
Abstract
The goal of this study was to investigate the possible role of reactive oxygen species (ROS) in signaling, in modulation of the cytoskeleton, and in differentiation of fibroblasts. For this purpose, we have applied a novel mitochondria-targeted antioxidant: plastoquinone conjugated with decyltriphenylphosphonium (SkQ1). This antioxidant at nanomolar concentration prevented ROS accumulation and cell death induced by H2O2 in fibroblasts. We found that scavenging of ROS produced by mitochondria activated the Rho/ROCK/LIMK signaling pathway that was followed by phosphorylation of cofilin and stabilization of actin stress fibers. The mitochondria-targeted antioxidant induced differentiation of human subcutaneous fibroblasts to myofibroblasts as revealed by expression of fibronectin isoform (EDA-FN) and smooth muscle actin (α-SMA). This effect was shown to be mediated by transforming growth factor β1 (TGFβ1), which was activated by matrix metalloprotease 9 (MMP9) in the culture medium. Scavenging of ROS stimulated secretion of MMP9 rather than its processing. The same effect was achieved by the nontargeted antioxidant Trolox at higher concentration, but the thiol antioxidant N-acetylcysteine (NAC) inhibited MMP activity and was not able to induce myofibroblast differentiation. The myofibroblast phenotype was supported due to autocrine TGFβ1-dependent stimulation after removal of SkQ1. It is concluded that ROS scavenging in mitochondria induces TGFβ1-dependent myofibroblast differentiation. Antioxid. Redox Signal. 13, 1297–1307.
Introduction
Transforming growth factor β1 (TGFβ1) is a key factor inducing myofibroblast differentiation during wound healing as well as in pathological fibrosis. Exogenous TGFβ1 induces formation of myofibroblasts in different fibroblast cultures. Myofibroblast differentiation is also regulated by other cytokines, chemokines, and adhesion molecules (EGF, FGF-2, IL-1, IFN-γ). When differentiation is induced by these factors, it probably also depends on active TGFβ1 (19). Expression of α-SMA and other specific myofibroblasts proteins (EDA-FN, collagen I, etc) depends on TGFβ1-induced stimulation of the SMAD pathway in combination with other pathways (13, 36). Moreover, RhoA pathways can be rapidly activated by TGFβ1 by SMAD-independent mechanisms (13). These pathways are known to be involved in cytoskeleton remodeling.
TGFβ1 is secreted by the cell as a large latent complex (TGFβ–LAP/LTBP) covalently bound to the ECM (3). Physiological activation of TGFβ1 can be induced by proteolytic cleavage of this latent complex by plasmin or metalloproteases (MMP2 and MMP9). However, cleavage-independent activation is also possible. It is mediated by the interaction of the complex with thrombospondin-1 or integrins, as well as low pH or oxidative stress (3, 39, 40).
Reactive oxygen species (ROS) function as second messengers for signal transduction induced by many cytokines (including TGFβ1, EGF, PDGF), leading to proliferation, differentiation, or cell death (10, 11, 37). Mitochondria are one of the major sources of ROS in the cell and can, in principle, contribute to regulation of various signaling pathways (11, 18, 29 –31), but it remains unclear whether mitochondrial ROS are specifically required for this regulation. Studies in this direction are greatly hindered by the absence of specific tools for measurement and modification of ROS production in mitochondria. Recently, mitochondria-targeted antioxidants were developed and applied to studies of cell signaling. The first mitochondria-targeted antioxidant MitoQ was synthesized and studied by Murphy and coworkers (23). A novel group of mitochondria-targeted antioxidants called SkQs was recently developed in our laboratory. SkQ1 [10-(6′-plastoquinonyl) decyltriphenylphosphonium] was found to be more efficient as an antioxidant than MitoQ in artificial membranes, isolated mitochondria, cells in culture, and some animal models (1, 4, 5, 7, 27, 32, 35). Using SkQ1, we have shown that ROS produced by mitochondria are critical for fragmentation of mitochondrial reticulum and apoptosis induced by exogenous hydrogen peroxide or by inhibitors of mitochondrial functions (4). It was found that mitochondrial ROS are involved in cytoskeleton remodeling in fibroblasts and epithelial cells during neoplastic transformation in vitro and in progression of some tumors in vivo (1). In the present study, we have applied SkQ1 and some other antioxidants to studies on fibroblast-to-myofibroblast differentiation. We show for the first time that scavenging of mitochondrial ROS strongly stimulates this differentiation.
Materials and Methods
Cells
Normal human subcutaneous diploid fibroblasts (HSCF, Cell Culture Collection, Institute of Medical Genetics, Russian Academy of Medical Sciences, Moscow) were used. Cells were cultured in DMEM (Dulbecco's Modified Eagle's Medium) (Gibco, Carlsbad, CA) supplemented with 10% FBS (HyClone, San Jose, CA) at 37°C with 5% CO2. Cells between 5–10 passages were used for experiments. It was found that the level of α-SMA and EDA-FN (markers of myofibroblasts) was very low in these cells. Some spontaneous differentiation of fibroblasts was observed at later passages.
Accumulation of SkQ1 in mitochondria
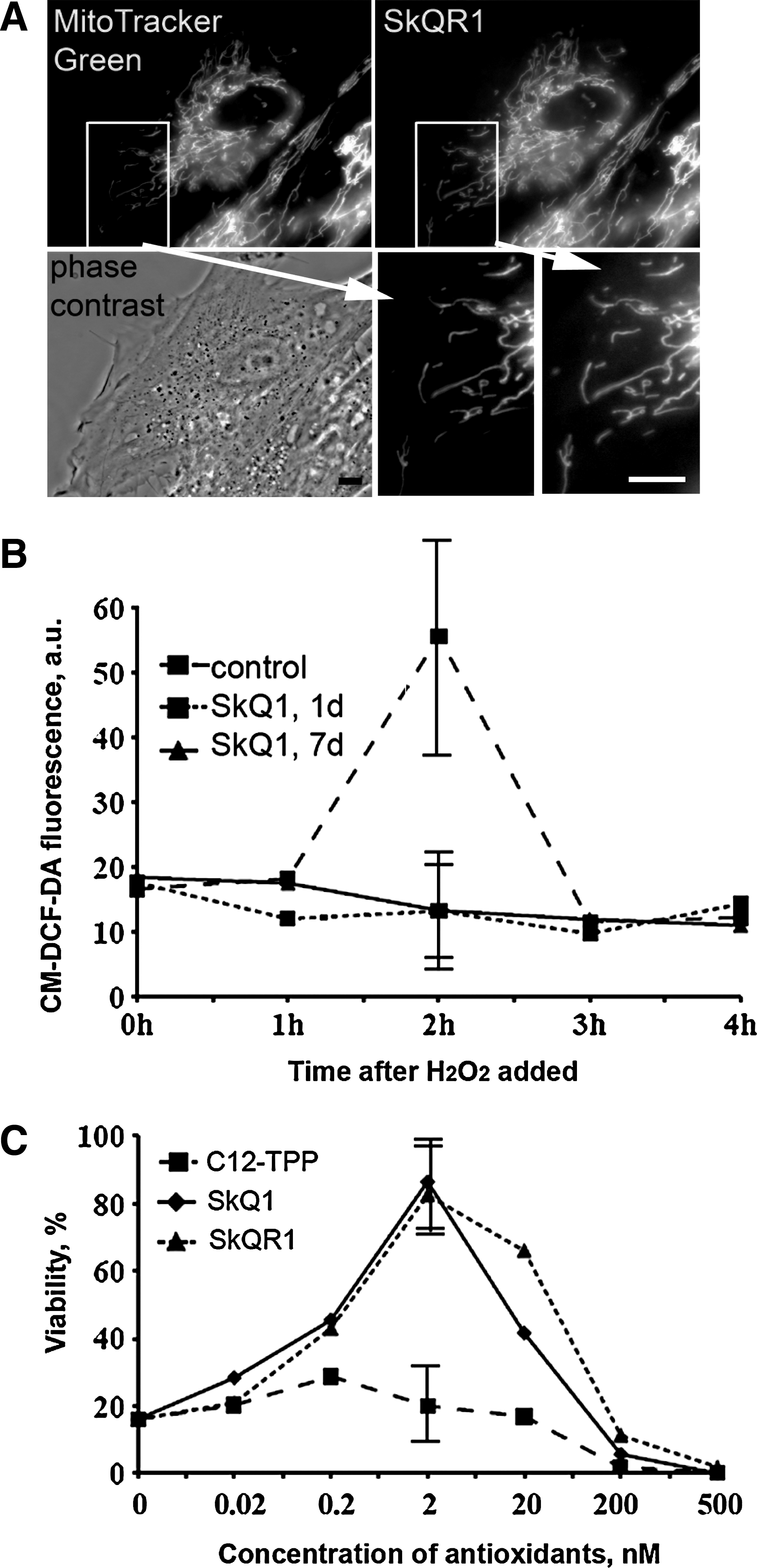
To study the localization of SkQ1 in cells, we used its fluorescent analog SkQR1 [10-(6′-plastoquinonyl) decylrhodamine-19], where the triphenylphosphonium cationic group is replaced by rhodamine-19 (4). Mitochondria were visualized with MitoTracker Green (Molecular Probes, Eugene, OR). Cells were treated with SkQR1 (20 nM, 2 h) and MitoTracker Green (300 nM, 30 min, 37°C) and analyzed using an Axiovert microscope(Carl Zeiss) equipped with a Neofluar 100 × NA 1.3 objective. The thickness of the optical slices was about 1 μm.
Measure of ROS production
SkQ1-treated cells were incubated with 200 μM H2O2 in the medium. ROS production was measured after loading the cells with 5 μM CM-DCF-DA (Molecular Probes) for 15 min at 37°C, as described earlier (1, 22). Cells were analyzed with an FC 500 cytofluorimeter (Beckman Coulter, Brea, CA).
Cell viability
After incubation with antioxidants, the cells were treated with H2O2 for 24 h, and cell viability was determined with CellTiterBlue reagent (Promega Corporation, Madison, WI). Intensity of fluoresce was measured with a Fluoscan Ascent fluorimeter (Thermo Electron Corporation, San Jose, CA).
Western blotting analysis
Cell extracts were prepared as described earlier (1), and protein concentration of the extracts was determined using the RC DC protein assay system (BioRad, Hercules, CA). Protein samples (5–10 μg) were separated on SDS-polyacrylamide gel and transferred to PVDF membrane (Amersham Biosciences, Piscataway, NJ). The membrane was probed with antibodies specific to α-SMA (Sigma), EDA-FN (a kind gift of Dr. Zardi (33)), SMAD2, phospho-SMAD2, and cofilin or phospho-cofilin (Cell Signaling Technology, Danvers, MA). Antibodies against α-tubulin or vimentin (Sigma) were used as a loading control. Membranes were treated with HRP-conjugated secondary antibody (Sigma) and developed with ECL chemiluminescence reagents (Amersham) according to the manufacturer's protocol. ImageJ software (National Institutes of Health,
Immunostaining and morphometry
Cells were fixed with 2% paraformaldehyde followed by 5 min treatment with methanol at −20°C for immunofluorescence staining or with 0.2% Triton X-100 for actin staining with phalloidin-TRITC (Sigma). Antibodies against α-SMA, actin, vinculin (Sigma), and EDA-FN were used. Goat anti-mouse antibodies conjugated with Alexa-488 (Molecular Probes) and TRITC-conjugated antibodies against rabbit immunoglobulins (Jackson Labs, Bar Harbor, ME) were used as secondary antibodies. Images were acquired using an Axiovert microscope equipped with objectives 20× (dry) and 100× (oil immersion Neofluar) (Carl Zeiss, Jena, Germany). The ImageJ software was used for morphometry. Elongation index was calculated as log2(major radii/minor radii) (16).
Zymography
Zymography of MMP activities were measured as described earlier (9, 24). Electrophoresis was performed on samples (30 μl culture medium per lane) at 4°C in 7.5% polyacrylamide gels containing 10% SDS and gelatin (1 mg/ml) under nonreducing conditions and without boiling of samples. The ImageJ software was used for densitometric analysis of bands.
ELISA
For TGFβ1 detection, culture medium was collected and immediately analyzed by sandwich ELISA as recommended by the manufacturer's instruction (R&D Systems, Minneapolis, MN). Latent TGFβ1 in the medium was activated by acidification to measure total TGFβ1. The recombinant human soluble receptor II of TGFβ1 (TGFβRII) (R&D Systems) was used as a capture reagent in combination with biotinylated TGFβ1 polyclonal detection antibody (R&D Systems) to measure active TGFβ1.
Statistical assay
Statistica 6.0 software was used for statistical analyses. Data presented as bar graphs are means ± SEM. Sample size: N ≥ 3 for Western blot analysis and zymography, N ≥ 500 for morphometry, N ≥ 15 for ELISA. Statistical analysis was performed using Student's t-test. The p values <0.05 (*), <0.01 (**), and <0.001 (***) are considered as statistically significant.
Results
SkQ1 inhibits oxidative stress and cell death in HSCF
SkQ1 has demonstrated very high antioxidant efficiency in model systems and isolated mitochondria (5). In experiments with HeLa cells, it was shown that SkQ1 and SkQR1 accumulated in mitochondria and prevented H2O2-induced apoptosis at nanomolar concentration (4). Cellular localization and antioxidant activity of SkQ were analyzed in the HSCF model to verify these observations. SkQR1, a fluorescent SkQ analog, was selectively accumulated in mitochondria of HSCF as shown by colocalization with MitoTracker Green (Fig. 1A). When oxidative stress was induced in HSCF by exogenous H2O2, a secondary delayed production of endogenous ROS was detected, which later decreased to the background level. Pretreatment of the cells with SkQ1 (2 nM) completely abolished this effect of H2O2 (Fig. 1B). SkQ1 and SkQR1 also prevented decrease in cell viability induced by H2O2, and the maximal protection level was observed at 2 nM concentration of these antioxidants (Fig. 1C). It was found here that SkQ1 induced activation of TGFβ1 (see below) which could resulted in some cell resistance against apoptogenic stimuli. However, in control experiments, 24 h incubation of fibroblasts with recombinant TGFβ1 did not affect H2O2-induced cell death (not shown). C12TPP, the SkQ analog lacking the antioxidant (plastoquinone) moiety, failed to preserve viability of H2O2-treated fibroblasts (Fig. 1C). The protonophorous uncoupler FCCP, which depolarizes the inner mitochondrial membrane, prevented accumulation of SkQR1 in mitochondria as well as the protective action of SkQ1 and SkQR1 (not shown). The extremely high efficiency of mitochondria-targeted antioxidant and the effect of FCCP indicate that under oxidative stress the mitochondrion is the major source of secondary ROS causing loss of cell viability.
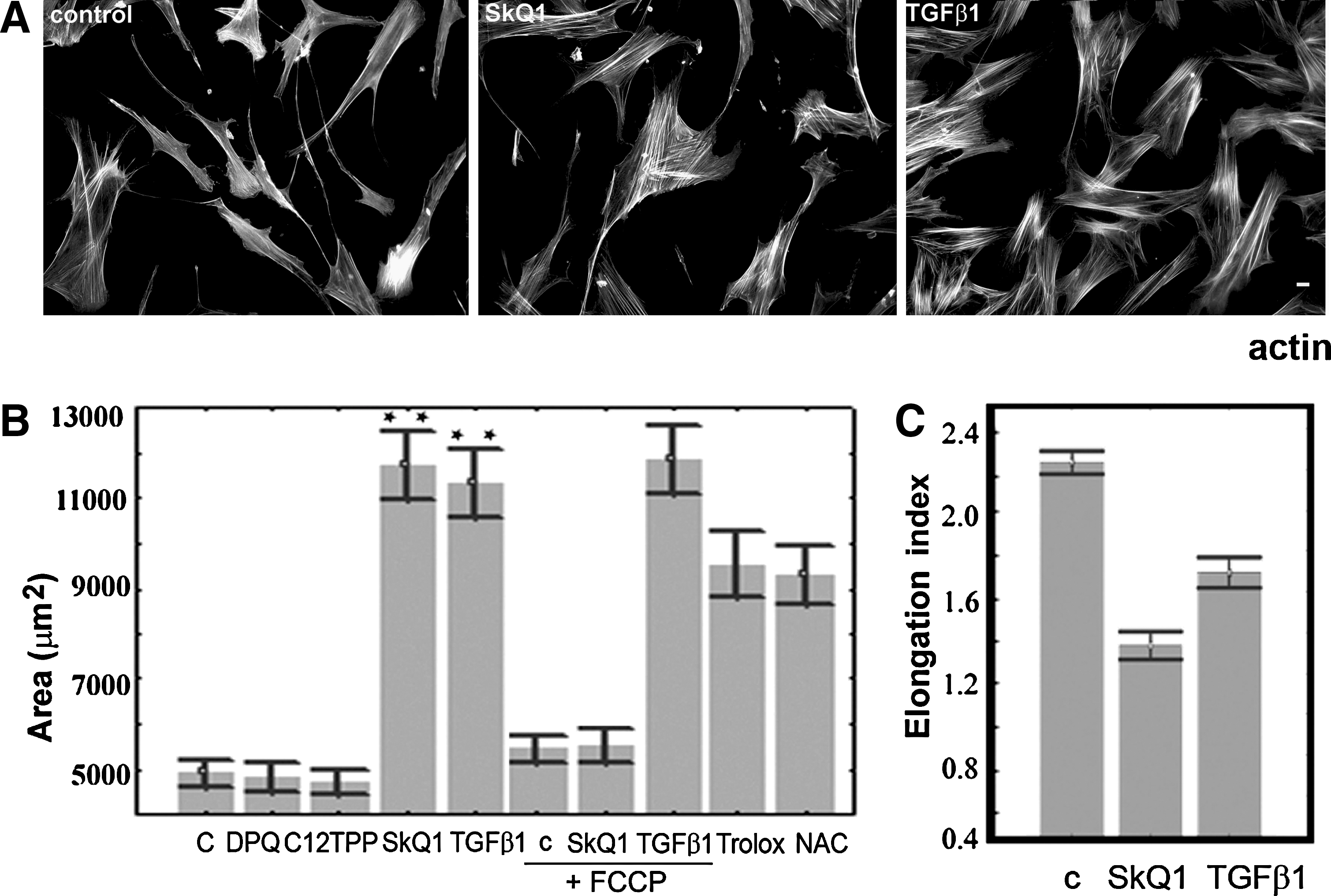
SkQ1 induces differentiation of fibroblasts into myofibroblasts
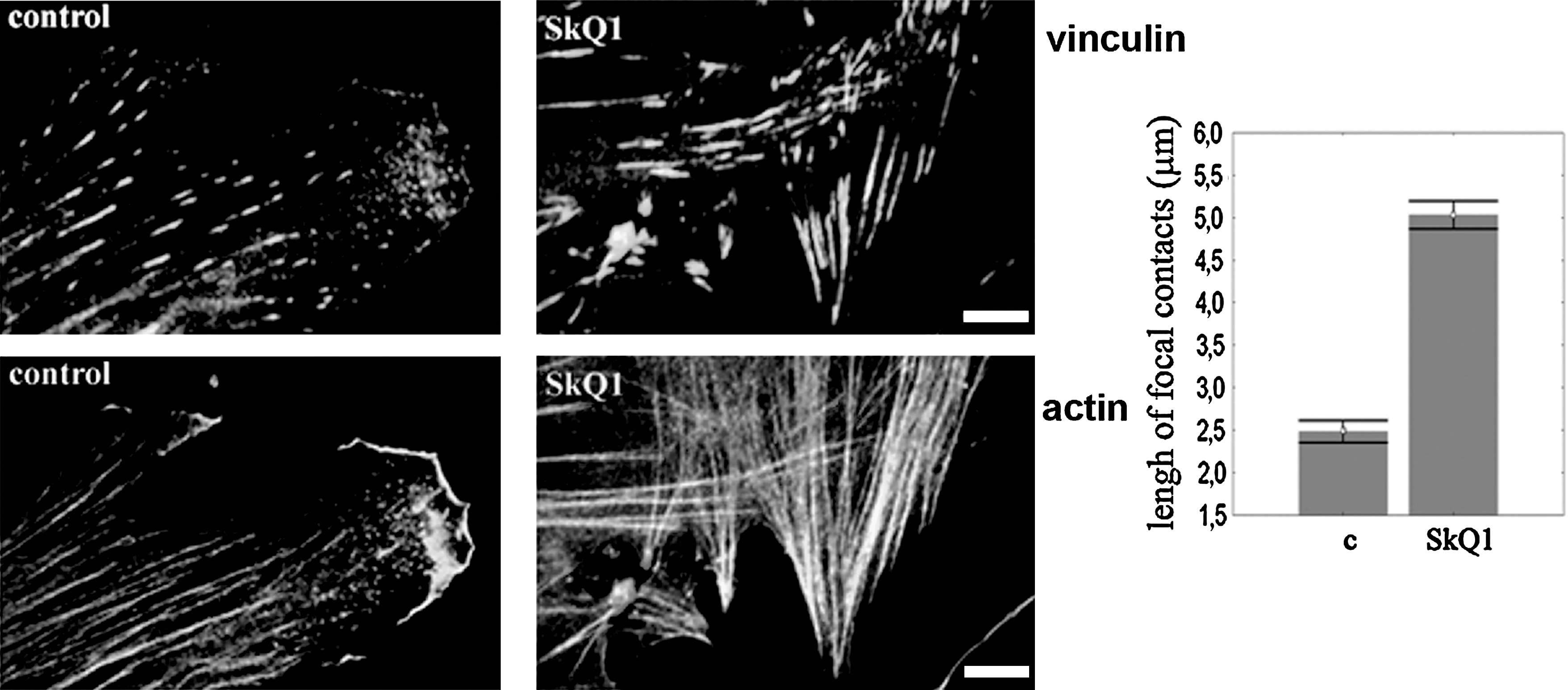
Prolonged incubation of HSCF with SkQ1 induced dramatic changes in cell morphology. The area occupied by cells on the substrate was doubled, while their elongation index was decreased (Fig. 2). The changes in morphology were accompanied by a significant increase in number and thickness of actin stress fibers (Fig. 2A). Accumulation of the stress fibers was detected at 12–24 h and continued for 3–5 days after addition of SkQ1. Lengthening of vinculin-positive focal contacts at the ends of actin fibers was observed, indicating maturation of the focal adhesions (Fig. 3). These effects on cell morphology remained for at least 7 days after removal of SkQ1. The effects of SkQ1 were mediated by its antioxidant activity, since C12TPP was completely ineffective (Fig. 2B). Accumulation of the antioxidant in mitochondria was critical for the observed action, and its uncharged analog decylplastoquinone (DPQ) was ineffective. Depolarization of the mitochondrial membrane induced by the protonophorous uncoupler FCCP prevented electrophoretic accumulation of SkQ1 in mitochondria and abolished its effects on cell morphology. In control experiments, FCCP did not induce significant changes in the fibroblast morphology and did not prevent spreading of the cells induced by TGFβ1 (the role of TGFβ1 is explained below), indicating that depolarization of mitochondria did not interfere with the transitions of fibroblast phenotype. The classical antioxidants Trolox (a water-soluble analog of vitamin E) and N-acetylcysteine (NAC) induced similar changes in morphology and cytoskeleton of fibroblasts (Fig. 2B), but only at much higher concentrations (0.1 mM and 5 mM, respectively), confirming the key role of mitochondrial ROS as determinants of fibroblast morphology.
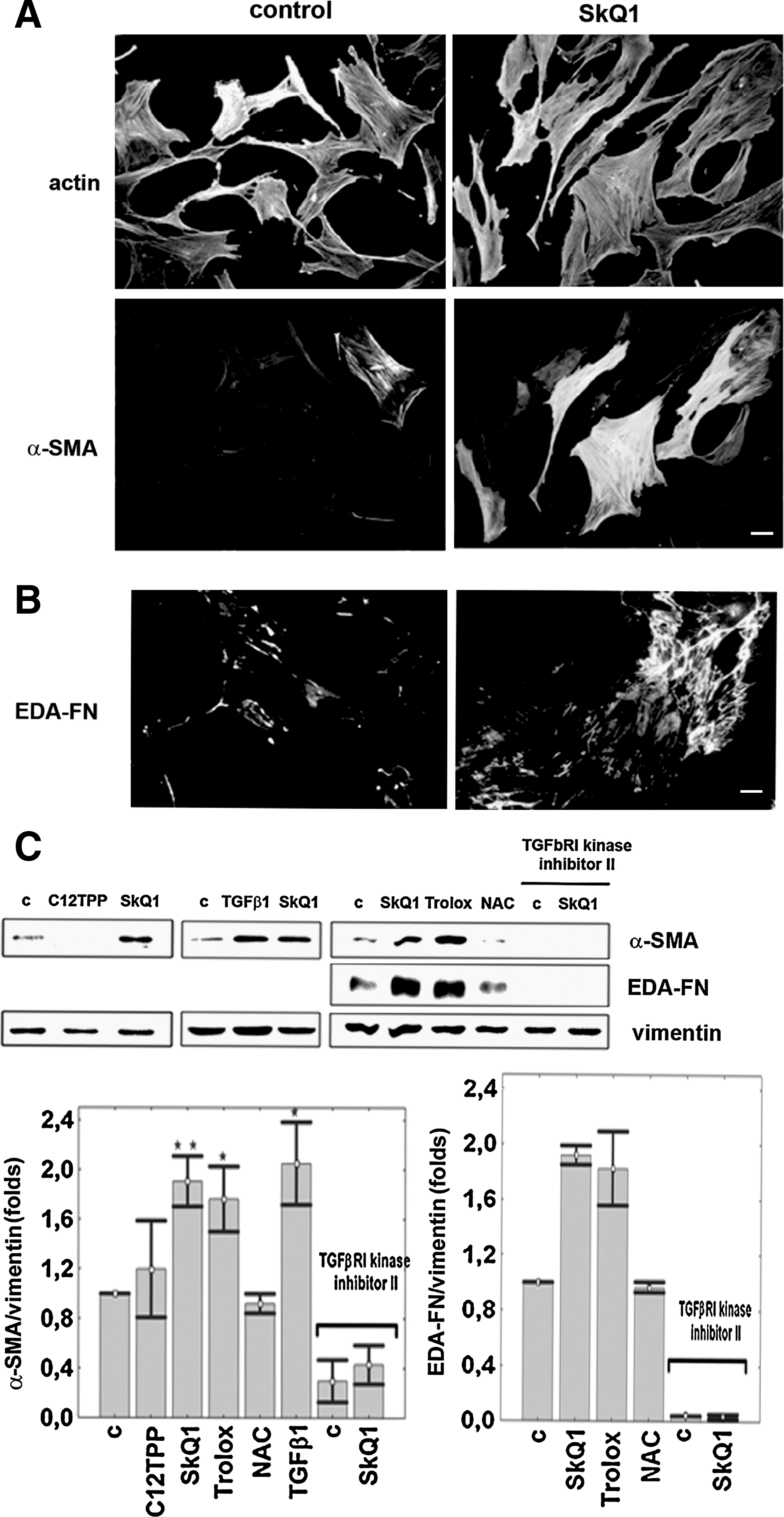
The changes in HSCF induced by SkQ1 made their phenotype similar to that ascribed to myofibroblasts (15, 19, 20). We analyzed expression of the major myofibroblast markers EDA-FN and α-SMA. The appearance of EDA-FN was more pronounced after 1–3 days of treatment with SkQ1. The expression of α-SMA became detectable on days 3–4 and well-pronounced on days 5–7 of the treatment (Fig. 4). EDA-FN formed a typical network in the extracellular matrix, and α-SMA was incorporated in stress fibers. The same effect of expression of myofibroblast differentiation markers was observed after incubation with 0.1 mM Trolox (Fig. 4C), but NAC was ineffective.
The major mechanism of stress fiber stabilization is related to phosphorylation and inactivation of cofilin (8). This protein interacts with F-actin, preventing formation of the fibrils. Incubation with 20 nM SkQ1 induced phosphorylation of cofilin (Fig. 5). A significant increase in cofilin phosphorylation was observed at 2–4 h and remained high on 12 h of treatment with SkQ1, when accumulation of the stress fibers was observed. Trolox and NAC also induced phosphorylation of cofilin, but at concentrations 10,000 or 250,000 times higher, respectively (Fig. 5). LIMK1/2 is the major kinase phosphorylating cofilin in fibroblasts, and this kinase can be activated via phosphorylation of Thr508/505 by Rho-dependent kinase ROCK (8, 38). We analyzed activation of LIMK1/2 using phospho-specific antibodies and demonstrated rapid phosphorylation of LIMK1/2 after treatment with SkQ1 (Fig. 5A). A specific inhibitor of ROCK, HA-1077, decreased the basal level of cofilin phosphorylation and inhibited phosphorylation of cofilin induced by SkQ1 and the other antioxidants (Fig. 5B). These data indicate that scavenging of ROS produced in mitochondria activates Rho-dependent signaling.

SkQ1-induced differentiation of fibroblasts is mediated by TGFβ1 activation
We suggest that the effects of SkQ1 are mediated by TGFβ1, a major inducer of myofibroblast differentiation. Exogenous TGFβ1 induced cell spreading, stress fibers formation, and expression of EDA-FN and α-SMA (Figs. 2B, 2C and 4C), in agreement with data in the literature (15). In our experiments, SkQ1 did not affect the level of TGFβ1 mRNA or the total amount of TGFβ1 either in cell lysates (not shown) or in the culture medium (Fig. 6A). However, we detected a transient increase in the amount of active TGFβ1 after 1 h of incubation with SkQ1, followed by return to the basal level in the next 2–3 h (Fig. 6A). The concentration of the active TGFβ1 in samples with SkQ1 was 0.6 ng/ml. When added to the same concentration, exogenous TGFβ1 induced the typical myofibroblast differentiation (not shown). We observed induction of α-SMA expression by SkQ1 and Trolox, while these antioxidants partially decreased the effect of exogenous TGFβ1 (Fig. 6B).

Further evidence for activation of TGFβ1 in the SkQ1-treated fibroblasts came from measurements of SMAD2 phosphorylation. SMAD2 is a signaling protein that is phosphorylated directly by TGFβ-receptor after TGFβ1 binding (13, 36). We showed that SMAD2 was phosphorylated 2–3 h after addition of SkQ1 (Fig. 6C). Trolox also induced phosphorylation of SMAD2, but NAC was ineffective, consistent with the absence of any NAC-induced expression of myofibroblast markers. To verify the role of TGFβ1 in the SkQ1-induced effects, we applied the TGFβ1 receptor I kinase inhibitor II. This inhibitor prevented the SkQ1-induced expression of myofibroblast markers (Fig. 4C).
To study the possible role of TGFβ1 in reorganization of actin cytoskeleton by the antioxidants, we applied the soluble receptor of TGFβ1 (TGFβRII). This receptor significantly decreased the basic level of phospho-cofilin and prevented phosphorylation of cofilin in response to SkQ1. Exogenous TGFβ1 also induced phosphorylation of cofilin, while inhibitor of the Rho pathway abrogated this effect (Fig. 5) in line with earlier observations (26, 38).These data indicate that activation of the Rho/ROCK/LIMK pathway by SkQ1 depends on TGFβ1. NAC induced cofilin phosphorylation and changes in cytoskeleton and morphology of fibroblasts that were similar to the effects of SkQ1 and Trolox (Figs. 2B and 5), but NAC did not induce TGFβ1 activation and signaling (Fig. 6C). This means that antioxidants can activate Rho by both TGFβ1-dependent and TGFβ1-independent mechanisms.
The culture medium where the SkQ1-treated fibroblasts were grown induced the same morphological changes and expression of myofibroblast markers as SkQ1 did (Figs. 7A and 7B). Addition of anti-TGFβ1 antibodies (Figs. 7A and 7B) into the conditioned medium completely abolished this effect. Thus, we conclude that self-maintaining of the myofibroblast differentiation initiated by SkQ1 is supported by autocrine stimulation of TGFβ1. This conclusion is consistent with an increase in the concentration of active TGFβ1 (but not total TGFβ1) in the conditioned medium. A similar effect was induced by Trolox, but not by NAC.
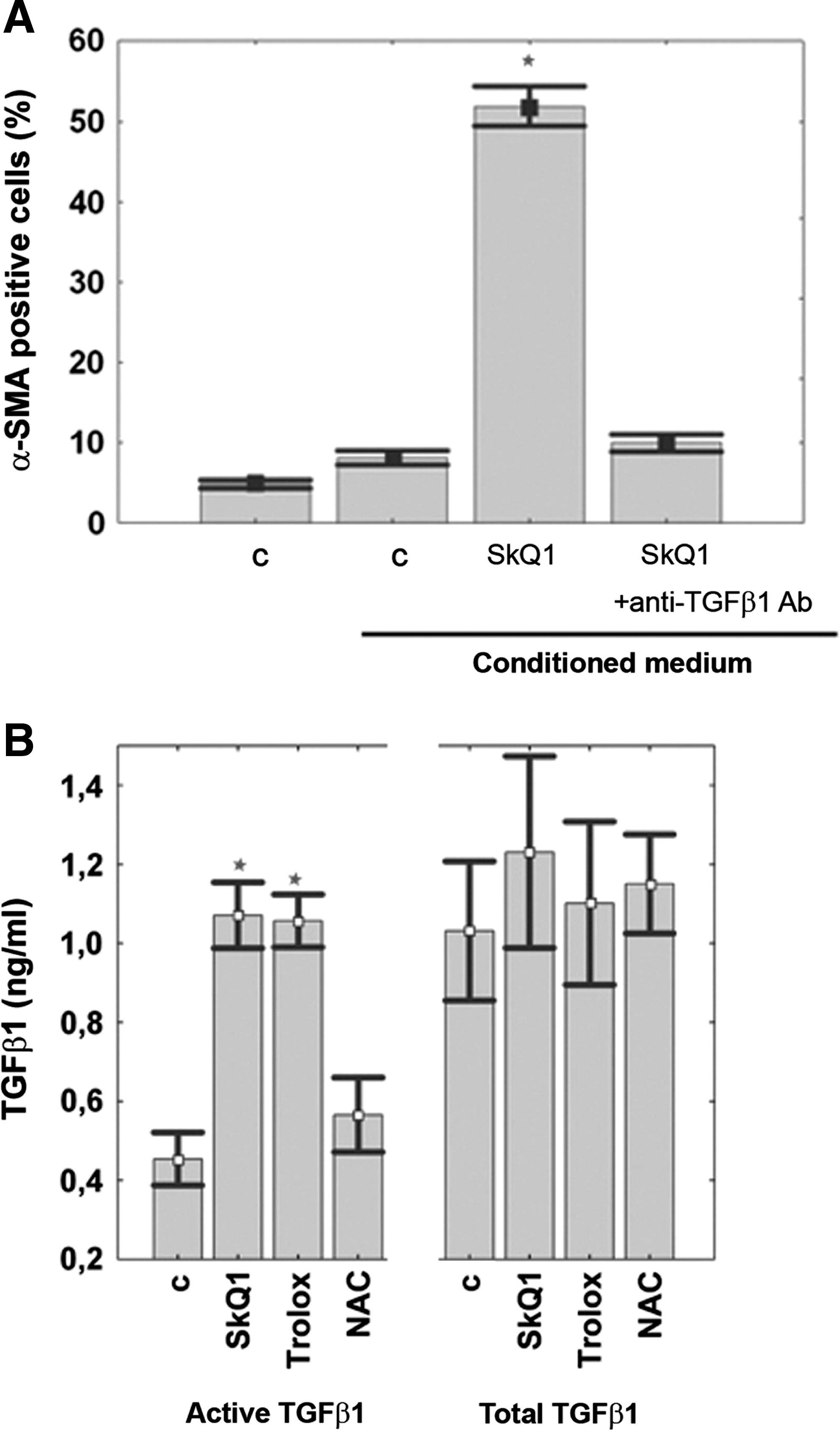
Activation of TGFβ1 induced by SkQ1 is catalyzed by matrix metalloprotease 9
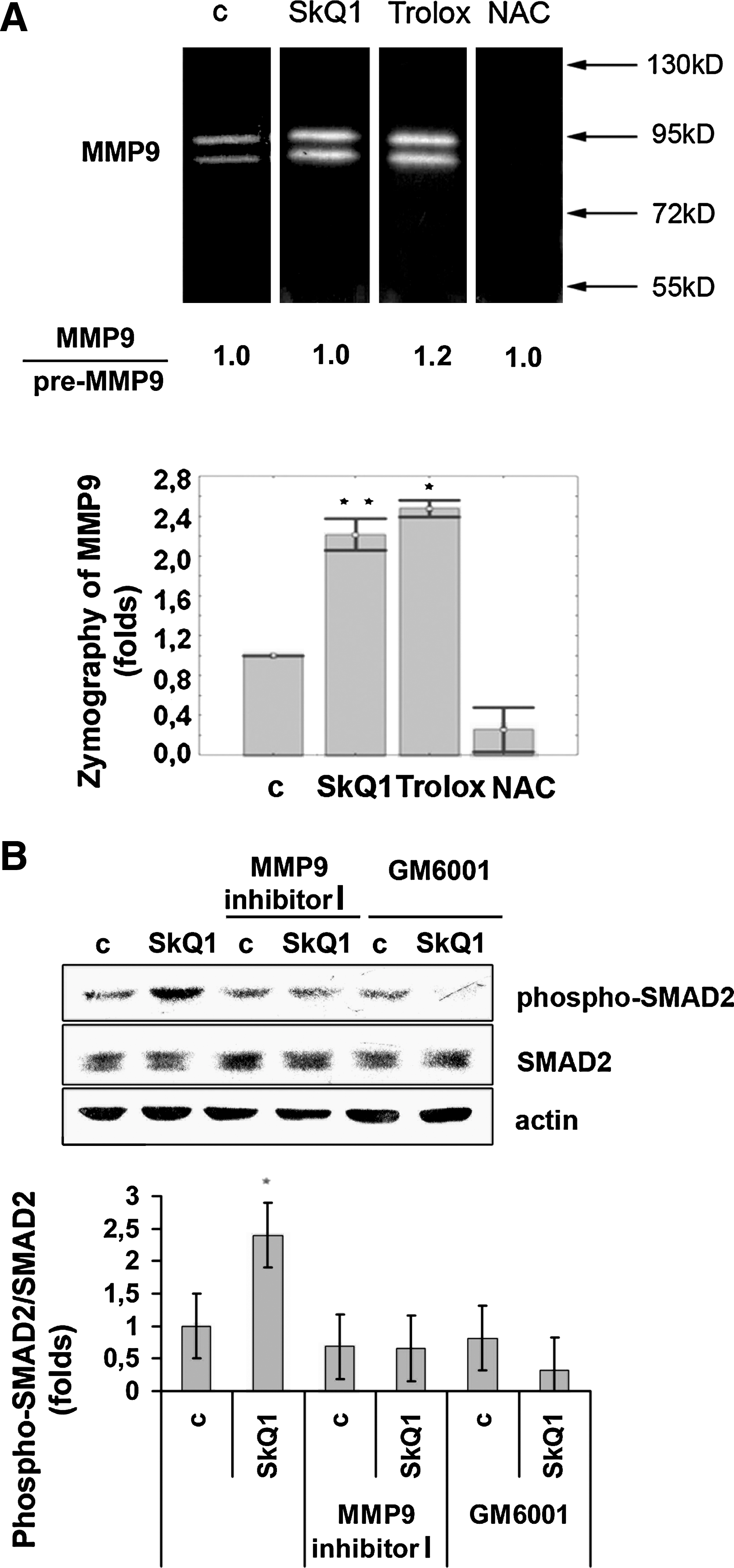
One of the mechanisms of activation of the latent TGFβ1 complex involves matrix metalloprotease 9 (MMP9) (3, 40). SkQ1 (20 nM) and 0.1 mM Trolox rapidly (in 15–45 min) stimulated MMP9 (but not MMP2) in fibroblasts (Fig. 8A). Incubation of HSCF with SkQ1 simultaneously increased the content of the active form of MMP9 and its precursor, as revealed by zymography. We did not observe significant changes in the ratio of these two forms, indicating that SkQ1 induces secretion of MMP9 rather than its processing. In contrast, 5 mM NAC inhibited the basal MMP9 level. The inhibitory effect of NAC on MMP9 activity was described earlier and attributed to docking to Zn2+ at the active site (9, 28). The absence of NAC-induced MMP9 increase correlates perfectly with the absence of TGFβ1 activation, SMAD2 phosphorylation, and myofibroblast marker expression by this antioxidant (Figs. 4C and 6C). To test the necessity of MMP in TGFβ1 activation, we used the inhibitor of the MMP group (MMP-1, −2, −3, −8, −9) GM6001 (10 μM, Millipore, Temecula, CA) and specific MMP9 inhibitor-I (100 nM, Merck, Whitehouse Station, NJ). Both agents prevented SkQ1-dependent stimulation of SMAD2 phosphorylation, indicating that MMP activity was critical for stimulation of TGFβ1 signaling (Fig. 8B). We suggested that MMP9 is a key effecter in our model, but we could not exclude the role of some other MMPs.
Discussion
Scavenging of ROS in mitochondria with the novel mitochondria-targeted antioxidant SkQ1 induced dramatic changes in the actin cytoskeleton, including stabilization of stress fibers and related focal adhesions (Figs. 2 and 3). These changes were accompanied by cell area enlargement and followed by expression of the major markers of myofibroblast differentiation EDA-FN and α-SMA (Fig. 4). The induction of myofibroblast differentiation is due to the antioxidant activity of SkQ1 in mitochondria. This follows from the facts that: (i) neither DPQ nor C12TPP leads to myofibroblast differentiation; (ii) the uncoupler FCCP, which prevents accumulation of SkQ1 in mitochondria, blocks the SkQ-dependent transition; (iii) the traditional antioxidant Trolox induces myofibroblast differentiation, but at much higher concentration than SkQ1. The antioxidant NAC, in line with data published earlier (2), induced the same remodeling of actin cytoskeleton as Trolox and SkQ1 (Fig. 2B). However, NAC did not induce expression of myofibroblast differentiation markers (Fig. 4C). These data indicate that the antioxidants activate two separate signaling pathways, both resulting in changes in cell morphology. The further experiments showed that this complex transition is mediated by (a) stimulation of Rho-dependent signaling and (b) activation of extracellular TGFβ1 (Fig. 9).

The mitochondria-targeted antioxidant SkQ1 induced stabilization of stress fibers mediated by Rho-dependent phosphorylation and inactivation of the actin-depolymerizing protein cofilin. We found that cofilin is phosphorylated by LIMK1/2. This kinase was activated by Rho-dependent kinase ROCK via phosphorylation at Thr508/505 (Fig. 5). Higher concentrations of NAC and Trolox induced the same effects. We suggest that mitochondrial ROS are preferentially responsible for modulation of Rho-dependent signaling. Similar observations were made in our previous study on mouse fibroblast cell line transformed by N-Ras (1). The mechanism of ROS-induced inhibition of Rho in Ras-transformed fibroblasts was recently described (34). This mechanism includes ROS-dependent oxidation and inactivation of protein phosphatase LMW resulting in inhibition of Rho-GAP. Inhibition of Rho in this model prevented ROCK-dependent activation of LIMK, which phosphorylated cofilin. Perhaps SkQ1 and other antioxidants in our experiments inhibited the similar ROS-dependent signaling. The effects of the mitochondria-targeted antioxidant indicate that mitochondrial ROS significantly contribute to Ras-induced changes of actin cytoskeleton and play the major role in regulation of actin dynamics in non-transformed fibroblasts. It seems possible that the effect of mitochondrial ROS is amplified by NOX1, since its expression can be activated by ROS produced in mitochondria (14).
The incubation of fibroblasts with SkQ1 resulted in expression of EDA-FN and α-SMA, indicating myofibroblast differentiation (Fig. 4). We suggest that the effect of SkQ1 is mediated by TGFβ1. We found that SkQ1 induces temporary activation of TGFβ1, followed by SMAD2 phosphorylation (Fig. 6). Activation of TGFβ1 was not accompanied by activation of its expression, since the total amount of this protein was not increased. It is known that TGFβ1 forms a large latent complex bound to the extracellular matrix. The mechanisms of its activation are investigated and intensively discussed (3). One of the major proteolytic activators of latent TGFβ1 is MMP9 (3, 40). SkQ1 and Trolox induced rapid accumulation of MMP9 in the culture medium (Fig. 8A). We did not observe a significant effect of SkQ1 on the ratio of the active form of MMP9 to its precursor, indicating that SkQ1 induces secretion of MMP9. These data suggest that the suppression of exocytosis by excessive ROS (25) was prevented by SkQ1 and Trolox. These antioxidants could stimulate general secretion, while the inhibitory effect of NAC on the MMP9 activity could be based on direct interaction with the metal in the active site of the MMP (9, 28). Using the specific inhibitor of MMP9, we confirmed that this protease activity was necessary for SkQ1-induced activation of TGFβ1-dependent signaling (Fig. 8B).
TGFβ1 is known to trigger a plethora of signaling events, both SMAD-dependent and independent. SMAD-dependent expression of EDA-FN and α-SMA was critical for myofibroblast differentiation induced by SkQ1. At the same time, SMAD-independent signaling is involved in activation of the RhoA/ROCK/LIMK1/2/cofilin pathway, leading to stabilization of stress fibers and related focal adhesions (13, 26, 38). Binding of TGFβ1 in the medium by soluble receptor TGFβRII significantly reduced cofilin phosphorylation induced by SkQ1, indicating the key role of TGFβ1 (Fig. 5). The same pathway was induced by NAC, which does not stimulate TGFβ1, indicating the direct activation of Rho by ROS scavenging.
The observed stimulation of TGFβ1 by SkQ1 had two phases: the first was accomplished 1 h after addition of SkQ1 (Fig. 6A), while the second developed during 7 days (Fig. 7B). The experiments with conditioned medium demonstrated that TGFβ1 activated in the second phase induced an autocrine loop that can support myofibroblast phenotype. The amount of MMP9 in the culture medium drops below the initial level 7 days after SkQ1 treatment (not shown), so the high level of active TGFβ1 in the culture was supported by another mechanism. Activation of TGFβ1 in myofibroblast culture could be associated with an increase in adhesion to ECM and mediated by a local integrin-dependent transition, as described earlier (6, 39), rather than by proteolysis.
The dynamics of myofibroblast differentiation induced by SkQ1 can be divided into the earliest activation of RhoA/ROCK/LIMK1/2-signaling, which depends on TGFβ1, and later actin stress fiber formation and maturation of focal adhesions. We observed an enhancement of EDA-FN expression after 2–3 days, and we detected α-SMA expression at day 4, with further increase after 7–9 days of incubation. All these events were described earlier in more detail for myofibroblast differentiation induced by TGFβ1 (15, 19, 21, 33). It seems important that the dynamics of these steps was very similar in our experiments with the antioxidants and in the studies of TGFβ1-induced differentiation. It was shown earlier that ROS are involved in TGFβ1-dependent signaling. In particular, a member of the NOX family, NOX4, has been implicated in cardiac myofibroblast differentiation from fibroblast precursors (12). In our model, antioxidants both induced activation of TGFβ1 and partially suppressed the effect of added TGFβ1 (Fig. 6B). Future studies on physiological functions of the cells differentiated by SkQ1 (such as cell contraction) would show the degree of myofibroblast maturation in vitro. The possible induction of myofibroblast differentiation by SkQ1 in vivo could contribute to stimulation of dermal wound healing, and these experiments are ongoing in our laboratory.
In general, we conclude that scavenging of ROS in mitochondria with the novel mitochondria-targeted antioxidant SkQ1 induces Rho-dependent reorganization of actin cytoskeleton and TGFβ1-dependent myofibroblast differentiation.
Footnotes
Acknowledgments
This work was supported by Russian Foundation for Basic Research grants 07-04-00335, 09-04-00667 and Mitotech LLC. We thank M. Domninskaya for technical assistance, Dr. R. Zinovkin and Dr. M. Skulachev for careful reading of the manuscript and discussion, and Dr. S. Golyshev for assistance with microscopy.
Author Disclosure Statement
No competing financial interests exist.