
Abstract
The cytoprotective mechanisms of ursodeoxycholic acid (UDCA) in primary biliary cirrhosis (PBC) have not been fully clarified. UDCA has some antioxidant properties. Nuclear factor-E2–related factor-2 (Nrf2) plays a critical role in protecting a variety of tissues against oxidative stress. Therefore, to investigate the potential antioxidant effects of UDCA in PBC, we determined the intracellular status of both oxidant stress and antioxidant defenses in paired pre- and posttreatment liver biopsies from 13 PBC patients by immunodetection of 8-hydroxydeoxyguanosine (8-OHdG), Nrf2-, and Nrf2-mediated antioxidant proteins. After UDCA treatment, the number of 8-OHdG-positive hepatocytes or bile duct cells decreased with improvement of hepatic injury. The hepatic levels of both total and phosphorylated Nrf2 protein were increased, along with upregulation of nuclear phosphorylated Nrf2 expression in bile duct cells. In addition, the levels of both thioredoxin (TRX) and thioredoxin reductase 1 (TrxR1) protein were increased in the liver after UDCA. The upregulation of hepatic TRX or TrxR1 protein expression positively correlated with that of total Nrf2 protein expression. In conclusion, UDCA treatment can enhance hepatic Nrf2 activation and upregulate hepatic TRX and TrxR1 protein expression. Hepatic Nrf2 activation may play a role in the therapeutic response to UDCA in PBC. Antioxid. Redox Signal. 13, 259–268.
Introduction
Oxidative stress activates a specific stress response, an adaptive mechanism aimed to protect cells against oxidative injury and to maintain tissue redox balance. This stress response includes enhanced expression of endogenous antioxidant molecules. Transcription of most antioxidant genes is regulated through the transcription factor nuclear factor-E2–related factor (Nrf2) and antioxidant response elements (AREs) in the promoter regions of these genes (26). Nrf2 is normally sequestered in the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1), an actin-anchored cytosolic protein that facilitates Nrf2 degradation by a ubiquitin-proteosome pathway (32). With oxidative stress, Nrf2 dissociates from Keap1 and translocates into the nucleus, where it activates ARE-mediated gene transcription and induces the coordinate transcription of ARE-regulated genes including γ-glutamylcysteine synthetase (γGCS) (27, 44), heme oxygenase 1 (HO-1) (1), peroxiredoxin (PRX) (13), glutathione peroxidase 2 (GPx2) (3), thioredoxin (TRX) (18), and thioredoxin reductase 1 (TrxR1) (40).
Recently, UDCA was shown to induce Nrf2 activation in cultured cells and mice (2, 33, 45). Therefore, our goal was to test the hypothesis that Nrf2 activation may contribute to the antioxidant effects of UDCA in PBC. This study focused on early-stage PBC in which UDCA treatment provides more beneficial effects, and the initial changes are more central to the pathogenesis of the disease than to late-stage PBC.
Materials and Methods
Patients
This study included 13 patients (one male and 12 female patients) with early PBC who were evaluated at the Hepatology Division, Department of Internal Medicine, Hamamatsu University School of Medicine. The inclusion of all PBC patients in this study was dependent on the availability of both pre- and post-UDCA treatment liver biopsies. Diagnosis of PBC was based on clinical, laboratory, and histologic findings. None of the patients received any specific treatment before the first liver biopsy. These patients underwent the second liver biopsy 36 ± 4 months (23–68 months) after starting treatment with UDCA (12–15 mg/kg/day, 600 mg/day). Normal liver tissue was obtained from surgical resection specimens of four patients with metastatic liver tumors. None of these patients received UDCA treatment before surgery. A serum sample was obtained from each patient at the time of liver biopsy and was tested for serum total bilirubin, aminotranferases, alkaline phosphatase (ALP), γ-glutamyltransferase (γGT), and immunoglobulin M (IgM). These parameters were assayed with standard techniques in clinical laboratories. In addition, the pre- and posttreatment serum samples were stored at −40°C until use.
All patients gave informed consent for liver biopsy. The study protocol conformed to the ethical guidelines of the Helsinki Declaration of 1975, as revised in 1983, and was approved by the Institutional Review Board.
Liver biopsies
Liver tissue was obtained by percutaneous needle biopsy. All liver biopsy specimens were fixed in formalin and were paraffin embedded before staining with hematoxylin and eosin and Azan-Mallory, and subsequent immunohistochemical analysis. A portion of each sample from 13 patients was immediately frozen in liquid nitrogen and stored at −80°C until use for protein extraction. Histologic staging was defined according to the Ludwig system (24). The main histologic features were semiquantitatively graded as follows: fibrosis, 0–3 (0, absent; 1, portal or periportal fibrosis; 2, presence of numerous septa; 3, cirrhosis); portal inflammation, 0–3 (0, absent; 1, mild; 2, moderate; 3, severe); periportal necroinflammatory lesions, 0–2 (0, absent or mild; 1, moderate; 2, severe); lobular necroinflammatory lesions, 0–2 (0, absent or mild; 1, moderate; 2, severe); and ductular proliferation, 0–2 (0, absent or mild; 1, moderate; 2, severe) (37, 38).
Immunohistochemistry
Immunohistochemical detection of 8-OHdG, Nrf2, and phosphorylated Nrf2 was performed by using a streptavidin-biotin complex peroxidase kit, according to the manufacturer's instructions (Nichirei, Tokyo, Japan) (43). In brief, deparaffinized sections (4 μm thick) were subjected to autoclave heating treatment in 10 mM citrate buffer (pH 6.0) for antigen retrieval. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol. The sections were treated with 10% normal rabbit serum to block nonspecific binding of antibodies, and then were incubated with mouse monoclonal anti-8-OHdG (Japan Institute for the Control of Aging, Shizuoka, Japan; 1:50 dilution), rabbit polyclonal anti-Nrf2 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:50 dilution), or rabbit monoclonal anti-phospho-Nrf2ser40 (Epitomics, Burlingame, CA; 1:100 dilution) at 4°C overnight. After the reaction, biotinylated rabbit anti-mouse or goat anti-rabbit IgG immunoglobulins (Dako, Glostrup, Denmark) and streptavidin conjugated with peroxidase were sequentially used, followed by color development with 3,3'-diaminobenzidine tetrahydrochloride and hydrogen peroxide. Nuclear staining was carried out with Mayer's hematoxylin. The specificity of the staining was confirmed by preincubating the antibody with an excess of specific antigen peptide (Santa Cruz Biotechnology, Santa Cruz, CA). For semiquantitative assessment of 8-OHDG, Nrf2, or phosphorylated Nrf2 expression, the number of immunoreactive cell nuclei was counted among 300 hepatocytes in at least three periportal and three perivenous zones or all bile duct cells in each section.
Immunoblotting
Frozen liver tissue from 17 patients (PBC, 13; normal, 4) was homogenized in protein extraction buffer (Santa Cruz Biotechnology) containing a complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). The lysates were diluted 1:1 with 2x Laemmli sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. The protein concentration of samples in SDS sample buffer was determined by using Peterson's modification of the micro-Lowry method (34). The protein extracts were subjected to SDS-PAGE on a 12.5% polyacrylamide gel. The resolved proteins were electrophoretically transferred to polyvinylidene difluoride membranes. The blots were blocked overnight at 4°C with TBS-T buffer (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.5% Tween 20) containing 10% nonfat dry milk, and were probed with specific polyclonal or monoclonal antibodies. The following antibodies were used: rabbit polyclonal anti-Nrf2 (1:200 dilution), rabbit monoclonal anti-phospho-Nrf2ser40 (1:5,000 dilution), rabbit polyclonal anti-HO-1 (Stressgen, Victoria, BC, Canada; 1:5,000 dilution), rabbit polyclonal anti-γGCS (Lab Vision Corp., Fremont, CA; 1:400 dilution), goat polyclonal anti-GPx2 (Abcam, Cambridge, UK; 1:250 dilution), rabbit polyclonal anti-PRX1 (Abcam, 1:2,000 dilution), mouse monoclonal anti-TRX antibody (Redox Bio Science Inc., Kyoto, Japan; 1:100 dilution), mouse monoclonal anti-TrxR1 (Santa Cruz Biotechnology, 1:100), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Trevigen, Gaithersburg, MD; 1:1,000 dilution) antibodies. Bound primary antibody was detected by using anti-rabbit, anti-mouse, or anti-goat IgG horseradish peroxidase conjugate (Santa Cruz Biotechnology; 1:5,000 dilution), and the blots were developed with chemiluminescence detection. The bands were quantified by densitometry to obtain an optical density value, which then was normalized with respect to the GAPDH value.
Serum TRX measurement
Serum was obtained from venous blood drawn at the time of the first and second liver biopsies. Blood samples were separated by centrifugation at 1,000 g for 15 min at room temperature and stored at −70°C. Serum TRX levels were measured with a sensitive sandwich enzyme-linked immunosorbent assay (ELISA) kit (Redox Bio Science Inc., Kyoto, Japan) (20). In brief, ADF 21-antibody–precoated 96-microwell plates were incubated for 2 h at room temperature with 20 μl of serum or standard solution (ADF; 0, 30, 60, 120 ng/ml) in the presence of 200 μl of 50 mM sodium phosphate buffer (pH 6.0) containing 150 mM NaCl, 1.0 mM MgCl2, 1.0% bovine serum albumin, and 0.1% NaN3. The plates were washed 5 times with 10 mM sodium phosphate buffer (pH 7.5) containing 0.05% Tween 20 and 150 mM NaCl (wash solution), and then 200 μl of the horseradish peroxidase–labeled anti-ADF antibody was added to the plates for incubation at room temperature for 2 h. After the plates were washed 5 more times with wash solution, 100 μl of 100 mM triethanolamine-succinate buffer (pH 4.4) containing 1.5 mM H2O2 and 0.13% 2,2'-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid was added, and it was incubated for 0.5 h at room temperature. The reaction was then stopped by the addition of 100 μl of 1% oxalic acid solution, and the optical density was measured at 450 nm with an EL340i microplate reader (Bio-Tek Instruments, Inc. Winooski, VT). All measurements were made in duplicate, and the average value was used.
Bile acid measurement
The level of serum total bile acids was measured with the 3α-hydoroxysteroid dehydrogenase method. Bile acid fractions were determined with specific liquid chromatography–electrospray mass spectrometry, by using an HPLC system (Agilent 1100 series, Agilent Technologies, Santa Clara, CA) equipped with a C18 cartridge (CAPCELL PAK C18 UG120A, Shiseido, Tokyo, Japan) and a mass spectrometer (Quattro Ultima, Micromass Technologies, Manchester, UK). These measurements were performed by a contract laboratory (SRL, Tokyo, Japan).
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) unless otherwise stated. Comparisons of mean values between groups were performed with the Wilcoxon paired signed-rank test or the Mann–Whitney U test, appropriately. Correlation coefficients were calculated by using linear correlation analysis. A p value of <0.05 was considered significant.
Results
Clinical characteristics of PBC patients
The clinical characteristics at the time of pre- and posttreatment liver biopsies are shown in Table 1. All patients were positive for anti-mitochondria antibody at the time of pretreatment liver biopsy. After UDCA treatment, the serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), ALP, γGT, and IgM levels significantly improved. No significant difference in serum total bilirubin levels and Mayo risk score was observed between before and after UDCA treatment. No significant improvement was found in histologic stages after UDCA treatment (Table 1). The degree of lobular necroinflammatory lesions improved after UDCA treatment, whereas no significant differences in the degree of fibrosis, portal inflammation, periportal necroinflammatory lesions, and ductular proliferation were found between pre- and posttreatment (Table 1).
Values are expressed as mean ± SEM. The p values refer to Wilcoxon paired signed-rank test.
Hepatic 8-OHdG expression
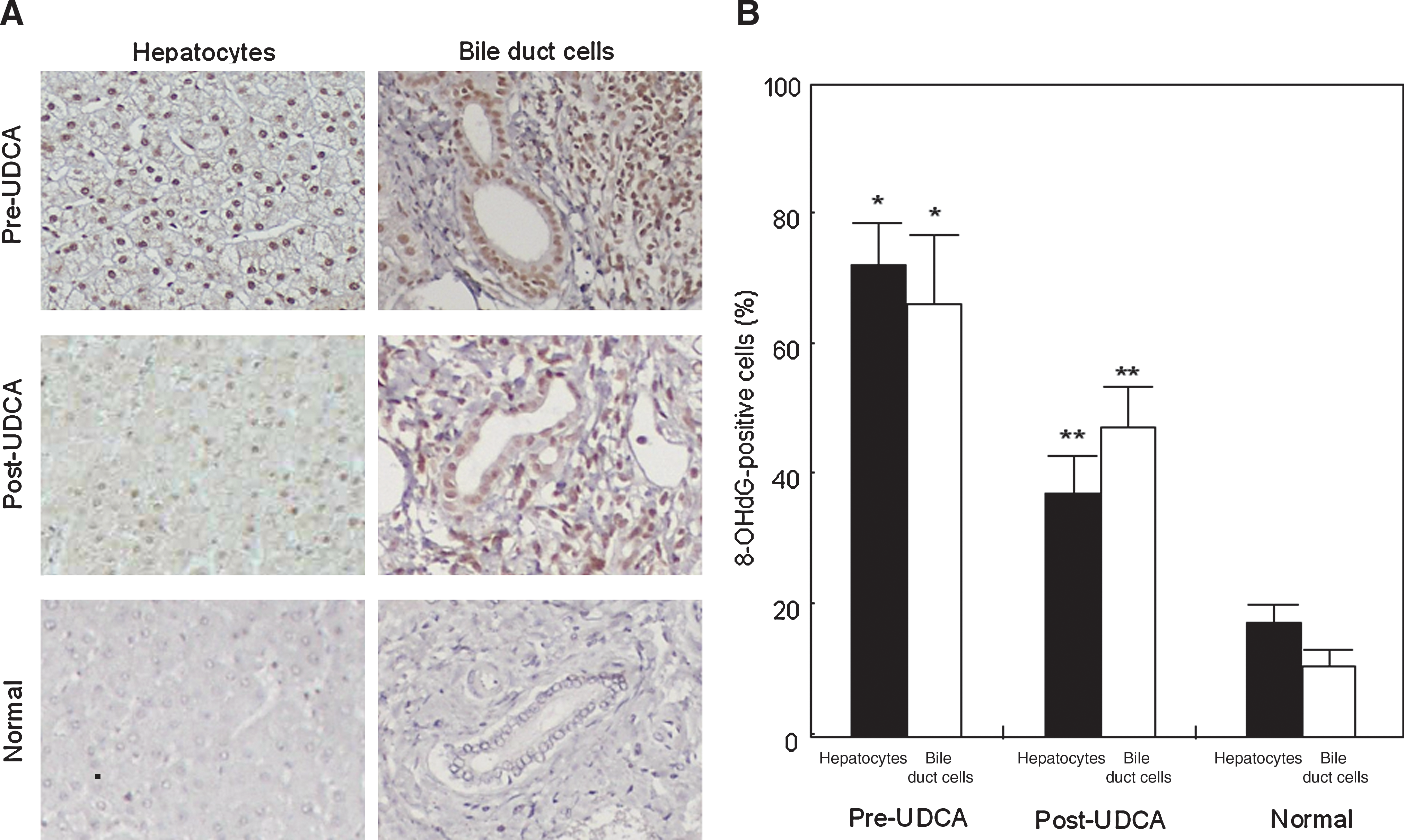
We examined oxidative stress in normal and PBC livers before and after UDCA treatment, by using immunohistochemistry for 8-OHdG. In liver specimens from PBC patients before UDCA treatment, positive staining for 8-OHdG was detected preferentially in the nuclei of hepatocytes, bile duct cells and portal inflammatory cells, and occasionally in the nuclei of sinusoidal cells, as shown in Fig. 1A. The percentage of positive hepatocytes or bile duct cells was significantly higher before UDCA treatment than after the treatment (hepatocytes: 73% pre-UDCA vs. 37% post-UDCA, p < 0.01; bile duct cells: 66% pre-UDCA vs. 48% post-UDCA, p < 0.01) (Fig. 1B). The percentage of positive hepatocytes or bile duct cells was significantly higher in pre-UDCA livers of PBC than in normal livers (hepatocytes: 73% pre-UDCA vs. 19% normal, p < 0.01; bile duct cells: 66% pre-UDCA vs. 11% normal, p < 0.01) (Fig. 1B).
Hepatic total and phosphorylated Nrf2 expression
We examined total and phosphorylated Nrf2 expression in normal and PBC livers before and after UDCA treatment with immunoblotting and immunostaining. Immunoblotting showed significant increases in hepatic total and phosphorylated Nrf2 protein after UDCA treatment (Fig. 2). Levels of total and phosphorylated Nrf2 protein were positively correlated with each other (r = 0.74; p < 0.001). No significant differences in levels of total or phosphorylated Nrf2 protein were observed between pre-UDCA and normal livers (Fig. 2).

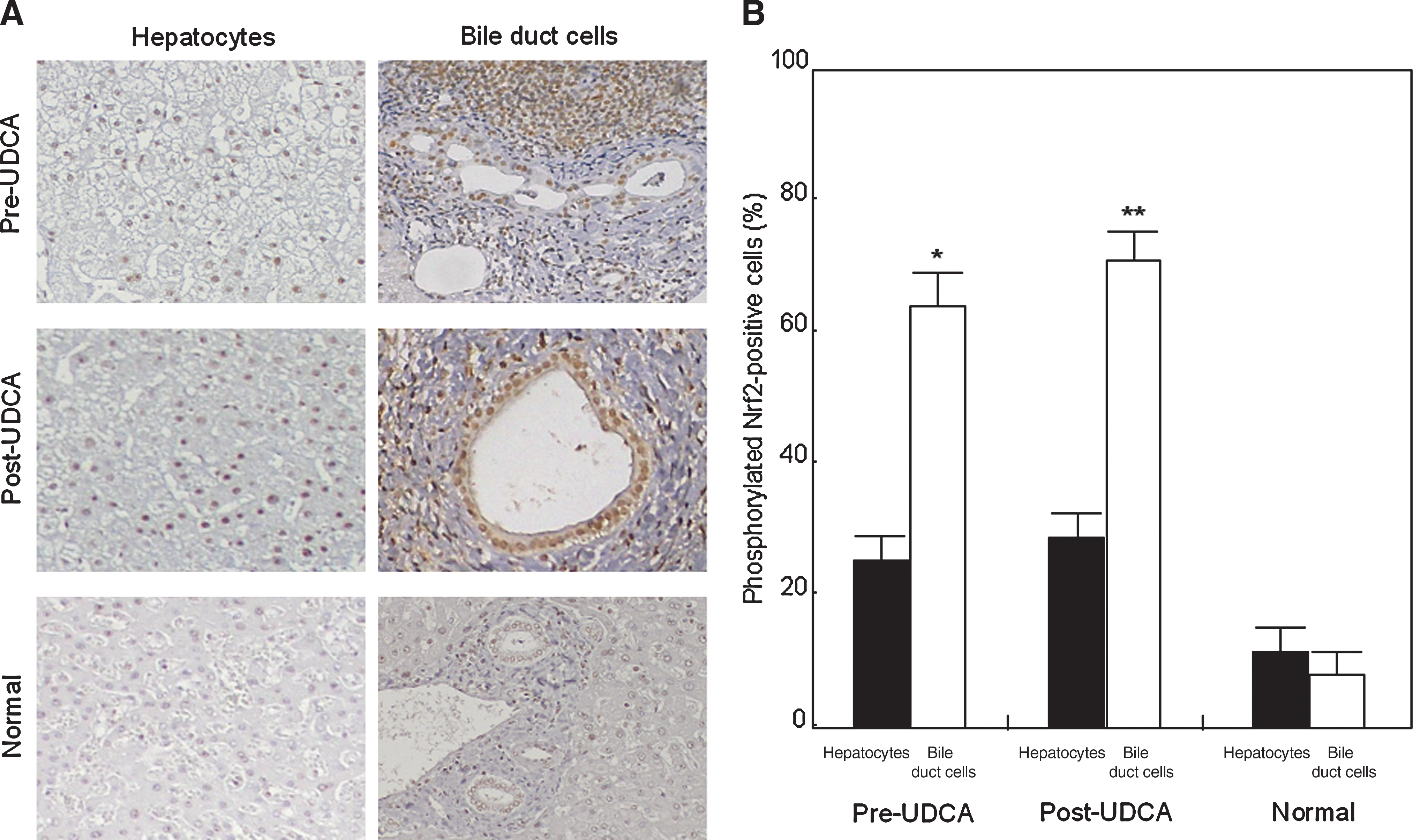
Immunostaining showed that almost all nuclei of hepatocytes and bile duct cells expressed total Nrf2 protein regardless of the presence or absence of UDCA treatment (data not shown). The expression of phosphorylated Nrf2 in hepatocytes did not differ significantly between pre- and post-UDCA treatment (Fig. 3). Nuclear expression of phosphorylated Nrf2 in bile duct cells was more frequently detected after UDCA treatment (62% pre-UDCA vs. 69% post-UDCA; p < 0.05) (Fig. 3). The expression of phosphorylated Nrf2 in hepatocytes did not differ significantly between pre-UDCA and normal livers (Fig. 3). The percentage of positive bile duct cells was significantly higher with pre-UDCA treatment than in normal livers (62% pre-UDCA vs. 7% normal; p < 0.05)
Hepatic and serum Nrf2-mediated antioxidant protein expression
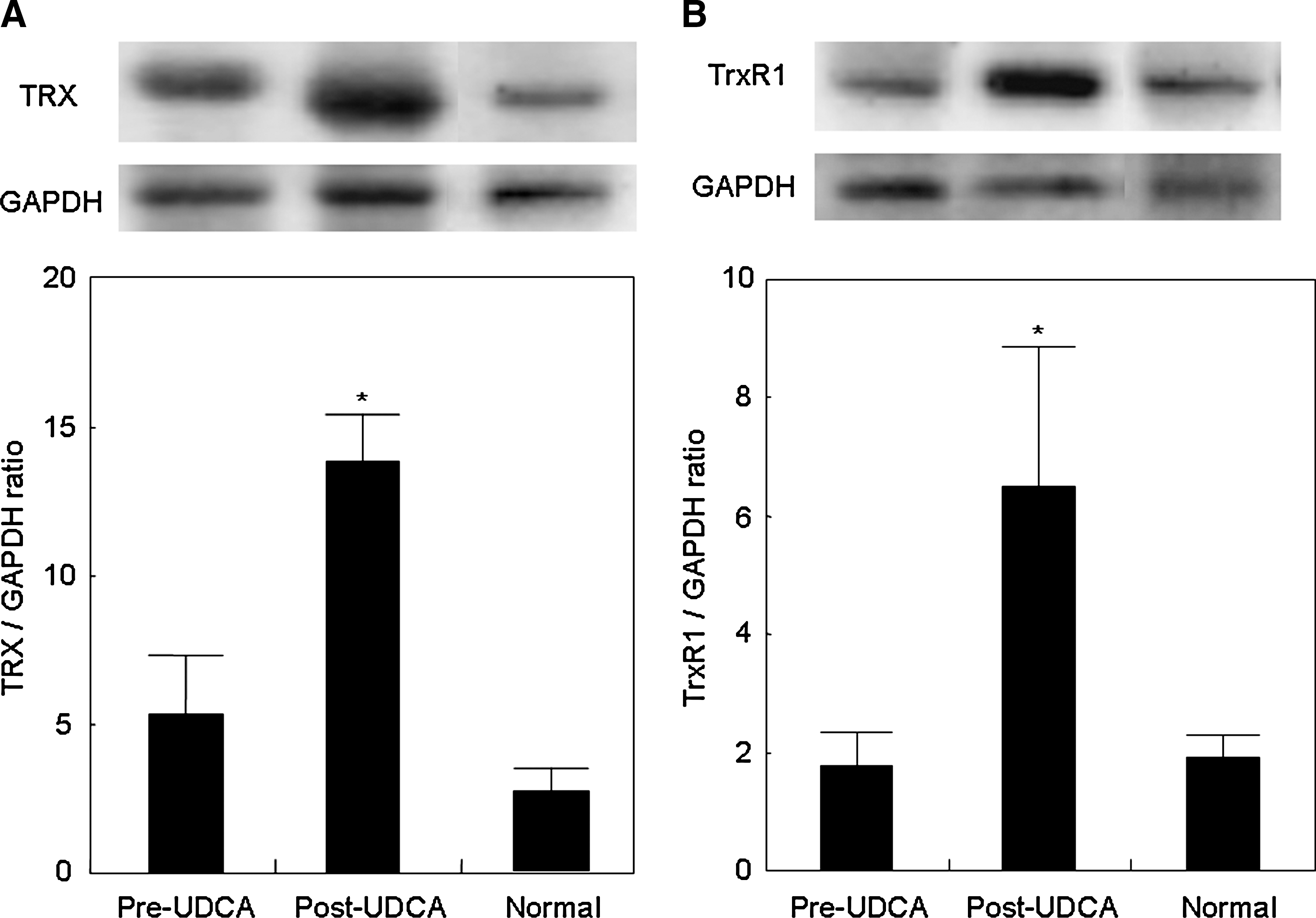
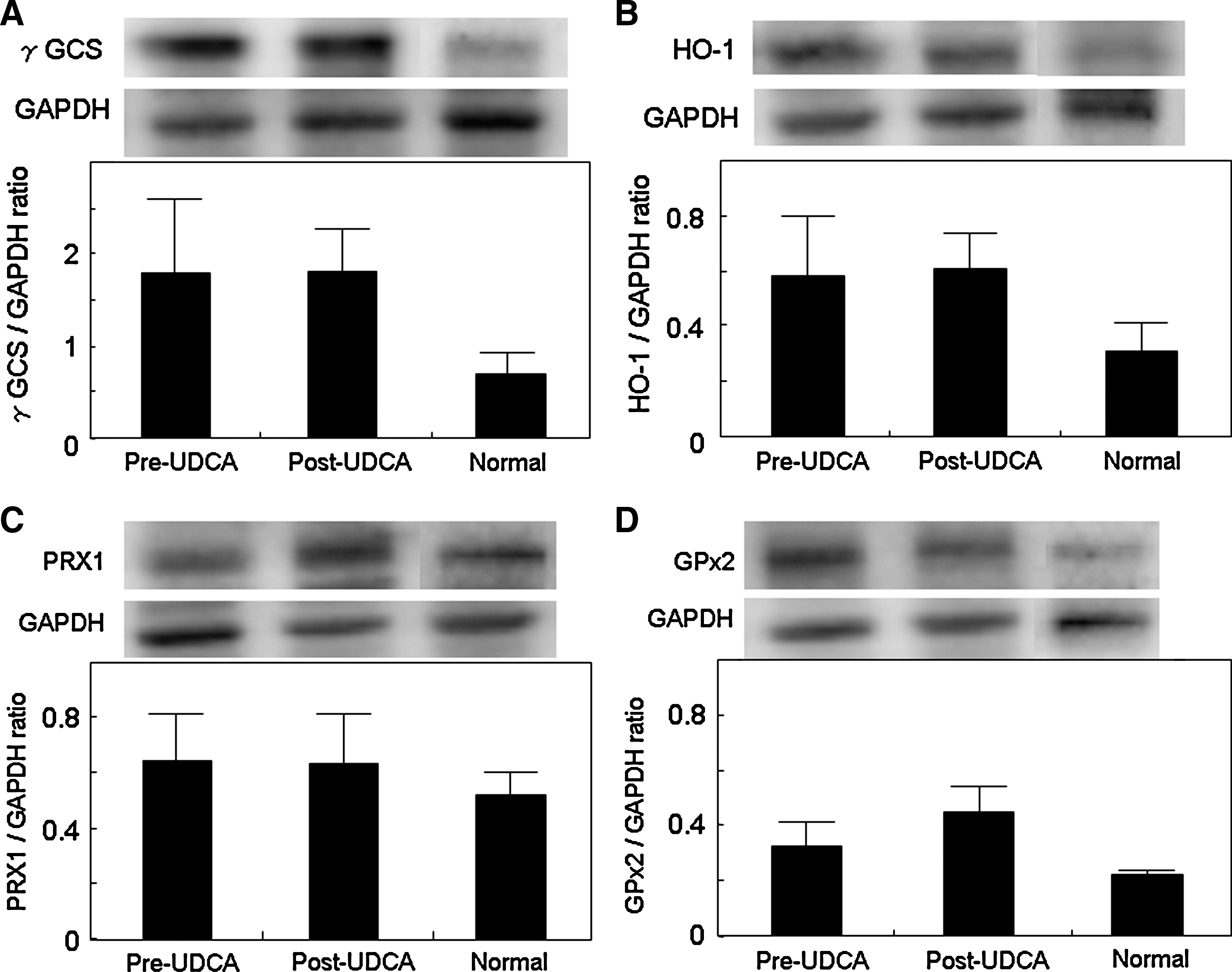
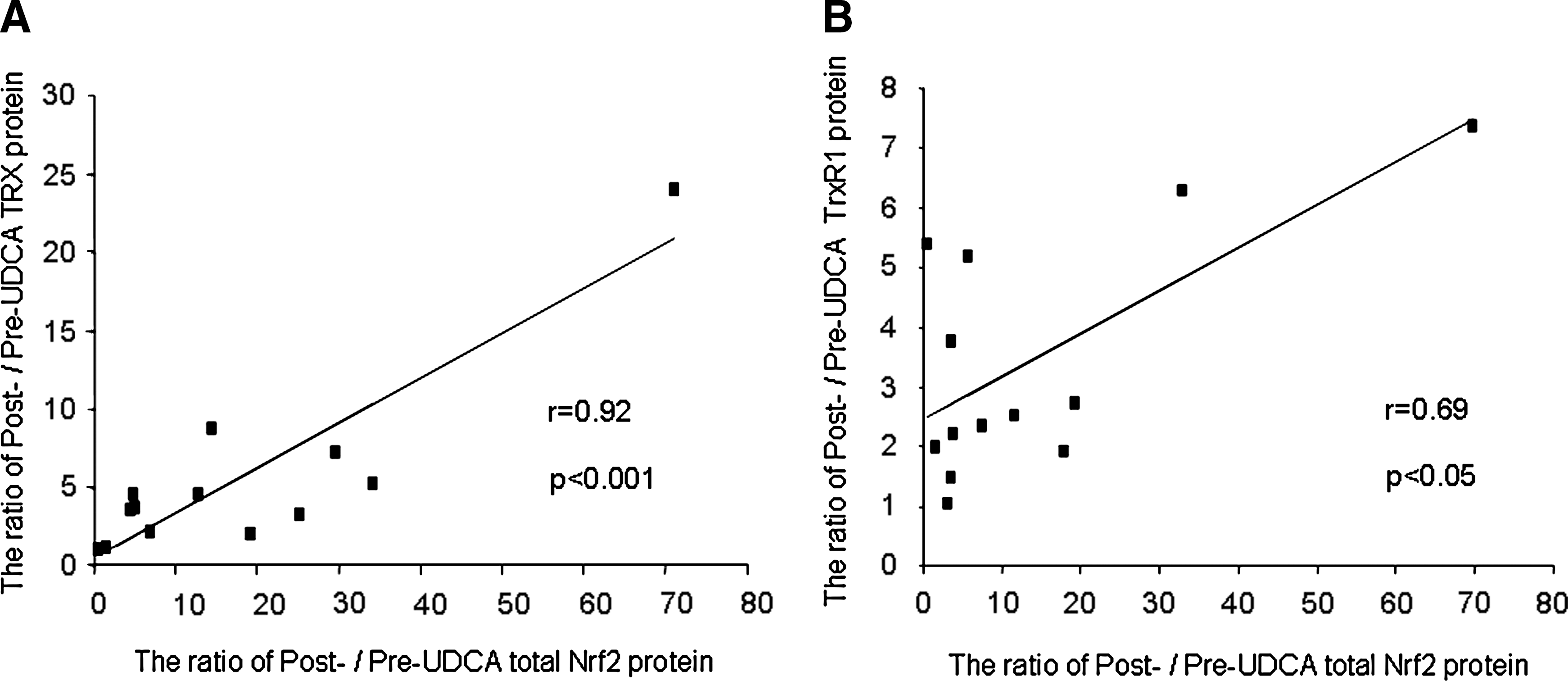
We examined hepatic expression of Nrf2-mediated antioxidant proteins (HO-1, γGCS GPx2, TRX, TrxR1, and PRX1) in normal and PBC livers before and after UDCA treatment by using immunoblotting. TRX and TrxR1 protein significantly increased after UDCA treatment (Fig. 4), whereas no significant change was observed for the others (Fig. 5). A significant correlation was found between the ratios of post- to pre-UDCA Nrf2 protein levels and those of either TRX or TrxR1 protein levels after UDCA treatment (Fig. 6). These Nrf2-mediated antioxidant protein expressions did not differ significantly between pre-UDCA and normal livers (Fig. 5).
We measured serum TRX levels before and after UDCA treatment. Serum TRX levels also significantly increased after UDCA treatment (15.8 ± 4.9 ng/ml before UDCA vs. 24.5 ± 6.3 ng/ml after UDCA; p < 0.05). No significant relation was noted between serum TRX levels and hepatic TRX, total Nrf2, or phosphorylated Nrf2 expression.
Serum bile acids
Total bile acid concentration and the ratio of UDCA to total bile acids in serum significantly increased after UDCA treatment. The proportions of chenodeoxycholic acid and deoxycholic acid were decreased significantly after UDCA treatment. No significant difference in the proportion of cholic acid and chenodeoxycholic acid was observed after UDCA treatment (Table 2).
Values are expressed as mean ± SEM. The p values refer to Wilcoxon paired signed-rank test.
Discussion
PBC is a slowly progressive, autoimmune cholestatic liver disease. UDCA is well known to be a beneficial agent for treating patients with PBC. Especially in early-stage PBC, a substantial proportion of the patients with UDCA treatment have a favorable outcome (38). This study also confirms the beneficial effects of UDCA in early-stage PBC. However, the mechanisms of UDCA action have not been fully clarified. It has been postulated that the inherently greater hydrophilic nature of UDCA prevents hepatocellular damage by replacing more hepatotoxic hydrophobic bile acids (36). The present study confirms the previous studies showing that UDCA therapy in PBC patients causes a marked enrichment in the proportion of UDCA, with a concomitant decrease in the proportion of chenodeoxycholic acid and deoxycholic acid in serum bile acids. Recently, it was suggested that hepatic oxidative stress is involved in the pathogenesis of PBC (14, 17, 19, 30). In addition, UDCA has been shown to exert some cytoprotection against oxidative stress (21, 23, 25, 42). Therefore, to investigate the mechanisms involved in the beneficial effects of UDCA in PBC, we focused on potential antioxidant effects of UDCA. We examined the intracellular status of oxidant stress and antioxidant defenses in the livers of PBC patients during UDCA treatment by using immunodetection of 8-OHdG, a useful marker of oxidative stress, Nrf2, an oxidative stress-sensitive transcription factor, and Nrf2-mediated antioxidant proteins. Our findings can be summarized as follows: (a) an increased number of 8-OHdG-positive hepatocytes or bile duct cells improved after UDCA treatment; (b) both hepatic total and phosphorylated Nrf2 protein levels increased, along with upregulation of nuclear phosphorylated Nrf2 in bile duct cells after UDCA treatment; and (c) both TRX and TrxR1 protein levels increased after UDCA treatment, whereas the expression of γGCS, HO-1, PRX, or GPx2 protein did not change.
In this study, increased nuclear expression of 8-OHdG in the hepatocytes and bile-duct cells was observed in PBC patients. This confirms previous studies demonstrating that enhanced hepatic oxidative stress exists in this disease (14, 17, 19, 30). In addition, this study showed for the first time that oxidative DNA damage is diminished in the hepatocytes and bile-duct cells of PBC patients with improvement of hepatic injury after UDCA treatment, raising the possibility that UDCA treatment can ameliorate hepatic oxidative stress in PBC. UDCA has direct and indirect antioxidant properties. The direct antioxidant mechanism is scavenging hydroxyradicals. A recent study showed pharmacologic inhibition of iron- and hydroxyradical-dependent oxidative damage by UDCA (21). The indirect antioxidant mechanism of UDCA is to induce endogenous antioxidant defenses. UDCA treatment upregulates γ-GCS expression and consequently increases the rate of glutathione (GSH) synthesis. Thus, the antioxidant defense mediated by GSH is enhanced by UDCA treatment (25). However, in this study, the ameliorated hepatic oxidative stress may also be a consequence of improved hepatic injury. UDCA has several beneficial effects, including protection against the cytotoxicity of hydrophobic bile acids, stimulation of hepatobiliary secretion, and protection of hepatocytes against bile acid–induced apoptosis.
In the present study, increased levels of hepatic Nrf2 protein were observed in PBC patients after UDCA treatment. This increase in Nrf2 protein is likely due to increased protein synthesis or decreased protein degradation or both. Some Nrf2-activating chemicals enhance transcription of Nrf2 mRNA, whereas others increase Nrf2 levels by interfering with the degradation of existing Nrf2 protein (32). Nrf2 can also regulate its own expression through direct binding to its promoter region. Previous studies reported that upregulation of Nrf2 gene expression at the transcription level and increased protein stability are important mechanisms for Nrf2 activation (31). In addition, increased expression of hepatic Nrf2 phosphorylated at Ser-40 also was found in the present study. Nrf2 phosphorylation is one of important mechanisms for the activation of Nrf2-ARE–mediated pathways. Nrf2 is phosphorylated at multiple sites by different protein kinases, including protein kinase C (PKC) (12), phosphatidylinositol 3-kinase (15), mitogen-activated protein kinase (46), ER-localized pancreatic endoplasmic reticulum kinase (8), and protein kinase CK2 (35). Phosphorylation of Nrf2 at Ser-40 through a PKC-based mechanism plays a critical role in the dissociation of Nrf2 from Keap1, which binds to unphosphorylated Nrf2 and stimulates its degradation (12).
During the activation of Nrf2, nuclear accumulation of Nrf2 is an essential mechanism for serving as a transcriptional activator. In this study, immunohistochemistry showed nuclear accumulation of total and phosphorylated Nrf2 in hepatobiliary cells of untreated PBC patients. This may be because of enhanced hepatic oxidative stress, because Nrf2 undergoes nuclear translocation during oxidative stress (16). After UDCA treatment, increased nuclear accumulation of phosphorylated Nrf2 was shown only in the bile duct cells. Arisawa et al. (2) showed nuclear accumulation of Nrf2 in HepG2 cells exposed to UDCA. Okada et al. (33) reported that UDCA increased nuclear Nrf2 protein in rat liver–derived RL34 cells and in mouse liver. Therefore, these findings raise the possibility that UDCA treatment may also stimulate hepatocellular nuclear accumulation of Nrf2 in PBC.
The mechanisms responsible for enhanced hepatic Nrf2 activation after UDCA treatment in PBC remain unclear. UDCA has been shown to activate multiple kinase-signaling pathways, such as protein kinase C (4), extracellular signal-regulated kinase 1/2 (9), mitogen-activated protein kinase (39, 41), p38 mitogen–activated protein kinase (41), phosphatidylinositol 3-kinase/Akt, or a combination of these (2, 9, 39, 41). These signaling pathways have been shown as well to influence the stability of the Nrf2-Keap1 complex. In addition, it was recently demonstrated that UDCA promoted nuclear accumulation of Nrf2 through activation of the PI3K/Akt pathway (2).
Nrf2 induces the coordinate transcription of ARE-regulated antioxidant genes, including γGCS, HO-1, PRX1, GPx2, TRX, and TrxR1. Recent experimental studies demonstrated that UDCA can activate Nrf2-ARE–mediated transcription (2, 33, 45). Examining hepatic expression of these antioxidant proteins during UDCA treatment in PBC patients, the present study showed that both TRX and TrxR1 protein levels increased after UDCA treatment, and that the enhancement of this protein expression positively correlated with that of Nrf2, suggesting that the upregulation of TRX and TrxR1 expression after UDCA treatment may be dependent on Nrf2. Conversely, γGCS, HO-1, PRX, or GPx2 protein levels were found to be unchanged after UDCA treatment. Multiple kinases alter transcription of Nrf2 target genes, and Nrf2-driven transcription is influenced by its several basic region leucine-zipper transcription factors including AP-1, small Maf, and large Maf proteins (22, 45). Therefore, hepatic Nrf2-responsive gene expression may be differentially regulated during UDCA treatment in PBC patients.
TRX, a small multifunctional protein, acts as an intracellular reductase by a dithiol/disulfide exchange reaction using cysteine residues in the conserved active-site sequence (11). TRX protects cells from stress-induced damage through antioxidative, antiapoptotic, and antiinflammatory effects. Inhibition or genetic suppression of TRX results in increased oxidant stress and apoptosis, and increased sensitivity to oxidants, whereas TRX overexpression enhances resistance to oxidant-induced apoptosis (5). In addition, exogenous TRX was recently shown to attenuate hepatic oxidative damage (6). Therefore, during UDCA treatment for PBC, the upregulation of TRX expression may reduce hepatic oxidative stress and inhibit apoptosis, leading to amelioration of hepatic injury. In this study, the expression of hepatic TRX before UDCA treatment in PBC patients failed to reflect an upregulation mechanism in response to oxidative stress. This may be explained by the recent demonstration that hepatic TRX had a transient increase at the early stage followed by downregulation at the later stage during ongoing cholestasis (10).
TrxR1, a selenocysteine-containing oxidoreductase, catalyzes the conversion of oxidized TRX to reduced TRX in an NADPH-dependent manner, enabling the active functioning of TRX (28). The coordinate expression of TRX and TrxR1 provides an efficient antioxidant defense system. Because the present study showed that a positive correlation exists between hepatic TRX and TrxR1 protein levels, hepatic TRX and TrxR1 expression is likely to be coordinately regulated in PBC patients.
In conclusion, this study indicates that UDCA treatment enhances hepatic Nrf2 and phospho-Nrf2 levels and upregulates hepatic TRX and TrxR1 protein expression in PBC patients. Hepatic Nrf2 activation may play a role in the therapeutic response to UDCA in PBC. This study provides the first step toward understanding the mechanisms of the beneficial effects of UDCA treatment in PBC. Further comparative analysis of hepatic Nrf2 activation between PBC patients with clinical response or nonresponse during UDCA treatment will allow us to understand better the therapeutic implication of hepatic Nrf2 in PBC.
Footnotes
Acknowledgment
We thank Robert S. Britton, Ph.D., Division of Gastroenterology and Hepatology, Saint Louis University School of Medicine, for critical reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.