
Abstract
Signaling cascades initiated or regulated by calcium (Ca2+), reactive oxygen (ROS), and nitrogen (RNS) species are essential to diverse physiological and pathological processes in vascular smooth muscle. Stimuli-induced changes in intracellular Ca2+ regulate the activity of primary ROS and RNS, producing enzymes including NADPH oxidases (Nox) and nitric oxide synthases (NOS). At the same time, alteration in intracellular ROS and RNS production reciprocates through redox-based post-translational modifications altering Ca2+ signaling networks. These may include Ca2+ pumps such as sarcoplasmic endoplasmic reticulum Ca2+-ATPase (SERCA), voltage-gated channels, transient receptor potential canonical (TRPC), melastatin2 (TRPM2), and ankyrin1 (TRPA1) channels, store operated Ca2+ channels such as Orai1/stromal interaction molecule 1 (STIM1), and Ca2+ effectors such as Ca2+/calmodulin-dependent protein kinase II (CaMKII). In this review, we summarize and highlight current experimental evidence supporting the idea that cross-talk between Ca2+ and ROS/RNS may represent a well-integrated signaling network in vascular smooth muscle. Antioxid. Redox Signal. 12, 657–674.
Introduction
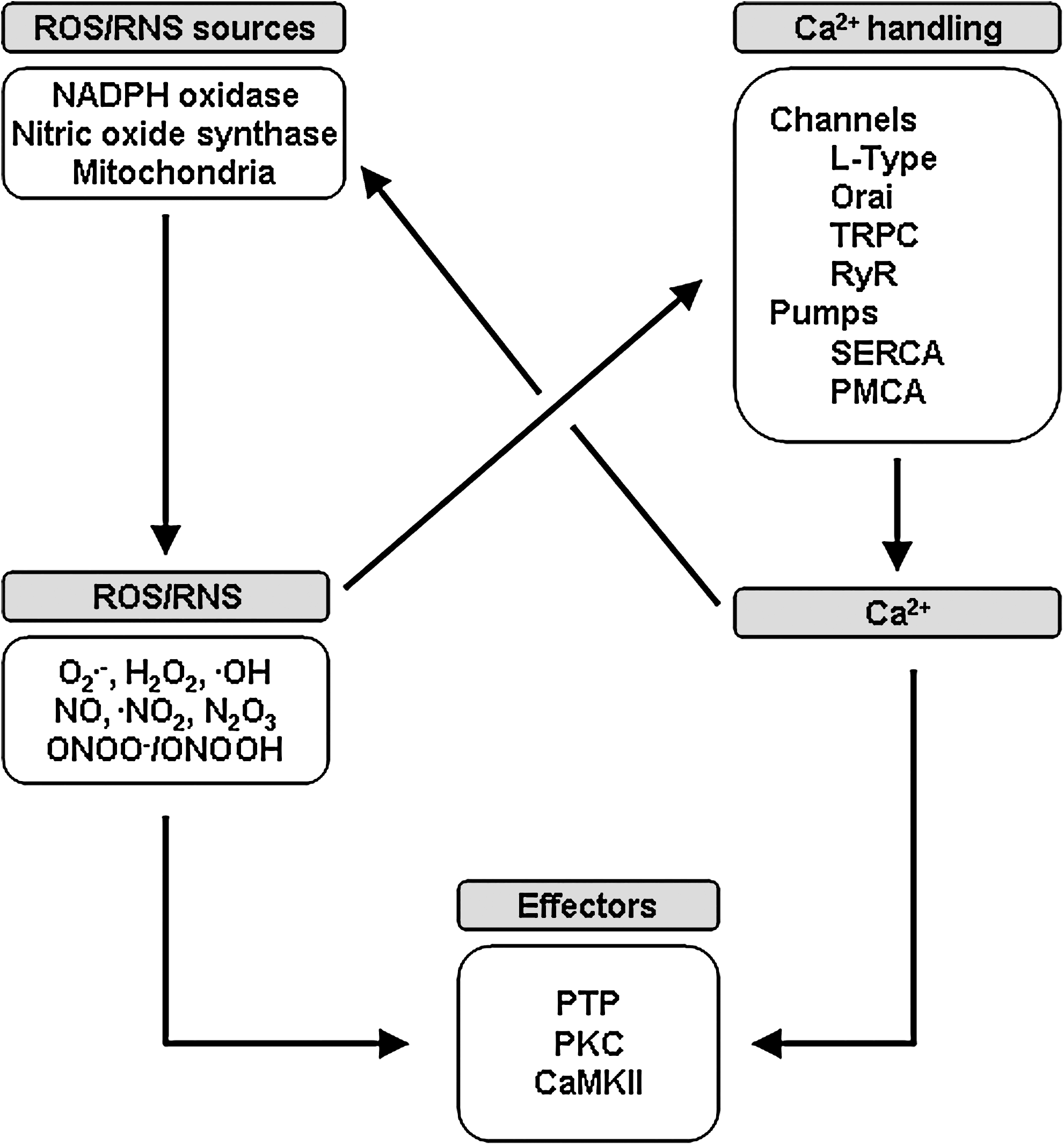
Reactive oxygen and nitrogen species (ROS and RNS) represent a large class of molecules regulating many aspects of VSMC biology, including contraction, proliferation, and migration. Primary sources of ROS and RNS in VSMC include the mitochondria, multiple isoforms of NADPH oxidases (NOX), and nitric oxide synthases (NOS), as well as storages pools of nitric oxide (23). Many of the enzymes involved in ROS and RNS synthesis are Ca2+-sensitive and changes in the amplitude and oscillatory patterns of intracellular Ca2+ signals in response to mechanical and chemical stimulations allow for rapid modulation of ROS/RNS production. Reciprocally, ROS/RNS regulate VSMC Ca2+ signaling through site-specific modifications of amino acid residues such as the oxidation, nitrosation, or nitration of cysteine and tyrosine residues. These alter the molecular components that directly regulate intracellular Ca2+ concentration (i.e., ion channels or transporters) or modify specific signaling molecules that are regulated by intracellular Ca2+. In this review, we discuss the evidence indicating crosstalk between Ca2+ and ROS/RNS signaling at multiple levels, lending credence to the paradigm that these represent well-integrated signaling systems in vascular smooth muscle (Fig. 1).
Calcium Signaling and Signaling Pathways in Vascular Smooth Muscles
Ca2+ and smooth muscle contractility
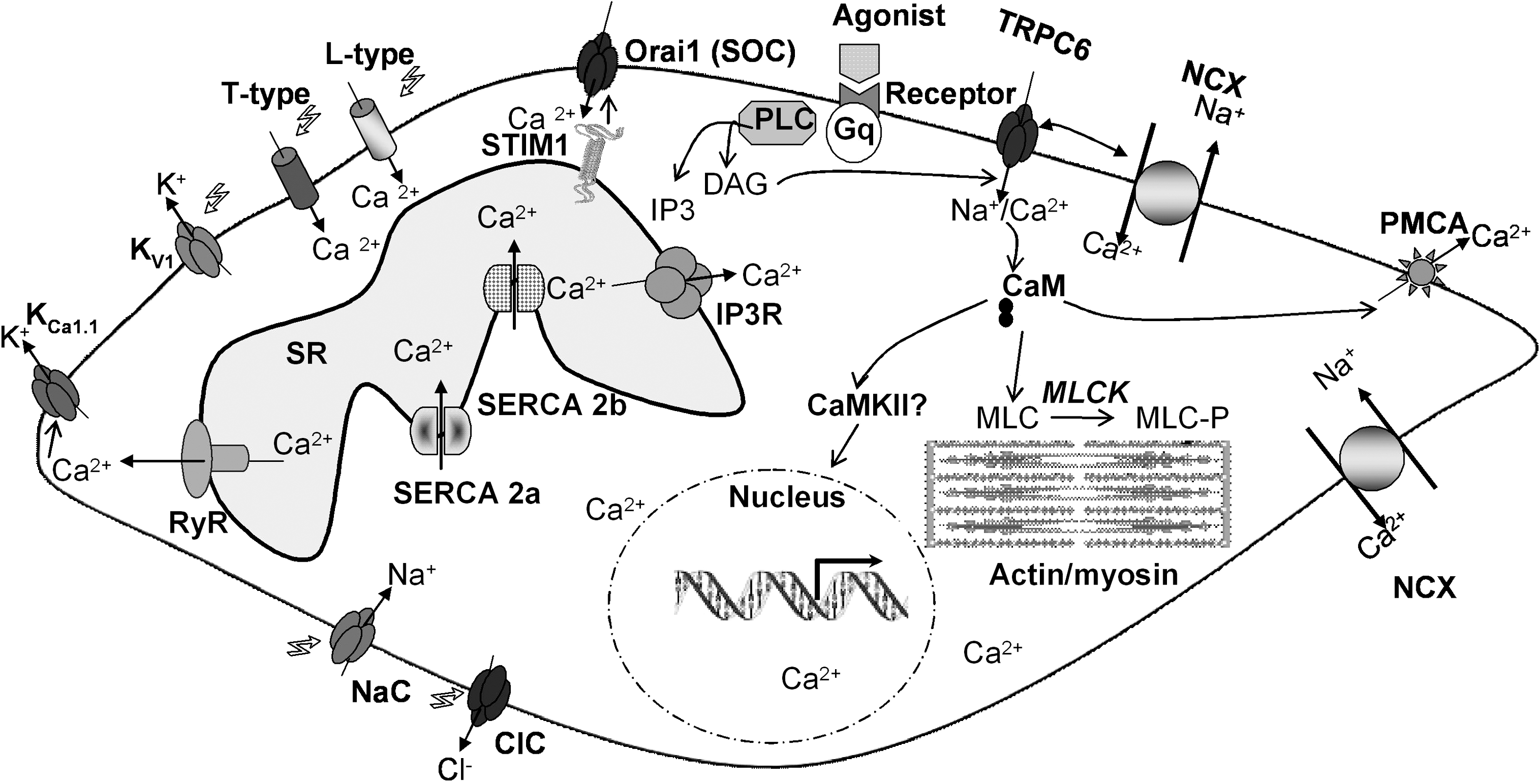
Unlike cardiac myocytes, VSMC are characterized by relatively slow contractions that are, in some instances, sustained over extended periods of time. This is presumably due to the ability of VSMC to utilize Ca2+ gradients at low global intracellular Ca2+ concentrations and thus maintain contraction. VSMC generate rhythmical contractions resulting from a complex interplay between ion channels, pumps, and transporters that increase global intracellular Ca2+ concentration in the form of a longitudinally traveling Ca2+ wave. The contractility of VSMC in vivo is under the control of a plethora of neuronal and humoral agonists that usually act through membrane receptors that couple to phosphoinositide-specific PLC to produce inositol-1,4,5-trisphosphate (IP3) and cause a series of Ca2+ transients due to Ca2+ release from the sarcoplasmic reticulum (SR) via the IP3 receptor (IP3R) (14). These agonist-induced Ca2+ transients or oscillations are necessary for the initiation of VSMC contraction. Since internal Ca2+ stores are finite, sustained increase in VSMC intracellular Ca2+ depends on Ca2+ entry through PM channels that are required to replenish the SR after each cycle of Ca2+ release. There is a wide variety of PM channels that have been implicated in this process; their relative importance and the extent of their contribution might depend on the VSMC type and the nature of the stimulus involved. Nevertheless, it is clearly established that the L-type high voltage-gated Ca2+ channels play an important role in increasing global Ca2+ levels during VSMC contraction (14). The PM depolarization necessary for activating L-type Ca2+ channels might be achieved through agonist-activated nonselective ion channels, such as isoforms of the transient receptor potential canonical channels (e.g., TRPC6) (157). Alternatively, discrete clusters of persistently active L-type Ca2+ channels operating in a high open probability mode, could contribute to steady-state Ca2+ entry into VSMC (14, 130). Another class of voltage-activated Ca2+ channels called T-type (named for the transient nature of their currents) has been suggested to contribute to global Ca2+ entry in airway smooth muscle (14). Ca2+ entry through PM channels activates, through the process of Ca2+-induced Ca2+ release (CICR), the ryanodine receptors (RyR) at the SR inducing further Ca2+ release. Further Ca2+ release might be contributed through the IP3R activated by agonist-induced IP3 production and by sensitization of the IP3R by Ca2+. These regenerative Ca2+ release cycles maintained by Ca2+ entry channels will cause global Ca2+ rise in the form of a traveling wave along the cell that leads to the initiation of the contractile response through activation of Ca2+/calmodulin-dependent myosin light chain kinase (MLCK). Ca2+ can activate Ca2+-dependent chloride (Cl−) channels, causing further depolarization and activation of L-type Ca2+ channels and subsequent Ca2+ release from the SR, ensuring the establishment of a positive feedback loop. Through gap junctions, depolarization can reach neighboring cells, activating their L-type Ca2+ channels and Ca2+ transients and helping synchronize the contractility wave along the vessel (14).
The reversal of VSMC contractility or vasorelaxation is achieved through mechanisms that limit Ca2+ entry through the PM in combination with cytoplasmic Ca2+ clearance. Discrete subplasmalemmal Ca2+ release through RyR, also called Ca2+ sparks, reduces Ca2+ entry through L-type Ca2+ channels by activating Ca2+-activated potassium (K+) channels and causing hyperpolarization (117). Ca2+ clearance from the cytoplasm depends on the action of sarcoplasmic-endoplasmic reticulum Ca2+-ATPase (SERCA) and PM Ca2+-ATPase (PMCA) that pump Ca2+ into the SR and outside the cell respectively. Figure 2 depicts the major ion channels and transporters controlling intracellular Ca2+ concentration in contractile VSMC.
Smooth muscle receptor-activated channels
Store-operated Ca2+ entry (SOCE)
VSMC functions are regulated by a wide variety of growth factors and vasoactive compounds that achieve their goal through binding to their specific receptors at the PM. These receptors typically couple to activation of PLC isoforms and production of second messengers IP3 and diacylglycerol (DAG) upon hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2; Fig. 2). The fall of the Ca2+ concentration within the lumen of the ER as a result of the action of IP3 on the IP3R activates a ubiquitous entry of Ca2+ across the PM originally recognized and termed capacitative Ca2+ entry (CCE) by Putney (138). The channels mediating this entry are called store-operated channels (SOC) (124, 138). The first and best characterized SOC channel is found in hematopoietic cells and conducts a highly Ca2+-selective, nonvoltage-gated, inwardly rectifying current first described in rat basophilic leukemia (RBL) mast cells and termed the Ca2+ release activated Ca2+ current (CRAC) (75). Major advances have been achieved recently regarding the molecular identity of SOC channels and the mechanisms linking internal Ca2+ store depletion to this Ca2+ entry in nonexcitable cells (140). An RNA silencing (siRNA)-based screen identified a Ca2+-binding protein, stromal interaction molecule 1 (STIM1) as the long sought Ca2+ sensor in the ER (98, 144) capable of oligomerization and reorganization into punctuate structures in defined ER–PM junctional areas upon store depletion to somehow signal the activation of the SOC channel at the PM (Fig. 3). Subsequently, genome-wide screens revealed that the membrane protein Orai1 is an essential component of SOC channels (45, 183) and that Orai1 is an essential pore forming unit of SOC channels. STIM1 and Orai1 alone can recapitulate most of the biophysical characteristics of CRAC currents. The mechanisms of STIM1/Orai1 coupling are beginning to emerge and appear to involve direct binding of a minimal, highly conserved ∼107-amino acid region of STIM1 to the N- and C-termini of Orai1 to open the CRAC channel (114, 125, 199). Nonetheless, it is clear that additional STIM1/Orai1 binding partners are involved (188). STIM1 is a type I transmembrane protein residing primarily in the ER, but can be found to a limited extent in the PM (∼10% of total proteins) (68). PM STIM1 is involved in the activation of the arachidonate-activated Ca2+ entry pathway (108). STIM1 has a closely related homologue, STIM2 (68), which is active at basal ER Ca2+ concentrations and can activate Ca2+ influx via Orai1 upon smaller decrease in ER Ca2+ content, compared to STIM1 (21). Orai1 has two homologs, Orai2 and Orai3, that exhibit distinct pharmacological, biophysical, and ion selectivity properties (32); the role of Orai2 and Orai3 in native Ca2+ entry pathways in different cell types including smooth muscle remains unknown. Nonetheless, Orai isoforms were shown to form heteromeric channels when ectopically expressed in HEK293 cells (99), and it is therefore conceivable that such heteromultimers might contribute to the diversity of Ca2+ entry pathways in different cell types by providing specific channels tailored to the cells' signaling needs.
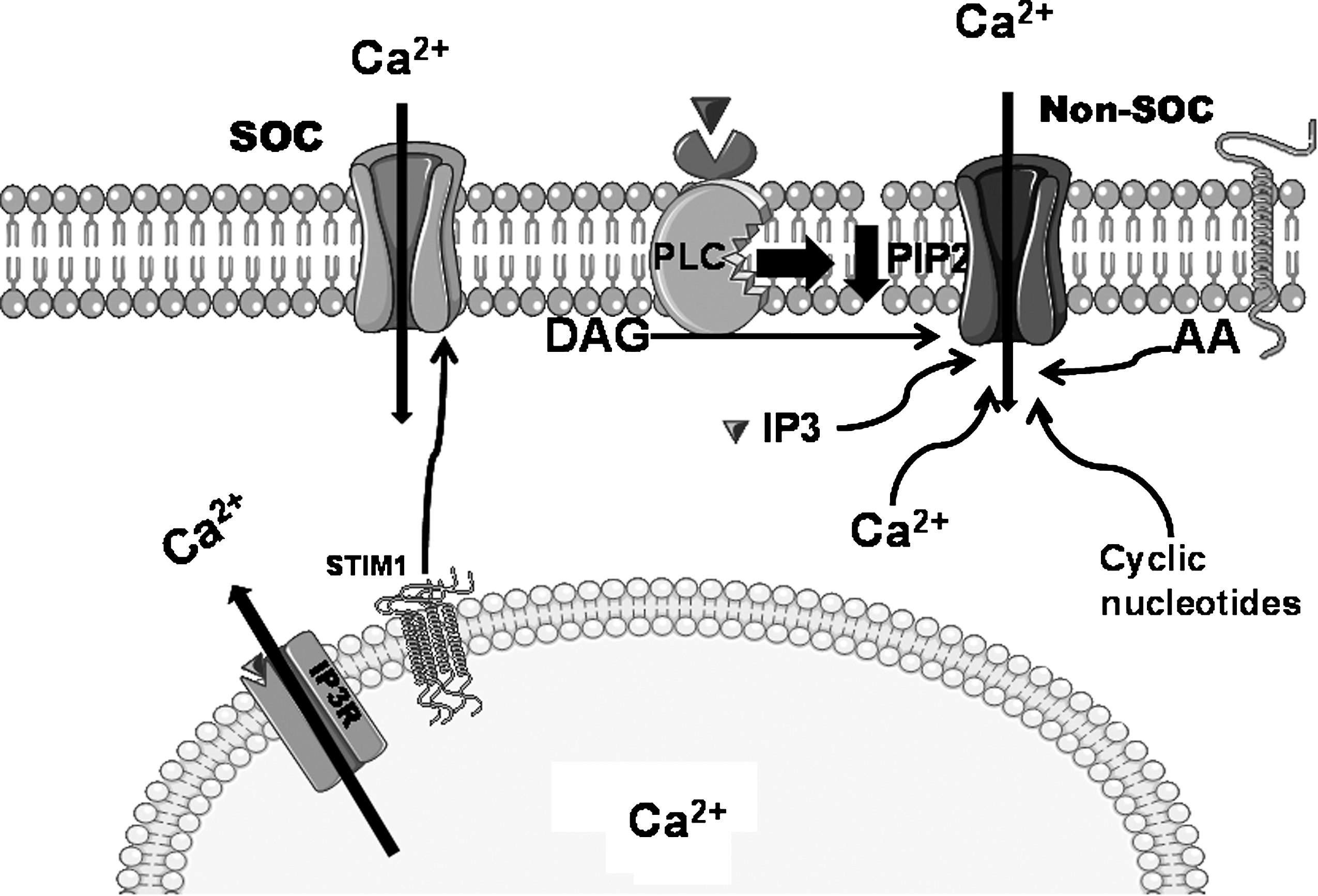
Store-independent Ca2+ entry pathways
A large number of pathways are known to contribute to the generation of Ca2+ signals in cells following receptor activation. The interplay between these different pathways generates a diverse and complex array of Ca2+ signals, which are required for VSMC function. The interplay between these Ca2+ entry pathways remains largely unknown. In addition to SOC, there are other modes of regulated Ca2+ entry across the PM. All of these routes of Ca2+ entry into cells that do not depend on the state of filling of internal Ca2+ stores are often referred to as noncapacitative or store-independent Ca2+ entry pathways (Fig. 3). Products of the PLC pathway (IP3 and DAG) are both involved in activating PM Ca2+ channels. In some instances, DAG in a PKC-independent manner activates PM Ca2+ channels (71, 95, 120, 175) and IP3 is also shown to act directly on IP3R located at the PM (168). Another lipid second messenger, arachidonic acid (AA), is known to activate PM Ca2+ channels (154). Other second messengers such as cyclic ADP-ribose (cADPr), cyclic GMP (cGMP), and Ca2+ itself were implicated in a variety of Ca2+ entry pathways (7). Shuttleworth and colleagues have described a role of PM-resident STIM1 in activating the AA-regulated Ca2+ (ARC) channels to which Orai1 and Orai3 contribute subunits (108, 109). However, the exact signaling mechanisms controlling other store-independent Ca2+ channels as well as the molecular identities of these channels are largely unknown (7).
TRPC channels
Canonical TRP (TRPC) channels constitute one of the major subfamily of the larger transient receptor potential (TRP) family of ion channels. To date, 28 mammalian TRPs have been identified that can be divided into six subfamilies: TRPC (Canonical), TRPM (Melastatin), TRPV (Vanilloid), TRPA (Ankyrin), TRPP (Polycystin), and TRPML (Mucolipin). Over the past decade, the seven TRPC members (TRPC1-7) have been the focus of intensive investigations in many cell types, including VSMC as potential candidates for SOC and non-SOC channels, by virtue of their activation by mechanisms downstream of the PLC pathway (156, 174). Patch clamp recordings in VSMC cell lines and primary VSMC cells from different species and different vascular beds have suggested that store depletion activates a nonselective SOC conductance, distinct from CRAC currents (22, 124, 178). In VSMC from rabbit aorta and portal vein, nonselective SOC currents have been described (178). These channels have an estimated conductance of 2–3 pS, were selective for cations but did not discriminate between monovalents and divalents. In particular, evidence from several laboratories suggested that TRPC1 is involved in SOCE in smooth muscle cell lines (22), and in VSMC from arteries and veins isolated from many species (124). A study in human pulmonary artery SMC showed that inhibition of TRPC1 expression using antisense oligonucleotides decreases SOC entry (164). However, the involvement of TRPC proteins in making SOC channels is a highly controversial topic at this time. Simultaneous knockdown using RNA interference of the three isoforms of TRPCs expressed in primary rat aortic VSMC (TRPC1/4/6) failed to affect the magnitude of SOCE (136). Dietrich et al. provided evidence that SOC currents in VSMC are intact in TRPC1 knockout mice (34) and internal Ca2+ store repletion was found to be normal in TRPC3 knockout mice and TRPC1/4/6 triple knockout mice (67). Furthermore, data from our group demonstrated the functional existence of CRAC currents encoded by STIM1/Orai1 in primary VSMC and the A7r5 cell line (136) as well as in endothelial cells (1), arguing that CRAC is a general mechanism for SOCE in all cells and casting doubt on TRPC channels as candidates for SOC channels. Two previous studies showed a role for STIM1 and Orai1 in thapsigargin-mediated SOCE in airway SMC (128, 129). However, the role of STIM2 and Orai2/3 proteins in VSMC Ca2+ signaling remains unknown.
A less controversial hypothesis is that of TRPCs generally encoding store-independent nonselective cation channels. Many groups have showed that TRPC3/6/7 subfamily members are activated by DAG when ectopically expressed in cell lines (176). Inoue et al. presented convincing evidence for the involvement of TRPC6 in the endogenous non-SOC DAG-activated cation entry controlled by α1-adrenergic receptors in rabbit portal vein smooth muscle (81). Data from Murayama et al. suggested that heteromultimeric TRPC6-TRPC7 channels contribute to vasopressin-activated, nonselective cation channels in A7r5 VSMC (103). Data from Van Breemen and colleagues described functional coupling of the Na+/Ca2+ exchanger (NCX) in its reverse-mode with receptor-activated store-independent TRPC6 in rat aortic smooth muscle cells following ATP stimulation. NCX reversal which contributes to Ca2+ entry was enhanced following stimulation with ATP and a DAG analog, consistent with the known properties of TRPC6 (132, 165). Earlier studies by Groschner and co-workers suggested both physical and functional coupling of TRPC3 to the cardiac-type Na+/Ca2+ exchanger (NCX1) in the control of Ca2+ homeostasis (145). Upon PLC-coupled receptor stimulation, Na+ entry through TRPC3 channels triggers Ca2+ entry by NCX operating in its reverse mode (39). Xi et al. described a store-independent mechanism of IP3/IP3R-dependent activation of TRPC3 channels in constriction of cerebral arteries (189).
Ca2+ channel modulation and vascular smooth muscle phenotypes
In response to various environmental stimuli, including growth factors, cytokines, mechanical influences, and various inflammatory mediators, the resident quiescent VSMC undergo transcriptional changes affecting both the downregulation of contractile proteins and concurrent upregulation of proteins supporting a proliferative phenotype (76, 185). This plasticity is believed to have evolved as a mechanism of vascular repair during injury and/or vascular adaptation to increasing demands by enabling VSMC to “switch” to a noncontractile, proliferative, and migratory phenotype. This phenotypic switch can, under certain circumstances, contribute to vascular disease such as atherosclerosis, hypertensive microvessel remodeling, vein graft failure, and restenosis following percutaneous intervention (26, 148). VSMC phenotypic change was shown to correlate with downregulation of voltage-activated L-type Ca2+ channels and increase in TRPC1 and TRPC6 mRNA and protein expression (12). Balloon dilatation of isolated human internal mammary artery showed a similar increase in TRPC1 and TRPC6 mRNA expression 48 h post injury (12). TRPC1 is upregulated following experimental vascular injury in mice and pigs, and is associated with the conversion of smooth muscle cells from a quiescent to a proliferative phenotype initiating neointimal formation (89). When TRPC1 function in saphenous vein organ culture was blocked with anti-TRPC1 antibody, neointimal growth in organ culture and VSMC proliferation were significantly inhibited (89). In pulmonary artery SMC, TRPC1 and TRPC6 expression increased following serum stimulation (53, 198). In patients with idiopathic pulmonary arterial hypertension (IPAH), a disease characterized by excessive pulmonary artery SMC proliferation, TRPC3 and TRPC6 were strongly upregulated (45) and siRNA against TRPC6 markedly attenuated pulmonary artery SMC proliferation in culture (197). Berra–Romani et al. showed that the protein levels of STIM1 and all Orai isoforms are dramatically increased in proliferative rat mesenteric VSMC, suggesting a role of these proteins in altered Ca2+ handling during vascular remodeling (13). Recent data from our group showed a similar increase of STIM1/Orai1 protein levels and SOCE in proliferative migratory rat aortic VSMC. Interestingly, knockdown of either STIM1 or Orai1 attenuated proliferation and migration of these VSMCs (136). More recently, two studies showed increased STIM1 expression in an in vivo model of vascular injury and inhibition of neointima formation upon in vivo knockdown of STIM1 (4, 60). Furthermore, Giachini et al. reported increased expression and activity of STIM1/Orai1 and SOCE in aorta from hypertensive rats (48). All these studies provide evidence that voltage-independent store-operated and store-independent Ca2+ channels contribute to vascular disease by controlling VSMC proliferation and migration during phenotypic modulation.
Calcium-Mediated Regulation of Primary Sources of ROS/RNS in Vascular Smooth Muscle
In VSMC, ROS and RNS are derived from multiple sources, which include several isoforms of NADPH oxidases and NO synthases. The biochemistry and physiological implications of Nox and NOS activity in VSMC have been extensively reviewed and we briefly summarize salient biochemical and functional features and highlight the Ca2+ requirements for several of these enzymes.
NADPH oxidases
Five NADPH oxidases (Nox) isoforms (Nox1–5) and two homologous oxidases (Duox1 and 2) all share the ability to generate O2 •− and/or H2O2 through reduction of molecular oxygen using NADPH as the electron source (91). Nox1, 2, 4, and 5 are found in VSMC and their expression varies between species, vascular beds, and normal and diseased vessels. Nox1 and Nox2 (gp91 phox) are probably the better characterized isoforms. Mainly, they require several cytosolic activators including Rac 1 and 2, p47phox, and p67phox (or Nox1 homologues NOXO1 and NOXOA1) that bind the membrane-associated subunits p22phox and Nox upon stimulation by growth factors and cytokines (92). A number of animal studies point to a role for Nox1 and 2 in cardiovascular diseases including hypertension (36, 104), atherosclerosis (8, 159), and restenosis (95, 172, 188), but their contribution to human diseases is unclear. Another isoform, Nox4, is abundantly expressed in VSMCs (29). Its activity is usually considered to be constitutive and only dependent on protein levels. In contrast to Nox1 and 2, little is known about the regulation of its activity other than interactions with p22phox (85, 102) and polymerase delta interacting protein 2 (Poldip2), (101). Functionally, Nox4 has been implicated in VSMC differentiation (29), polyploidy (105), and response to TGFβ (162), 7-ketocholesterol (127), and TLR-4 signaling (126).
An interesting issue derived from the study of Nox4 is the realization that not all Noxs may in fact produce O2 •−. Nox activity is classically believed to be derived from electron transfer from cytosolic NADPH to FAD, the two nonidentical hemes, and finally to molecular oxygen to form O2 •−, H2O2 being derived from O2 •− dismutation (44). However, recent results obtained from complementary approaches to detect O2 •− and H2O2 indicate that the major detectable product of Nox4 is H2O2 and not O2 •− (151). Although the underlying mechanism for the distinctive activity of Nox4 among the other Noxs is unknown, it has been rationalized that the interaction of O2 •− at the active site of Nox4 may be stable enough to allow for direct sequential electron transfer to generate H2O2 (35).
The subcellular localization of Noxs is essential to rationalize the integration of ROS production to cell signaling, and some of the most detailed studies dealing with this issue have been performed on cytokine-stimulated VSMC. Both TNF-α and IL-1β induce the translocation of Nox1 and 2 into endosomal structures to produce O2 •− into the lumen of the endosome (94, 110). The anion transporter ClC-3 is required to allow charge neutralization of the electron flow produced by Nox1 (110) and superoxide dismutase 1 is recruited to the endosome to facilitate H2O2 gradients (66). This results in the spatially segregated production of ROS to control the recruitment of tumor necrosis receptor associated factor (TRAF) molecules to their cognate receptor, leading to downstream activation of the transcription factor NFκB (94). Nox1 also colocalizes with caveolae while Nox4 is associated with focal adhesions in VSMC (70) and in the endoplasmic reticulum in endothelial cells (27).
Among the 5 Noxs, Nox5 is the only isoform that is directly Ca2+ sensitive. It is not expressed in rodents but has been found in human VSMC and its expression is increased in VSMC in human atherosclerotic coronary arteries (61). There is little known regarding the functional implications of Nox5. In ovarian VSMC of Drosophila melanogaster, Nox5 participates in Ca2+ signal transduction linked to muscle contraction and ovulation (142). Nox5 also positively regulates PDGF-induced proliferation in human aortic smooth muscle cells through stimulation of the JAK/STAT pathway (83). Unlike the other Noxs, Nox5 does not require p22phox or the cytosolic subunits to function but instead contains four canonical EF-hands on its N-terminal end that provide direct sensitivity to Ca2+ (6). A binding site for calmodulin is located in the NADPH-binding domain (169), and PKC-mediated phosphorylation of Ser/Thr residues in the FAD-binding domain may confer increase Ca2+ sensitivity to Nox5 (82). Similarly, Duox1 and 2 contain EF-hands that may provide regulation by Ca2+ but their expression and function in VSMC have not been characterized (91).
In addition to its direct interaction with catalytic subunits, Ca2+ may also control Nox activity by regulating cytosolic components of Nox1 and 2. For example, in VSMC from human small arteries, serine phosphorylation of p47phox is required for ROS production by angiotensin II (173). One of the kinases responsible for p47phox phosphorylation is protein kinase C, although the specific isozyme responsible has not been identified. Similarly, in rat aortic VSMC, angiotensin II stimulation of Nox1 is partly regulated by a PKC (152). These observations—although incomplete—suggest that conventional PKC isozymes (α, βI, βII, and γ) which require Ca2+ for activation might participate in Nox activation in VSMC.
Nitric oxide synthase
Mammalian cells synthesize NO through the five-electron oxidation of one of the two-guanidino nitrogen of L-arginine. Three classes of nitric oxide synthases (NOS) have been described: neuronal-NOS (nNOS, type I), inducible-NOS (iNOS, type II), and endothelial-NOS (eNOS, type III) (112). All three isoforms are expressed in the vasculature and all require oxygen, NADPH, 5,6,7,8-tetrahydrobiopterin, and Ca+2/calmodulin as cofactors. In contrast to eNOS and nNOS, the Ca2+-independence of iNOS activity is due to calmodulin tightly bound to the enzyme (146).
All three NOS may generate other reaction products than NO itself, including nitroxyl (NO−) (150), O2 •−, and peroxynitrite (ONOO−/ONOOH) (181). The latter two products may be formed when electron transfer within the active site of NOS becomes independent of L-arginine oxidation. Decreased bioavailability of L-arginine and H4B contributes to this uncoupling of NOS activity, which has been implicated in a variety of vascular disorders such as hypertension and atherosclerosis (111).
In the healthy vessel, the endothelium serves as the main source of NO through eNOS activity to maintain vascular tone, and regulate platelet aggregation and leukocyte adhesion (87, 112, 139). The activation of soluble guanylate cyclase by diffusible NO in VSMC to produce the second messenger cGMP is the primary transduction pathway responsible for eNOS-dependent vasorelaxation. Cyclic GMP acts directly or indirectly—via activation of cGMP-dependent protein kinase (PKG)—on many effector proteins to lower intracellular Ca2+ levels (97). Effector proteins include the type 1 IP3R, and phospholamban in the SR, the smooth muscle Ca2+-activated K+ channels, and L-type Ca2+ channels. Another potential mechanism is through the PKG-dependent phosphorylation of threonine and serine residues on TRPC proteins including TRPC3 and possibly TRPC6 and 7 (90, 195). As a consequence, the NO-cGMP pathway reduces the activity of these channels providing an additional feedback mechanism to regulate intracellular Ca2+ (195).
Neuronal NOS is expressed in VSMC upon stimulation by growth factors (115), statins (116), and shear stress (122). The upregulation of nNOS contributes to the compensatory mechanism associated with adaptive chronic hypoxia (186) and nNOS VSMC is vasculoprotective (113) and atheroprotective (88). In a fashion similar to eNOS in the endothelium, the generation of NO by VSMC nNOS is coupled to Ca2+ because nNOS requires Ca2+/calmodulin for maximum activity (112). Based on overexpression studies, the PDZ domain of nNOS has been found to interact with the C-terminal end of the human plasma membrane Ca2+ ATPase 4 (hPMCA4), resulting in a decrease in NO production due to a local decrease in Ca2+ concentration (55). The significance of these findings to VSMC function is unclear but studies by Gros et al. indicate that PMCA-dependent regulation of nNOS activity may regulate smooth muscle vascular reactivity (55).
Modalidities of Signaling Through ROS/RNS
Proteins may be modified by ROS and RNS through a plethora of reactions. Examples of resultant modifications include the carbonylation and deamination of amino acid side chains, the oxidation of methione residues to sulfoxides, the nitration of tyrosine residues, and the oxidation and nitrosation of cysteine residues. Functionally, these result in increase susceptibility of proteins to degradation, increase protein fragmentation, or alteration in protein function.
Reactive oxygen species (ROS)
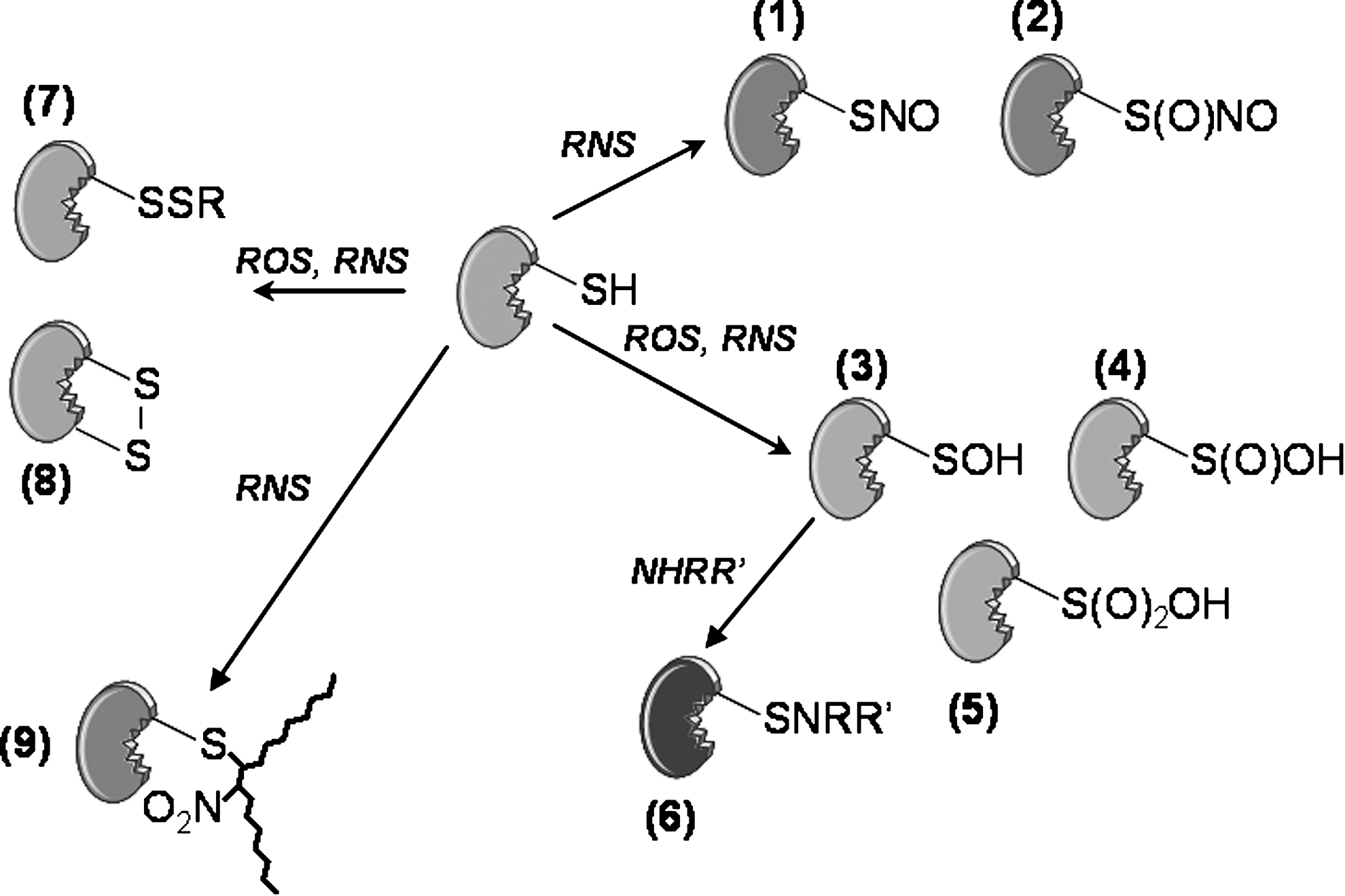
For Nox-derived ROS, it is generally assumed that H2O2 regulates signaling pathways through reactions with amino acid residues in proteins, principally cysteine. This residue is one of the most reactive and carries out disulfide formation, metal-binding, electron donation, hydrolysis, and redox catalysis (49). The oxidative modifications include disulfide bridges, or the formation of oxidation products of increasing oxidation states, including sufenic (Cys-SOH), sulfinic (Cys-SO2H), and sulfonic (Cys-SO3H) acid (Fig. 4). The protein tyrosine phosphatase (PTP) family contains a canonical CXXXXXR sequence, in which the conserved cysteine residue acts as a nucleophile to form a thiol phosphate intermediate during catalysis (33). The local environment of the active site contributes to the low pKa (4.5–5.5) of the cysteine such that the thiolate anion (RS−) is the dominant form. This increases the susceptibility of cysteine to reversible oxidation by H2O2 to sulfenic acid with the consequential reversible inhibition of phosphatase activity (106). As such, PTP by H2O2 is one of the most common mechanisms for the effect of H2O2 on signaling networks (141). Oxidation of the thiolate to higher oxidation state products may also occur, and in the case of the H2O2 scavenging enzyme peroxiredoxin, this may provides a mean to regulate intracellular H2O2 concentrations (188).
Rather than a direct reaction of H2O2 with active sites of proteins containing cysteine residues, it is possible that specificity might depend on intermediate protein transducers such as thiol peroxidases. In this case, a specific thiol peroxidase would reversibly oxidize cysteine residues in binding protein partners and would serve to signal and propagate local changes in ROS gradients (31). For example, peroxiredoxin 2 (PRX2) associates with the PDGF receptor in human and mouse VSMCs to regulate the phosphorylation of specific tyrosine residues on the receptor in response to an endogenous source of H2O2 (28).
Reactive nitrogen species (RNS)
Reactive nitrogen species (RNS) represent a variety of chemical intermediates among which the prototypical molecule is NO. In general, RNS may be formed through secondary reactions of NO with ROS or as products of enzymatic activities such as peroxidases and the three NOS isozymes (54). The detection of nitrated tyrosine residues has been used in many studies as evidence for RNS formation because the reaction requires peroxynitrite derived from the diffusion-limited reaction of NO with O2 •− (10) or the oxidation of nitrite (NO2 −) to nitrogen dioxide (•NO2 •) by peroxidases (40). Increase immunostaining for nitrotyrosine has been determined in many cardiovascular conditions, including atherosclerosis and restenosis, and in some cases has been localized to smooth muscle cells (11, 123). The daunting task for many of these studies is to understand the functional significance of such modifications under conditions where multiple oxidative, nitrative, and nitrosative hits are likely to occur on the same pool of macromolecules. This is further complicated by the steady increase in the number of modifications that are being characterized, which now include also nitroalkylated and N-nitrosated proteins (Fig. 4) (9, 23).
The S-nitrosation of cysteine residues in proteins is a post-translational modification with important implications regarding cell signaling (160). The term nitrosation is often used interchangeably with “nitrosylation” in reference to other post-translational modifications such as phosphorylation. In many cases, a role for cysteine nitrosation has been demonstrated by mutation of the relevant cysteine residue and evidence of coincidental decrease in nitrosation and protein function or activity. However, absolute quantitation of nitrosated protein levels relative to activity is usually difficult to assess. Most of all, the direct mutation of a cysteine residue does not dissociate the effect of nitrosation from other alternate roles of the residue (electron donation, hydrolysis, or others). To this end, decoupling of the effect of nitrosation may be achieved through mutational modifications of the immediate environment of the cysteine residue rather than the nitrosated cysteine itself (17).
Nitric oxide alone reacts only very slowly with cysteine residues and proposed mechanisms of protein nitrosation include the transnitrosation of cysteine with low molecular weight S-nitrosothiols, the reaction of thiols with dinitrosyl iron or iron–nitrosyl complexes, direct reaction of thiols with NO in the presence of an electron acceptor, copper-catalyzed nitrosation, reaction of thiols with either peroxynitrite, or autoxidation products of NO (59). The reaction of NO with thiols in oxygenated environments does not lead exclusively to the formation of S-nitrosothiols but a large fraction of products may rather represent oxidized forms of cysteine including glutathionylated residues and disulfide bridges (84, 147). Alternatively, RNS and ROS-modified cysteine residues may only serve as transient intermediates as in the case of the regulation of small GTPase proteins including p21(Ras) (140). Finally, the importance of molecular oxygen in promoting nitrosation has been recently challenged by new studies showing that intracellular S-nitrosation is independent of oxygen (19) and that oxygen in fact promotes inactivation of NO through dioxygenation to nitrate (NO3 −) (62).
Ca2+ Signaling Through ROS/RNS
Although little is known concerning the role of ROS/RNS in VSMC Ca2+ signaling, an emerging paradigm for the action of ROS/RNS on Ca2+ signaling from a variety of cell types seems to involve the inhibition of Ca2+ pumps and activation of Ca2+ release and entry channels thus increasing the overall cytoplasmic Ca2+ concentration (69, 172).
SERCA and PMCA pumps
Through ATP hydrolysis, SERCA and PMCA actively clear Ca2+ from the cytoplasm and thus play a key role by maintaining cytoplasmic Ca2+ homeostasis. Quiescent VSMC express two splice variants of the SERCA2 gene, 2a and 2b and multiple isoforms of PMCA1 and 4 (3, 76, 170). The role of ROS/RNS in modulating SERCA activity remains controversial. SERCA inhibition in vitro through modification of sulfhydryl groups either by ROS or RNS has been reported in several cell types (69). Treatment of coronary arteries denuded of endothelium with peroxynitrite leads to inhibited contractile responses to angiotensin II or SERCA pump inhibitors such as thapsigargin (57, 58). Treatment of SR vesicles with NO donors and peroxynitrite induces S-nitrosylation of cysteines on SERCA (69). However, earlier reports suggested that NO-mediated SERCA activation in VSMC increases Ca2+ uptake into the SR, thus inhibiting store-operated Ca2+ entry and causing vasorelaxation (30, 177). An explanation for this apparent discrepancy might be that small concentrations of ROS/RNS are stimulatory while elevated concentrations cause inhibition. Similarly, the PMCA pump has been reported to be reversibly inhibited by H2O2, O2 •−, •OH, and ONOO−/ONOOH (69).
Ca2+ release channels: IP3R and RyR
Several studies reported stimulation of IP3R-mediated Ca2+ release from internal stores in response to ROS (69). The modification of cysteine residues in the IP3R by the thiol reagent, thimerosal, causes Ca2+ spikes through increase IP3R sensitivity to resting levels of IP3 (18). Subsequent reports showed that physiological levels of cellular ROS produced through membrane-associated NOXs could increase the sensitivity of the IP3R to IP3 (79). Another major Ca2+ release channel, RyR, has been shown to be modified by compounds affecting sulfhydryl groups with consequences on channel function (38, 69, 190). RyR isoforms can be activated by S-nitrosylation (163, 192). Exogenous addition of redox compounds as well as endogenous production of ROS/RNS modify RyR on cysteines residues and cause their activation. Experimental evidence in skeletal muscle suggested that Cys3635 is involved in RyR1 redox sensing; the modification of this residue cause RyR1 activation and protection from calmodulin binding-mediated inhibition at high Ca2+ concentrations (133, 200). Furthermore, S-nitrosylation of Cys3635-containing the calmodulin binding domain is responsible for RyR1 activation by NO at physiologically relevant oxygen tensions (163). Other studies suggested that endogenous ROS produced via NOX enzymes activates Ca2+ release through RyR (42).
Ca2+ entry channels
While several reports have shown that oxidation increases the activity of voltage-gated Ca2+ channels, others reported inhibition of channel activity by oxidizing agents (69). This might be explained by the concentrations of ROS/RNS involved. Alternatively, different ROS/RNS might have different effects on ion transport mechanisms or different cell types might display different sensitivity to various modifiers. In addition to their effects on Ca2+ release channels, ROS such as H2O2 and O2 •− were shown to stimulate Ca2+ entry through L-type and T-type voltage-gated channels in VSMC (167). Other groups reported increased L-type channel activity by ROS (80), and a role of H2O2-activated L-type Ca2+ channels in mitochondria-derived superoxide production (184). In smooth muscle and cardiac myocytes, NO seems to have an inhibitory effect on L-type Ca2+ channels through S-nitrosylation (25, 135). Hool reported that hypoxia in cardiac myocytes can inhibit the basal L-type Ca2+ current and increases the sensitivity of this current to beta-adrenergic receptor stimulation (72). The effects of hypoxia could be mimicked by decreasing cellular H2O2 with catalase, arguing for a role for ROS in the sensitization of L-type Ca2+ channels (73). However, Hudasek et al. found a role of endogenous H2O2 production in activating L-type Ca2+ channels, but failed to detect an effect of either hydrogen peroxide or NOX in the hypoxia-dependent regulation of the these channels (80).
While pharmacological compounds revealed that H2O2 induced Ca2+ mobilization in pulmonary artery smooth muscle through multiple pathways involving release from the internal stores and entry from the outside (96), information regarding the involvement of ROS/RNS in modulating voltage-independent Ca2+ channels such as store-operated and receptor-operated Ca2+ channels in different cell types in general and VSMC in particular remains scarce. Studies on TRPC3 and TRPC4 cation channels expressed in HEK293 cells showed they formed redox-sensitive ion channels that could be activated by exogenous application of hydrogen peroxide (5, 56). Whether TRPC channels are directly modified by ROS remains unknown but redox activation of TRPC3 was proposed to occur through ROS-induced tyrosine phosphorylation and activation of PLC (56). More recently, the same group presented evidence for existence of redox-sensitive TRPC3/TRPC4 heteromeric channels in endothelial cells (134). In this study, TRPC3/4 heteromeric channels were activated by cholesterol oxidase, suggesting that ROS activation of TRPC3/4 channels might be mediated by cholesterol oxidation. NO-mediated cysteine nitrosylation have been shown to cause activation of several members of the wider TRP family, TRPC1, TRPC4, TRPC5, TRPV1, TRPV3, and TRPV4 (196). A cytoplasmic Cys553 along with Cys558 in TRPC5 are nitrosylated and native TRPC5 is nitrosylated in response to G-protein coupled receptor stimulation, thus eliciting Ca2+ influx in endothelial cells (196).
Hydrogen peroxide was shown to evoke Ca2+ entry through activation of the widely expressed melastatin-related TRP channel, TRPM2 (65). Yamamoto et al. showed that TRPM2 activation controls ROS-induced chemokine production in monocytes. Hydrogen peroxide-activated Ca2+ influx through TRPM2 was essential for downstream signaling and production of interleukin-8. Monocytes from TRPM2-deficient mice showed impaired hydrogen peroxide-activated Ca2+ influx and production of the mouse functional homolog of interleukin-8 (193). Based on studies on a splice variant of TRPM2 with a deletion in its C-terminus, which is sensitive to hydrogen peroxide but not to ADP-ribose, Wehage et al. proposed that oxidative stress-induced gating of TRPM2 is distinct from that of ADP-ribose (187). However, Perraud et al. suggested that oxidative and nitrosative stress-induced TRPM2 activation occurs through stimulation of ADP-ribose production by mitochondria. ADP-ribose interaction within a binding cleft in the C-terminal NUDT9-H domain of TRPM2 would activate the channel (131). Another TRP channel, TRPA1, can be activated by thiol reactive compounds of diverse structure via reversible covalent modification of cysteine residues within the cytoplasmic N terminus of the channel (179). TRPA1 channels were shown to be strongly activated by hypochlorite and H2O2 in primary sensory neurons of the airway, suggesting that TRAP1 acts as an oxidant sensor in the airway (15). A recent report demonstrated a role for TRPA1 in mediating airway inflammation and hyperreactivity in asthma (24). Other ion transport mechanisms not discussed here are also targets of ROS/RNS, such Na+/Ca2+ and Na+/H+ exchangers, Na+, Cl− and K+ channels, ion co-transporters and other pumps such as the Na+, K+ ATPase (69, 90). Modification of the newly discovered components of the SOC pathway (STIM and Orai) by ROS/RNS has not been reported. Nevertheless, STIM1 proteins possess several cysteines residues that could be targets for modification. Similarly, all three Orai isoforms possess predicted extracellular and intracellular cysteines.
ROS and RNS Signaling Through Regulation of Ca+2-Dependent Effectors: The Example of Ca2+/Calmodulin-Dependent Protein Kinase II
As described in the previous sections, the reciprocal regulation of intracellular Ca2+ and ROS/RNS concentrations is an essential integration point of Ca2+ and ROS/RNS signaling. It is also evident that many Ca2+-dependent effectors are themselves potential effector molecules for ROS and RNS. This has been already discussed in the context of phosphatases and we argued earlier for an important role for protein kinase C in redox-dependent cytokine signaling in VSMC. Other examples in the context of smooth muscle include ion channels and transporters, transcriptional regulators, and kinases. In this regard, Ca2+/calmodulin-dependent protein kinase II (CaMKII) is of particular interest because of its known roles in mediating VSMC signaling pathways and its ability to mediate transcriptional events. Recent evidence would indicate that it is also a direct effector of ROS and RNS. Below, we summarize CaMKII signaling in vascular smooth muscle and detail its regulation by ROS and RNS as a prototypical example of Ca2+-dependent kinase, which activity is also directly impacted by ROS and RNS.
Four CaMKII isoforms (α, β, γ, and δ) have been identified with each isoform having multiple splice variants (171). These isoforms are capable of forming homo-or heteromultimers (155). Recent structural information suggests that CaMKII exists intracellularly as a dodecamer containing two rings of six CaMKII subunits CaMKIIγ and CaMKIIδ are the predominant CaMKII isoforms expressed in VSMC. Along with the dynamic changes in Ca2+ handling that occur as VSMC undergo phenotypic change, the content of CaMKII in VSMC also changes. CaMKIIγ is most abundantly expressed in differentiated, contractile VSMC with at least six splice variants having been identified (47, 155). In the synthetic VSMC, which are proliferative and migratory, CaMKIIδ2 is the major CaMKII isoform expressed. CaMKIIδ3, a splice variant of CaMKIIδ2 that localizes to the nucleus has also been identified in synthetic VSMC (149) albeit at much lower expression levels than CaMKIIδ2. Recently, we reported that the dramatic shift in expression from CaMKIIγ in differentiated VSM to CaMKIIδ in synthetic VSM occurs not only in vitro when VSMC are removed from intact blood vessels and cultured but also in response to vascular injury (77). VSMC taken from the neointima of injured carotid arteries were shown to primarily express CaMKIIδ and suppression of CaMKIIδ with shRNA prevented neointimal formation (77). In synthetic VSMC, CaMKIIδ2 is an important mediator of cell proliferation (75) and migration (107), suggesting that the shift in expression from CaMKIIγ to CaMKIIδ is an important component of a blood vessel's response to injury. Additional functions of CaMKIIδ in VSMC include regulation of Ca2+-dependent tyrosine kinase activity (52) and G protein coupled receptor (GPCR) - and adhesion-dependent activation of ERK (2, 51, 100).
The activation of CaMKII is multifaceted involving several steps (Fig. 5). Upon association with Ca2+/CaM, CaMKII's autoinhibition is relieved, allowing full catalytic activation. This is followed rapidly by intersubunit, intraholoenzyme auto-phosphorylation of Threonine 286/287 which markedly increases CaM binding affinity (63) and also enables the retention of kinase activity in the absence of high intracellular Ca2+(64). The multimeric structure of CaMKII allows for inter- and intrasubunit phosphorylations which further contributes to the means that CaMKII can be regulated (64). Serine/threonine phosphatases, most notably PP2A and PP1 (20, 161), are also important components in the regulation of CaMKII activity. Both PP2A and PP1 have the capability to dephosphorylate CaMKII and suppress its activity (161). There is increasing evidence that these phosphatases physically interact with CaMKII and mediate both basal and stimulated CaMKII activity (161). The specific phosphatase that mediates CaMKII may be CaMKII isozyme and cell type dependent.

While it is well-established that regulation of CaMKII activity is dependent upon intracellular Ca2+, there is some evidence that CaMKII activity is also mediated by ROS such as H2O2 (118) and NO (158) (Fig. 5). Recent evidence strongly suggests that increases of CaMKII activity can be measured in endothelial cells (118) and cardiomyocytes (191) in response to H2O2 and result in CAMKII-dependent activation of p38MAPK, cytoskeletal rearrangements, and abnormal heart function. The mechanisms by which CaMKII is activated in response to increases in ROS such as H2O2 have not been completely elucidated. Nor is it clear if ROS other than H2O2 are capable of modulating CaMKII. More detailed studies have revealed that CaMKII can be modulated in a direct manner by ROS or indirectly through ROS-dependent regulation of CaM (16) or phosphatase activity (194).
CaMKIIδ contains two methionine residues in its regulatory domain. A study performed in cardiomyocytes demonstrated that these methionine residues are oxidized in response to angiotensin II, and this oxidation enables CaMKIIδ to retain kinase activity in the absence of Ca2+/CaM (41). Interestingly, this study also showed that oxidation of cysteine residues on CaMKII has no impact on CaMKIIδ activity. In rat pituitary tumour GH3 cells, nNOS-dependent increases in NO resulted in the S-nitrosylation of cysteine residues on CaMKIIα suppressing CaMKIIα's activity and function (158). A study done with mouse synaptosomes revealed that CaMKII is oxidized under ischemic conditions and that this oxidation attenuated CaMKII activity by inducing disulfide- and nondisulfide-dependent aggregations (153). Taken together, these reports indicate that CaMKII is susceptible to direct oxidation that has both positive and negative consequences. These studies also indicate that whether methionine or cysteine residues are oxidized and/or which CaMKII isozyme is being affected is important in determining the overall effects of ROS-dependent modification of CaMKII. Because of the paucity of studies examining the consequences of direct oxidation of CaMKII, it is premature to make any generalizations as to its physiological relevance.
CaMKII can also be modulated in an indirect manner in response to increasing ROS concentrations. One interesting way is through the oxidation of CaM (46), a mechanism that is potentially important for ion channels and pumps that are regulated by CaM such as voltage-gated Ca2+ channels and TRP channels. The oxidation of methionine residues reduces CaM's ability to bind Ca2+ and to interact with its target proteins. Robison et al. showed that oxidation of CaM's methionine residues resulted in a decrease of CaMKII activity, reduced CaMKII's ability to interact with cellular receptors, and attenuated CaM's ability to mediate CaMKII's association with cytoskeletal proteins (143). It is interesting to note that while methionine oxidation of CaM may reduce CaMKII activity, methionine oxidation of CaMKII itself enhances CaMKII activity (41). This apparent contradiction further emphasizes the need to more thoroughly examine the relationship between increasing ROS levels and CaMKII activation. Another mean by which ROS may mediate CaMKII is through regulation of phosphatase activity. In Jurkat cells, stimulation with PMA, a potent PKC activator, resulted in the activation of CaMKII without increasing intracellular Ca2+. Catalase, an H2O2 scavenger, was able to attenuate this PMA-induced CaMKII activity. Further analysis revealed that stimulation with PMA also resulted in the inactivation of the serine/threonine phosphatase PP2A, leading the authors to conclude that stimulation with PMA increased ROS concentration that resulted in the induced loss of PP2A activity and resultant gain in CaMKII activity (78). Although this is the only report showing that CaMKII may be activated in this manner, several studies have shown that ROS mediate PKC activity through regulation of phosphatase activity (50).
Concluding Remarks
It is evident (and has been known for a long time for NOS) that Ca+2 activates various Nox and NOS. An important issue that still needs to be addressed is the molecular identity of the Ca+2-mobilizing stimuli in VSMC that regulate Nox and NOS. In addition, in spite of constant progresses in the identification of specific ROS/RNS intermediates through electron paramagnetic resonance or other analytical approaches, the technology to resolve spatially and temporally intracellular ROS and RNS concentration is still inadequate. This is clearly lagging when compared to the resolution attained with regard to Ca+2 imaging of cellular microdomains and sophisticated patch clamp protocols for direct measurements of ion channel conductances. Advancements in ROS/RNS measurements are bound to improve with the development of a new generation of redox sensitive protein-based detectors (37).
As illustrated in this review, recent studies have focused on oxidative and nitrosative modifications of amino acid residues in proteins as a mean to regulate Ca2+ signaling. It is important to note that the oxidation of polyunsaturated fatty acids may also represent an important process by which Ca2+ signaling may be regulated by ROS. This is because many lipid messengers such as phosphoinositides (PIP, PIP2, PIP3, etc.), DAG, and arachidonid acid are important regulators of Ca2+ channels and transporters, such that lipid peroxidation may indirectly impact the activity of these proteins. Although the implications of ROS-mediated lipid peroxidation in the context of tissue injury have been documented for some time now (119), evidence for a role for lipid peroxidation in Ca2+ signaling remains scarce.
Overall, the mechanism by which specificity is determined through ROS/RNS signaling is still poorly understood, even more so in the context of human diseases. Deregulation of Ca2+, ROS, and RNS homeostasis are hallmarks of many cardiovascular diseases in which VSMC proliferation is a burden. As such, the processes that result in atherosclerotic and restenosis lesions involve dramatic increases in NOX and NOS activities and changes in Ca2+ handling and effectors in smooth muscle. We have illustrated potential direct effects of ROS/RNS on Ca2+ release and entry channels, transporters, and effectors such as CaMKII. An intriguing possibility is the role that ROS and RNS may play in processes of neointimal formation that depend on increased Ca2+ concentrations and activation of effector molecules controlling VSMC proliferation and migration. Although not yet reported, it is certainly reasonable to think that in these ROS/RNS rich environments, certain ion channels along with downstream effectors such as CaMKII might be enriched and where their activity and function may be dramatically affected.
Footnotes
Acknowledgments
We thank Dr. Klaus Groschner (University of Graz, Austria) for expert review and discussion of the manuscript. Work in the authors' laboratories was supported by grants from Philip Morris USA, Inc. and Philip Morris International (to DJ) and NIH grant K22ES014729 (to MT). Figures were produced using Servier Medical Art (