
Abstract
Cultured neurons tolerate low H2O2 concentrations (≤50 μM) through the activity of constitutive antioxidant response elements (ARE). At H2O2 levels (≥100 μM), neurons increase expression of the gene encoding for inducible hemoxygenase-1 while superoxide dismutase-2 and catalase remain unchanged. Despite this adaptive response, the endogenous antioxidant systems are overwhelmed, leading to decreased viability. Elevating the neuronal cell content of human neuroglobin (Ngb) prior to insult with 100 or 200 μM H2O2 enhanced cell viability and this resulted in a significant decrease in oxidative stress and an increase in the intracellular ATP concentration, whereas in parental cells exposed to the same H2O2-insult, oxidative stress and ATP increased and decreased, respectively. The mechanism for this increase in ATP involves sustained activation of the mito-KATP channel and an increase in phosphoinositide-3 kinase (PI3K)-mediated phosphorylation of Akt. Pharmacological inhibitors directed toward PI3K (wortmannin and LY294002), or the mito-KATP channel (glybenclamide) inhibited the H2O2-mediated increase in ATP in cells overexpressing human Ngb and consequently cell viability decreased. Neuroglobin's ability to bolster the intracellular pool of ATP in response to added H2O2 is central to the preservation of cytoskeletal integrity and cell viability. Antioxid. Redox Signal. 13, 769–781.
Introduction
Neuroglobin (Ngb) is a heme-protein from the globin superfamily and is expressed in the brain (62), central nervous system, and retina (16). To date, the exact function of Ngb has yet to be elucidated, but there are several proposed mechanisms. Similar to hemoglobin (Hb) and myoglobin (Mb), Ngb reversibly binds oxygen (10) and may participate in oxygen transport, storage, and release. This may involve formation of a disulfide bond within the protein backbone that introduces subtle changes to the distal cavity region (21). Despite this knowledge, the biological function of Ngb remains unclear. Previous studies have implicated Ngb to be involved in scavenging reactive oxygen species (ROS) (24), modulating nitric oxide (•NO) (19) and intracellular metal ion homeostasis (13), and/or serving as an oxygen sensor (9, 56).
Changes in Ngb expression correspond to the severity of histological and functional deficits after stroke (52). However, questions remain as to whether neuroprotection is conferred by Ngb (27). Enhanced Ngb expression is linked to neuroprotection in some (51, 52) though not all models of ischemic stroke (43). This conflict in the available literature indicates further studies are required to establish whether Ngb can protect neurons from ischemic insult, and if so, the mechanism(s) involved in this biological activity.
The loss in blood supply to oxygen-sensitive brain tissue leads to cerebral oxidative stress (2). Enhanced oxidative stress promotes production of oxidants, including superoxide radical anion (O2•−) and its dismutation product hydrogen peroxide (H2O2) (24)— both promote brain injury following stroke (36). The abundance of mitochondria in the ischemic brain are a major source of oxygen-centered radicals, and induction of mitochondria dysfunction leads to overproduction of O2•− with parallel increases in H2O2 within the affected cerebral tissue (50). For example, experimental forebrain ischemia reperfusion yields [H2O2] up to 100 μM in the rat brain (28).
Scavenging of ROS has been identified as one mechanism of action for Ngb in cultured human neuronal cells overexpressing the hemoprotein (17, 34). We also identified alternate secondary mechanisms that contribute to its protective activity in response to experimental ischemia (13), including maintenance of intracellular metal ion homeostasis and cytoskeletal integrity. Herein, we expand on our initial observations and determine whether Ngb's activity extends to protecting neuronal cells challenged with H2O2 as a model for free radical formation in the brain after ischemic insult.
Materials and Methods
Cell culture materials and general chemicals were from Sigma-Aldrich (Sydney, Australia) and were the highest quality available. Buffers were prepared in MilliQ water.
Human Ngb construct
Cloning of the Ngb open reading frame was performed with recombination-based Gateway technology (Invitrogen, Sydney, Australia), as described elsewhere (13). The construct containing the (hexa-HIS and V5) flanking peptides was confirmed using BigDye Terminator Cycle Sequencing (SUPAMAC, Sydney, Australia) and was used in all cell manipulations.
Cell transfection
The human Ngb plasmid DNA was transfected into the neuroblastoma cell line SH-SY5Y using Lipofectamine 2000 (Invitrogen). Transfection was achieved with high efficiency (∼90%–95%), as described previously (13). Cells transfected with the PDEST40 blank vector yielded the parental (control) cells. Accumulation of the human Ngb-fusion protein in differentiated cells was verified by Western blot analysis with an antibody raised against a V5-epitope (Supplemental Fig. 1A; see
Cell culture
The neuroblastoma cell line SH-SY5Y transfected with the blank vector (referred to hereafter as parental cells) or with human Ngb (Ngb-transfected cells) were inoculated at a density of ∼1 × 104 cells and subcultured in Dulbecco's Modified Eagle's Medium/Ham's F12 (JRH Biosciences, KA) containing 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2.5 mM L-glutamine, and 5 ml of nonessential amino acid solution, at 37°C in an atmosphere of 5% CO2(g). Studies were performed with cells grown to ∼70%–80% confluence yielding ∼5 × 106 to ∼5 × 109 cells/ml.
Cells were induced to differentiate into a neuronal phenotype by treatment with 50 ng/ml of 12-o-tetradecanoylphorbol-13-acetate (TPA) over a period of 3 days (exchanging media every day of subculture). The neuronal phenotype was verified by monitoring mRNA expression of the neuronal cell marker growth-associated protein (GAP-43) with quantitative RT-PCR (Supplemental Fig. 2; see
Cell model
Differentiated neurons were prepared for challenge with H2O2 by substituting growth media with HEPES-buffered physiological saline solution (HPSS) containing: 22 mM HEPES (pH 7.4), 124 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 0.16 mM HPO4, 0.4 mM H2PO4, 5 mM NaHCO3, and 5.6 mM D-glucose. Next, vehicle (phosphate buffered saline; PBS, pH 7.4) or a bolus of H2O2 was added (10, 50, 100 to 200 μM) and the cells re-cultured at 37°C for 2 h. Cells were harvested by scraping and cell pellets were obtained by centrifugation (∼56 g). In some cases, cell pellets were immediately frozen in liquid nitrogen and stored at −80°C for use in biochemical studies.
Flow cytometry
Cell viability
To determine oxidative stress response in neurons, cells were pretreated with 50 μg/ml nonfluorescent dihydrorhodamine (DHR-123), then exposed to H2O2 (0–200 μM). Oxidation of DHR-123 yields the fluorescent product R-123. Mean fluorescence intensity was used as a surrogate marker for oxidative stress and was measured by flow cytometry (FACSCalibur BD Biosciences, Sydney, Australia) with excitation (Ex) 488 nm and Emission (Em) 540 nm.
Similarly, apoptosis and necrosis was determined using flow cytometry with a commercial Annexin V-FITC detection kit (BD Biosciences). Binding of FITC-conjugated annexin V (Ex 488 nm; Em 520 nm) and the counterstain propidium iodide (PI) (Ex 488 nm; Em 620 nm) was analyzed as described previously (32). To assess the involvement of mito-KATP channels or phosphoinositide-3 kinase (PI3K), complementary studies were performed with parental cells incubated with a selective mito-K ATP channel opener (57) (diazoxide; 100 μM, 10 min, 37°C), or an activator of PI3K (23) (human leptin; 100 nM, 30 min, 37°C) then challenged with H2O2.
Ca2+ content
Cells were then challenged with H2O2 (0–200 μM) for 2 h, the HPSS replaced with Dulbecco's buffered phosphate saline (DBPS; deficient in Mg2+ and Ca2+cations) and intracellular Ca2+ was assessed with flow cytometry (Ex 506 nm; Em 526 nm) as described previously (60). Esterified Fluo3AM enters cells where esterases yield the active intracellular Ca2+ probe. An advantage for this probe is that it is not necessary to monitor two emission wavelengths to determine Ca2+ flux. The possibility of unrestricted Ca2+ uptake during cell harvest was nullified by using DBPS deficient in multiply charged cations.
Caspase activation
The activity of effector or executor caspases-3 and -7 was measured using a commercial kit (Caspase-GloTM 3/7, Promega, Australia). Briefly, cells were treated with 0–200 μM H2O2 and incubated for 2 h. Growth supplement was replaced with PBS and Caspase-GloTM 3/7 reagent added (1:1 v/v; 20°C for 40 min) and total caspase 3/7 activity assessed by measuring luminescence with a Victor III Multi-label plate reader (Perkin Elmer, Sydney, Australia). Data sets were analyzed and expressed as a fold-increase in activity relative to the controls.
Intracellular ATP and mitochondrial function
The ratio of intracellular ATP/ADP (Perkin Elmer) and the absolute levels of ATP (ATPlite®, Perkin Elmer) were determined with commercial kits (47). Neuronal cells were cultured in 96-well plates, treated with H2O2 (0–200 μM), incubated for 2 h and then the samples were analyzed with a Victor III Multi-label plate reader according to the manufacturers instructions. Cellular ATP was normalized against total cell protein to account for any differences in cell density. Cell protein was determined using the BCA assay (Sigma).
Mitochondrial depolarization (a surrogate for mitochondrial dysfunction) was determined by flow cytometry. All samples were incubated with 200 μM MitoProbe, JC-1© (Molecular Probes, Eugene, OR) for 20 min at 37°C. Cell suspensions were centrifuged, the pellet washed and re-suspended in 500 μl PBS and then analyzed. Emissions monitored at 525 and 620 nm were used to determine the shift of green to red fluorescence expressed as a fold-change in mean fluorescence intensity compared to the control.
Total ATP was also assayed in cells pre-incubated with chemical mediators. Cells were pre-incubated with 10 μM glybenclamide, a mito-KATP channel inhibitor (3), 250 nM wortmannin or 50 μM LY294002, 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride, as inhibitors of PI3K (42), for 20 min at 37°C prior to H2O2-insult. The wortmannin concentration employed was selected as specificity is lost at higher doses (49). Intracellular ATP was also assessed in parental cells treated separately with 100 μM diazoxide (10 min, 37°C) or 100 nM human leptin (30 min, 37°C), prior to H2O2 insult (2 h, 37°C).
Chromatographic determinations of ATP
Changes in ATP were verified using complementary HPLC measurements. After H2O2 treatment, both parental and Ngb-transfected cells were lysed with ice cold NaClO4 (400 μM). The samples were centrifuged (3000 g, 5 min), the supernatant placed on ice and treated with ice cold 1 M NaHCO3, centrifuged again (13,000 g, 3 min) and the supernatant was analyzed by reversed-phased (LC-18) HPLC with gradient elution. Solvents consisted of (A) 0.15 M NH4H2PO4 (pH 6.0) and (B) a mixture of acetonitrile/methanol (1/1, v/v). Analysis was performed using a (1 ml/min) linear gradient from 100% A to 85% A and 15% B as described (61). The column was equilibrated with buffer A for 10 min before sampling continued. Cellular ATP was monitored at 260 nm and was quantified by peak area comparison. Authentic ATP (purity 99%) eluted at ∼5 min. Intracellular ATP was expressed as fold-change relative to the control (assigned unitary value).
Gene and protein regulation
After exposure to 0–200 μM H2O2, cells were lysed and total RNA extracted with a Total RNA Kit (Sigma) as per manufacturer's instructions. Complementary cDNA was constructed by reverse transcriptase-polymerase chain reaction (RT-PCR) using BioScript Rnase H Minus (Bioline, Sydney, Australia) and an Eppendorf Master-Cycler. Reaction mixtures contained: 2 μl total RNA, 1 μl oligo (dT), and 9 μl diethyl-pyrocarbonate (DEPC) treated Nanopure water. Reactions were then heated (70°C, 5 min) and then chilled (4°C, 5 min). Mixtures were further treated with 1 μl RnaseOut (an RNAase inhibitor), 1 μl dNTP Mix (PCR Grade), 4 μl of 5 × reaction buffer (supplied by the manufacturer), 1.75 μl DEPC-treated nanopure water, and 0.25 μl Bioscript Rnase H Minus (Bioline) to a total volume of 20 μl. All samples were heated to 42°C for 60 min and then to 70°C for 10 min to stop the reaction.
Gene-specific PCR amplification was carried out using Biomix Master Mix (Bioline, Sydney, Australia) with an Eppendorf Master-cycler system. Primer sets were synthesized by Proligo (Table 1). Cycling consisted of activation (94°C, 5 min), 45 cycles of denaturation (94°C, 30 s), annealing for 30 s, elongation (72°C, 1 min), and then another elongation step (72°C, 10 min). Amplified cDNA was resolved on 2% agarose containing 2 μg/ml ethidium bromide. Gels were imaged using a BioDocAnalyzer (Biometra, Gottingen, Germany) and transformed to TIF for manipulation with Microsoft Power Point (2000). Semiquantitative densitometry was performed with ImageJ v1.30 software (
All primer sequences were designed using the Primer 3 sequencing program (available on the Internet). Sense and anti-sense primers were synthesized by Proligo (Lismore, NSW Australia). Annealing temperature was 60°C for all primers.
Western blot analysis
Western blot studies were performed as described in detail elsewhere (39). For studies on the HO-1 protein, microsomal isolates (see below) were first adjusted for protein content prior to loading onto gels. After separation on SDS-PAGE, detection of HO-1 was achieved with a rabbit polyclonal antibody raised human HO-1 (final dilution 1:2000 v/v) and a goat anti-rabbit IgG-HRP conjugate (final dilution 1:10,000 v/v).
Detection of total Akt protein (and its phosphorylated form) was performed by Western blotting and subsequently validated with a Ser473-p-AKT/t-AKT dual label CELISA kit according to manufacturer's instructions (Millipore, Sydney Australia). Cell lysates were separated on SDS-PAGE, proteins transferred to nitrocellulose, and the blots assessed for total Akt (t-Akt) (dilution 1:1000 v/v) and phosphorylated Akt (Ser473-p-Akt) (dilution 1:1000 v/v) (∼60 Kda) using appropriate antibodies (Cell Signalling Technology, Brisbane, Australia). As a positive control, shrimp alkaline phosphatase (SAP) (Quantum Scientific, Sydney, Australia) was used in parallel samples to dephosphorylate proteins before visualization with enhanced chemiluminescence detection (WEST-ZOL™ Biotech., Gyeonggi-do, Korea) (39). A loading control was performed on a parallel blot using a polyclonal anti-human β-actin (Sapphire Bioscience, Sydney, Australia). Images were captured (KODAK Imaging system) and converted to “tagged image files” for further manipulations with Microsoft Office 2007. Where required, CELISA plates were analyzed with a FLUOstar Multilabel plate reader (BMG Labtech, Sydney Australia) with fluorescence parameters: Akt–Ex 500 nm; Em 550 nm; p-Akt–Ex 360 nm; Em 460 nm.
Hemoxygenase activity
Total HO activity was assessed in microsomal preparations prepared as described elsewhere (47). Assays were performed using 400–600 μg microsomal protein. Total HO activity was determined by monitoring bilirubin (BR) formation (product of heme catabolism) with gradient reversed-phase HPLC (47). The detection limit for BR under these conditions was ∼15 pmol with authentic BR eluting at ∼8.5 min. Finally, activity was expressed as a fold-increase in BR compared to the corresponding control.
Assessment of cell structure and receptor-mediated transport
Immunostaining
Cells were grown onto coverslips in DMEM/HAM's F12 media, then differentiated for 3 days using TPA as described previously (13). Next, the media was replaced with HPSS containing H2O2 (concentrations indicated in the figure legends) and cells subcultured in 5% CO2(g) at 37°C. After 2 h, media was aspirated, cells were fixed in methanol, and blocked with 1% w/v BSA at 37°C. After 30 min, the blocking buffer was removed and primary antibody (see below) was added and incubated at 20°C. After 1 h, cells were washed (3 × PBS), incubated with the secondary antibody for 1 h, and finally visualized.
Cytoskeletal structure
Staining of cytoskeletal β-actin was performed with a monoclonal antibody raised against human β-actin (dilution 1:100 v/v), along with an anti-mouse–FITC conjugate secondary antibody (dilution 1:2500 v/v) that was visualized with Ex 530 nm.
Receptor-mediated endocytosis
A fluorescently labeled transferrin isoform (conjugated to Alexa Fluor 488, Invitrogen) was employed to assess receptor-mediated endocytosis, both before and after exposure of neurons to H2O2 (range 0–200 μM). Cells were incubated in 50 μg/ml of labeled transferrin (10 min, 37°C) prior to H2O2 challenge. Next, excess transferrin was removed by thorough washing in 0.2 M acetic acid containing 0.5 M NaCl (pH 2.8; 10 min at 4°C). Cells were fixed by incubating in 4% w/v paraformaldehyde (pH 7.5; 15 min; 20°C) and visualized with an Axiovert 200 inverted fluorescent microscope. Images were false-colored subsequent to acquisition using AxioVision software (AxioVision v4.5, Carl Zeiss, Australia).
Statistical analyses
Analyses were performed with Prism Software (v3.0, GraphPad Software Inc). Data were analyzed using One-way ANOVA with Bonferroni's post-hoc analysis or by Student t-test using Welch's correction for unequal variances. Statistical significance was set at p < 0.05.
Results
Cell viability
Initial studies demonstrated that apoptosis increased significantly in parental cells when measured 24 h post-insult yet cells transfected with human Ngb were significantly protected from apoptosis at all H2O2 concentrations investigated (Supplemental Fig. 3; see
Exposure of cultured parental cells to 10 μM H2O2 for 2 h elicited no change in the extent of apoptosis and necrosis as compared to the vehicle-treated controls (Fig. 1A). This suggested that the endogenous neuronal antioxidant capacity was sufficient to cope with this level of insult. However, necrosis increased significantly and dose-dependently in parental cells exposed to 50–200 μM H2O2 for 2 h, reaching ∼3-fold higher levels than the control. Under these conditions, the proportion of apoptotic cells remained unchanged. Cells overexpressing human Ngb showed comparable resistance to 10 μM H2O2 insult. However, when exposed to 50–200 μM H2O2 cells overexpressing human Ngb showed a continued tolerance to H2O2 insult with consistently lower levels of necrosis and no measurable change in apoptosis relative to controls.

Caspase activation
Activity of the effector caspases 3/7 increased signficantly in a H2O2 dose-dependent fashion in parental cells exposed to 50–200 μM H2O2 (Fig. 1B). Cells transfected with the human Ngb–fusion protein showed marked resistance to caspase activation with the extent of caspase activity only reaching significance at the highest H2O2 dose tested. Despite being unable to detect apoptosis 2 h after insult (as judged by annexin V staining), neuronal cells exposed to H2O2 (final concentrations ≥ 10 μM) showed increased effector 3/7 activation that was inhibited by the overexpression of human Ngb.
Oxidative stress in neuronal cells exposed to H2O2
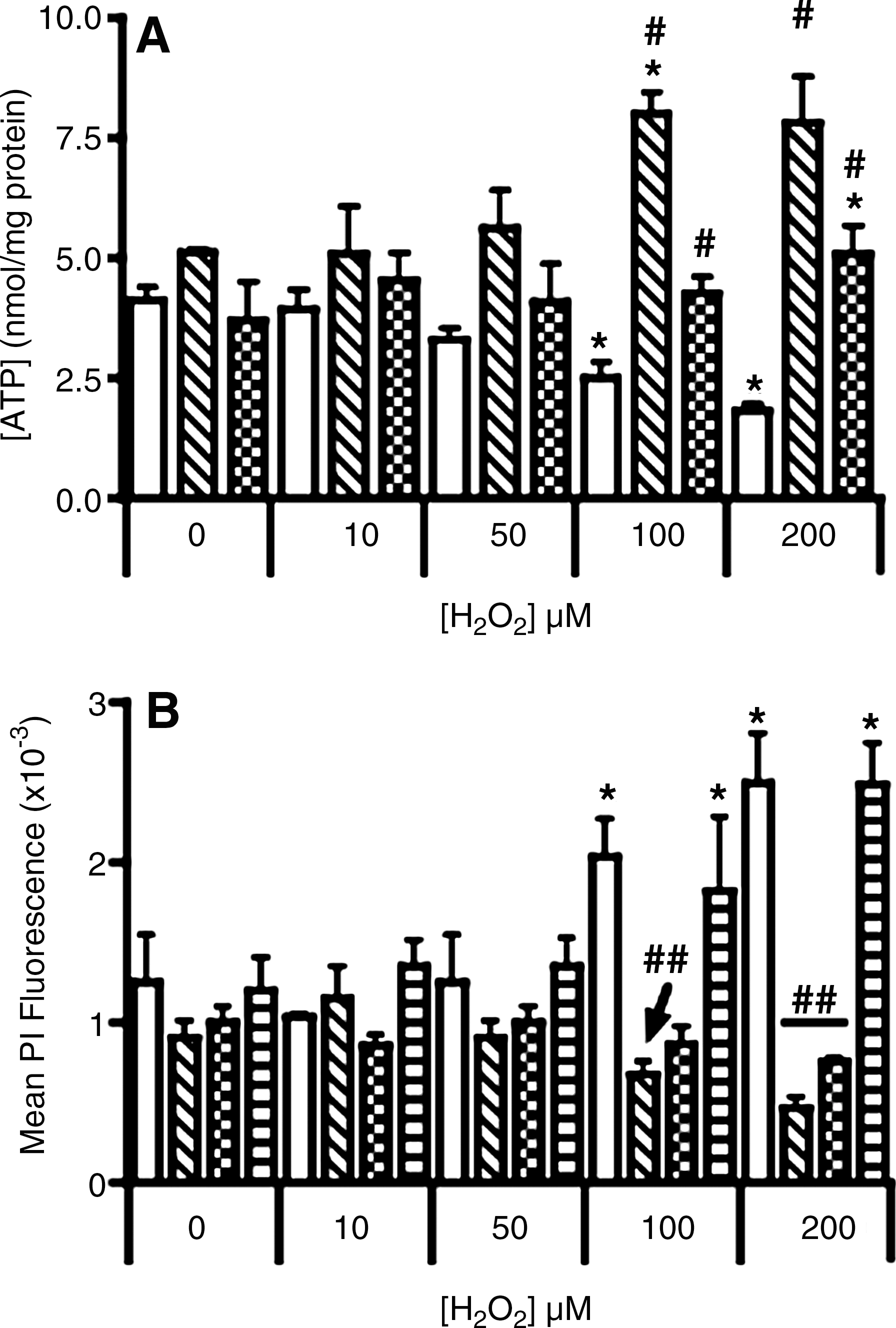
Treating cultured neuronal cells with 0–200 μM H2O2 for 2 h resulted in a dose-dependent increase in R-123 fluorescence, reaching more than 15-fold greater than that in control cells treated with vehicle alone (Fig. 2A). Cells overexpressing human Ngb displayed significantly lower fluorescent intensity than the parental cells at all corresponding H2O2 doses.
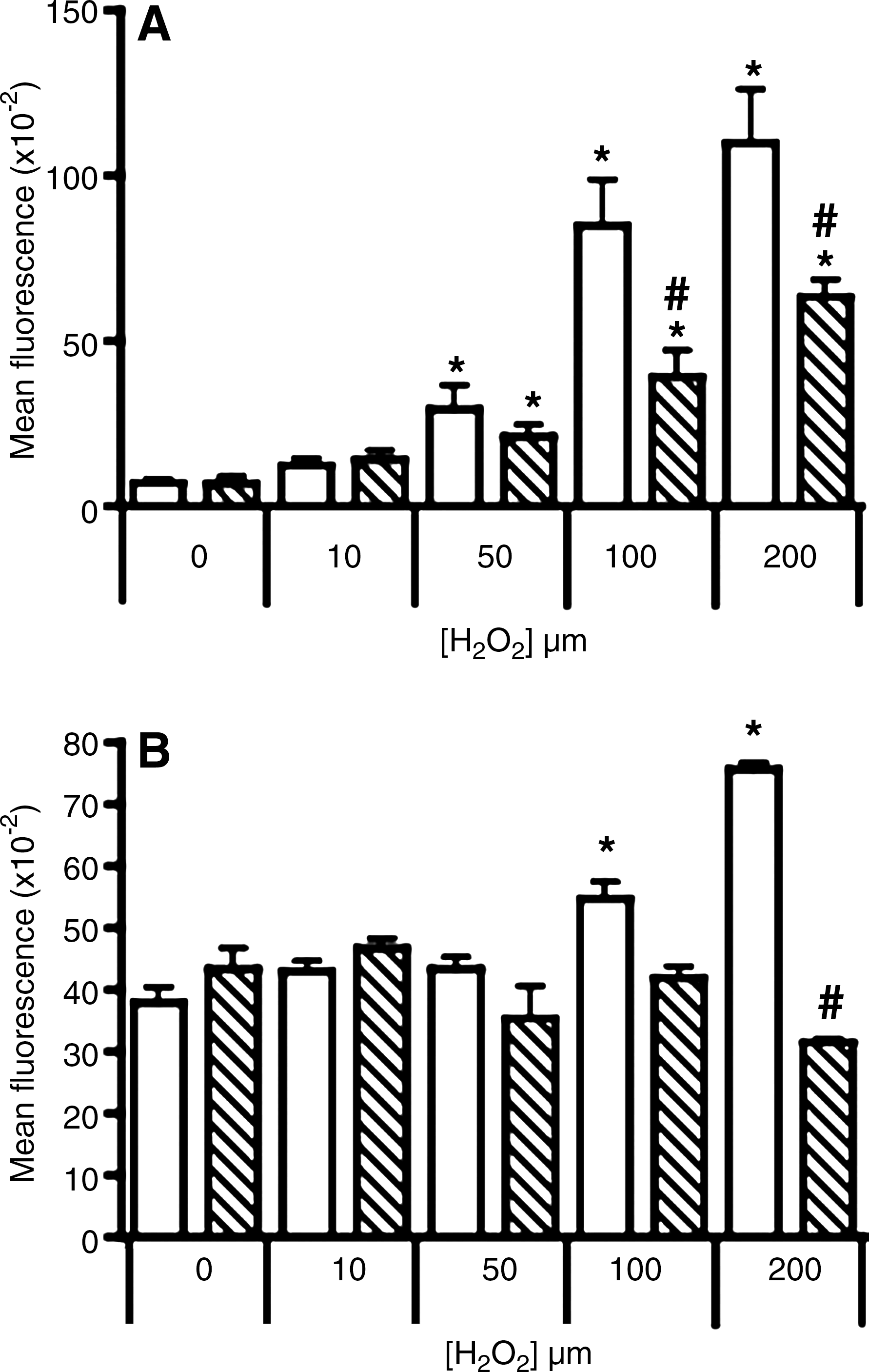
Mitochondrial membrane potential
Parental and Ngb-transfected neuronal cells exhibited similar resistance to mitochondrial dysfunction induced by 10 or 50 μM H2O2 (Fig. 2B). Mitochondrial depolarization increased significantly in parental cells exposed to 100 or 200 μM H2O2. However, mitochondrial function was preserved in Ngb-transfected cells at all the H2O2 concentrations investigated.
Changes to intracellular calcium
Prior to treatment with H2O2, parental and Ngb-transfected cells showed near identical baseline levels of free Ca2+ (Fig. 3). However, a Ca2+-influx was apparent in parental cells exposed to increasing dose of H2O2 and this was inhibited by Ngb. Parallel imaging studies with cells overexpressing human Ngb indicated these cells were largely resistant to Ca2+-influx (not shown).
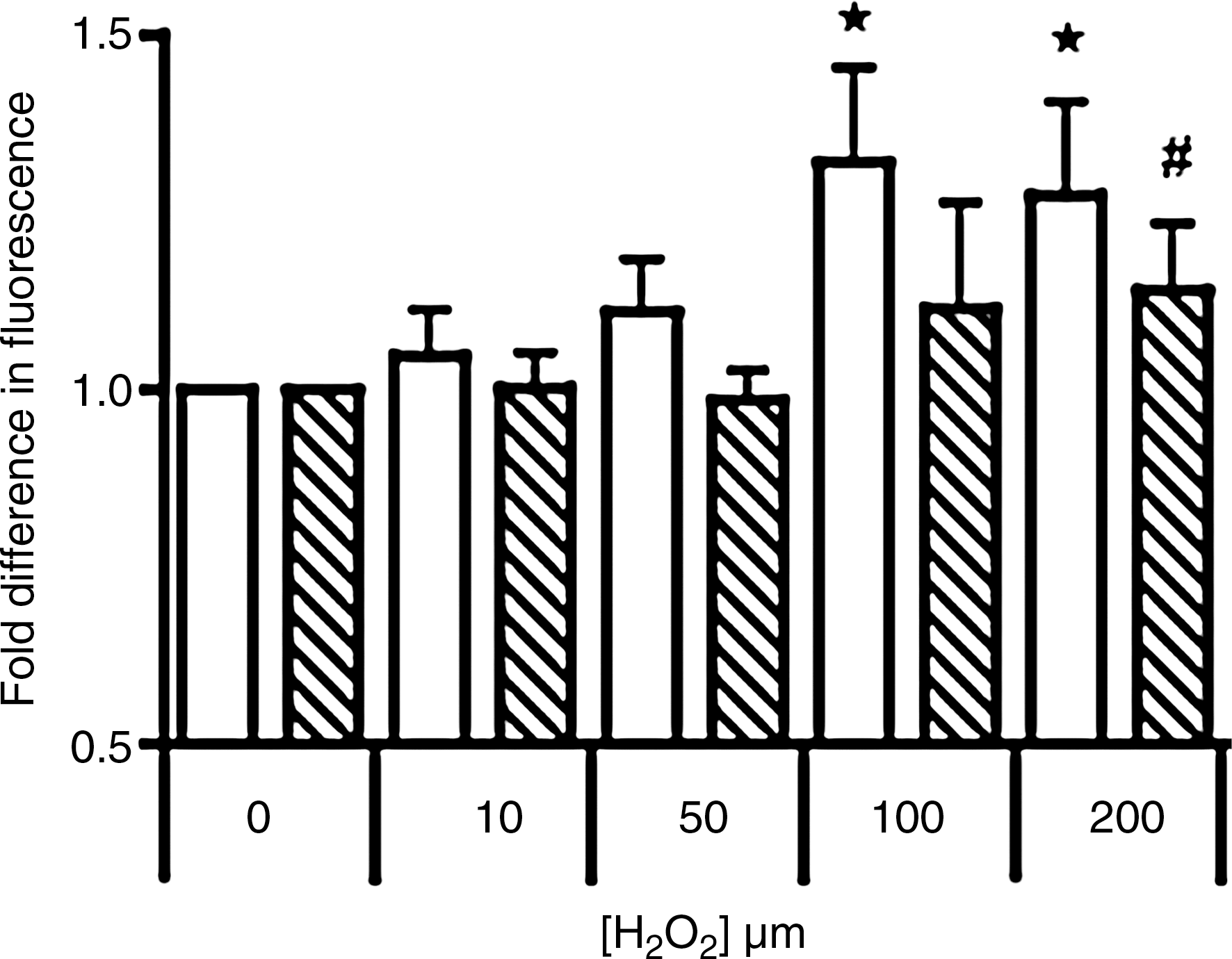
Intracellular ATP
Initially we determined the ratio of ADP/ATP in response to H2O2-insult (Fig. 4A).Unaltered ADP/ATP ratios were detected in both parental cells and cells containing the human Ngb–fusion protein when exposed to H2O2 ≤ 50 μM. However, increasing the peroxide challenge to 100 or 200 μM induced a marked increase in the ADP/ATP ratio in parental cells, whereas unexpectedly the ratio decreased below baseline levels in cells transfected with human Ngb. The latter may indicate an increase in ATP or a decrease in the pool of available ADP. To determine whether ATP increased, intracellular ATP was examined independently. Similar to the ADP/ATP ratio, ATP content in parental cells was unaffected by exposure to ≤ 50 μM H2O2 (Fig. 4B). Thereafter, levels of ATP decreased in a dose-dependent fashion reaching significance at the highest H2O2 dose tested. Similarly, Ngb-transfected cells were resistant to 10 or 50 μM H2O2. However, ATP increased markedly (∼2-fold) in cells transfected with human Ngb following treatment with 100 or 200 μM H2O2 (Fig. 4B). Parallel ATP assessment with liquid chromatography confirmed intracellular ATP increased in Ngb overexpressing cells (Figs. 4C and 4D). The lower fold-increase in ATP levels detected by HPLC likely reflects the extraction procedure that may have degraded labile ATP.

Gene regulation
Oxidative stress can elicit an antioxidant gene response in neuronal cells (13, 47). Parental and Ngb-transfected cells were probed for expression of HO-1, CAT, and MnSOD as these genes are associated with the stress response (25). Of the genes studied, HO-1 increased substantially after insult (Fig. 5A and Table 2) and was sustained for up to 12 h (not shown). Cells transfected with human Ngb were resistant to H2O2-induced HO-1 regulation (Table 2).
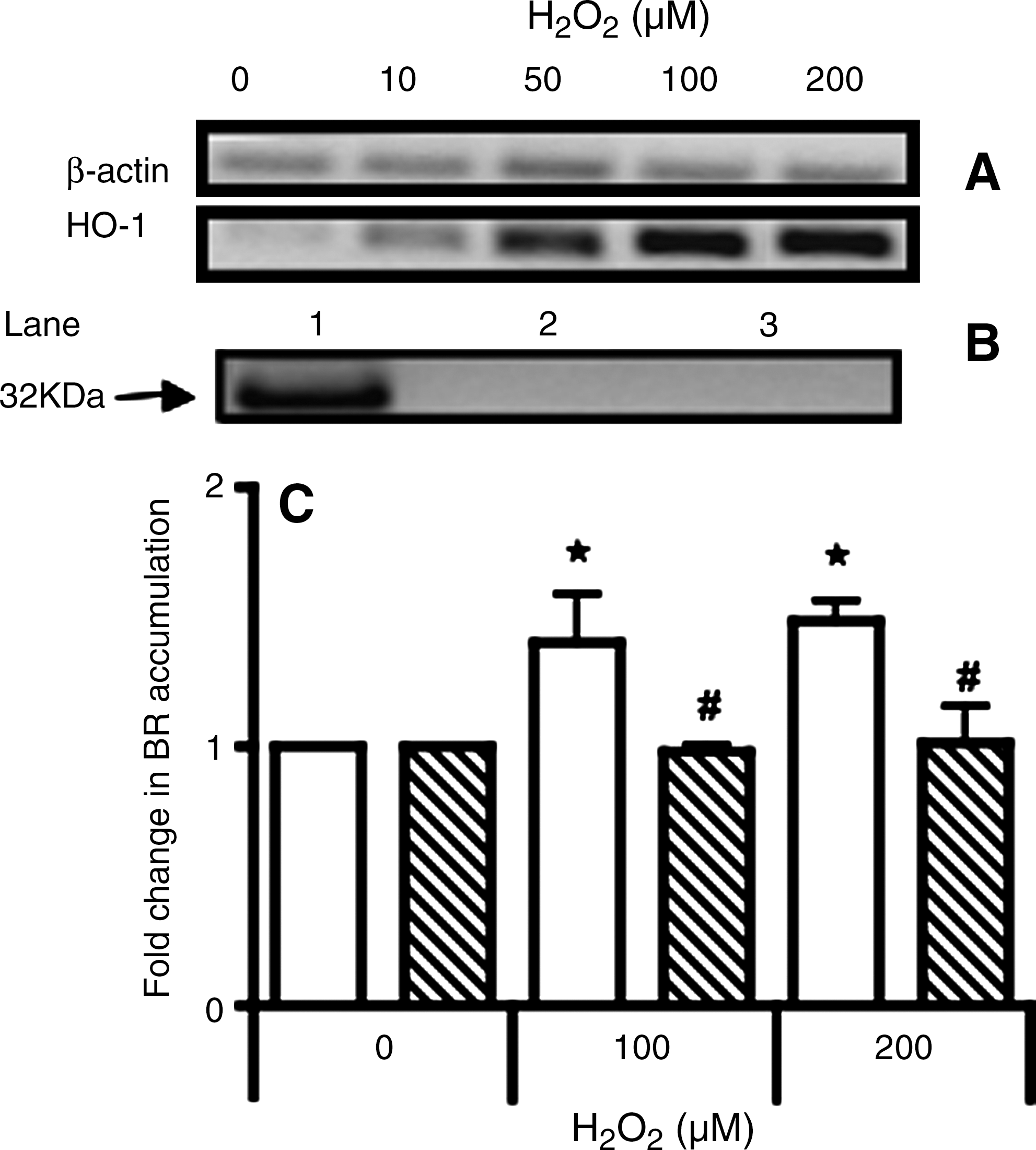
Expression of early antioxidant response genes in parental and Ngb-transfected cells were assessed before and after 2 h insult with H2O2 (10–200 μM). Data represent mean fold-change relative to vehicle treated controls ± (SD) from n = 3 independent experiments which have been normalized to the corresponding β-actin house-keeping gene.
Parental cells exposed to 200 μM H2O2 showed a parallel accumulation of HO-1 protein as demonstrated by Western blot (Fig. 5B). However, no increase in HO-1 protein was detected in Ngb-transfected cells exposed to the same insult. In agreement with the gene and protein studies, total HO activity increased in parental cells when exposed to 100 or 200 μM H2O2 (as judged by the extent of bilirubin (BR) accumulation) whereas, HO activity in Ngb-transfected cells was unchanged (Fig. 5C).
H2O2-mediated changes in receptor-mediated transport and cytoskeletal structure
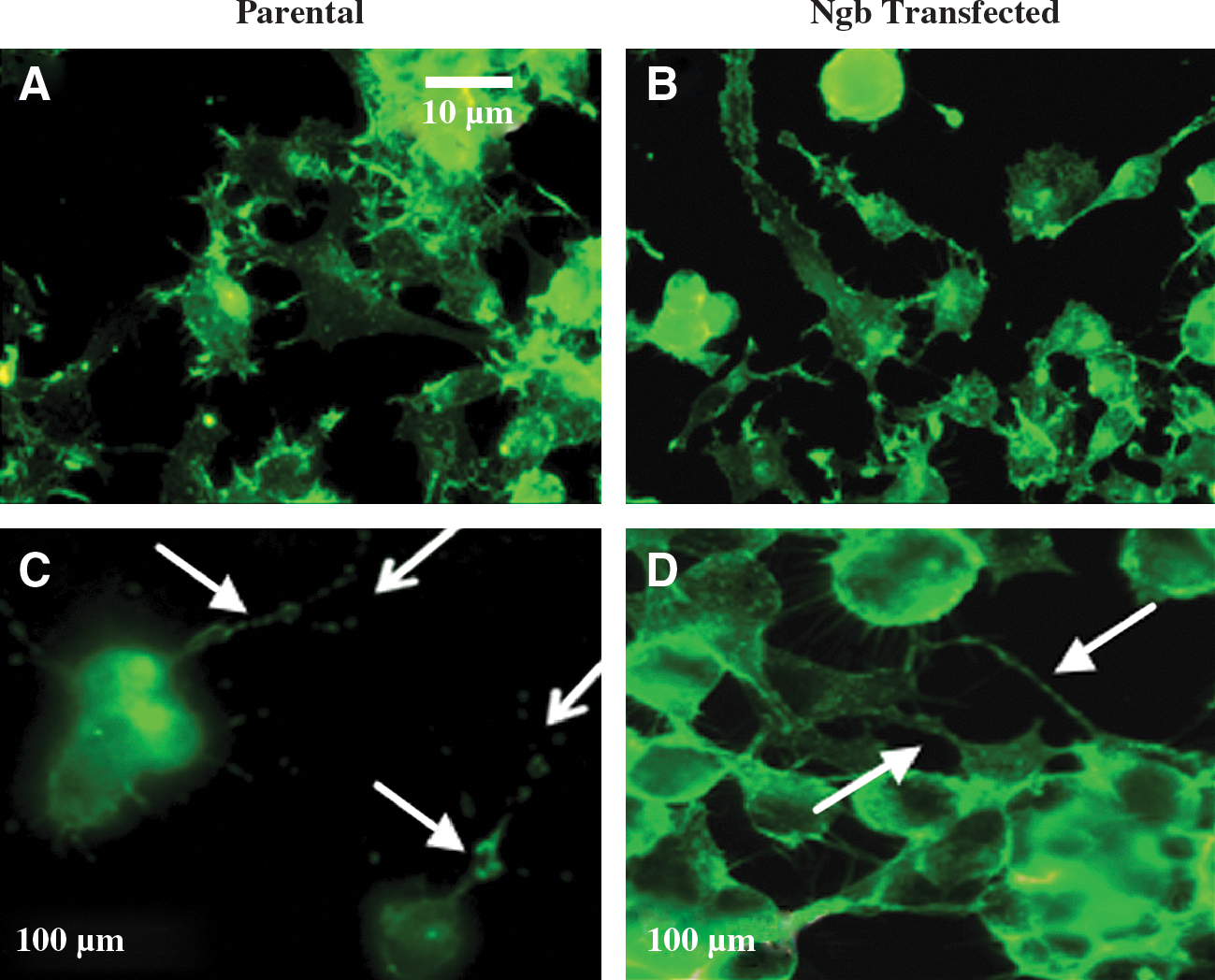
In the absence of H2O2, parental cells and cells overexpressing human Ngb showed similar up-take of fluorescent-labelled transferrin (compare Figs. 6A and 6B). Upon challenge with increasing doses of H2O2, parental neurons exhibited a decrease in total fluorescence compared to control and this was more marked for cells exposed to 100 or 200 μM H2O2 (compare Figs. 6A and 6C). Under identical experimental conditions, cellular fluorescence remained unchanged in Ngb-transfected cells, indicating that receptor-mediated up-take of labeled transferrin was not altered by H2O2 insult (compare Figs. 6B and 6D).

Immuno-staining for the structural protein β-actin indicated little change in the fluorescent distribution for cells exposed to ≤50 μM H2O2 (not shown). Parental neurons displayed a pattern of condensed actin filaments, particularly along axonal and dendritic processes when challenged with 100 or 200 μM H2O2 (compare Figs. 7A and 7C). Reflecting the protective action of human Ngb (Fig. 1), the extent of actin condensation was less evident in Ngb-transfected cells under identical H2O2 insult (Figs. 7B and 7D). These data suggest that human Ngb acts to maintain cell membrane integrity and this may assist in maintaining receptor-mediated binding of targeted ligands (Fig. 6).
Assessing the mechanism for Ngb-mediated increase in cellular ATP
The observation that ATP increased in neurons overexpressing human Ngb after 100 or 200 μM H2O2 insult (Fig. 4) led to pharmacologic inhibitor studies probing the involvement of the mito-KATP channel in this process. In the absence of H2O2, ATP concentration was ∼5.7 ± 1.6 nmol/mg cell protein (mean ± SD; n = 3), indicating that treatment with the inhibitors did not alter baseline concentrations of ATP. Treatment of human Ngb overexpressing cells with glybenclamide prior to H2O2 insult inhibited the increase H2O2-mediated in ATP (Fig. 8A). Phosphorylation of protein within the mito-KATP channel by PI3K is implicated in regulating the mitochondrial pore (45). Thus, we sought to inhibit PI3K using two structurally unrelated inhibitors (wortmannin and LY294002) to determine the effect on intracellular ATP. Similar to glybenclamide, wortmannin and LY294002 inhibited increases in ATP upon (100 or 200 μM) H2O2 insult (Fig. 8A). At least in the case for LY294002, the reversal in ATP accumulation coincided with increased mitochondrial depolarization (Supplemental Fig. 3B), indicating that mitochondrial dysfunction contributed to the ATP loss. Parallel studies on cells pre-incubated with LY294002 showed that cell viability was largely resistant to peroxide insult (Supplemental Fig. 3C). These data indicate an involvement of PI3K in the protective activity of Ngb and that primarily mitochondrial dysfunction contributes to the decreased pool of ATP in Ngb-transfected cells exposed to H2O2.
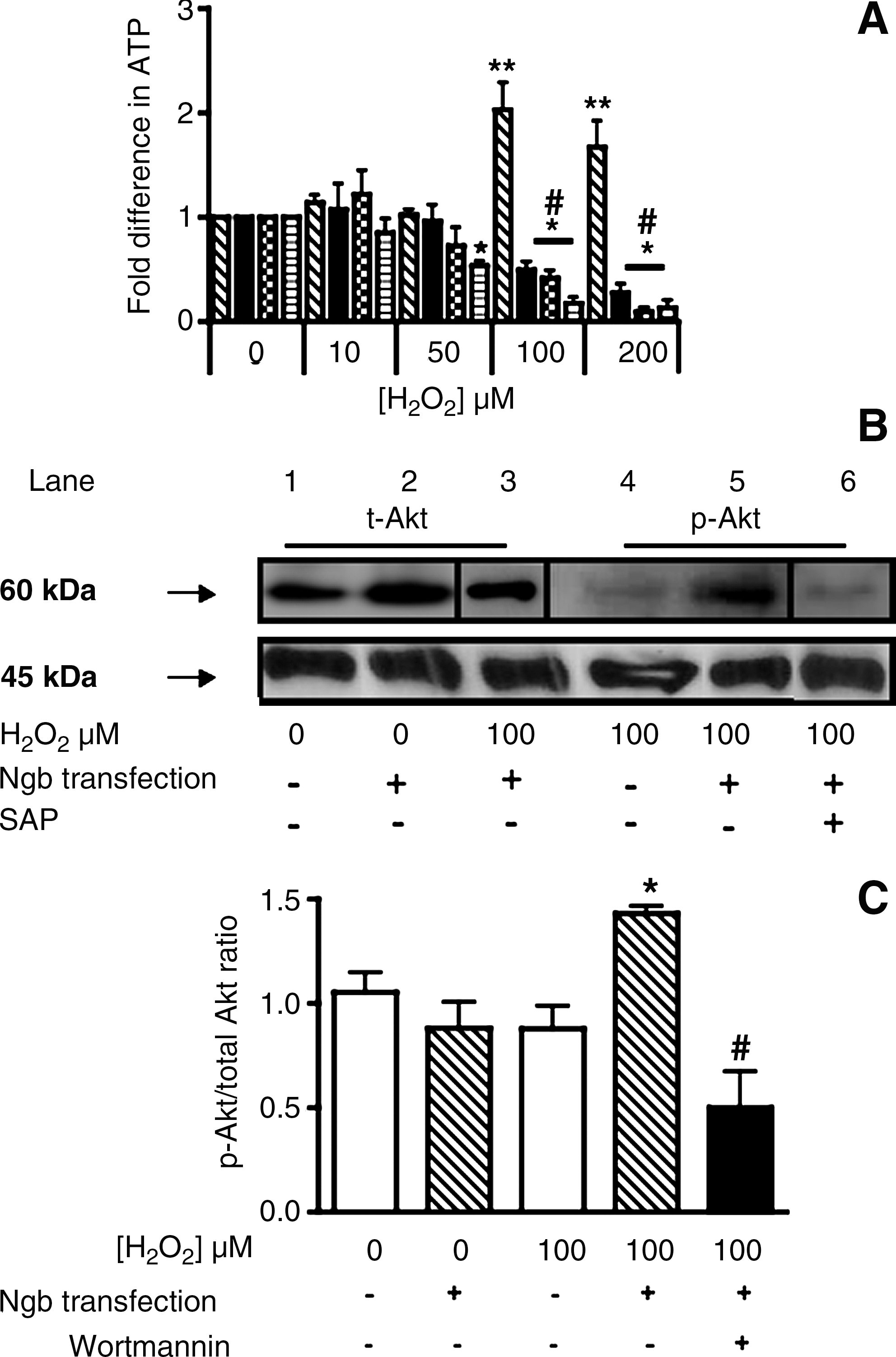
To validate that a PI3K-dependent mechanism was involved in regulating ATP in cells overexpressing Ngb, we monitored total Akt and p-Akt in parental and Ngb-transfected cells before and 2 h after insult with 100 μM H2O2 by using Western blotting; we anticipated enhanced Akt phosphorylation (Fig. 8B). In the absence of H2O2, total Akt was marginally elevated in Ngb-transfected cells compared to parental controls while baseline levels of p-Akt remained below the detection limit (not shown). When challenged with 100 μM H2O2, a dose that increased intracellular ATP, p-Akt markedly increased in Ngb-transfected cells, while in parental cells p-Akt was detected at lower concentrations under the same conditions. However, total Akt was unchanged in both cell types at these concentrations (not shown). As anticipated, the addition of SAP significantly diminished the level of p-Akt in Ngb-transfected cells exposed to 100 μM H2O2 thereby, confirming our assignment of p-Akt to the respective protein bands.
To verify the changes in p-Akt, we next determined the ratio of p-Akt/total Akt under the experimental conditions described above (Fig 8C). In the presence of 100 μM H2O2, parental cells showed a marginal decrease in the p-Akt/total Akt ratio. Consistent with the Western blot data, the p-Akt/total Akt ratio ratio increased in cells overexpressing human Ngb and this was inhibited by wortmanin (Fig. 8C).
Pharmacologic activation of PI3K in neuronal cells
To support the idea that Ngb-transfected cells maintained cell viability in response to H2O2 challenge via a PI3K dependant pathway, we sought to recapitulate this pathway in parental neurons by administering diazoxide—a selective mito-KATP channel opener, or leptin—a selective short-term PI3K activator, prior to H2O2 insult. Cells pretreated with diazoxide maintained intracellular ATP at all H2O2 doses tested, whereas cells exposed to leptin showed an immediate and significant increase in ATP (Fig. 9A) that mimicked that seen in Ngb-transfected cells (Fig. 4).
Parallel assessment of cell viability demonstrated that cells incubated with leptin were highly resistant to necrosis across all H2O2 doses, whereas diazoxide-treated cells revealed a trend to lower viability that became significant at the highest H2O2 (Fig. 9B). These data indicate that maintenance of the mito-KATP channel alone is not sufficient to confer protection against necrosis. Notably, pretreatment of parental cells with LY294002 prior to leptin reversed the protective effect of leptin, further reinforcing a role for PI3K in enhancing cell survival in the presence of H2O2 insult (Fig. 9B).
Discussion
Parental cells were able to tolerate a relative low level of peroxide insult with functional parameters remaining unchanged at ≤50 μM H2O2 challenge likely through the action of endogenous antioxidants. However, cells exposed to H2O2 at concentrations greater than 50 μM experienced marked deficits in viability with parallel increases in mitochondrial dysfunction and oxidative stress. Consistent with previous reports (17, 34), overexpression of Ngb inhibited oxidative stress in neuronal cells, thereby supporting the proposition that Ngb acts as a ROS scavenger (37). Cells overexpressing human Ngb also showed decreased mitochondrial dysfunction and Ca2+-influx, muted caspases 3/7 activation, enhanced cell viability, and maintenance of receptor-mediated endocytosis and cytoskeletal integrity.
The ability of Ngb to protect neuronal cells from H2O2 challenge through an antioxidant mechanism is established (17) and this may involve preserving mitochondrial function (37). Our data recapitulate the Ngb-mediated protection afforded to mitochondria in neurons exposed to H2O2 with a consequential decrease in oxidative stress yielding enhanced cell viability. Furthermore, our data demonstrate a H2O2-stimulated increase in total ATP within cells overexpressing Ngb. Thus, by preventing an energy deficit (i.e., through increasing the intracellular ATP during oxidative insult), Ngb may also prevent the breakdown of ion homeostasis, release of excitatory neurotransmitters, and excitotoxic stimuli that promote neuronal cell death (6).
Mitochondrial dysfunction leads to the overproduction of O2•− that is central to promoting enhanced apoptosis in neuronal cells (58). Maintenance of mitochondrial function may be crucial to cell survival. Therefore, a mechanism whereby Ngb stimulates ATP production during oxidative stress may be critical to maintaining neuronal cell viability and function. Phosphorylated Akt targets to the mitochondria and inhibits the release of apoptosis-inducing factor and cytochrome c, thereby inhibiting apoptosis (7, 54). Our results therefore add to the previously identified ROS-scavenging action of Ngb and reveal an involvement for Ngb in peroxide-stimulated PI3K activation and opening of the mito-KATP channels to preclude apoptotic sequelae associated with acute oxidative stress. Interestingly, the antioxidant catapol inhibits H2O2-mediated endothelial cell apoptosis through a mechanism involving activation of the PI3K/Akt signaling pathway (26). Thus consistent with our observations for Ngb in cultured neurons, antioxidants may induce PI3K and preserve cell viability. Alternately, opening of the mito-KATP channels represents a powerful contributor to neuroprotection (11). Precisely how Ngb is able to modulate PI3K activity and why this activity requires stimulation by added H2O2 at doses >50 μM warrants further investigation. Nevertheless, our data reveal that the antioxidant action of Ngb is linked to maintaining cellular energetics and viability during experimental oxidative stress.
Sustained opening of the mito-KATP channel is associated with cerebral preconditioning against ischemia reperfusion injury (63). Phosphorylation of protein components that comprise the mito-KATP channel are important in opening of the pore that sustains ATP release. Both PI3K (45) and PKC epsilon (46) are implicated in this regulatory process. In this study, we provide evidence to implicate Ngb in regulating PI3K activity that, in turn, affects the mito-KATP channel and increases the proportion of p-Akt relative to total Akt. This mode of action for Ngb in the face of challenge with pathological concentrations of H2O2 was recapitulated with a pharmacologic activator of PI3K and suggests that Ngb may enhance neuronal cell survival after acute cerebral ischemia, where increased generation of H2O2 through uncontrolled production of O2•− is implicated (55). Such a link between Ngb and pAkt in neuronal cells may explain the involvement of Ngb in “resetting levels of apoptosis”, albeit through mechanisms that do not necessarily involve a direct interaction with cytochrome c (8, 15).
How Ngb interacts with H2O2 to stimulate PI3K activity is not known. Maintenance of actin cytoskeletal integrity is necessary for PI3K activation (41), however it remains unclear from our data as to whether Ngb-mediated stabilization of actin in turn activates PI3K or whether PI3K is activated directly by Ngb during excessive oxidative stress. In addition, a continuous availability of ATP may underpin PI3K activation. Nevertheless, our studies with leptin or diazoxide show that activation of PI3K and opening of the mito-KATP channel are potentially useful strategies to protect cultured neurons from H2O2 insult.
Interestingly, the mito-KATP channel is reported to be redox sensitive with the oxidation of key proteins involved in pore opening (18). However, ROS activation of the mito-KATP channel does not seem to be relevant in our experimental system since overexpression of human Ngb downregulated oxidative stress yet enhanced ATP release through the opening the mito-KATP channel. In addition to redox regulation, S-nitrosylation of cysteine residues in the SUR1 subunit activates the mito-KATP channel (14, 30). Whether human Ngb plays a role in facilitating S-nitrosylation of cysteine residues in the SUR1 subunit is not known and further studies investigating this potential relationship are warranted.
Given the impact of oxidative stress on neuronal function, it was reasonable to consider the expression levels of natural antioxidants, such as HO-1, SOD, and CAT in our cell model (31). The selective induction of HO-1 compared with other antioxidant enzymes has been reported previously (5). Induction of the ARE HO-1 is regulated via transcription factors such as HIF-1α, Nrf2 (29), and AP1 (22), which themselves may be under redox regulation. HO-1 expression is triggered by a variety of stress-inducing stimuli including hypoxia, UV irradiation, H2O2, and NO (53). Protective effects of HO-1 are linked to signal transduction and activators of transcription (STAT) proteins (including PI3K) [e.g., STAT3 (64) acting via cytokines IL-6 and IL-10 (12, 34)]. Upstream STAT/JAK signaling is regulated by an influx of Ca2+ (40), which is consistent with the observed Ca2+-influx in parental cells exposed to 100 or 200 μM H2O2 that corresponded to a induction of HO-1 gene and protein with parallel increases in activity.
The pathological relevance of this study depends on whether neuronal cells are exposed to the H2O2 levels used here. Under normal physiological conditions [H2O2] is strictly regulated and ranges 0.13–0.25 μM (20). By contrast, [H2O2] up to 100 μM are reported in rat striatal tissue following forebrain ischemic injury (28). Furthermore, following cerebral ischemia, infiltrating polymorphonuclear neutrophils (PMN) may be a source of H2O2 in affected brain tissues (4). In humans the density of circulating PMN reaches ∼1.5-6 × 106 cells/ml. At this cell density 0.08–0.48 mM H2O2/h is generated upon exogenous chemical activation (38). Moreover, H2O2 is diffusible and less reactive than other ROS; therefore, it is plausible that neurons are exposed (locally) to high micromolar [H2O2] in the setting of cerebral ischemia and that H2O2-doses of ∼100 μM may be pathological. Another consideration is the levels of endogenous Ngb versus the 4-fold higher levels achieved through overexpression of the protein here (Supplemental Fig. 1B).
Neuroglobin has been implicated in oxygen transport (44), as an oxygen sensor that initiates the activation of other proteins with regulatory functions (33), or even as a dioxygenase that limits nitrosative stress (59). Though not yet completely understood, Ngb can act to preserve cell viability through multiple protective mechanisms that involve regulation of metal ion homeostasis in neuronal cells (13). The present study expands on these potential activities and links Ngb with the activation of both PI3K and the mito-KATP channel and an increase in the p-Akt/total Akt ratio in response to an oxidative insult that overwhelms the endogenous antioxidant capacity. Notably, activation of the PI3K/Akt signaling pathway is also implicated in regulating the pro-apoptotic JNK pathway (1). The authors also acknowledge the possibility that H2O2 itself may induce Ngb expression (35). However, at least in our hands, no change in the expression of the human Ngb–fusion protein was evident upon H2O2 insult (Supplemental Fig. 1A). Studies with Ngb-deficient mice may yield more information on the role for Ngb during cerebral ischemia.
Footnotes
Acknowledgments
This work was supported by the Australian Research Council (DP0343325 and DP0878559) and National Heart Foundation Grants (G07S30435 to PKW and Scholarship PB08S4123 to STA).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.