
Abstract
Proteins are main targets for oxidative damage that occurs during aging and in oxidative stress situations. Since the mitochondria is a major source of reactive oxygen species, mitochondrial proteins are especially exposed to oxidative modification, and elimination of oxidized proteins is crucial for maintaining the integrity of this organelle. Hence, enzymatic reversal of protein oxidation and protein degradation is critical for protein homeostasis while protein maintenance failure has been implicated in the age-related accumulation of oxidized proteins. Within the mitochondrial matrix, the ATP-stimulated mitochondrial Lon protease is believed to play an important role in the degradation of oxidized protein, and age-associated impairment of Lon-like protease activity has been suggested to contribute to oxidized protein buildup in the mitochondria. Oxidized protein repair is limited to certain oxidation products of the sulfur-containing amino acids cysteine and methionine. Oxidized protein repair systems, thioredoxin/thioredoxin reductase or glutaredoxin/glutathione/glutathione reductase that catalytically reduce disulfide bridges or sulfenic acids, and methionine sulfoxide reductase that reverses methionine sulfoxide back to methionine within proteins, are present in the mitochondrial matrix. Thus, the role of the mitochondrial Lon protease and the oxidized protein repair system methionine sulfoxide reductase is further addressed in the context of oxidative stress and aging. Antioxid. Redox Signal. 13, 539–549.
Introduction
Indeed, ROS are highly reactive components that cause oxidative damages to macromolecules. Mitochondrial proteins are specially exposed to mitochondrial ROS and represent targets for oxidative modifications and loss of function. Indeed, ROS can readily react either directly with the side chains of amino acid residues and the peptidic backbone of proteins or with lipids and carbohydrates, the oxidized forms of which can generate lipid peroxidation and glycoxidation adducts on proteins, respectively (5, 6). Mitochondrial dysfunction, together with elevated production of ROS, has been associated with numerous diseases and with the nonpathological aging process (58, 62, 97).
Elimination of damaged protein has been recognized as a critical function for maintenance of cellular homeostasis and survival (87). Increasing experimental evidence indicates that failure of protein maintenance (i.e., degradation and repair), is a major contributor of age-associated accumulation of oxidatively modified proteins (26) (Fig. 1). Repair is limited to few protein oxidative modifications, such as cysteine and methionine oxidation. Interestingly, cysteine and methionine are among the amino acids the most susceptible to oxidation. Methionine oxidation can be reversed by the methionine sulfoxide reductase (Msr) enzymes, MsrA and MsrB, which reduce the two diasteroisomers of the oxidized methionine, methionine-S-sulfoxide and the methionine-R-sulfoxide, within proteins, respectively (73, 76). Importantly, MsrA and MsrB are both present in mitochondria, and the methionine sulfoxide reductase system has been implicated in longevity and resistance to oxidative stress in different cell types and model organisms (56, 74, 95). Oxidized protein degradation is mainly achieved by the proteasomal system in the cytosol and the nucleus while the Lon protease has been implicated in oxidized protein degradation within the mitochondrial matrix (10). It is now well established that proteasomal function is generally impaired with age while recent data argue for oxidative stress and age-associated alterations of Lon protease function (22, 79, 80, 103). This review will address the implication of the mitochondrial Lon protease and the oxidized protein repair system methionine sulfoxide reductase in the maintenance of mitochondrial protein homeostasis in the context of oxidative stress and aging.
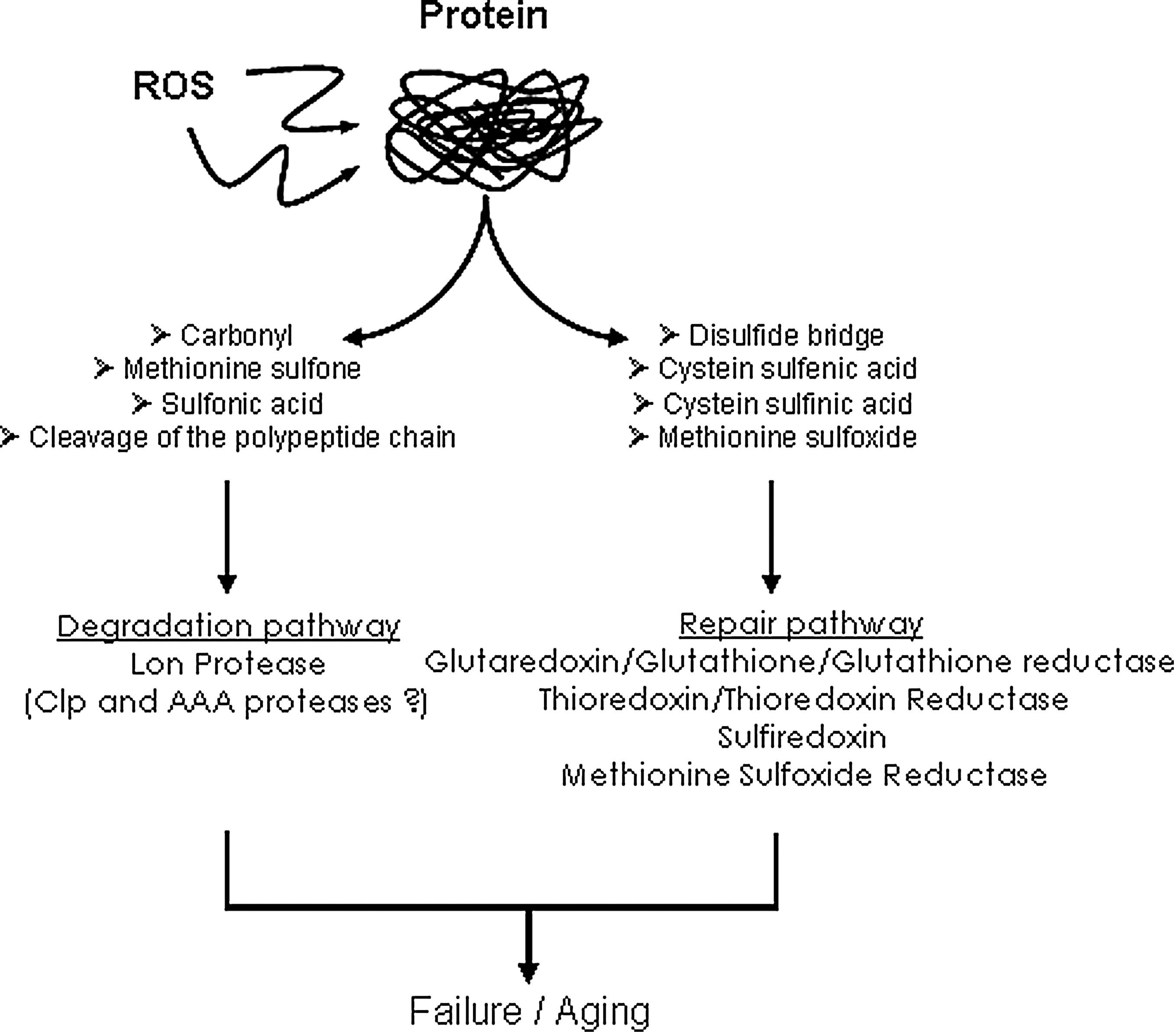
Protein Oxidative Damage
Oxidative modifications can induce reversible or irreversible damages on proteins. Irreversible modifications are caused by the formation of hydroxyl and carbonyl groups but also by the conjugation of lipid peroxidation products such as malondialdehyde and 4-hydroxy-2-nonenal or the formation of glycoxidation adducts like carboxymethylysine and pentosidine (5). The formation of such carbohydrate oxidation and lipid peroxidation adducts may bring carbonyl groups and generate intramolecular crosslinks. Levels of protein oxidative modification in biological samples have been assessed using immunochemical approaches (64) and more recently stable isotopic dilution-based mass spectrometry analysis (93) that allow detection and quantification of such irreversible modifications. Irreversible damages generally cause function impairment or complete inactivation of the modified protein. Irreversibly altered proteins are eliminated through degradation pathways that include the proteasome in the cytosol or the Lon protease in mitochondria (12, 38, 98). Nevertheless, some modifications altering sulfur-containing amino acids can be repaired by specific enzymatic systems like thioredoxin/thioredoxin reductase and methionine sulfoxide reductase (87). Together with aromatic amino acids, sulfur-containing amino acids, cysteine and methionine, are the most sensitive to oxidation. Cysteine oxidation leads to the formation of sulfenic acid that can react with other cysteine residues to form disulfide bridges. This sulfenic acid may also be further oxidized and produce sulfinic and sulfonic acids. Thioredoxin and glutaredoxin are ubiquitous members of the thiol/disulfide oxydoreductase family. The thioredoxin/thioredoxin reductase system is involved in the reduction of disulfide bridges and sulfenic acid, while the glutaredoxin/glutathione/glutathione reductase system reduces disulfide bridges and low molecular mixed disulfides (43). Sulfiredoxin has been shown to reduce cysteine sulfinic acids in peroxiredoxins (7, 82, 114). The catalytic site of thioredoxin and glutaredoxin is based on two cysteines that can be oxidized and then form a disulfide bridge. Such oxidized thioredoxin and glutaredoxin species are subsequently reduced by the thioredoxin reductase and glutathione/glutathione reductase, respectively. These reductase systems, both located in the cytosol and in the mitochondria of mammalian cells, have been associated with cellular protection against oxidative stress and redox signalling pathways (43, 44).
Repair of Oxidized Proteins by the Methionine Sulfoxide Reductase System
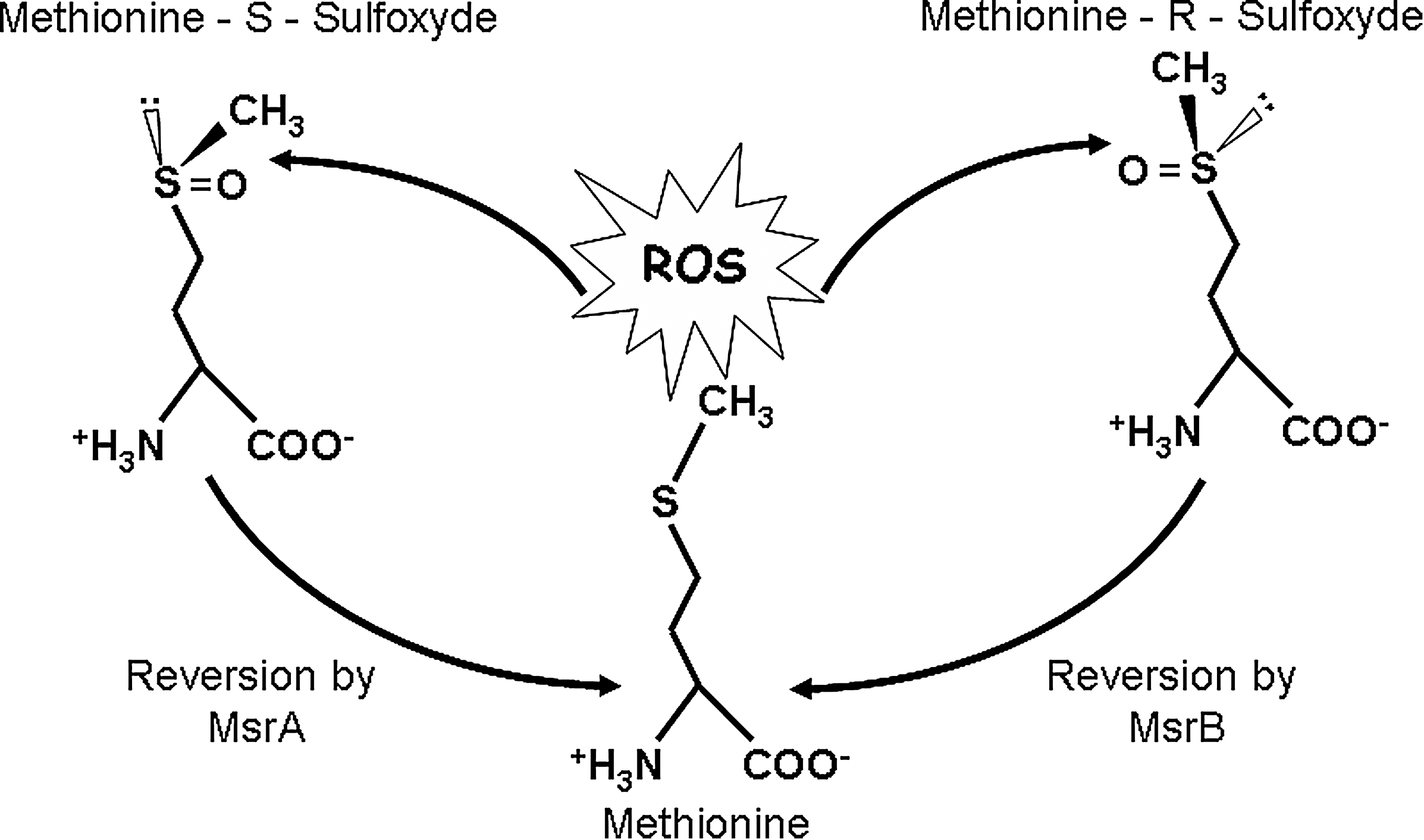
Oxidation of methionines can induce the formation of two diastereoisomers of methionine sulfoxides, the Met-S(O) and Met-R(O) forms. These sulfoxide residues can be further oxidized by ROS and generate methionine sulfones. Methionine oxidation has been associated with the impairment of protein function in various cases (3, 13, 19, 25, 35, 49, 110), mostly by disturbing the interacting properties of the targeted protein. While the formation of methionine sulfones is an irreversible damage, methionine sulfoxides can be reversed by a repair system, the methionine sulfoxide reductases (Msrs). The S and R diastereoisomeric forms of methionine sulfoxide are enzymatically reduced back to methionine by MsrA and MsrB, respectively (Fig. 2). As this reduction induces the recovery of some proteins function (73, 76), repair of oxidized methionines may be involved in the regulation of protein activities. More generally, accumulation of oxidized methionines and impairment of Msr has been linked with aging, neurodegenerative diseases, and oxidative stress.
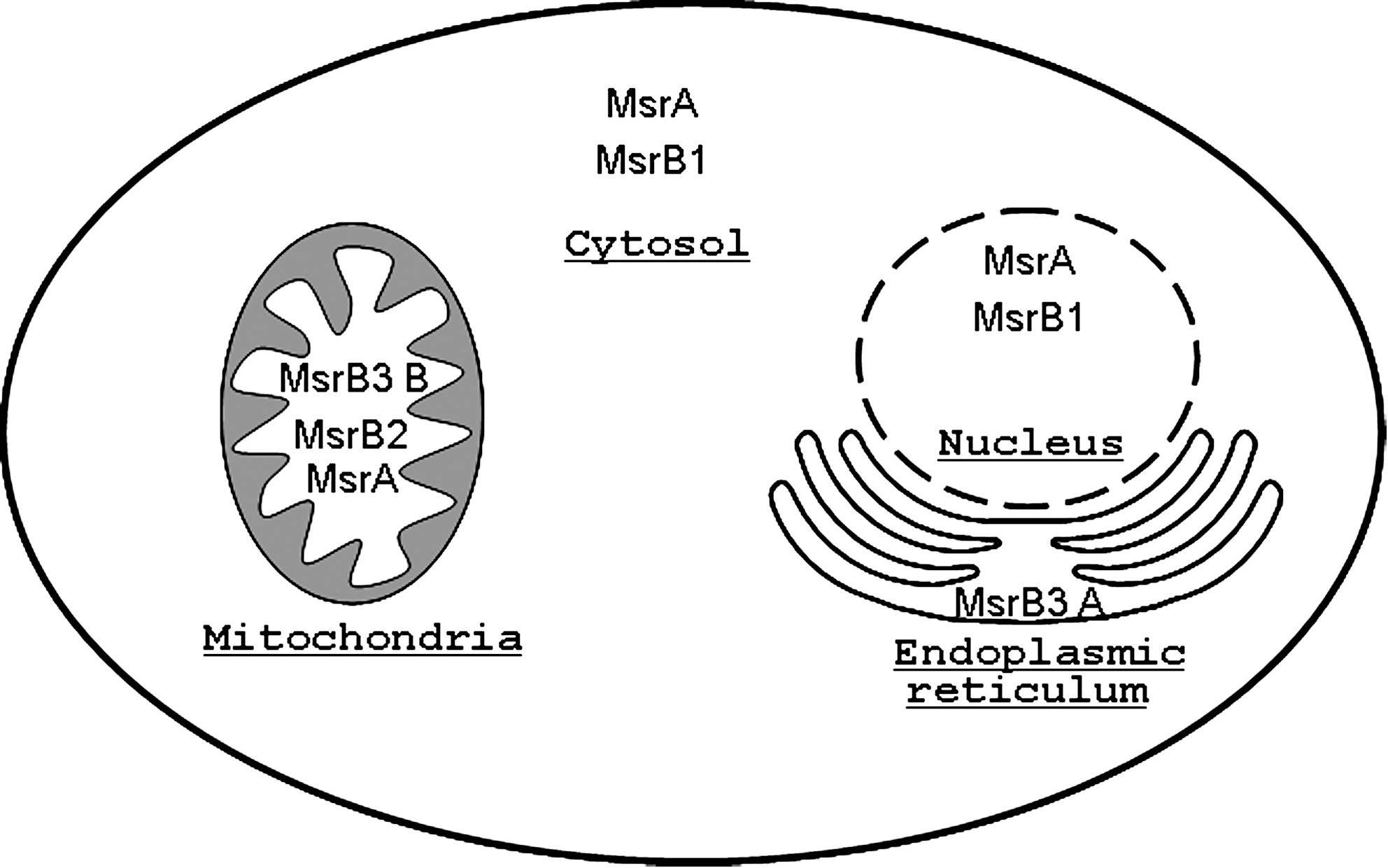
Msr distribution and regulation
Msr genes are found in a wide range of organisms, from bacteria to mammals. However, their number differs from one organism to another. Single MsrA and MsrB genes exist in Escherichia coli, Caenorhabditis elegans, and Drosophila megalonaster, while multiple copies are found in Arabidopsis thaliana or Homo sapiens (23). MsrA and MsrB share little similarity in their sequence and structure (67, 105).
In mammals, a single copy of the MsrA gene is present in the genome with different isoforms being expressed in the cytosol and mitochondria of almost all analyzed cell types except for leukemia cell lines (41, 111). A MsrA isoform was also found in the nucleus of mouse cells (54). The human MsrA expression has been shown to be under the control of a promoter containing a CCAA box negative regulatory region (20).
In contrast, the different forms of MsrB come from three distinct genes in mammals. The primary identified Msr, MsrB1, also called SelX or SelR, is a selenoprotein found in the nucleus and the cytosol; MsrB2, reported first as CBS-1, is present in the mitochondria; the MsrB3 gene encodes for two splice variants, MsrB3A and MsrB3B, localized in the endoplasmic reticulum and the mitochondria, respectively (40, 52). The MsrB enzymes are present in all eukaryotic tissues but show different levels of expression (40). It has been shown that different factors affect the transcription of the human MsrB1 gene: the transcription factor Sp1 may regulate its expression while methylation of the promoter may also be an important factor to control the promoter activity (21). An analysis of MsrA-knockout mice also suggests that the expression of MsrB1 is influenced by MsrA and by selenium (78). In addition, many MsrBs have been characterised as zinc-containing enzymes. These MsrBs bind a zinc atom to cysteine residues organized in Cysteine-X-X-Cysteine motifs in which X refers to any amino acid. The precise role of zinc still has to be elucidated, but it has been suggested to be involved in the structural function of the enzymes (57).
It is interesting to note that both MsrA and MsrB are found in different compartments of the cell (Fig. 3), most likely to reduce the two diastereoisomeric forms produced by methionine oxidation. Still, the endoplasmic reticulum possesses only an MsrB3 enzyme form, suggesting the putative existence of an epimerase in this compartment to allow the reduction of the Met-S(O) form.
Catalytic mechanism of Msrs
The catalytic mechanism of MsrA and MsrB is based on three steps (9). First, the Msr catalytic cysteine residue interacts with the methionine sulfoxide substrate, inducing the formation of a sulfenic acid intermediate and the release of the reduced methionine. Then, the sulfenic acid intermediate is attacked by a recycling cysteine residue, leading to the formation of an intramolecular disulfide bond. Finally, the disulfide bond is reduced by an electron donor, the NADPH-dependent thioredoxin/thioredoxin reductase system in vivo, leading to the regeneration of the Msr active site (Fig. 4).
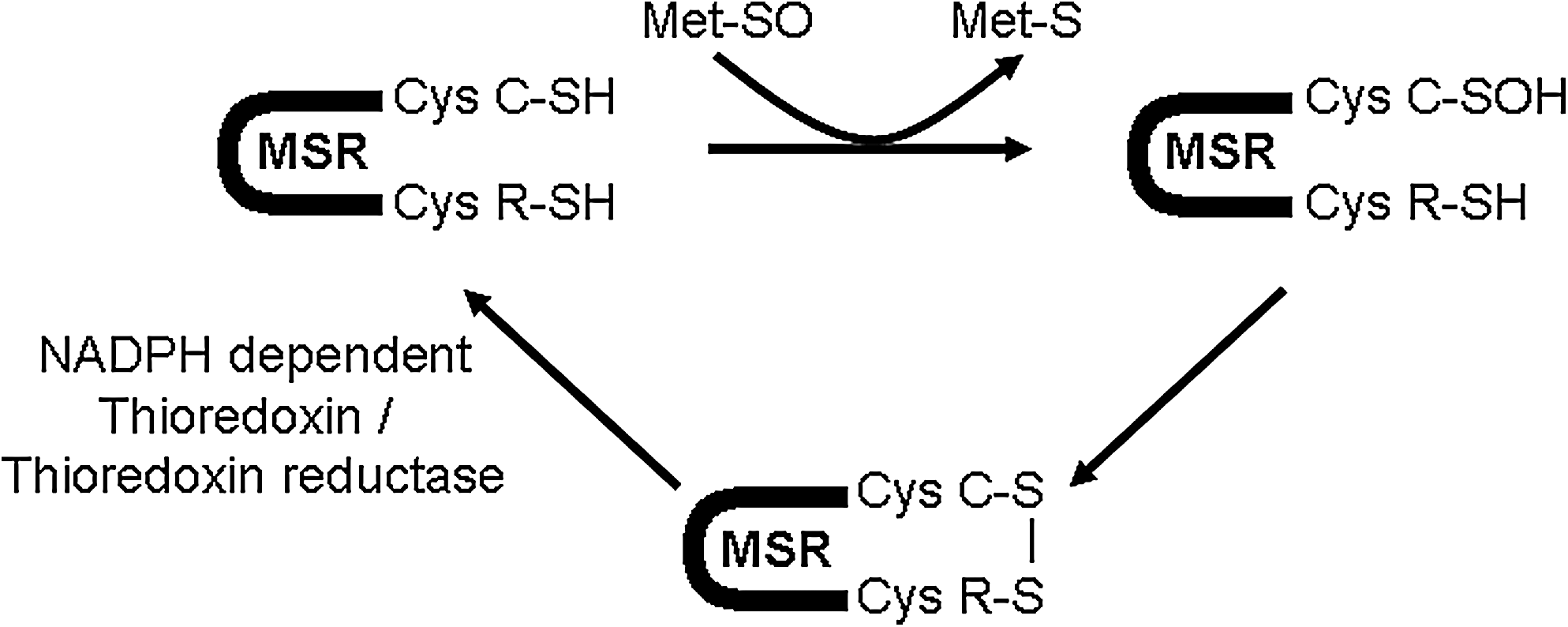
However, some variations are observed in the catalytic mechanism of the different Msrs, particularly in the number of recycling cysteines involved in this process. Indeed, MsrA can have one recycling cysteine residue in their active site like in Neisseria meningitidis but two recycling cysteines can also be implicated in the catalytic mechanism, as observed in Escherichia coli and bovine MsrAs. In the latter case, two successive disulfide bonds are formed, between the catalytic and a recycling cysteine residue and then amid the two recycling residues. However, some MsrAs appear not to have any recycling residue, suggesting that the sulfenic acid intermediate is directly reduced by an electron donor. This trait has been observed in Chlamydomonas MsrA (51). Similarly, MsrBs can be divided in two subclasses, the ones that possess one recycling cysteine residue (approximately 60% of the MsrBs) as seen in Neisseria meningitidis and the ones that lack this residue (the remaining 40%) like in the three mammalian MsrBs. However, the selenoprotein MsrB1 is believed to use an alternative recycling cysteine residue located in a different position than expected (53). Otherwise, in the absence of a recycling cysteine, the sulfenic acid intermediate may be reduced by alternative mechanisms (24).
The role of Msrs in oxidative stress conditions
The main role for Msrs is to repair oxidized methionines. From this characteristic, various functions have been allocated to the Msr system. It has been shown that Msrs act as an effective antioxidant for cells. Indeed, surface-exposed methionines are easily oxidized by ROS and are subsequently reduced back by the different Msrs. The successive oxidation/reduction reactions are believed to trap the ROS, the Msr system is then acting as a potent ROS scavenger (63). Furthermore, by preferentially attacking the surface-exposed methionines and not the crucial residues located in the catalytic site of the peptides, the ROS will not damage important protein functions. Such antioxidant property is crucial to maintain protein homeostasis in cells.
In addition, the successive methionine oxidation/reduction has also been proposed to act as a protein regulating system, supporting a role of potential regulator of cellular function (46). Similarly to the phosphorylation/dephosphorylation process, the activity of a protein will be dependent on the level of oxidation of specific methionines. Oxidation of methionine residues causing an impairment of function has been described in various proteins that are Msr substrates in vitro, such as human alpha-1-proteinase inhibitor (49), apolipoproteins AI and AII (35), HIV-2 protease (19), Ffh (25), calmodulin (3, 110) and mitochondrial cytochrome c (13).
A multitude of analyses have demonstrated the role of Msrs in antioxidant defense. Cells treated with different doses of hydrogen peroxide enhance their expression levels of MsrA and MsrB2, two Msrs found in the mitochondria compartment (90, 101). Overexpression of MsrA in human fibroblasts and of MsrA or mitochondrial MsrB2 in human leukemia cells induces resistance to oxidative stress, maintains a lower level of intracellular ROS, and prevents accumulation of protein oxidative damage (16, 17, 92). MsrB2 overexpression also proves to be effective to prevent the loss of mitochondrial membrane potential under oxidative stress conditions (16). Mitochondrial damage was also prevented by overexpression of MsrA in lens and PC12 cells (71, 115). However, silencing of the different Msrs in human lens cells decreases protection against oxidative stress and alters viability of the cells (71, 72). In addition, MsrA silencing in human lens cells induces a loss of mitochondrial membrane potential (71).
Msr enzymes may also be affected by oxidative stress as their activity was shown to decrease during cardiac ischemia-reperfusion and UV irradiation, events that are associated with increased ROS production and protein oxidative damage (89, 91). Furthermore, MsrA is prone to inactivation by hydrogen peroxide in vitro (59).
The role of Msrs in aging and age-associated neurodegenerative diseases
The free radical theory of aging proposed that the accumulation of macromolecular damages, as the consequence of the increase of ROS production, is one of the main causes of aging (42). Consequently, resistance to oxidative stress is believed to be a key determinant of lifespan prediction. Therefore, the role of the Msr system in aging and longevity has been tested.
Msr activity was shown to decrease during aging in different rat organs and in mouse liver (83, 88), while replicative senescence, a model of cellular aging, revealed that Msr activity and the expression levels of MsrA and mitochondrial MsrB2 are reduced in senescent cells (90). The decreased efficiency of these repair enzymes may contribute to the accumulation of oxidative protein damage. In yeast, under aerobic conditions, deletion of the MsrA gene induces a lifespan shortening of 26% while its overexpression increases it by 25% (56). Little effects were observed when MsrB was deleted or overexpressed alone. However, the double mutant msraΔmsrbΔ has a shortened lifespan compared to the single msraΔ mutant (56), suggesting that both enzymes play a role in yeast longevity. Drosophila overexpressing bovine MsrA show a resistance to paraquat-induced oxidative stress and a 70% increase in median lifespan (95). However, transgenic fruit flies overexpressing Drosophila MsrB or mouse mitochondrial MsrB2 appear to have a lifespan and an oxidative stress resistance similar to control flies, suggesting different effects of MsrA or MsrB on Drosophila aging (99). In Caenorhabditis elegans, deletion of the msra-1 gene shortens the lifespan by 30% and causes a decrease in resistance to paraquat stress (74). Initially, the absence of MsrA was thought to reduce lifespan in mice (77), but recent findings suggested that the lack of MsrA has no effect on mouse longevity, even though it increases sensitivity to oxidative stress (96). Therefore, further experiments need to be conducted to evaluate the precise role and function of the different Msrs in the aging process.
The Msr system seems to play a role in age-associated neurodegenerative disorders such as Parkinson's and Alzheimer's diseases. Indeed, the brain of Alzheimer's disease patients showed a significant decline in Msr activity (34). Furthermore, the amyloid β-peptide plays a key role in the neurodegeneration observed in Alzheimer's disease brains. Oxidation of the Met-35 residue located in its C-terminal region appears to be critical to induce such deleterious effects (47). MsrA knockout mice also display enhanced neurodegeneration associated with high levels of β amyloid deposition in brain hippocampus (85). Oxidation of the α-synuclein protein has been associated with the pathogenesis of Parkinson's disease. α-Synuclein does not contain tryptophan or cysteine residues, therefore exposure to mild oxidative stress induces oxidation of the four methionine residues, preventing fibril formation (107). Fruit flies overexpressing human α-synuclein in the nervous system showed Parkinson's disease traits and have been used since as a model to investigate such disorder (27). Overexpression of MsrA inhibits development of the locomotor and circadian rhythms deficiencies observed in this Drosophila model. A dietary supplementation with S-methyl-L-cysteine, a methionine analog, also prevents those Parkinson's-like symptoms (113). The homozygous deletion of DJ-1, a gene encoding for an ubiquitous protein whose precise biochemical function is unclear, is also associated with recessively transmitted Parkinson's disease (8). Furthermore, brains of Parkinson and Alzheimer patients show oxidative damages in DJ-1. In addition to carbonyl formation and cysteine oxidation, unsuspected methionine oxidation was identified in DJ-1 (18). Even if mutations or post-translational modifications of DJ-1 are strongly linked with early onset familial Parkinsonism, its role in this pathogenesis still remains unclear.
Degradation of Oxidized Protein by the Mitochondrial Lon Protease
Mammalian mitochondria contain three major ATP-dependent proteases that are involved in matrix protein degradation, Lon, Clp-like, and other AAA proteases (ATPases associated with a variety of cellular activities). AAA and Clp-like proteases are hetero-oligomeric complexes located in the inner mitochondrial membrane and matrix, respectively (50, 108). These proteases have chaperone activity and participate in the degradation of misfolded and damaged proteins and/or maintenance of mitochondrial genome stability (65). Both chaperone and proteolytic functions have been implicated in the prevention of the accumulation of aggregated proteins. In addition, the Lon protease seems to play a critical role in the removal of oxidized proteins since oxidized aconitase has been reported to be a specific substrate of the Lon protease (10). Therefore, the Lon protease is expected to play a critical role for eliminating oxidized proteins in the mitochondrial matrix, hence maintaining mitochondrial functional and structural integrity.
Structure and function of the Lon protease
The Lon protease is encoded by the nuclear genome like most of the mitochondrial proteins and the lon gene encodes a 963 amino acids protein which is homologous to the bacterial protease La (112). Indeed, the human Lon protease is a highly conserved protein with homologs present in bacteria, archae, and eukaryotes, that displays 25% sequence similarity with the Lon protease from E. coli. The Lon protease is composed of three domains (48, 75) (Fig. 5). The amino-terminal domain is involved in oligomerization and protein substrate binding in concert with the AAA+ module which constitutes the ATPase domain. The AAA+ module is composed of two subdomains, the first one involved in ATP binding (α/β domain) and the second one involved in ATP hydrolysis (α domain). Stimulation by ATP is an important characteristic of the Lon protease while ADP acts as an inhibitor of the protease (70, 106). Moreover, misfolded protein substrates have been reported to stimulate both the ATPase and protease activities of the E. coli Lon protease (36). The carboxy-terminal domain, also called P domain, carries the serine-lysine catalytic dyad (1). When isolated, the P domain does not degrade protein substrates such as casein but can still degrade small peptides such as melittin (94).

The Lon protease is a serine protease that forms an acyl-enzyme intermediate (84). There are few Lon protease inhibitors including the nonspecific serine inhibitor phenylmethyl sulfonyl fluoride (PMSF), the peptide boronate MG262, and certain coumarinic derivatives (4). Although there is no specific peptide consensus sequence, the cleavage specificity would be similar to chymotrypsin with a preference for hydrophobic residues at P(-1) and P(+1) positions. Hydrophobic loops exposed at the surface of proteins have been proposed to be essential features for substrate recognition and, like other ATP-dependent proteases, the Lon protease would degrade its protein substrates processively (81, 84). The Lon protease is active as an homo-oligomer composed of six monomers in E. coli and seven monomers in yeast (86, 102). The homo-oligomeric enzyme is a ring-shaped complex reminiscent of other ATP-dependent proteolytic machineries such as the 26S proteasome. However, Lon is structurally distinct from other two-component ATP-dependent proteases since, unlike these proteases, both proteolytic and ATPase activities are carried by the same polypeptide chain. In yeast, the absence of Lon results in the accumulation of electron dense aggregates and mitochondrial DNA deletion (104, 109).
Interestingly, in addition to its proteolytic activity, the mammalian Lon protease has also been shown to display chaperone properties and to bind mitochondrial DNA, both in vitro and in cultured cells, as well as to interact with mitochondrial DNA polymerase γ and the Twinkle helicase (32, 65). Human Lon has been shown to bind preferentially the noncoding region of the mitochondrial DNA, also referred as the control region that carries the heavy- and light-strand promoters required for mitochondrial DNA transcription and replication (68). DNA binding-related roles of the Lon protease include degradation of specific factors involved in transcription and/or replication, remodeling of the mitochondrial nucleoids, and maintenance of the integrity of mitochondrial DNA (61).
Lon protease and oxidized protein degradation
The human Lon protease has been implicated in the removal of oxidized protein since aconitase, an iron–sulfur cluster enzyme that belongs to the tricarboxylic acid cycle and which is highly susceptible to oxidative inactivation, has been shown to be degraded by the Lon protease when aconitase is inactivated after exposure to oxygen radicals (10). Indeed, the matrix proteolytic activity was found to copurify with the Lon protease after size exclusion and affinity chromatography of the mitochondrial matrix extracts. In addition, the proteolytic activity responsible for the degradation of oxidized aconitase was stimulated by ATP and inhibited by the serine protease inhibitor PMSF. Oxidized aconitase was degraded by the purified Lon protease in the presence of ATP with a specific activity of 2 mg degraded oxidized aconitase per mg of Lon protease per hour. Moreover, decreased Lon protease activity and content induced by anti-sense oligonucleotides treatment of WI-38 human lung embryonic fibroblasts resulted in the accumulation of oxidized aconitase (10).
It has also been further shown that chronic knockdown of the Lon protease results in disruption of mitochondrial structure, impairment of mitochondrial function, decreased mitochondrial mass and cell death with a majority of cells undergoing caspase 3 activation and apoptosis within 4 days (11). Aberrant mitochondrial morphology and the presence of electron-dense inclusion bodies of aggregated proteins in the mitochondrial matrix have been evidenced by electron microscopy in these Lon deficient cells.
Conversely, the upregulation of Lon protease content and/or activity has been associated with different stress situations such as endoplasmic reticulum stress and hypoxia (45). In the adaptive response to hypoxia, the Lon protease function would be implicated in the remodeling of the cytochrome oxidase holoenzyme through the degradation of the Cox4-1 isoform of the Cox4 subunit, hence allowing its replacement by the Cox4-2 isoform (33, 45). More recently, in a muscle creatine kinase mutant mouse heart model for Freidrich ataxia, a progressive increase in protein level of the ATP-dependent Lon and also ClpP mitochondrial proteases was observed in the heart (39). This upregulation was followed by an increase in proteolytic activity and a simultaneous loss of mitochondrial Fe–S proteins with no substantial change in their mRNA level, suggesting that Fe–S proteins are potential targets of Lon protease. Moreover, ATP-stimulated mitochondrial matrix proteolytic activity was increased during cardiac ischemia-reperfusion, a pathophysiological process associated with the transient production of mitochondrial reactive oxygen species (14). This increased proteolytic activity that occurred at early times of reperfusion, was accompanied with a concomitant decrease of oxidized protein but was not associated with an upregulation of the Lon protease content. Hence, these findings suggest that the Lon protease may be regulated by pro-oxidants in a manner consistent with the removal of oxidized proteins (15). Finally in rhabdomyosarcoma cells, it has been shown that the expression of Lon protease is induced by multiple stressors such as heat shock, serum starvation, and hydrogen peroxide-generated oxidative stress (80). Induction of the Lon protease expression by pretreatment of the cells with low-level stress was found to protect against oxidative damage, diminished mitochondrial function, and loss of cell proliferation induced by toxic levels of hydrogen peroxide. In contrast, when Lon induction was prevented by treatment with siRNA, no protection was observed, pointing out the important role of the Lon protease in protection against oxidative damage and for the maintenance of mitochondrial integrity (80). Taken together, these findings underscore the critical role of the Lon protease in mitochondrial protein homeostasis and protection against mitochondrial dysfunction in such deleterious situations as oxidative stress.
Oxidized proteins and the Lon protease in aging
As already stated above, mitochondria is a major source of reactive oxygen and also nitrogen species, inflicting oxidative damage on mitochondrial macromolecules such as DNA and proteins. It is now well documented that mitochondrial production of reactive oxygen species increases with age while accumulation of oxidized protein represents a hallmark of cellular aging. Oxidatively modified protein buildup with age results, at least in part, from the increase of reactive oxygen species and other toxic compounds coming from both cellular metabolism and external factors including environment (100). Increasing experimental evidence has also indicated that failure of protein maintenance (degradation and repair) is a major contributor to the age-associated accumulation of damaged proteins (28). Oxidized protein degradation is mainly achieved by the proteasomal system in the cytosol and in the nucleus while the Lon protease has been implicated in oxidized protein degradation within the mitochondrial matrix (38, 79).
Age-related decline of Lon gene expression was first documented in mouse skeletal muscle (60). Interestingly, the 4-fold decrease in Lon mRNA level observed in aged mice was completely prevented in aged mice subjected to dietary restriction, the only known intervention that slows down aging in mammals. Subsequently, the Lon protease activity and protein level were found to be reduced about 5-fold in skeletal muscle of old mice as compared to young mice, while a high level of oxidized proteins, including mitochondrial aconitase, was evidenced in old animals (12). We have previously reported that the age-related accumulation of oxidized and glycoxidized proteins in rat liver mitochondrial matrix is associated with an alteration of the ATP-stimulated Lon-like protease function (2). In this study, the effects of aging were monitored at the levels of mitochondrial respiration, oxidatively modified protein content, and ATP-stimulated proteolytic activity in the mitochondrial matrix. No change in oxygen consumption was observed between old and young rats. The Lon-like protease activity was decreased by 2.5-fold in old rats, although the Lon protein level remained the same in the mitochondrial matrix of young and old rats, suggesting that Lon protease is inhibited and/or inactivated. The decline in Lon protease activity was associated with a decreased activity of mitochondrial aconitase (2). Increased amount of oxidatively modified proteins in the mitochondrial matrix may interfere with degradation of others proteins by the Lon protease, as it has been observed for the proteasome in the cytosol (31, 37). Indeed, highly modified proteins such as proteins crosslinked with the lipid peroxidation product 4-hydroxy-2-nonenal can bind and inhibit its proteolytic activity (30). Alternatively, accumulation of inactive Lon protease may result from its decreased turnover or from free radical-mediated inactivation. Such a free radical-mediated inactivation of both ATPase and proteolytic activities of the Lon protease has been recently reported upon treatment of isolated rat brain mitochondria with the reactive nitrogen species peroxynitrite (103). Such inactivation was found to occur prior to electron transport chain dysfunction and to be partially restored upon reduced glutathione addition, suggesting that the Lon protease represents a sensitive target for oxidative inactivation and may be subjected to redox regulation. Moreover, using transgenic mice underexpressing mitochondrial superoxide dismutase as a model of oxidatively challenged animals, Bota et al. (12) have previously reported a decrease in Lon protease level and activity that was associated with increased content of oxidized proteins. In contrast to what we observed in rat liver and what was previously shown in mouse skeletal muscle, the ATP-stimulated Lon-like protease activity was not affected with age in rat heart mitochondria (22). Nevertheless, the Lon protease expression was increased 5-fold at the protein level in the heart of old animals compared to young ones, suggesting compensation for loss in specific activity. Taken together, these results indicate that the Lon protease undergoes age-related alterations that are organ specific, and that are likely to contribute to the age-associated accumulation of oxidatively modified and inactivated proteins such as mitochondrial aconitase. Finally, although no age-related decline of Lon protease abundance and activity could be evidenced in the Podospora anserina fungus, it has been recently reported that constitutive overexpression of the Lon protease in this fungus results in extended lifespan without impairment of vital functions such as respiration, growth, and fertility (69). As expected, the transgenic strains exhibited increased mitochondrial ATP-stimulated protease activity together with a lower level of oxidized and glycoxidized proteins and they also showed reduced production of hydrogen peroxide and higher resistance to oxidative stress. This last finding strongly suggests that improved mitochondrial protein maintenance represents a key element in longevity and may therefore represent a promising anti-aging strategy.
Conclusion
Accumulation of oxidized protein is a hallmark of cellular aging. Increased ROS production, especially by the mitochondria, and decreased removal of oxidized proteins have been implicated in this process (26, 87). Indeed, failure of protein maintenance represents a critical factor of aging and age-associated increased susceptibility to oxidative stress. Although mitochondrial dysfunction has been implicated in aging and degenerative diseases, much attention has been given to mitochondrial DNA mutations and deletions rather than to alterations of mitochondrial protein and their maintenance systems. Within the mitochondrial matrix, the Lon protease and the methionine sulfoxide reductase enzymes are responsible for elimination of oxidized protein through degradation and repair, respectively (29, 55, 79). Both systems, present in almost all organisms, seem to be essential for aerobic life and have been associated with longevity and resistance to oxidative stress (56, 69, 74, 95). Overexpression in different cellular models of either MsrA in the cytosol and the mitochondria and MsrB2 exclusively in the mitochondria resulted in enhanced protection against oxidative stress and a decreased accumulation of oxidized proteins (16, 92). Conversely, knockdown or knockout of either Msr or Lon protease in either cellular models or model organisms resulted in increased sensitivity to oxidative stress and/or impairment of mitochondrial function (56, 74, 95). In addition, both Msr and Lon protease are themselves targets for inactivation by a variety of oxidative stress, which may have an early implication in mitochondrial disorders associated with increased ROS production (91, 103). Hence, conditions that affect the removal of mitochondrial oxidized proteins and the mechanisms by which the mitochondrial protein degradation and repair systems, such as the Lon protease and methionine sulfoxide reductase, are impaired with aging need to be better defined. Indeed, deciphering these events will provide valuable information that may be helpful for defining strategies aimed at protecting or rescuing their crucial functions that are expected to be relevant for promoting healthy aging.
Footnotes
Acknowledgments
The authors wish to acknowledge the support of grants from the EU Framework Programmes 6 & 7 (IP 518230 Proteomage and CP 200880 Mark-Age), the French National Research Agency (ANR-MRAR –039-03) and the CNRS Interdisciplinary Programme on Aging and Longevity.