46). The inhibition of MAPK by RAAS blockade may also offer additional benefit in db/db mice. In contrast, RAAS blockade did not reverse the reduced phosphorylation of eNOS at Ser1177 in db/db mouse aortas (Supplemental Fig. 4), implicating that chronic RAAS blockade increased NO bioavailability by reducing oxidative stress rather than enhancing the NO production from eNOS (Supplemental Fig. 4). Although eNOS phosphorylation is known to decrease with prolonged oxidative stress (23), Ang II is reported to exert different effects, either increasing or decreasing eNOS phosphorylation (38, 39, 48). However, we observed that RAAS blockade does not affect eNOS phosphorylation. The present findings further support the primary role of RAAS-dependent oxidative stress in endothelial dysfunction in diabetic mice.
The overproduction of ROS in diabetic mouse aortas, as reflected by increases in nitrotyrosine formation and DHE fluorescence intensity, is reversed by RAAS blockade. Similar to previous findings of eNOS uncoupling in diabetes (22, 31), we also confirmed this by showing that ACh stimulates further increase of ROS only in diabetic but not in nondiabetic mouse aortas, which is blocked by L-NAME or endothelium removal. More relevantly, we demonstrate that blockade of RAAS and associated oxidative stress by losartan, apocynin, or tempol, greatly reduces the ROS production upon stimulation of ACh. These results indicate that ROS derived from NAD(P)H oxidases is likely required for stimulation of eNOS uncoupling to further increase intracellular ROS generation. In addition, we show that the release of ROS was dependent on the presence of extracellular Ca2+ ions which is in accordance with Guzik et al. who showed Ca2+ as an important intracellular activator of NAD(P)H oxidases (18).
More significantly, we demonstrate a critical role of AT1R-mediated ROS in impaired endothelium-dependent dilatations of human renal arteries. Renal arteries from diabetic patients have higher AT1R expression than nondiabetic control. Similar to db/db mouse aortas, the impaired dilatations in human arteries from diabetic patients can also be effectively rescued by acute treatment with losartan, thus favoring the use of AT1R blockers for reversing endothelial dysfunction in patients with diabetes. In summary, the present study has provided scientific basis with novel evidence in support of clinical application of selective AT1R blockers for the prevention and treatment of diabetes-related vascular dysfunction.
Footnotes
Acknowledgments
This study was supported by Hong Kong Research Grant Council (CUHK 4653/08M and HKU 2/07C), CUHK Focused Investment Scheme, and CUHK Li Ka Shing Institute of Health Sciences.
Author Disclosure Statement
The authors have no competing financial interests to disclose.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.