
Abstract
Hydrogen sulfide (H2S) plays an important role in cardiovascular, central nervous, and gastrointestinal systems. Being the third gaseous mediator, H2S has been shown to act as a vasodilator. In recent times, more and more attention has been paid to the biological functions of H2S in inflammation. Substance P is an 11 amino acid neuropeptide that is released from nerve endings in many tissues. Subsequent to its release, substance P binds to neurokinin-1 (NK-1) receptors on the surface of effector cells and, in addition to being a mediator of pain, it plays an important role in many inflammatory states including asthma, immune-complex-mediated lung injury, experimental arthritis, and inflammatory bowel disease. Substance P has been shown to increase microvascular permeability and promote plasma extravasation. Using animal models of inflammation of different etiologies such as acute pancreatitis, sepsis, and burns, studies in our laboratory have recently shown an important role of the pro-inflammatory action of H2S and substance P. Antioxid. Redox Signal. 12, 1191–1202.
Introduction
It is, however, now apparent that H2S is also synthesized naturally in the body from L-cysteine mainly by the activity of two enzymes, cystathionine-γ-lyase (CSE, EC 4.4.1.1) and cystathionine-β-synthase (CBS, EC 4.2.1.22) (Fig. 1). Both enzymes are pyridoxal phosphate-dependent and are expressed in a range of mammalian cells and tissues. Of these, CBS seems to be the main H2S-forming enzyme in the central nervous system, whilst CSE is the main H2S-synthesizing enzyme in the vasculature. H2S has been shown to act as a potent vasodilator (3, 23). Nowadays, more and more evidence suggests that H2S performs a wide range of physiological and pathological functions, although much less is known about its physiological and pathological roles compared to its two counterparts, NO and CO. For example, H2S opens ATP-dependent potassium (K+ ATP) channels in vascular smooth muscle cells, gastrointestinal smooth muscle cells, cardiomyocytes, neurons, and pancreatic ß-cells, therefore regulating vascular tone, intestinal contractility, myocardial contractility, neurotransmission, and insulin secretion (3, 23). In the nervous system, H2S promotes hippocampal long-term potentiation (LTP) by enhancing the sensitivity of N-methyl-D-aspartic acid (NMDA) receptors to glutamate and plays a role in neurodegenerative diseases (3, 23). In addition, H2S may scavenge reactive nitrogen species (RNS), peroxynitrite (OONOO-), oxygen free radicals, and lipid peroxidations, resulting in cardiovascular protection and neuron protection. As the end product of CBS- and CSE-catalyzed cysteine metabolism, H2S exerts a negative feedback effect on the activity of these enzymes (23).
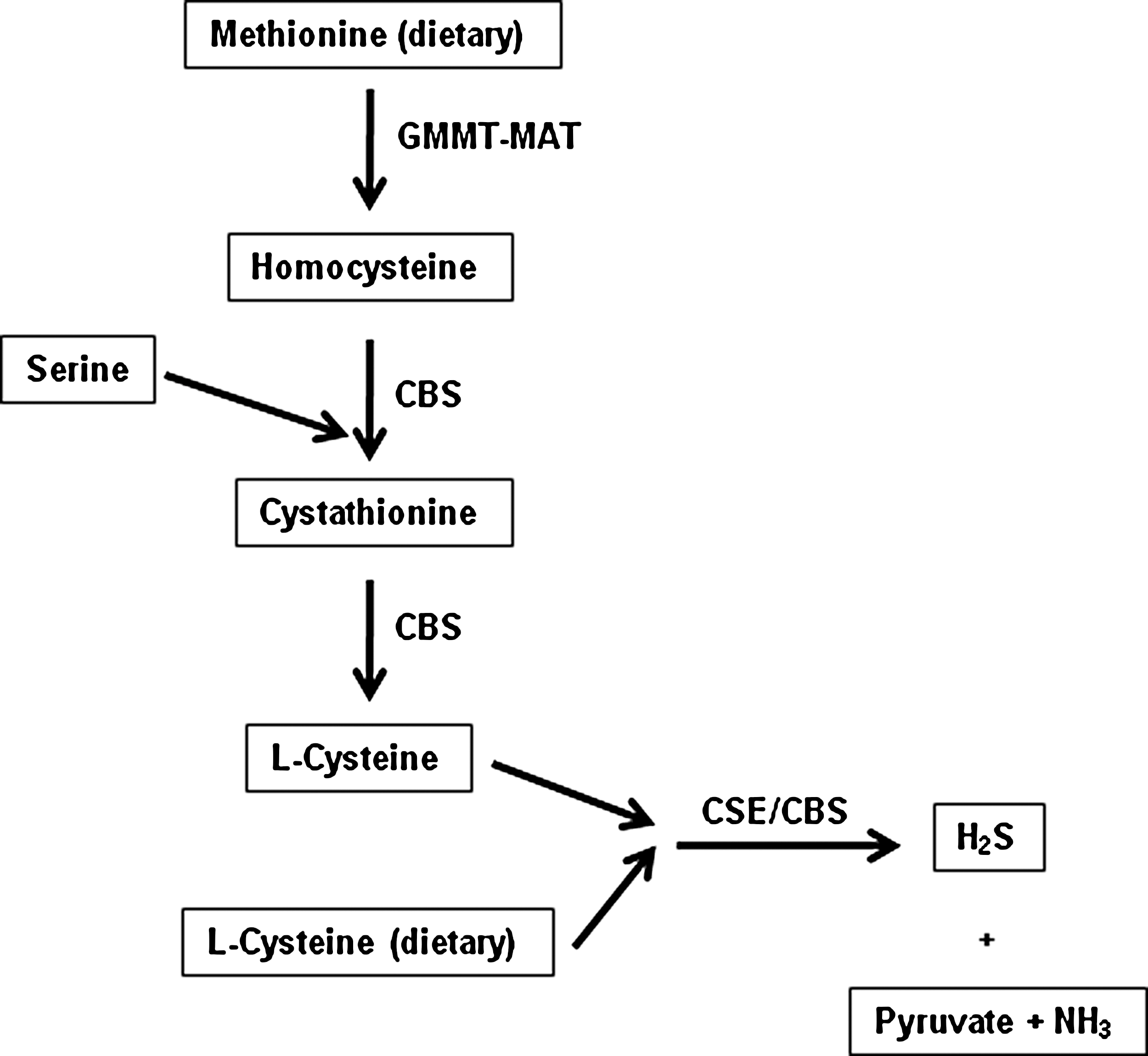
H2S in vivo is metabolized by oxidation in mitochondria or by methylation in the cytosol. H2S can be scavenged by methemoglobin (2, 48) or metallo- or disulfide-containing molecules such as oxidized glutathione (38). H2S is excreted mainly by the kidney as free or conjugated sulfate (48). The interaction of hemoglobin and H2S calls for special attention. Hemoglobin may well be the common “sink” for CO in forming scarlet carboxyhemoglobin (50), for NO in forming nitrosyl-hemoglobin, and for H2S in forming sulfhemoglobin (1).
Tachykinins constitute a family of peptides which share the common C-terminal sequence Phe-Xaa-Gly-Leu-Met-NH2. This sequence plays a key role in their interaction with specific receptors and is important for producing most of their biological effects. However, other domains of the tachykinin peptide sequence, such as the N-terminal sequence have also been shown to be important for producing certain effects such as mast cell degranulation. In 1931, Von Euler and Gaddum discovered that an extract from horse intestine and brain caused intestinal smooth muscle contractions; they named the active compound “substance P” (for “preparation”) (22). In subsequent decades, related nonmammalian peptides such as eledoisin and physalaemin were discovered, and more recently a group of substance P-like mammalian peptides were found in the central nervous system, including neurokinin-A (previously termed substance K) and neurokinin-B (previously termed neuromedin-K). After several years, the 11 amino acid sequence of substance P was finally discovered in 1970 by Chang and Leeman (13). The family of tachykinins includes the peptides substance P, neurokinin-A, neurokinin-B, and two higher molecular weight forms of neurokinin-A: neuropeptide-γ and neuropeptide-K. The amino acid sequences of these peptides are given in Fig. 2.
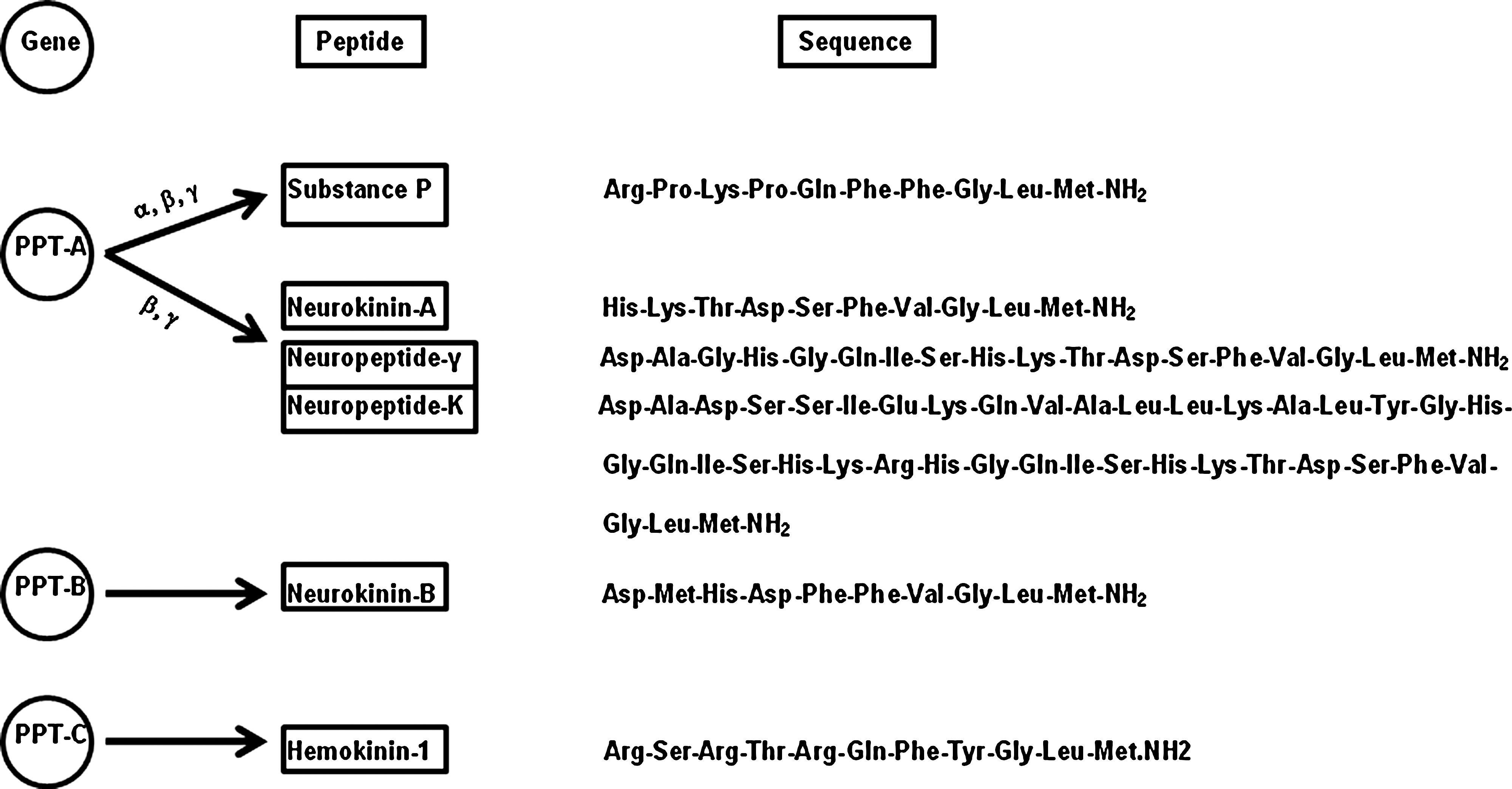
In mammals, two genes have been shown to code for peptides of the tachykinin family: the preprotachykinin-A gene (PPT-A or PPT-I), which encodes both substance P and neurokinin-A, and the preprotachykinin-B gene (PPT-B or PPT-II), which encodes neurokinin-B (Fig. 2). The primary ribonucleic acid (RNA) transcript of the PPT-A gene is spliced to yield three different forms of messenger RNA (mRNA) termed α, β, and γ forms (25, 4). α, β, and γ PPT-A mRNAs code for the synthesis of substance P whereas β and γ PPT-A mRNAs code for the synthesis of both substance P and neurokinin-A. In addition, β and γ PPT-A mRNAs encode the synthesis of two N-terminally extended forms of neurokinin-A (neuropeptide-K and neuropeptide-γ). Both these peptides are capable of producing full biological responses (43). Expression of the PPT-A gene has been detected in both central and peripheral nervous system, in enteric neurons of the gut, and in various cells of the immune system. In contrast, the PPT-B gene is expressed almost exclusively in the central nervous system. Recently, the molecular cloning of a further preprotachykinin gene (termed PPT-C) has been described from cDNA of murine hematopoietic cells (56). PPT-C mRNA was detected exclusively in pro- and pre-B lymphocyte cells from bone marrow. The novel tachykinin encoded by PPT-C gene (termed hemokinin-I) may act as an autocrine factor for the growth of hematopoietic cells. It has also been suggested that hemokinin-I may stimulate a specific receptor, distinct from any other known tachykinin receptor (56). However, further experimentation is needed to clearly define the role of hemokinin-I in mammals. Owing to the lack of PPT-B gene expression in peripheral tissues, substance P and neurokinin-A are the only tachykinins detected in appreciable amounts in these tissues. In normal, noninflamed tissues, tachykinins originate from neuronal cells, being present in (a) peripheral endings of capsaicin (a sensory neuron excitotoxin—a chemical derivative of vanillyl amide and the pungent agent in red pepper) -sensitive primary afferent neurons and (b) enteric neurons of both submucosal and myenteric plexuses innervating all layers of the gut. In addition, certain immune cell types synthesize and possibly release tachykinins during inflammation, thus representing a non-neuronal source of tachykinins in inflamed tissues (13). An inflammatory response that results from the release of substances from primary sensory nerve terminals is termed “neurogenic inflammation”. Figure 3 illustrates the interaction of substance P with various immune cells in neurogenic inflammation.

In this review, recent evidence that points to a key role of H2S and substance P as novel mediators of inflammation is discussed.
Systemic Inflammatory Response Syndrome (SIRS)
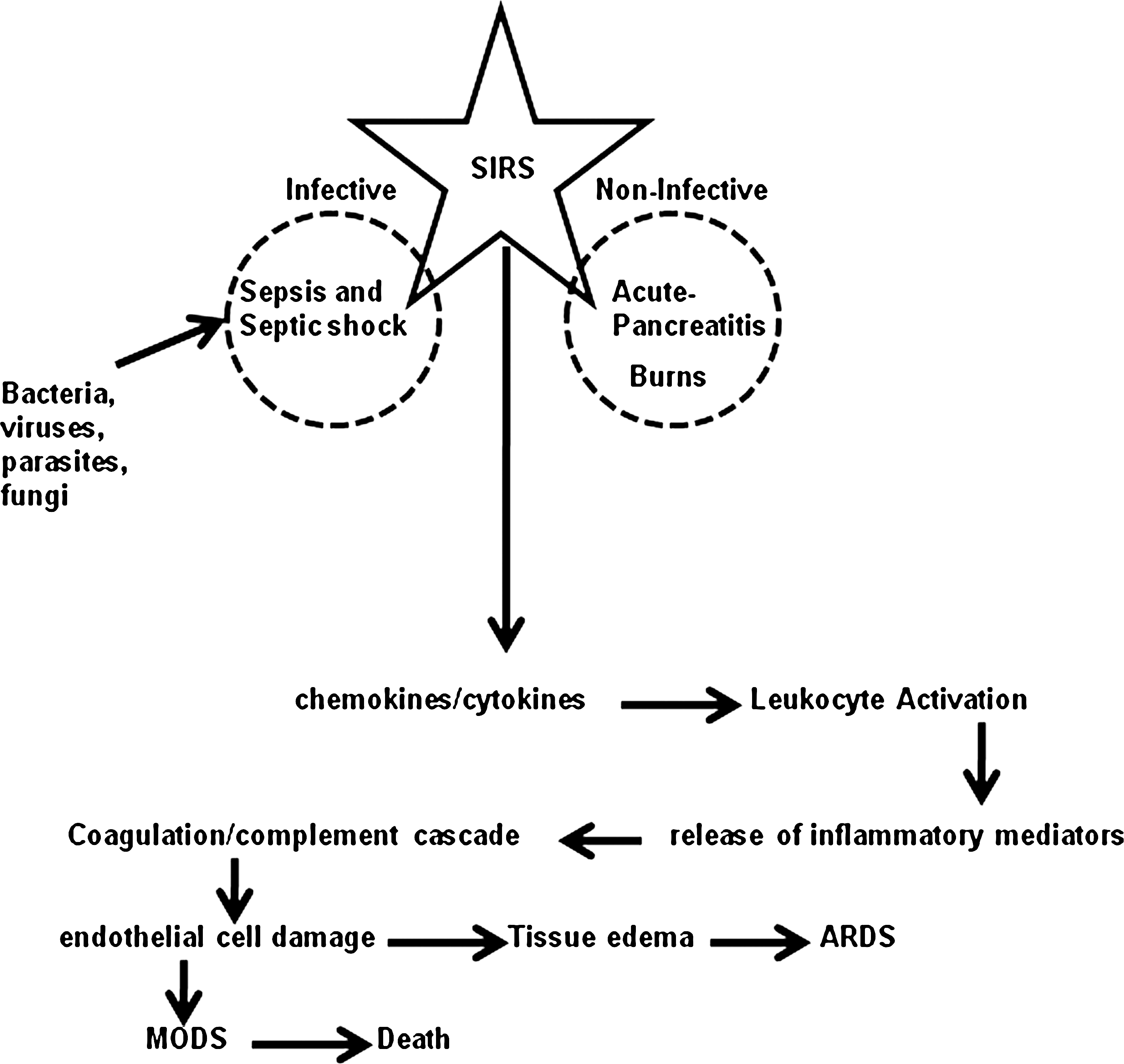
In general terms, SIRS is an entirely normal response to injury. However, cytokine-mediated systemic leukocyte activation, a direct consequence of SIRS, if excessive, can lead to multiple organ dysfunction syndrome (MODS) and multiple organ failure (5). As a consequence of an overactive SIRS response, leukocytes become activated within the general circulation and some then lodge within the pulmonary microcirculation. As the condition develops, leukocytes migrate into the pulmonary interstitium and increased endothelial permeability leads to tissue edema. The leukocytes in the lungs both respond and contribute to the inflammatory process in acute respiratory distress syndrome (ARDS) (5). Several infective and non-infective causes of SIRS are recognised (Fig. 4). Infective causes of SIRS include sepsis and septic shock, infection caused by bacterial pathogens, viruses, fungi, and parasites. Sepsis is defined as a SIRS in which there is an identifiable focus of infection. Noninfective causes of SIRS include acute pancreatitis and burns.
Patients with an attack of ARDS who survive the initial inflammatory insult may die following a relatively minor second event that would not normally be life-threatening. According to the two-hit hypothesis, the initial overactive SIRS somehow primes the inflammatory response. Recovery is possible if no further insult occurs. A relatively minor secondary event such as a line infection or chest infection, will, however, lead to an exaggerated secondary inflammatory response and possibly death (5).
Etiologies of SIRS
Recent work has focused on the identification of inflammatory mediators that play a key role in the pathogenesis of SIRS of different etiologies, and could serve as potential therapeutic targets. Three different etiologies of SIRS—acute pancreatitis, sepsis, and burns (Fig. 4)—are described below.
Acute pancreatitis is a common clinical condition whose incidence has been increasing over recent years (10). In United States alone, >300,000 patients are hospitalized annually with acute pancreatitis, leading to 3,200 deaths. Acute pancreatitis is a contributing factor in an additional 4,000 deaths annually. It also inflicts a heavy economic burden; the direct cost in the United States alone is >$2 billion annually (10). In a majority of patients the condition is mild but ∼25% of patients suffer a severe attack and between 30–50% of these will die (10). Most cases are secondary to biliary disease or excess alcohol consumption. The events that regulate the severity of acute pancreatitis are, for the most part, unknown. The exact mechanisms by which diverse etiological factors induce an attack are still unclear, but once the disease process is initiated common inflammatory and repair pathways are invoked. There is a local inflammatory reaction at the site of injury, which if marked leads to SIRS, and it is this systemic response that is believed to be ultimately responsible for the majority of the morbidity and mortality (10).
Sepsis is defined as the presence of bacteria or their toxins in blood or tissue and the systemic response that follows. Sepsis leading to at least one organ failure characterizes severe sepsis and septic shock is defined by severe sepsis accompanied by hypotension unresponsive to fluid resuscitation. Severe sepsis and septic shock are one of the leading causes of mortality among intensive care unit and postoperational care patients (31). The incidence of sepsis in North America has been reported to be 3.0 per 1,000 population, which transforms into an annual number of 750,000 cases, with 210,000 of them being fatal and a large socioeconomic burden (31). The incidence of mortality due to sepsis is increasing and the most likely causes are the increased incidence of resistant pathogens and the advances of medical and surgical procedures that save the lives of many patients but leave them immunocompromised and in a state highly susceptible to death from severe sepsis and septic shock (31). Sepsis and the events that follow are stages in a progressive condition—a systemic response to infection brought about by various inflammatory mediators, such as cytokines and chemokines. A relationship between cytokine cascades and sepsis has long been established but new evidence suggests that adhesion molecules also play a key role.
Secondary organ damage, and acute lung injury in particular, is a common complication in patients with extensive burns in which the burned area exceeds 30% of the total body surface area. Inflammatory mediators play a key role in the pathogenesis of SIRS and consequent remote organ injury. Skin, a highly immunocompetent organ, reacts to burn injury via a complex inflammatory response. Although dermal inflammation is an important physiologic part of wound healing, excessive local wound inflammation may activate systemic inflammation. Severe burn injuries are known for ongoing and uncontrolled hyperactivation of various host defense mechanisms resulting in SIRS, multiorgan failure, and high patient mortality. The local dermal inflammatory process acts as a lasting trigger, stimulating SIRS via production of cytokines and other inflammatory mediators, activation of neutrophil trafficking, and potentially neural proinflammatory stress signaling. In this scenario, dampening excessive inflammation can be a warranted strategy to limit injurious host response to a noninfectious process. Burn injury robustly induces the dermal production of proinflammatory mediators, resulting in ongoing wound inflammation and tissue edema. In addition to local inflammation, severe dermal burns are known to induce SIRS, which correlates with a high risk of end organ failure as seen in burn-induced acute lung injury (ALI). In the absence of inhalational injury, burn wounds are the inflammatory source triggering SIRS. Several pathophysiological mechanisms are responsible for ALI and remote organ damage after local injury, such as the systemic release of proinflammatory mediators, neutrophil attraction, activation of polymorphonuclear cell (PMN) trafficking, as well as potential activation of sympathetic inflammatory signaling. ALI and ARDS top the list of early burn-induced complications and are associated with high mortality (15).
H2S: A Novel Mediator of Inflammation in SIRS
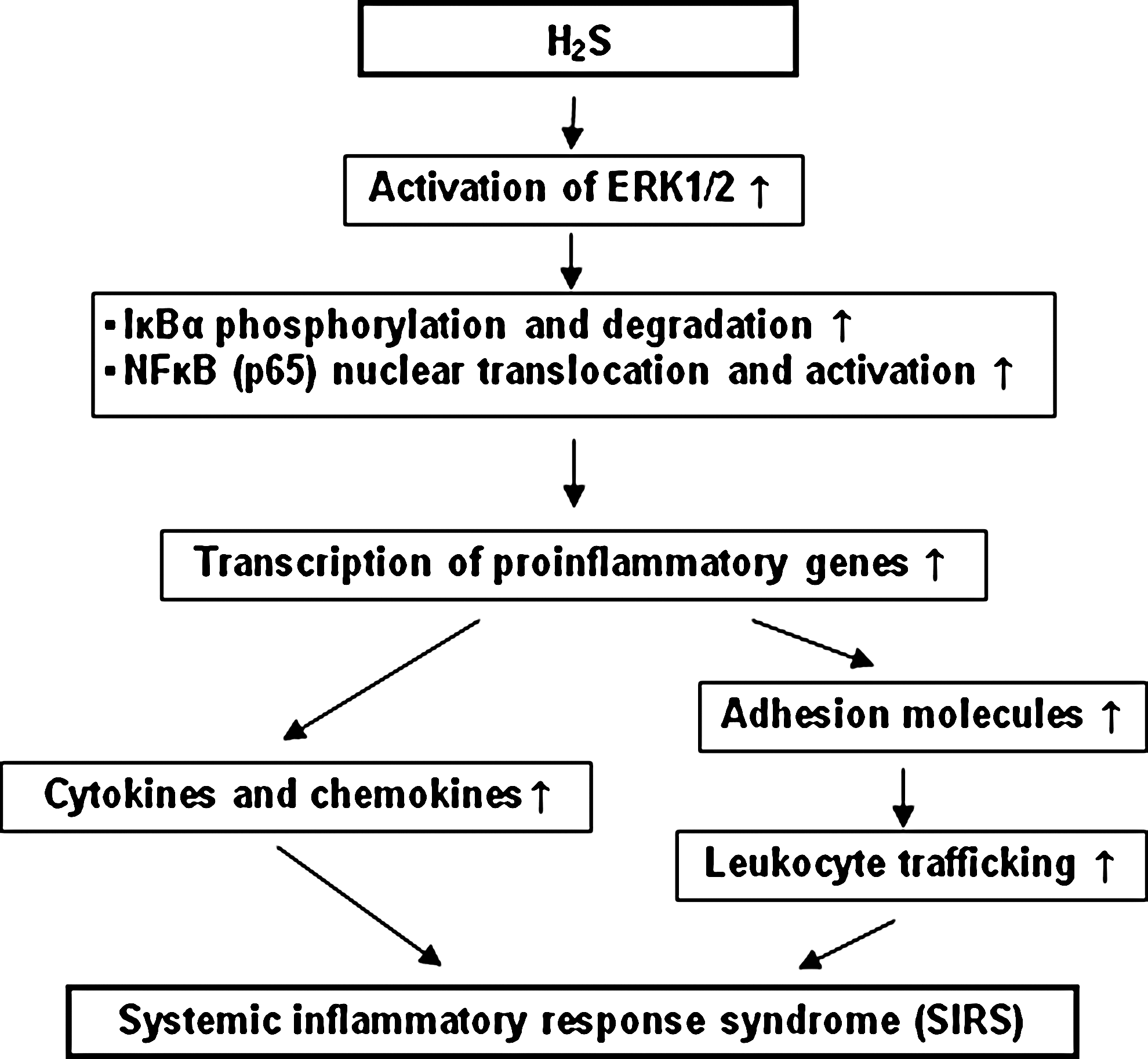
H2S dose-dependently activates human monocytes, thus upregulating the production of proinflammatory cytokines, at least partially via the activation of extracellular signal-regulated kinase (ERK)-NF-κB signaling pathway (57). Pretreatment with NF-κB inhibitor or extracellular signal-regulated protein kinase kinase 1 (MEK) antagonist significantly inhibited H2S induced activation of NF-κB and secretion of proinflammatory cytokines (57). In addition, H2S in vitro provoked the short-term survival of granulocytes via inhibition of caspase-3 cleavage and p38 mitogen-activated protein kinase (MAPK) activation and therefore contributed to the bactericidal activity of neutrophils (36).
H2S in acute pancreatitis
In the first article showing the role of H2S as a mediator of inflammation, we reported the presence of H2S synthesizing enzyme activity and CSE (as determined by mRNA signal) in the pancreas. Also, prophylactic, as well as therapeutic, treatment with the CSE inhibitor, DL-propargylglycine (PAG), significantly reduced the severity of caerulein-induced pancreatitis and associated lung injury (11). These effects of CSE blockade suggested an important proinflammatory role of H2S in regulating the severity of pancreatitis and associated lung injury and raise the possibility that H2S may exert similar activity in other forms of inflammation (11).
In a more recent study (44), we investigated the role of chemokines in H2S-mediated inflammation in acute pancreatitis, using both in vitro and in vivo approaches. In this study we observed a novel interaction between H2S and chemokines. Blockade of H2S biosynthesis with PAG, ameliorates the development of inflammatory process in caerulein-induced acute pancreatitis and acts as an anti inflammatory agent through downregulation of chemokine expression.
H2S in sepsis
Using a clinically relevant model of cecal ligation and puncture (CLP)-induced sepsis, we have shown that H2S acts as a mediator of inflammation in this condition (55). CLP-induced sepsis significantly increased the plasma H2S level and the liver H2S synthesis 8 h after CLP compared with sham operation. Induction of sepsis also resulted in a significant upregulation of CSE mRNA in liver. On the other hand, prophylactic as well as therapeutic administration of PAG significantly reduced sepsis-associated systemic inflammation, as evidenced by decreased myeloperoxidase (MPO) activity and histological changes in lung and liver, and attenuated the mortality in CLP-induced sepsis. Injection of NaHS, an H2S donor, significantly aggravated sepsis-associated systemic inflammation. Therefore, the effect of inhibition of H2S formation and administration of NaHS suggests that H2S plays a proinflammatory role in regulating the severity of sepsis and associated organ injury. Subsequent studies have shown the mechanism by which H2S contributes to inflammation in sepsis. For example, in one study (53) both prophylactic and therapeutic administration of PAG significantly reduced the mRNA and protein levels of IL-1β, IL-6, TNF-α, MCP-1 (CCL-2), and MIP-2 (CXCL-1) in lung and liver, coupled with decreased nuclear translocation and activation of NF-κB in lung and liver. Inhibition of H2S formation also significantly reduced lung permeability and plasma alanine aminotransferase activity. In contrast, injection of NaHS significantly aggravated sepsis-associated systemic inflammation and increased NF-κB activation. In addition, H2S-induced lung inflammation was blocked by the NF-κB inhibitor BAY 11-7082. Therefore, H2S upregulates the production of proinflammatory mediators and exacerbates the systemic inflammation in sepsis through a mechanism involving NF-κB activation. In another study (54), using intravital microscopy, we found that in sepsis, prophylactic and therapeutic administration of PAG reduced leukocyte rolling and adherence significantly in mesenteric venules coupled with decreased mRNA and protein levels of adhesion molecules (ICAM-1, P-selectin, and E-selectin) in lung and liver. In contrast, injection of NaHS upregulated leukocyte rolling and attachment significantly, as well as tissue levels of adhesion molecules in sepsis. Conversely, in normal mice given NaHS (10 mg/kg, i.p.) to induce lung inflammation, NaHS treatment enhanced the level of adhesion molecules and neutrophil infiltration in lung. These alterations were reversed by pretreatment with BAY 11-7082. Moreover, expression of CXCR2 in neutrophils obtained from H2S-treated mice was upregulated significantly, leading to an obvious elevation in MIP-2-directed migration of neutrophils. Therefore, H2S acts as an important endogenous regulator of leukocyte activation and trafficking during an inflammatory response. In a more recent study (52), we have shown that H2S regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. In this study, CLP-induced sepsis resulted in a time-dependent increase in the synthesis of endogenous H2S. Maximum phosphorylation of ERK1/2 and degradation of IκBα in lung and liver were observed 4 h after CLP. Inhibition of H2S formation by PAG significantly reduced the phosphorylation of ERK1/2 in lung and liver 4 h after CLP, coupled with decreased degradation of IκBα and activation of NF-κB. In contrast, injection of NaHS significantly enhanced the activation of ERK1/2 in lung and liver, therefore leading to a further rise in tissue NF-κB activity. As a result, pretreatment with PAG significantly reduced the production of cytokines and chemokines in sepsis, whereas exogenous H2S greatly increased it. In addition, pretreatment with PD98059, an inhibitor of MEK-1, significantly prevented NaHS from aggravating systemic inflammation in sepsis. This study, therefore, showed that H2S may regulate systemic inflammatory response in sepsis via the ERK pathway (Fig. 5). A similar pro-inflammatory action of H2S was also observed in lipopolysaccharide (LPS)-induced endotoxemia (21). Our current understanding of the actions of H2S in these models of inflammation is summarized in Fig. 5.
Substance P: A Novel Mediator of Inflammation in SIRS
Substance P has been shown to upregulate chemokine and chemokine receptor expression in primary mouse neutrophils (39). Substance P primed neutrophils for chemotactic responses not only to the CXC chemokine MIP-2 but also to the CC chemokine MIP-1α (CCL-3). The activating effect of substance P on neutrophils was further evidenced by upregulation of the CD11b integrin, the activation marker of neutrophils. SP induced both the mRNA and protein expression of the chemokines MIP-1α and MIP-2 in neutrophils and upregulated the chemokine receptors CC chemokine receptor (CCR)-1 and CXC chemokine receptor (CXCR)-2. This stimulatory effect on chemokine and chemokine receptor expression in neutrophils was further found to be neurokinin-1 receptor (NK-1R) specific. Pretreatment with selective NK-1R antagonists inhibited substance P-triggered activation of neutrophils and chemokine and chemokine receptor upregulation. Moreover, substance P-induced chemokine upregulation was NF-κB dependent. Also, substance P enhances NF-κB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways (42). In this study (42), we studied the effect of substance P on the murine monocyte/macrophage cell line RAW 264.7, as well as isolated primary macrophages. The data in this article showed that substance P, at nanomolar concentrations, elicited selective chemokine production from murine macrophages. Among the chemokines examined, MIP-2 and MCP-1 are the two major chemokines that were synthesized by macrophages in response to substance P. Furthermore, substance P treatment strongly induced the classic pathway of IκB-dependent NF-κB activation and enhanced DNA binding as well as transactivation activity of the transcription factor. Substance P-evoked transcriptional induction of chemokines was specific, since it was blocked by treatment with selective NK-1R antagonists. Moreover, substance P stimulation of macrophages activated the ERK1/2 and p38 MAPK but not JNKs. Blockade of these two MAPK pathways with specific inhibitors abolished substance P-elicited nuclear translocation of phosphorylated NF-κB p65 and NF-κB-driven chemokine production, suggesting that the two MAPKs lie in the signaling pathways leading to the chemokine response (40). In a more recent study (41), substance P/NK-1R has been shown to activate two convergent proinflammatory signaling pathways, PKCs and phosphoinositide 3-kinase (PI3K)-Akt, resulting in ERK1/2 and NF-κB activation and chemokine production in mouse macrophages. It was found that the conventional PKCα and novel PKCδ and ɛ were selectively activated by substance P via NK-1R on the cells. Activation of these PKC isoforms mediated the activation of downstream ERK1/2 and NF-κB, which drove the transcription of inducible chemokines in macrophages. Additionally, PI3K-Akt was also activated by substance P/NK-1R in macrophages. Inhibition of PI3K-Akt pathway attenuated ERK1/2 and NF-κB activation, suggesting it also played a part in substance P-induced cellular inflammatory response. Kinetic analysis indicated that PKC isoforms induced early ERK1/2 activation, while PI3K-Akt contributed to the pathway at later time points. It was further demonstrated that PKC and PI3K-Akt were activated independent of each other.
In light of the reported role of substance P in some, but not other inflammatory conditions (summarized in ref. 4), its contribution to inflammation in SIRS was investigated.
Substance P in acute pancreatitis
We first investigated the role of substance P and NK-1R in acute pancreatitis and associated lung injury (6). We have found that, in normal mice, substance P levels in the pancreas and NK-1R expression in the pancreatic acinar cells are both increased during secretagogue-induced experimental pancreatitis. To evaluate the role of substance P, pancreatitis was induced by administration of 12 hourly injections of a supramaximally stimulating dose of the secretagogue caerulein in mice that genetically lack NK-1R. During pancreatitis, the magnitude of hyperamylasemia, hyperlipasemia, neutrophil sequestration in the pancreas, and pancreatic acinar cell necrosis were significantly reduced in NK-1R-/- mice when compared with wild-type NK-1R+/+ animals. Similarly, pancreatitis-associated lung injury, as characterized by intrapulmonary sequestration of neutrophils and increased pulmonary microvascular permeability, was reduced in NK-1R-/- animals. These effects of NK-1R deletion indicate that substance P, acting via NK-1R, plays an important proinflammatory role in regulating the severity of acute pancreatitis and pancreatitis-associated lung injury. In a subsequent study (9), the effect of PPT-A gene deletion on the severity of acute pancreatitis and associated lung injury was investigated. The rationale for this study was that substance P acts primarily (but not exclusively) via the NK-1 receptor and neurokinin-A acts primarily via the NK-2R. Also, NK-1Rs bind other peptides in addition to substance P, not all of which are derived from the PPT-A gene. Deletion of PPT-A almost completely protected against acute pancreatitis-associated lung injury, with a partial protection against local pancreatic damage. These results show that PPT-A gene products are critical proinflammatory mediators in acute pancreatitis and the associated lung injury. This work led to the study on treatment for acute pancreatitis with pharmacological intervention against NK-1R using a receptor antagonist CP-96,345 (20). In this study, substance P levels in plasma, pancreas, and lungs were found to be elevated in a caerulein dose-dependent manner. Mice treated with CP-96345, either prophylactically, or therapeutically, were protected against acute pancreatitis and associated lung injury as evident by attenuation in plasma amylase, pancreatic and pulmonary MPO activities, and histological evidence of pancreatic and pulmonary injuries. Pulmonary microvascular permeability was also reduced as a result of CP-96345 treatment. These results point to a key role of NK-1 receptors in acute pancreatitis and associated lung injury as a potential therapeutic target.
Further studies on the mechanism by which substance P contributes to inflammation in acute pancreatitis showed a differential regulation of inflammation in the pancreas and lungs by substance P. In one study (19), the effect of NK-1R blockage on the expression of preprotachykinin genes and neurokinin receptors in acute pancreatitis was investigated. In the pancreas, CP-96,345 treatment resulted in suppression of the elevation of substance P concentration, PPT-A mRNA expression, and NK-1R mRNA and protein expression. In the lungs, the antagonist was found to suppress the increase in substance P concentration, PPT-A mRNA expression and PPT-C mRNA expression. However, the antagonist treatment further promoted the accumulation of pulmonary NK-1R mRNA and protein expression. NK-2R mRNA expression was not detected in normal pancreas. However, upregulated expression of the mRNA for this receptor was observed during acute pancreatitis and treatment with CP96,345 further increased this expression. Pulmonary NK-2R mRNA expression was found to be reduced during acute pancreatitis and CP96,345 treatment normalized this reduction. NK-3R mRNA expression was absent in both pancreas and lung. In another study (18), the effect of a specific NK-1R antagonist, CP-96,345, on the regulation of the expression of adhesion molecules ICAM-1, VCAM-1, E-selectin, and P-selectin as well as leukocyte recruitment during acute pancreatitis was investigated. mRNA expression of the four adhesion molecules was upregulated in the pancreas during acute pancreatitis. Treatment with CP-96,345 effectively reduced the mRNA expression of P-selectin and E-selectin but not ICAM-1 and VCAM-1. In the lung, ICAM-1, E-selectin, and P-selectin mRNA expression increased during acute pancreatitis; antagonist treatment suppressed this elevation. Similar expression patterns were seen in the immunohistochemical stainings. Intravital microscopy of the pancreatic microcirculation revealed the effect of CP-96,345 on leukocyte recruitment. This study provided important information on the relationship between NK-1R activation and the regulation of adhesion molecules. Also, these two studies pointed to the differential regulation of inflammation in the pancreas and lung with acute pancreatitis.
Isolated pancreatic acini were used as the experimental system to demonstrate a stimulation of chemokine synthesis by substance P (30). Exposure of mouse pancreatic acini to substance P significantly increased synthesis of MCP-1, MIP-1α, as well as MIP-2. Furthermore, substance P also increased NF-κB activation. The stimulatory effect of substance P was specific to chemokine synthesis through the NF-κB pathway, since the increase in chemokine production was completely attenuated when pancreatic acini were pretreated with the selective NF-κB inhibitor, NF-κB essential modulator-binding domain peptide. This study showed that substance P-induced chemokine synthesis in mouse pancreatic acinar cells is NF-κB dependent. Furthermore, substance P-induced chemokine synthesis is dependent on MAPKs- ERK1/2 and JNK, as well (32). A time-dependent activation of ERK1/2, JNK, NF-κB, and AP-1 was observed when pancreatic acini were stimulated with substance P. Moreover, substance P-induced ERK1/2, JNK, NF-κB, and AP-1 activation as well as chemokine synthesis were blocked by pretreatment with either MEK1 inhibitor or JNK inhibitor. In addition, substance P-induced activation of ERK1/2, JNK, NF-κB, and AP-1-driven chemokine production were attenuated by CP-96345 in pancreatic acinar cells. These results suggested that substance P-NK-1R induced chemokine production depends on the activation of MAPKs-mediated NF-κB and AP-1 signaling pathways. In a subsequent study (33), a key role of PKC-δ on substance P-induced chemokine synthesis in pancreatic acinar cells has been shown. In this study, we showed that substance P stimulated an early phosphorylation of PKC-δ followed by increased activation of MAPKKK, MEKK1, and MAPK ERK and JNK, as well as transcription factor NF-κB and AP-1 driven chemokine production. Depletion of PKC-δ with its inhibitor rottlerin or the specific PKC-δ translocation inhibitor peptide dose dependently decreased substance P-induced PKC-δ, MEKK1, ERK, JNK, NF-κB, and AP-1 activation. Moreover, rottlerin as well as PKC-δ translocation inhibitor inhibited substance P-induced chemokine production in a concentration-dependent manner. PKC-δ activation was attenuated by CP-96345, thus showing that PKC-δ activation was indeed mediated by substance P in pancreatic acinar cells. A crucial role of calcium in substance P-induced chemokine synthesis in mouse pancreatic acinar cells has also been demonstrated (35). In this study, substance P-induced chemokine production in pancreatic acinar cells resulted from PLC-induced elevated intracellular calcium and PKCα/βII activation, subsequently leading to the activation of MAPKs (ERK and JNK) and transcription factors NF-κB and AP-1. More recently, a critical role of SRC family kinases (SFKs) in substance P-induced chemokine production in mouse pancreatic acinar cells and its significance in acute pancreatitis has been demonstrated (34). Using primary preparations of mouse pancreatic acinar cells as the model system, we have shown that substance P/NK-1R induced the activation of SFKs. SFKs mediated the activation of MAPKs (ERK, JNK), transcription factors [signal transducer and activator of transcription (STAT) 3, NF-κB, AP-1], and production of chemokines in pancreatic acinar cells. We further tested the significance of the SFK signaling pathway in acute pancreatitis. Our results show, for the first time, that treatment of mice with a potent and selective SFK inhibitor, PP2 [4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo [3,4-D] pyrimidine], but not its negative inhibitor PP3 (4-amino-7-phenylpyrazol [3,4-D] pyrimidine), reduced the severity of pancreatitis. This was proven by significant attenuation of hyperamylasemia, pancreatic MPO activity, chemokines, and water content. Histological evidence of diminished pancreatic injury also confirmed the protective effect of the inhibition of SFKs. Moreover, treatment with CP-96345 attenuated acute pancreatitis-induced activation of SFKs, ERK, JNK, STAT3, NF-κB, and AP-1. In light of the studies summarized above, it would be reasonable to propose that drugs targeting the substance P-mediated signaling pathways could prove beneficial in improving treatment efficacy in acute pancreatitis.
Substance P in sepsis
Substance P has been shown to play a key pro-inflammatory role in sepsis as well. In order to investigate the role of PPT-A gene products in lung injury in sepsis, polymicrobial sepsis was induced by cecal ligation and puncture in PPT-A gene-deficient mice (PPT-A-/-) and the wild-type control mice (PPT-A+/+). PPT-A gene deletion significantly protected against mortality, delayed the onset of lethality, and improved the long-term survival following cecal ligation and puncture-induced sepsis. PPT-A-/- mice also had significantly attenuated inflammation and damage in the lungs. These data (29) suggested that deletion of the PPT-A gene may have contributed to the disruption in recruitment of inflammatory cells resulting in protection against tissue damage, as in these mice the sepsis-associated increase in chemokine levels is significantly attenuated. Similarly, PPT-A gene products have been shown to play a key role in multiple organ injury in LPS-induced endotoxemia (26). Administration of LPS to WT mice caused a significant increase in circulating levels of SP as well as in liver, lung, and kidney. PPT-A gene deletion significantly protected against liver, pulmonary, and renal injury following LPS-induced endotoxemia, as evidenced by tissue MPO activities, plasma alanine aminotransferase, aspartate aminotransferase levels, and histological examination. Furthermore, PPT-A-/- mice had significantly attenuated chemokines, proinflammatory cytokines, and adhesion molecule levels in the liver, lung, and kidney. These results showed that PPT-A gene products are critical proinflammatory mediators in endotoxemia and the associated multiple organ injury. In addition, the data suggest that deletion of the PPT-A gene protected mice against organ damage in endotoxemia by disruption in neutrophil recruitment. Furthermore, treatment with an NK-1R antagonist (SR140333) protected mice against lung injury in polymicrobial sepsis (17). SR140333 injected 30 min before or 1 h after CLP significantly attenuated the increased lung MPO activity and levels of MIP-2, MCP-1, IL-1β, IL-6, ICAM-1, and E- and P-selectin compared with CLP-operated mice injected with the vehicle. Histological evaluation of the lung sections further supported the beneficial effect of SR140333 on lung inflammation. Therefore, SP receptor antagonism can be a potential therapeutic target in polymicrobial sepsis, and this effect is brought about via reduction in leukocyte recruitment.
Substance P in burns
Substance P has recently been shown to act as a critical mediator of burn-induced acute lung injury (37). The results in this study show that burn injury in male BALB/c mice subjected to 30% total body surface area full thickness burn augments significant production of substance P, PPT-A gene expression, and biological activity of substance P-NK-1R signaling. Furthermore, the enhanced substance P-NK-1R response correlates with exacerbated lung damage after burn as evidenced by increased microvascular permeability, edema, and neutrophil accumulation. The development of heightened inflammation and lung damage was observed along with increased proinflammatory IL-1β, TNF-α, and IL-6 mRNA and protein production after injury in lung. Chemokines MIP-2 and MIP-1α were markedly increased, suggesting the active role of substance P-induced chemoattractants production in trafficking inflammatory cells. More importantly, administration of L703606, a specific NK-1R antagonist, significantly disrupted the substance P-NK-1R signaling and reversed pulmonary inflammation and injury. These findings have shown for the first time the role of substance P in contributing to exaggerated pulmonary inflammatory damage after burn injury via activation of NK-1R signaling. More work is obviously needed in order to clearly define the role of substance P in SIRS associated with burn injury, but these early results point to the promise of substance P as a therapeutic target for burns and associated SIRS. Our current understanding of the actions of substance P in different models of inflammation is summarized in Fig. 6.
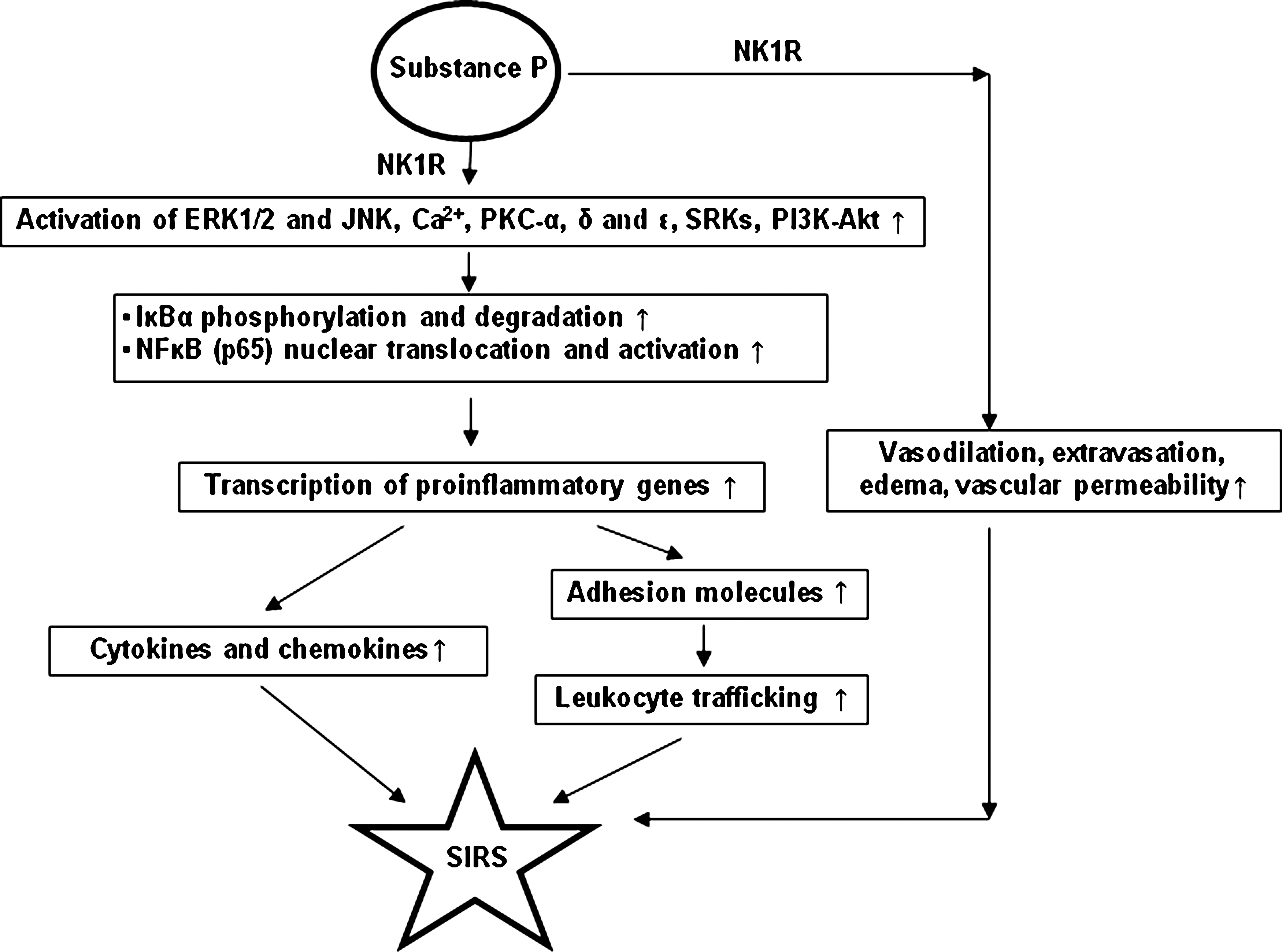
H2S and Substance P in Inflammation: Are They Doing Anything Together?
In view of the pro-inflammatory actions of H2S and substance P in inflammation, it has been interesting to investigate interaction between these two mediators. In the first report on the direct interaction between H2S and substance P (12), we found that intraperitoneal administration of NaHS, an H2S donor, to mice caused a significant increase in circulating levels of substance P in a dose-dependent manner. H2S, by itself, could also cause lung inflammation, as evidenced by a significant increase in lung MPO activity and histological evidence of lung injury. The maximum effect of H2S on substance P levels and on lung inflammation was observed 1 h after NaHS administration. At this time, a significant increase in lung levels of TNF-α and IL-1β was also observed. In PPT-A-/- mice, H2S did not cause any lung inflammation. Furthermore, pretreatment of mice with CP-96345 protected mice against lung inflammation caused by H2S. However, treatment with antagonists of NK-2, NK-3, and CGRP receptors did not have any effect on H2S-induced lung inflammation. Depleting neuropeptide from sensory neurons by capsaicin significantly reduced the lung inflammation caused by H2S. In addition, pretreatment of mice with capsazepine, an antagonist of the transient receptor potential vanilloid-1 (TRPV-1), protected mice against H2S-induced lung inflammation. These results demonstrated a key role of substance P and neurogenic inflammation in H2S-induced lung injury in mice.
This interaction between H2S and substance P is seen in SIRS of different etiologies as well. For example, in acute pancreatitis, PAG, given prophylactically as well as therapeutically, significantly reduced substance P concentrations in plasma, pancreas, and lung (8). Furthermore, prophylactic as well as therapeutic administration of PAG significantly reduced PPT-A mRNA expression and NK-1R mRNA expression in both pancreas and lung when compared with caerulein-induced acute pancreatitis. These results suggested that the pro-inflammatory effects of H2S may be mediated by SP-NK-1R pathway in acute pancreatitis. In isolated pancreatic acini (45), inhibition of endogenous production of H2S by PAG significantly suppressed caerulein-induced increase in substance P concentration, PPT-A expression, and NK-1R expression. To determine whether H2S itself provoked inflammation in acinar cells, the cells were treated with NaHS, that resulted in a significant increase in substance P concentration and expression of PPT-A and NK-1R.
Furthermore, H2S acts through substance P in sepsis as well. In a study that showed evidence on this interaction (51), PAG pretreatment or post-treatment significantly decreased the PPT-A gene expression and the production of substance P in lung, whereas administration of NaHS resulted in a further rise in the pulmonary level of substance P in sepsis. PPT-A gene deletion and pretreatment with L703606 prevented H2S from aggravating lung inflammation. In addition, septic mice genetically deficient in PPT-A gene or pretreated with L703606 did not exhibit further increase in lung permeability after injection of NaHS. These findings showed that in sepsis, H2S upregulates the generation of substance P that contributes to lung inflammation and lung injury mainly via activation of the NK-1R. Other studies have also suggested that H2S may regulate the release of SP in guinea pig airways and rat urinary bladder via the TRPV1 on sensory nerve endings (28, 46).
Transition to the Clinic
Many of today's medical illnesses can be attributed directly or indirectly to problems with inflammation. SIRS contributes to the majority of morbidity and mortality in several, as yet incurable clinical conditions such as acute pancreatitis, sepsis, and severe burns.
It is only recently that H2S has been identified as a mediator of inflammation and the role of substance P in SIRS is also just beginning to be understood. Clinical studies have just been started to investigate these mediators in clinical inflammatory conditions, and the early results are promising. For example, H2S has recently been shown to be elevated in chronic obstructive pulmonary disease (COPD), and its alteration in level may be connected with disease activity and severity (14). In yet another study, H2S levels have been shown to be elevated in the exhaled breath of chronic pancreatitis patients (24). Very recently, we have shown that circulating levels of substance P and H2S are elevated in patients with severe acute pancreatitis as against those with mild disease (16, 49). These early results are indeed promising, and point to the clinical potential of research on the role of H2S and substance P in inflammation. It is important to also note that, although the subject matter of this review has been inflammation involving SIRS, H2S, and substance P have also been shown to play an important role in other inflammatory conditions, such as joint inflammation/arthritis (7, 27).
More basic research into the mechanisms by which H2S and substance P contribute to inflammation in SIRS is, however, needed before one could take these studies to the clinic. Active research in this direction will facilitate a transition of our knowledge from bench to bedside.
Footnotes
Acknowledgments
The author would like to acknowledge grant support from National Medical Research Council and Biomedical Research Council.