
Abstract
Reactive oxygen species (ROS) have been implicated in many intra- and intercellular processes. High levels of ROS are generated as part of the innate immunity in the respiratory burst of phagocytic cells. Low levels of ROS, however, are generated in a highly controlled manner by various cell types to act as second messengers in redox-sensitive pathways. A NADPH oxidase has been initially described as the respiratory burst enzyme in neutrophils. Stimulation of this complex enzyme system requires specific signaling cascades linking it to membrane-receptor activation. Subsequently, a family of NADPH oxidases has been identified in various nonphagocytic cells. They mainly differ in containing one out of seven homologous catalytic core proteins termed NOX1 to NOX5 and DUOX1 or 2. NADPH oxidase activity is controlled by regulatory subunits, including the NOX regulators p47phox and p67phox, their homologs NOXO1 and NOXA1, or the DUOX1 or 2 regulators DUOXA1 and 2. In addition, the GTPase Rac modulates activity of several of these enzymes. Recently, additional proteins have been identified that seem to have a regulatory function on NADPH oxidase activity under certain conditions. We will thus summarize molecular pathways linking activation of different membrane-bound receptors with increased ROS production of NADPH oxidases. Antioxid. Redox. Signal. 13, 467–487.
Introduction

In fact, NOX family members are also responsive to activation of membrane-bound receptors and subsequent generation of ROS appears to be an important part in receptor-mediated signalling. However, apart from the molecular pathways linking receptor activation to stimulation of the neutrophil oxidase, the regulatory mechanisms controlling nonphagocyte NADPH oxidase activities have only been partially elucidated. In this review, we will thus summarize current knowledge about activation mechanisms of NADPH oxidases and link them to receptor-mediated signalling cascades.
The Family of NADPH Oxidases
The phagocyte NADPH oxidase is comprised of the membrane-bound proteins NOX2, formerly known as gp91phox, and p22phox, both forming the cytochrome b558, as well as of several regulatory proteins termed p47phox, p67phox, p40phox, and of the GTPase Rac (Fig. 2). They are localized in the cytosol under resting conditions (61, 139, 178, 201), thus preventing unwanted generation of O2 •- that can be toxic for the cells. The main function of these regulatory proteins is to bring NADPH in close proximity to the enzyme FAD in order to facilitate the electron flow. This is accompanied by an internal refolding in order to transfer the electron from the FAD to the heme and later on to oxygen (38).
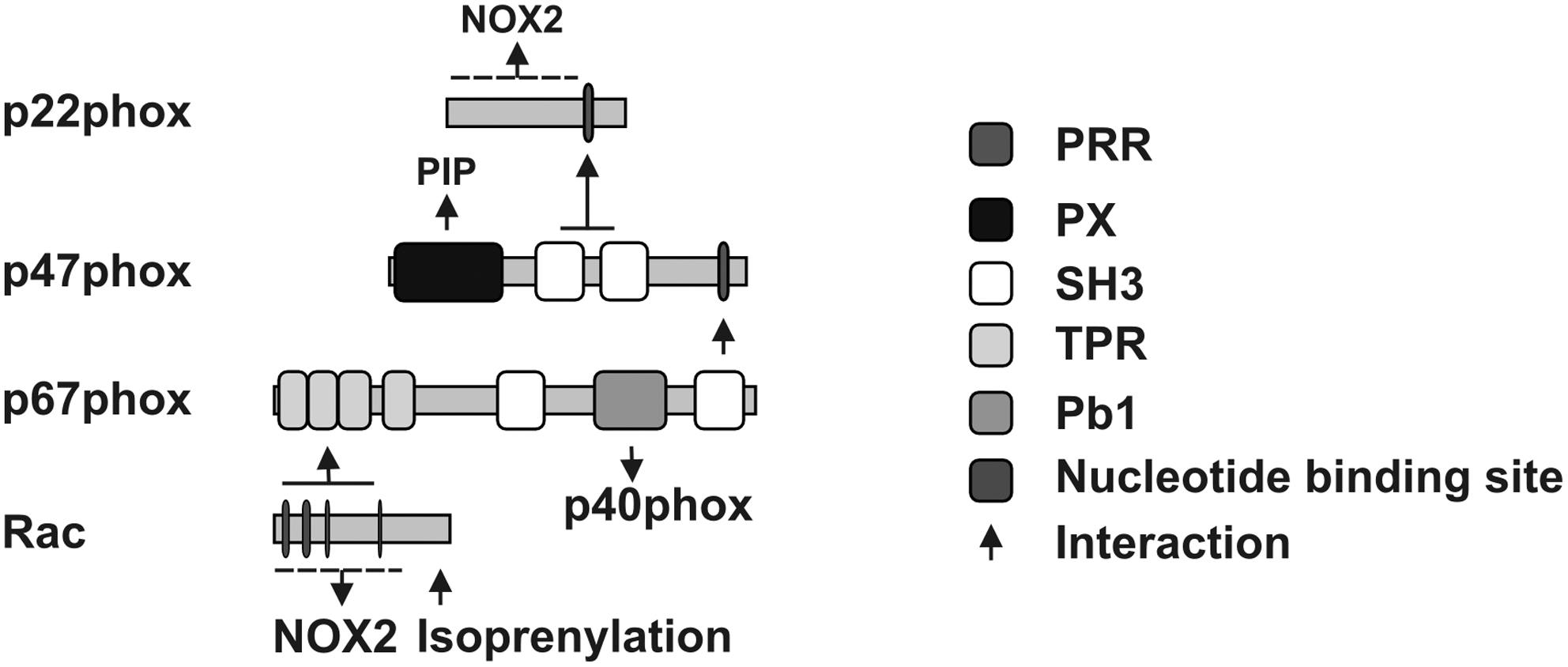
Activation of the NOX2-containing NADPH oxidase requires translocation of p47phox, p67phox, and Rac to the membrane and association of these proteins with the cytochrome b558, which then allows p67phox to promote the electron flow in the NOX2-containing NADPH oxidase via its activation domain by regulating the reduction of FAD (182). In addition, the cytosolic protein p40phox has been shown to either positively or negatively affect NADPH oxidase function (25, 133, 151). Finally, the small monomeric GTPases Rac1 and Rac2 are essential for full activation of the neutrophil NADPH oxidase (16).
In addition to neutrophils, O2 •- and other ROS play a role as signaling molecules in nonphagocytic cells (205, 260). NOX2 and other components of the phagocyte oxidase were identified and subsequently proofed to be active in nonphagocytic cells (81, 104, 112, 250). In contrast to the phagocyte NADPH oxidase, the NADPH oxidases in nonphagocytic cells produce only small amounts of ROS considered to act as second messenger molecules (26, 66, 88, 260). In nonphagocytic cells, localization of NOX2 was variable in different cell types. It has been identified in the plasma membrane (2, 196, 197), especially in caveolae (246), but also in lamellipodia (107, 246), endosomes (148, 184), and the endoplasmic reticulum (ER) (196, 253). In addition to the nonphagocytic NOX2-dependent NADPH oxidases, homologous proteins to NOX2 and subsequently also to other factors have been identified in nonphagocytic cells.
The first homolog of NOX2, initially termed MOX1 or NOH-1, now termed NOX1 (227), has been cloned from colon cDNA with a similarity of 56% to NOX2. Subsequently, NOX3, NOX4, NOX5, and DUOX1 and DUOX2 have been identified (29, 41). Whereas NOX3 and NOX4 appear to be structurally similar to NOX2 with a similarity of 58% (NOX3) and 37% (NOX4), NOX5 (27% similarity to NOX2) and DUOX 1/2 (47% similarity to NOX2) contain additional calcium-binding EF hands in the N-terminus (29, 41, 42). DUOX1 and DUOX2 have additional putative peroxidase domains and seem to perform a two electron reduction yielding H2O2, although it remains to be elucidated whether H2O2 synthesis by DUOX1/2 has O2 •- as an intermediate product (Fig. 1) (173).
Intriguingly, the NOX proteins have different preferences towards different cytosolic factors. NOX1 to NOX4 all need p22phox for full activity, whereas p22phox seems to be dispensable for NOX5 and DUOX1/2 function (13, 139, 256). In addition to NOX2, Rac has been shown to be involved in the regulation of NOX1 and NOX3 (114), whereas Rac dependency of the other family members is under debate.
Homologous forms of p47phox and p67phox, termed NOXO1 and NOXA1, were identified and shown to contribute to the regulation of NOX1 and NOX3 (Fig. 3) (12). The regulatory proteins DUOXA1/2 have been initially identified as maturation factors of DUOX1/2 (87), but have recently been shown to also contribute to DUOX1/2 activity (Fig. 3) (152, 173).

Regulation of p22phox
The p22phox subunit has been considered to be ubiquitously expressed (3, 52). Except for NOX5 and DUOX1/2, it is required for activation and stabilization of all NOX proteins. p22phox has been shown to be phosphorylated by phospholipase C(PLC)/protein kinase C or by phospholipase D (PLD) and the level of phosphorylation of p22phox correlated with NADPH oxidase activity in neutrophils (203). Although p22phox has been described as a plasma-membrane bound protein in neutrophils, a recent report showed that upon overexpression in HEK293 cells, p22phox was present at intracellular reticular cytosolic and nuclear membrane structures (241). However, when NOX1 or NOX3 were co-expressed, p22phox was localized at the plasma membrane. Similarly, GFP-tagged p22phox was only present in the plasma membrane when it was co-expressed with CFP-tagged NOX2 in HeLa cells (unpublished observation), suggesting that NOX1, NOX2, and NOX3 are able to guide p22phox to the plasma membrane, although the underlying mechanisms regulating this transition are still unclear.
Recently, two novel interaction partners of p22phox were identified, polymerase (DNA-directed) delta-interacting protein 2 (PoldiP2) (153), and protein disulfide isomerases (PDI) (110, 212). PoldiP2 has been described as a regulator of cell division (124, 149). Using a yeast-two-hybrid screen with the C-terminal, proline-rich region (PRR) of p22phox as bait, PoldiP2 was identified as a potential binding partner of p22phox (153). Furthermore, in vascular smooth muscle cells (VSMCs), PoldiP2 colocalized with NOX4 at focal adhesions and induced NOX4-dependent ROS generation and migration (153). PoldiP2 was also shown to interact with NOX1, although the relevance of this observation is still not resolved.
PDIs are chaperones usually residing in the ER where they help to promote protein folding by modulating the formation of disulfide bridges built by cysteines in the ER (62), but they have also been described to be present in the cytosol (238).
PDIs have been shown to colocalize and to interact with p22phox at the plasma membrane in phagocytes and VSMCs (110, 212). However, since mutation of all cysteines at the active site did not affect interaction of PDI with p22phox (65, 110), the role of PDIs for NADPH oxidase function seemed to be independent of their chaperone function. In VSMCs, interaction between PDI and p22phox—and also between NOX1 and NOX4, respectively, was associated with increased NADPH oxidase activity upon angiotensin-II stimulation (65, 110). In macrophages, PDIs formed a complex at the plasma membrane together with NADPH oxidase, and PDI appeared to be required for redox-dependent phagocytosis of Leishmania (212).
It has been described that p22phox is regulated at the transcriptional level in vascular cells or macrophages in response to different stimuli including insulin, thrombin, urotensin-II, or intermittent hypoxia (55, 56, 71, 183).
Regulation of NOX2
Activation by regulatory cytosolic proteins
The phagocyte NOX2 is a highly glycosylated protein residing in the plasma membrane in phagocytes. Activation of NOX2 requires membrane translocation of p47phox, p67phox, p40phox, and Rac, subsequently leading to the formation of the active oxidase complex (Fig. 2). Since p47phox is dispensable for NOX2 activation in a cell-free system in the presence of excess amounts of p67phox and Rac, it has been suggested that p47phox functions as an organizer, whereas p67phox serves as a direct activator of NOX2 (228).
In the resting state, the two SH3 domains of p47phox are inaccessible, as they are masked via an intramolecular interaction with the C-terminal flanking region termed the autoinhibitory region (AIR). Upon activation, p47phox undergoes stepwise phosphorylation at multiple serine residues in the C-terminus, several of which are present in the AIR. This leads to a conformational change which unmasks the SH3 domains, allowing these domains to interact with the PRR of p22phox as well as binding of the phox domain (PX) of p47phox to phosphatidylinositol (3,4)-bisphosphate (PI3,4P2) and phosphatidic acid (PA). This latter interaction is required for membrane localization of the complex (4, 12, 60, 61, 90, 228). Phosphorylation of p47phox in response to phorbol-12-myristate-13-acetate (PMA) or N-formylmethionyl-leucyl-phenylalanine (fMLP) can be mediated by various protein kinases including PKCζ, PKCβ, PKCδ, p21-activated kinase-1 (PAK1), extracellular signal-regulated kinases 1/2 (ERK1/2), and protein kinase B (PKB or AKT) (Fig. 4). In addition, partial phosphorylation of p47phox by ERK1/2 or mitogen-activated protein kinase p38 (p38MAPK) has been implicated in neutrophil priming by granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor α (TNFα), and subsequent stimulation of NADPH oxidase assembly. On the contrary, phosphorylation of p47phox by protein kinase A (PKA) or casein kinase II (CKII) may have an inhibitory effect on the enzyme (4, 12, 60, 61, 90, 228).
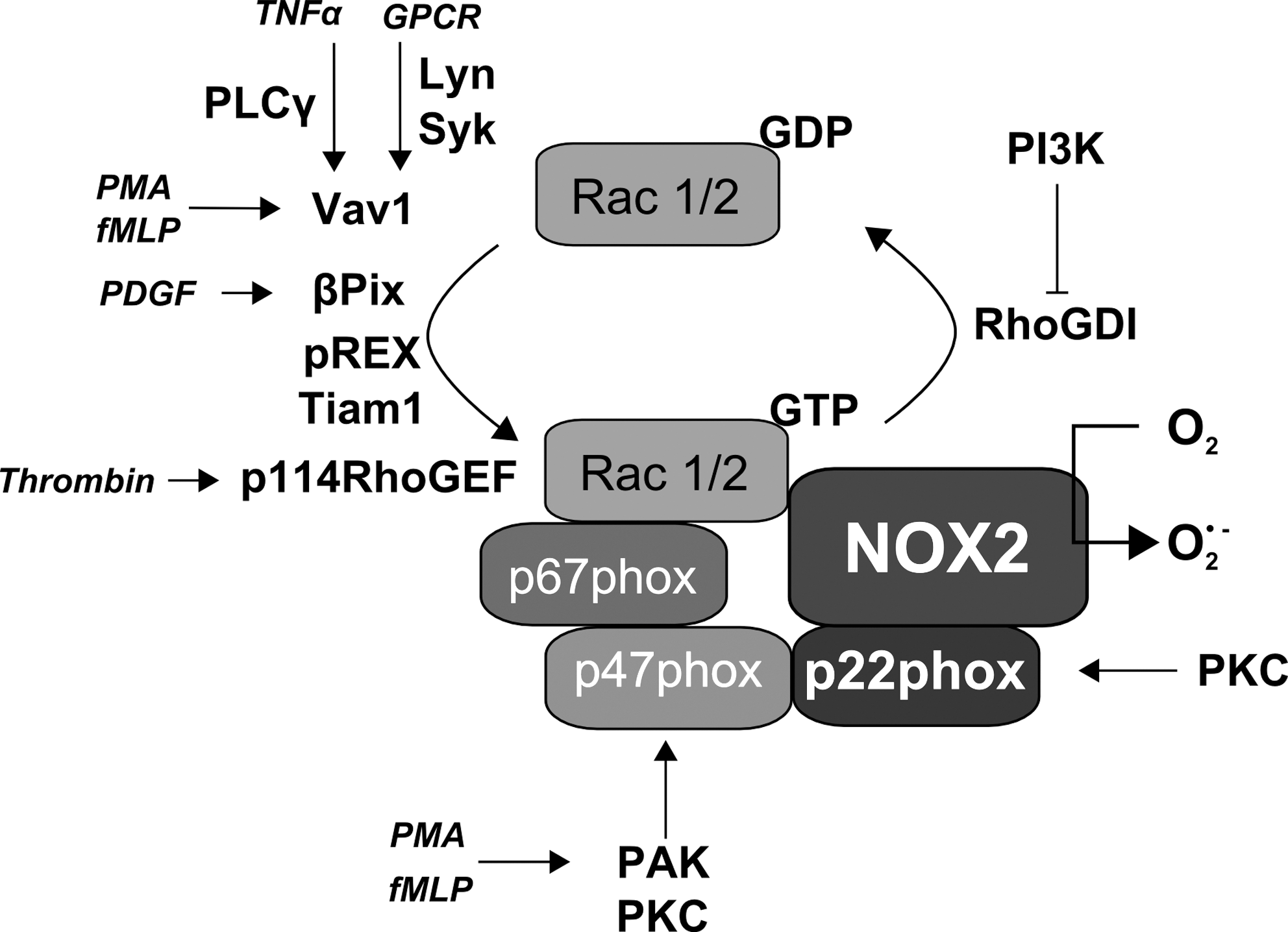
The presence of p47phox at the membrane promotes translocation of the “activator subunit” p67phox whereby the C-terminal SH3 domain of p67phox binds to the C-terminal PRR of p47phox. Subsequently, p67phox can bind to NOX2, and p40phox can be attracted to the complex (90, 228). Finally, active Rac interacts with NOX2 and with p67phox (46, 125). Once assembled, the complex is active and generates superoxide by transferring an electron from NADPH in the cytosol to oxygen on the luminal or extracellular space (16, 54, 88, 210).
Under resting conditions, Rac1 or Rac2 are bound to GDP in a complex containing a member of the guanosine nucleotide dissociation inhibitor (GDI) RhoGDI family. Activation of Rac1 or Rac2 is controlled by guanidine nucleotide exchange factors (GEF) which exchange GDP to GTP (see below) (53).
Activated Rac binds to p67phox at four tetratrico-peptide repeat (TPR) motifs (53, 125). This interaction seems to be dependent on the residues A27, G30, D38, Y40 of Rac (199) and necessary for translocation of p67phox to the membrane (168, 199, 213). There is also evidence that Rac can facilitate membrane translocation of p47phox in the absence of p67phox (199), although other studies could not provide evidence for a requirement of Rac for membrane translocation of p47phox (241).
Binding of Rac to NOX2 was first demonstrated with a fluorescent GTP analogue, which colocalized with Rac2 and purified flavocytochrome b558. This interaction was dependent on GTP and the so-called insert domain of Rac2 (46). However, the latter requirement was recently questioned since a Rac mutant where the insert domain was substituted with the corresponding region of Ras did not affect activation of NOX2 nor NOX2 localization to phagosomes (167).
Rather recently it was shown by pulldown assays using recombinant proteins that Rac2 interacts with the FAD binding domain of NOX2 at the residues aa 419–430, and for this interaction the residues K421, Y25, and K426 that are conserved between NOX1, NOX2 and NOX3, but not present in NOX4 or NOX5, are needed (114). Mutations of these residues resulted in a loss of Rac-dependent activation of NOX1 and NOX2, although the effects on the activity of NOX3, NOX4 or NOX5 were not investigated in that study. NOX2 was further shown to be phosphorylated at the cytosolic carboxy-terminal flavoprotein domain in neutrophils by PKC in response to PMA or fMLP, resulting in enhanced interaction with p47phox, p67phox and Rac2, and enhanced diaphorase activity by this domain (Fig. 4) (200).
Activation of Rac
Activation of Rac is associated with several GEF members including βPIX (205), P-Rex1 (258), Tiam1 (169, 199), Trio (169), p114RhoGEF (Fig. 4) (100), the Vav family (207) and SOS-1 (271). In most cases the GEFs become activated by phosphorylation by different kinases, dependent on the stimulus. For activation of Vav and βPIX, binding of phosphatidylinositols such as phosphatidylinositol (4,5)-bisphosphate (PI4,5P2) and phosphatidylinositol (3,4,5)-trisphosphate (PI3,4,5P3) via their pleckstrin homology domain is also necessary (145).
Activation of Rac by the Vav family in PMA- and fMLP-stimulated phagocytes promoted p47phox phosphorylation by PAK1 (167, 207). Interestingly, in fMLP-stimulated neutrophils, p67phox also binds to Vav1 and Rac2, but not to Rac1, activating the nucleotide exchange of Rac2 which enhances in turn the interaction between p67phox and Vav1 (165). Activation of Vav1 occurs via T-cell- and B-cell-antigen receptor, growth factor receptors, integrins, and chemokine receptors via tyrosine phosphorylation (239, 240). Activation of Rac2 in neutrophils upon Fcγ-receptor stimulation was dependent on Vav3 and phosphatidyl-inositol-3-kinase (PI3K) as well as on phosphorylation of p40phox (251).
The importance of Vavs for NADPH oxidase activation was shown in triple knock-out mice lacking Vav1, 2, and 3 (85), where ROS generation after TNFα and CD18 stimulation was impaired in neutrophils. In these neutrophils, translocation of p40phox and p47phox to the membrane was diminished, indicating that the translocation of the cytosolic subunits is dependent on Vav proteins. Upon reconstitution of the NADPH oxidase in COS cells (COSphox), a constitutively active Vav1 variant was the most potent GEF to increase ROS generation, although constitutively active Vav2 and Tiam1 also enhanced the levels of RacGTP (199). Tyrosine phosphorylation of Vav possibly involving lyn and syk kinases resulted in the activation of Rac1 in response to fibrillar β-amyloid-receptor complex stimulation in neonatal primary microglial cells (259). In adherent neutrophils, integrinα1β1 decreased Rac1-dependent ROS generation by increasing the activity of tyrosine phosphatases, leading to decreased phosphorylation of Vav1 at Y174, while phosphorylation of Vav1 by Syk was not affected (28, 270).
In contrast to the Vav family, which does not seem to be activated by G-protein-coupled-receptors (GPCR), the GEF P-Rex has been associated with GPCR signaling (245, 257). P-Rex-deficient neutrophils exhibited decreased Rac2 activation and ROS generation (258) and LPS-stimulated ROS generation was impaired in these neutrophils. In contrast, Rac1 activation was only marginally affected in unprimed and TNFα-primed cells, further indicating the specificity of this GEF in GPCR signaling and ROS production. Furthermore, the GEF βPIX has been shown to be involved in platelet-derived growth factor (PDGF)-induced ROS generation and to be physically associated with a not further specified NADPH oxidase complex (205). Rac2 can also be activated by PI3K in HL60 cells stimulated with 1-oleoyl-2-acetyl-sn-glycerol (OAG), a membrane-permeant analogue of diacylglycerol, or with fMLP. However, the GEF involved was not investigated (19). PI3,4,5P3 not only activates GEFs, but also enhances the dissociation of RacGDP from RhoGDI in the presence of specific liposomes, containing PI3,4,5P3, GTP, and the GEFs Trio or Tiam1 (243, 244). Trio and Tiam1 can form complexes with Rac at the membrane of phagocytes together with a membrane-localized nucleoside diphosphate kinase (NDPK) (169). In this complex, activation of Rac is even possible in the absence of GTP as long as ATP levels are sufficient since the recovery of GTP via GDP phosphorylation was mediated by NDPK which was activated by the GEFs Trio or Tiam1, thereby increasing GTP levels (Fig. 4).
Regulation of NOX1
NOX1 has been discussed in various signaling cascades. It seems to play an important role in the signaling of angiotensin-II, TNFα, and growth factors such as PDGF, epidermal growth factor (EGF), or basic fibroblast growth factor (bFGF). NOX1 has been found at the plasma membrane inside of caveolae, but recently NOX1 has been described inside early endosomes or even in the nucleus (102, 164).
NOX1 has been first considered as a subunit-independent enzyme showing low or no O2 •- production (227). Subsequently, homologues of p47phox and p67phox termed NOXO1 and NOXA1 for NOX organizer and activator, respectively, were identified (Fig. 3) and are required for full activation of NOX1, although p47phox and p67phox can enhance NOX1 activity probably in a cell type- and stimulus-dependent manner (7, 78, 233).
Although the amino acid sequence of human NOXO1 shares just 23% identity with human p47phox, the overall domain structure is similar (7, 231). Both have an N-terminal PX domain followed by two SH3 domains and an N-terminal PRR (Figs. 2 and 3). The AIR is missing in NOXO1, even though it has been suggested that the C-terminal PRR can interact with the SH3 domains similar to the AIR present in p47phox (264). In fact, blocking of the two SH3 domains of NOXO1 by interaction with the internal PRR domain prevented interaction with the PRR domain of p22phox (233, 243, 266) and inhibited interaction of NOXO1 via its PRR domain with the SH3 domains of NOXA1 (1, 233, 243). NOXO1 has been shown to be constitutively recruited to the plasma membrane by binding over its PX domain to phosphatidylinositol (3,5)-bisphosphate (PI3,5P2) and interacting there with the NOX1/p22phox protein complex (123, 232, 244). Interestingly, several splice variants of NOXO1 have been described and termed NOXO1α, β, γ, and δ. They contain an alternatively spliced exon 3 resulting in changes in their PX domain (32, 244). NOXO1α and δ were described to be present in internal membranes and not able to activate NOX1. NOXO1β was exclusively present in the plasma membrane and NOXO1γ was also found inside the nucleus (242) and both proteins were able to activate NOX1. NOXO1 is needed for the recruitment of NOXA1 to the NOX1-containing complex. NOXA1 binds to NOXO1 via its SH3 domain in order to recruit Rac1 (Fig. 5) (168, 231, 252). NOXA1 can then be phosphorylated by PKA at S172 and S461 leading to interaction with 14-3-3 and sequestering of NOXA1 from NOX1 thus keeping NOX1 in an inactive state (121).
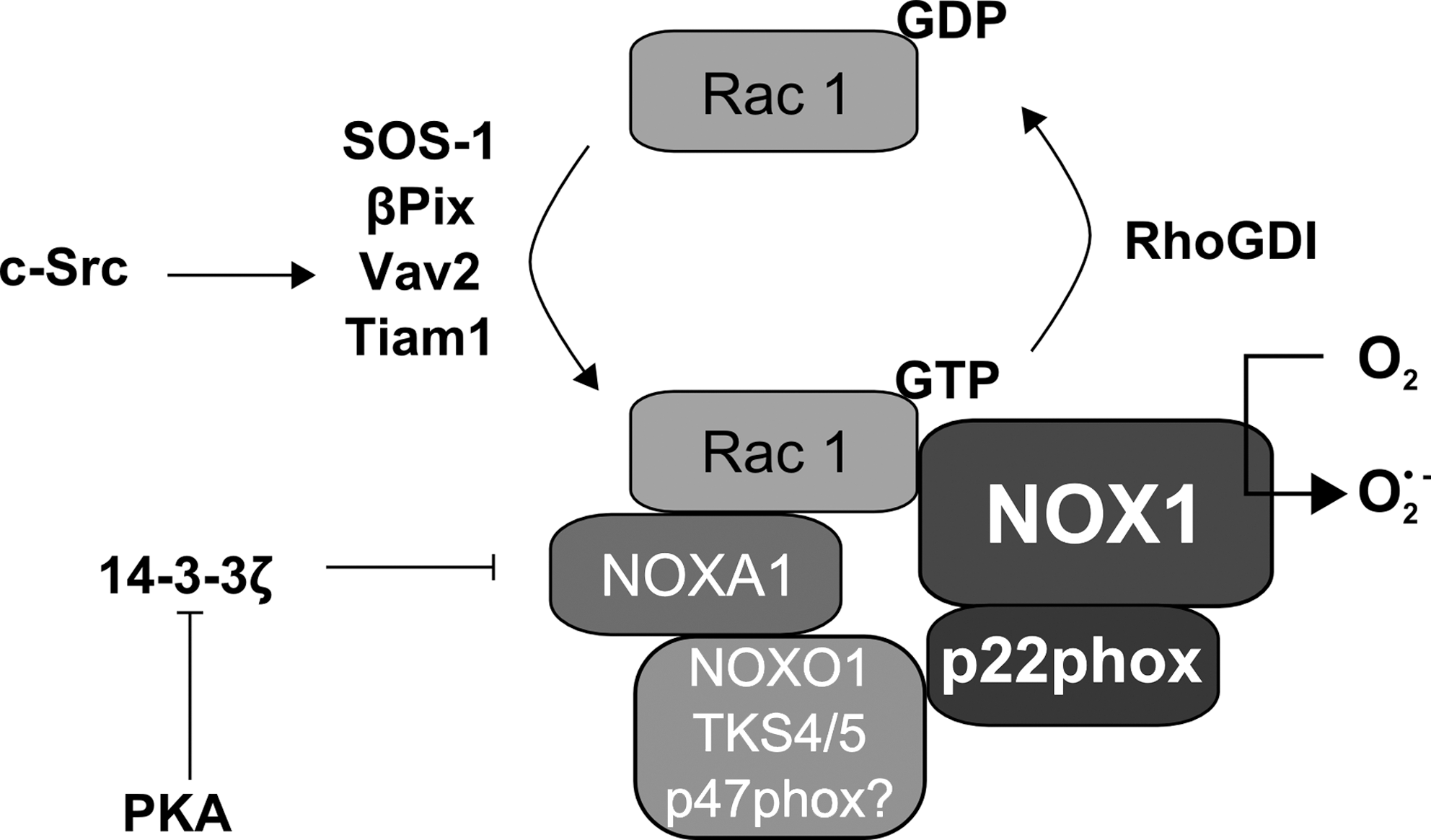
In addition to NOXO1 and NOXA1, the NOX1-dependent NADPH oxidase needs active Rac1 (Fig. 5) (30, 116, 231). NOX1 has been shown to directly interact via its C-terminus with Rac1 and to stably interact with βPIX (189). In addition, Vav2 activation by c-Src resulted in activation of NOX1 via Rac1 (76). Furthermore, the GEF SOS-1 has been discussed to be involved in NOX1 activation after angiotensin-II stimulation (271), whereas Tiam1 was involved in NOX1-dependent prevention of apoptosis in keratinocytes (209). Subsequently, Rac1 was shown to be required for NOXA1 recruitment to the plasma membrane since a fusion protein of NOXA1 with the polybasic and the isoprenylation region of Rac1 could recruit NOXA1 to the membrane and activate NOX1, although wild-type Rac1 transfected together with wild-type NOXA1 was more potent than this fusion protein (31, 170, 243).
Recently, Tks4 and Tks5 have been described to regulate NOX1 (46, 78). Both proteins are structurally related to p47phox and NOXO1. In contrast to p47phox and NOXO1, they have 4 or 5 SH3 domains, respectively (Fig. 3). These proteins were able if overexpressed in HEK293T cells together with NOX1, NOXA1 or p67phox and constitutively active Rac1, to induce NADPH oxidase activity to the same extent than the classical organizers NOXO1 or p47phox. Tks4 could also maintain NADPH oxidase activity in a human colon cancer cell line which only expressed NOX1 and NOXA1 but not NOXO1 (77). So far no specific SH3 domain could be assigned for the interaction of Tks5 and NOXA1 since mutation of all 5 SH3 domains was required to interrupt this interaction. Moreover, Tks5 also interacted with p22phox via its first two SH3 domains, which are highly similar to the two SH3 domains present in p47phox (Fig. 5) (45). Thus, Tks4 and Tks5 appear to be new regulators which could be involved in the organization and activation of NADPH oxidases, although their specific way of action has to be further elucidated.
Regulation of NOX3
NOX3 was initially described as being expressed in fetal tissues, and was found in the inner ear where it is involved in otoconia formation (138, 187). However, NOX3 has also been described in HepG2 cells, the mouse macrophage cell line RAW264.7 and in lung endothelium of mice (21, 214, 269).
NOX3 interacts with the proline-rich region of p22phox, thereby stabilizing p22phox and guiding it to the plasma membrane. This interaction is required for NOX3-dependent ROS production (177), which can be enhanced by NOXO1 or p47phox as well as by NOXA1 or p67phox (Fig. 6) (166). Although ROS generation by NOX3 was increased by p47phox and p67phox, it was not affected by the V204A mutation of p67phox which limits ROS generation by NOX2 (32). NOXO1 alone was able to induce ROS by NOX3 (32). A recent study showed that the residue I155 in NOXO1, which corresponds to I152 in p47phox, is needed for activation of NOX3-generated ROS (232), although PMA only enhanced ROS levels when NOX3 was co-expressed with p47phox. NOX3 also contains a conserved Rac-binding motif similar to NOX1 and NOX2, although the role of Rac for NOX3-dependent ROS production is still controversial (114, 120, 177, 241).

Overexpression of RhoGDI inhibited endogenous activation of Rac1 and prevented NOX3-mediated ROS generation upon NOX3 coexpression with p67phox or NOXA1. Consistently, mutation of the Rac-binding domain of both p67phox and NOXA1 decreased ROS generation in COS7 and CHO cells, although this effect was not observed in HeLa cells. In HEK293 cells, constitutively active Rac1 was able to enhance ROS generation of NOX3 in the presence of either p67phox or NOXA1. Apparently this effect was not dependent on the insert region of Rac1 which has been associated with p67phox binding and activation of ROS generation (167). Therefore, it is likely that Rac1 is able to regulate NOX3, but in cooperation with other regulative subunits in the complex with NOX3. Similar to NOX1, a role of Tks4 and Tks5 in regulating NOX3 NADPH oxidase has been recently suggested (77). As almost all studies concerning the regulation of NOX3 were performed in overexpression model systems, the elucidation of endogenous NOX3 regulation awaits further investigation.
Regulation of NOX4
In contrast to NOX1, NOX2, and NOX3, NOX4 does not seem to require interaction and activation by cytosolic regulatory subunits (119). Thus, many studies report that increased ROS generation by NOX4 is mediated by upregulation of NOX4 rather than by activation via a receptor-coupled signaling pathway.
NOX4 is mainly considered to be localized in the ER (27, 100, 195, 198, 228, 255), however, NOX4 was also detected in the nucleus, especially of differentiating cells (136, 196, 265), as well as in the plasma membrane (102) and even in the mitochondria (14).
Four additional splice variants of NOX4 termed NOX4B, NOX4C, NOX4D, and NOX4E (in this context, prototype NOX4 was termed NOX4A) have been described that all show an intracellular localization with an enrichment in the perinuclear area in A549 cells (84). NOX4A and NOX4B proteins could be shown in the nuclear fraction as well as in the post-nuclear supernatant of human coronary arterial endothelial cells, and on the mRNA level also NOX4C and NOX4D, but not NOX4E were detected in these latter cells (267). NOX4B is lacking either part of the NADPH oxidase binding domain, whereas NOX4C lacks the complete C-terminus including FAD and NADPH binding domains, and both variants decreased ROS generation upon overexpression. NOX4D lacks most of the transmembrane helices but still is able to increase ROS levels. NOX4E which is lacking, in addition to the domains missing in NOX4D, a part of the NADPH binding domain as NOX4B, did not increase ROS generation (84). However, further investigations are necessary to confirm the presence and the function of these splice variants of NOX4.
NOX4 has been shown to be dependent on p22phox (Fig. 7) (158). It apparently binds to a dual tryptophane motif in the N-terminus of p22phox at aa 6-11, although, in contrast to the situation with NOX1, NOX2, and NOX3, the C-terminus of p22phox seemed to be dispensable for the interaction with NOX4 (150). There is evidence that a functional NOX4/p22phox complex is present in focal adhesions at the plasma membrane of smooth muscle cells (36, 102). In addition, a functional NOX4/p22phox complex was found in the human lung epithelial carcinoma cells—which lack endogenous p22phox—when NOX4 and p22phox were overexpressed (150).

Although NOX4 has been considered to be constitutively active and not to depend on regulatory cytosolic subunits or Rac1 (119, 158), regulation of NOX4 by Rac has been suggested upon stimulation with angiotensin-II in mesangial cells, as NOX4 antisense oligonucleotides abolished Rac-induced ROS generation (80). However, the role of NOX1, which is also expressed in mesangial cells (198), was not investigated. In addition, in human aortic endothelial cells stimulation with oxidized phospholipids resulted in VEGF receptor-2 activation and recruitment of Rac to the plasma membrane for NOX4 activation, although no direct link between NADPH oxidase activation and Rac activation was shown in this study (144).
Recently, Tks5 has been suggested to be able to regulate NOX4 and to be important for invadopodia formation (Fig. 7) (45), although Tks5 failed to induce NOX4-dependent NADPH oxidase activity when overexpressed in HEK293T cells (77).
Thus, in contrast to other NOX forms that are controlled by the interaction with various activating or inhibiting factors, NOX4-dependent ROS productions seem to be mainly regulated at the level of expression. In this regard, NOX4 has been shown to be upregulated by transforming growth factor β (TGFβ) in hepatocytes and human airway smooth muscle cells, as well as in embryonic stem cells where NOX4 promoted their differentiation into smooth muscle cells (40, 178, 228, 265). NOX4 expression can also be induced by urotensin-II in pulmonary artery smooth muscle cells (55) and TNFα in endothelial cells (10, 225, 267). Surprisingly, NOX4 mRNA, but not protein, was shown to be upregulated after serum starvation, whereas serum induced a translationally initiated mRNA destabilization program (195).
Regulation of NOX5
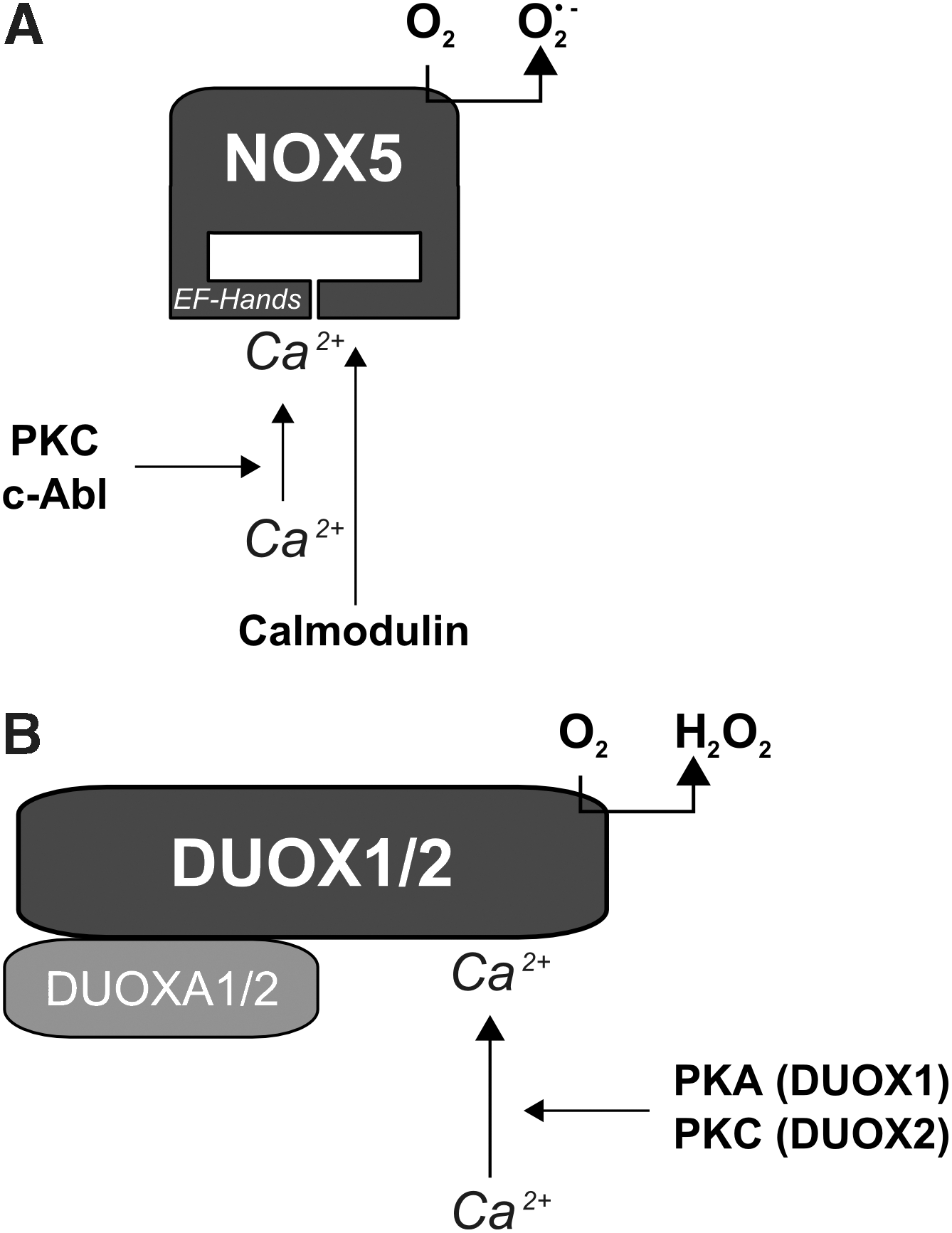
NOX5 was initially described as being expressed in human testis, spleen, and lymph nodes, and was also identified in endothelial cells and smooth muscle cells (6, 13, 30, 93, 113). In contrast to the other NOX homologues, NOX5 seems to be absent in rodents (117). NOX5 was found to be localized in the ER in endothelial cells (13). In contrast to other NOX proteins, NOX5 has 4 N-terminal EF hands which bear calcium-bindings domains (Fig. 8a). Four splice variants within this EF hand region have been described, NOX5α to NOX5δ, which are all sensitive to Ca2+(6, 8). Interestingly, different cell types seem to express different subsets of NOX5 variants. Endothelial cells expressed NOX5β and NOX5δ, colon carcinoma cells expressed NOX5α and NOX5γ, whereas smooth muscle cells expressed all NOX5 variants (13). In addition to these four NOX5 variants, also a smaller variant, lacking the EF hands, and thus resembling the other NOX proteins has been described as NOX5S (6, 13). NOX5S was shown to be functionally active in human endothelial cells, where it was involved in the angiogenic response towards thrombin (13). It was also transcriptionally upregulated in Barrett's esophageal adenocarcinoma cells (72, 219).
NOX5 was shown to bind to PI4,5P2 via a polybasic region downstream of the EF hands, thereby allowing its recruitment to the plasma membrane (120, 221).
NOX5 seems to be mainly regulated in a Ca2+-dependent manner. The binding of Ca2+ to the EF hands induces a structural rearrangement leading to binding of the N-terminus to its own C-terminus and this rearrangement activates NOX5 (8). Interestingly, a calmodulin binding domain is present in the C-terminus of NOX5, and calmodulin binding appears to increase the affinity of NOX5 for Ca2+ (234). However, whether the structural rearrangement is required for calmodulin binding or may allow recruitment of other proteins, like it is known for other Ca2+ binding proteins, is unclear to date.
NOX5 can also be phosphorylated by PKC at S486, S490, and T494 (109) which decreases the Km of the EF hands and thus enhances activation of NOX5 (Fig. 8a). At least two of these sites need to be phosphorylated for full activity of NOX5 (109). In addition to PKC, the tyrosine kinase c-Abl was shown to phosphorylate NOX5 thus enhancing its calcium sensitivity (59).
Interestingly, in contrast to other NOX proteins, p22phox does not seem to be required for NOX5 function although NOX5 is able to bind p22phox (13, 122). In addition, none of the classical cofactors of the NADPH oxidases, including Rac1, has been described to regulate NOX5 to date (13, 116, 122). However, the NOX5 heterolog in rice, RbOH1, is regulated by Rac1 (261). Subsequent studies are required to further delineate the regulatory networks allowing activation of NOX5.
Regulation of DUOX1 and DUOX2
In addition to the “classical” NOX proteins, the DUOX proteins (DUOX1/2) have at the N-terminus a unique peroxidase-like domain which is linked to the rest of the protein by two canonical EF hands which render the proteins sensitive towards Ca2+ (Fig. 1) (41). Due to their peroxidase domain, DUOX1 and DUOX2 are considered to generate H2O2 although it is not clear whether they also produce superoxide. These enzymes have been primarily identified in the thyroid where H2O2 is needed for iodination in the course of the synthesis of T3/T4 (42). DUOX2 transcripts have been found at higher levels compared to DUOX1 in the thyroid (185), and a mutation in the DUOX2 gene is accompanied by hypothyroidism (160, 174), implying that DUOX2 is the enzyme involved in basal T3/T4 synthesis. DUOX1/2 also seem to play a role in the maintenance of the airway fluid balance (67), in innate immunity in the gastrointestinal tract (93), and in the recruitment of monocytes after wounding in the zebrafish (180). Expression of DUOX1/2 alone retained the enzyme in the ER in an inactive state (43, 173, 174, 258).
The maturation factors DUOXA1 and DUOXA2 (Fig. 3) are thus required for translocation of DUOX proteins from the ER to the plasma membrane in order to build an active H2O2 producing system (87). Several splice variants of DUOXA1 termed DUOXA1α, β, γ, and δ have been described, although only DUOXA1α and γ seem to be able to function as maturation factors for DUOX1 and to secure localization of DUOX proteins in the plasma membrane (173). DUOXA1α and γ are also able to form an active complex with DUOX2, which does not result in an H2O2-producing system but in an O2 •- -producing one (173).
Interestingly, it has been suggested that DUOX1 and DUOX2 form heterodimeric complexes with their respective DUOXA maturation factors, which are dependent on heme incorporation into the NOX-homology domain (Fig. 8b). This seems to be similar to the situation with the cytochrome b558 which results from heterodimerization of NOX2 with p22phox as an associated subunit (152). In this regard it is of interest that DUOX proteins were able to interact with p22phox, but that this interaction was not, similar to the situation with the thioredoxin binding protein EFP1, required for ROS generation (43, 70, 122, 258). In addition, DUOX proteins have been shown to interact with NOXA1, thus leading to inhibition of DUOX function (186).
Although both DUOX proteins are Ca2+ sensitive due to the presence of the internal EF hand, they are differently activated (206). Whereas DUOX1 can be activated by forskolin and phosphorylated by PKA, DUOX2 is activated by PMA and phosphorylated by PKC (Fig. 8b).
It has been speculated that thyroid stimulating hormone (TSH) binding to its receptor may lead to activation of the heterotrimeric Gs or Gq protein which subsequently would lead to increased intracellular levels of cAMP, DAG, and Ca2+ (206). An involvement of Gq in the pathway leading to active DUOX protein has been shown in Drosophila where Gq activated PLCγ, thus increasing calcium levels which activated DUOX to ensure gut immunity (92).
Receptor-Linked Activation of NADPH Oxidases
Reactive oxygen species have been considered to act as signaling molecules in a wide variety of cells and under various conditions (57).
In neutrophils, ROS generation by NADPH oxidases by stimulation with bacterial toxins has been shown to be a tightly regulated process, involving distinct signaling cascades upon activation of receptors by LPS and other toxins. These receptor-mediated signaling cascades have been considered to protect the cells and tissues from unwanted activation of NADPH oxidases and subsequent toxic accumulation of ROS (43).
Although in nonphagocytic cells the amounts of generated ROS are much lower, the concept that ROS act as signaling molecules in response to various stimuli also implies the involvement of receptor-dependent signalling cascades in the specific and timely activation of NADPH oxidases in the cells. In the following we will summarize current knowledge about the mechanisms leading to receptor-mediated activation of NADPH oxidases.
The G-protein-coupled receptors
G-protein-coupled receptors (GPCR) were linked very early to activation of nonphagocytic NADPH oxidases since one of the first known stimuli of nonphagocytic NADPH oxidases has been the vasoactive peptide angiotensin-II (AT-II) (89).
The G-protein-coupled receptors (GPCR) are a big family of around 800 genes. All contain seven transmembrane helices and a cytoplasmic C-terminal tail that is able to bind and activate small heterotrimeric G-proteins (15). The heterotrimeric G-proteins are composed of three subunits α, β, and γ. Gα-proteins can bind GDP. To date, four types of Gα-proteins have been described, Gαi, Gαq, Gαs, and Gα12/13, whereas the Gβγ-complex is composed of unique proteins.
Ligand binding induces a conformational change of the GPCR leading to coupling and activation of the heterotrimeric G-proteins either by directly acting as a GEF of the receptor or by binding of a GEF. The GDP bound to the Gα-protein becomes exchanged against GTP, thus leading to the dissociation of the Gα-subunit from the Gβγ-complex. Both the GTP-Gα and the Gβγ-complex act as signaling molecules by activating adenylyl cyclases, G-protein-coupled receptor kinases (GRK), and phospholipases. The GTPase activity of the Gα-subunits leads, with the assistance of G-protein activating proteins (GAP), to the cleavage of GTP to GDP and Pi which inactivates the Gα-subunit allowing the formation of a heterotrimeric G-protein complex (105).
Angiotensin-II
Angiotensin-II (AT-II) can bind to two different GPCR, the angiotensin receptor type-I (AT1R) and II (74). In vascular cells and many other cells, AT-II preferentially binds to AT1R which is able to bind heterotrimeric G-proteins of the Gαq and Gαi family (101). AT-II has been described to elicit a biphasic increase in ROS production in VSMC, with a first response peaking within minutes, and the second response peaking within several hours with both responses requiring the presence of p22phox (89, 250).
Whereas the second peak appears to also involve the upregulation of NADPH oxidase expression, the first peak involved an AT1R-mediated signaling cascade by activating PLD and PKC, leading to ROS generation and redox-sensitive activation of matrix metalloproteinases (MMPs) which then can release membrane-bound EGF to activate the EGF receptor, which can lead to further activation of NADPH oxidase via activation of Rac through PI3K (102, 145, 222, 226, 256).
In large arteries, the AT-II response appears to be mainly mediated by NOX1 (51, 76, 145, 163), whereas NOX2 has been suggested to mediate AT-II signaling in small resistance vessels, in the heart and in the kidney (96, 105, 239).
AT-II stimulation can also lead to phosphorylation of p47phox by Src and translocation of p47phox and p67phox to the membrane-bound NADPH oxidase complex in endothelial cells and VSMCs (149, 238, 239). In parallel, AT-II can activate Rac1 and NOXA1, even though the activation of NOXA1 has only been described in AT1R-reconstituted HEK cells (33). This fast ROS production can be prevented by inhibitors of PKC, PI3K, or Src (220, 236). Binding of AT-II to AT1R also activated, via Gαq/11, tyrosine phosphorylation of the AT1R through c-Src thus leading to binding and activation of PLCγ and downstream activation of PKC and Vav2, respectively (34, 194, 206, 223, 237). PKC activated by AT-II can either directly phosphorylate p47phox or activate Rac by stimulating Vav2, which is also activated in a Src-dependent manner (76). Interestingly, the rapid activation of NADPH oxidases by AT-II seems to take place in caveolae (149, 250) where NOX1 has been found (102). Moreover, depletion of caveolin1 inhibited Rac activation and plasma membrane translocation and subsequently NADPH oxidase activation in VSMC by AT-II (271). Furthermore, AT-II has been described to promote the ER-residing chaperone PDI to the plasma membrane, thus mediating the interaction between PDI and p22phox, and also between NOX1 and NOX4, respectively, which seems to contribute to NADPH oxidase activity upon AT-II stimulation (65, 110).
Thrombin
Thrombin has been shown to act as potent stimulus of ROS production particularly in vascular cells and in platelets (100). Thrombin can activate the proteinase-activated receptors (PAR1, PAR3, and PAR4) which are members of the seven-transmembrane G-protein-coupled receptors (100). The best studied member is PAR1, which activates several heterotrimeric G-proteins such as Gαi, Gαq, and Gα12/13 (100).
In fact, thrombin leads, via PAR1 and Gαi, to the activation of Rac in VSMC (47, 56) which subsequently stimulates NADPH oxidase-dependent ROS production and induction of various signalling pathways, including activation of the transcription factors NFκB and HIF-1α and induction of plasminogen activator inhibitor-1 (PAI-1), vascular endothelial growth factor (VEGF) or tissue factor (9, 18, 47, 82, 99). Rac activation by thrombin has been associated with activation of PAK1 in several cell types, including smooth muscle cells (49, 83). PAK1 can phosphorylate p47phox resulting in activation of NADPH oxidases and ROS production (157). Indeed, thrombin has been shown to increase p47phox phosphorylation and to promote its translocation to the plasma membrane (18, 191). Activation of Gαi has also been linked to a decrease in cyclic AMP accumulation (39, 101).
Interestingly, thrombin can activate phosphodiesterase 2 (PDE2) in endothelial cells which hydrolyses cyclic AMP, and this activates Rac and subsequent ROS generation by NOX2 (48). This novel mechanism of NADPH oxidase activation has been linked to endothelial proliferation and angiogenesis and may also play a role in endothelial dysfunction.
Furthermore, thrombin can signal via Gβγ leading to the activation of p114RhoGEF, which can activate Rac and promote translocation of p67phox to the plasma membrane in NIH3T3 fibroblasts (183).
In hippocampus neurons and microglia cells, thrombin stimulation resulted in translocation of p47phox and p67phox and activation of ROS production, as well as in upregulation of NOX2, p47phox, and p67phox expression, leading to increased cell death of neuronal cells due to enhanced protein oxidation (35, 192).
Thrombin has also been shown to increase p22phox expression and ROS generation involving p38MAPK and PI3K (54). Whereas PAR1 can activate MAPK by Gαq and PLC (100), PI3K can be activated by Gβγ (38, 102). In addition, NOX2 and NOX5 protein levels, as well as Rac1 and PAK1 have been shown to be elevated after thrombin stimulation (13, 49).
Other G-protein-coupled receptors
In addition to AT1R and PAR1, various other G-protein-coupled receptors have been associated with increased ROS generation by NADPH oxidases. The vasoactive peptide urotensin-II, a ligand of GPCR14, which is now termed urotensin-II receptor, is able to induce p22phox- and NOX4-dependent ROS generation in pulmonary artery smooth muscle cells (55). In endothelial cells, atrial natriuretic peptide (ANP) has been shown to activate Rac and NOX2 by decreasing cAMP levels (48), thereby leading to stimulation of mitogen-activated protein kinase phosphatase-1 (73).
Also, activation of the LPA receptor induced NADPH oxidase-dependent ROS generation that resulted in transactivation of the Met receptor as well as of the EGF receptor (68). Activation of the β2-adrenergic receptor can activate Rac1 in a β-arrestin-1-dependent manner, leading to enhanced ROS generation (79). Bradykinin-mediated vasodilation has been associated with NOX2-dependent ROS generation (142). In retinal microvessels, endothelin-1 activation of the endothelin receptor A increased ROS generation by NADPH oxidases involving PKC (162).
This shows that activation of NADPH oxidases by G-protein-coupled receptors is a widely observed event, although the precise pathways, linking receptor activation, G-protein coupling and NADPH oxidase activation are only partially understood.
Receptor tyrosine kinases
A variety of growth factors that activate receptor tyrosine kinases (RTK), including VEGF, PDGF, and bFGF, but also insulin and angiopoietin, have been associated with increased levels of ROS. Ligand binding occurs at an extracellular binding domain resulting in receptor dimerization and activation of its intracellular tyrosine kinase domain. Dimerization of the receptor results in its autophosphorylation and the recruitment of several transmitter proteins which are also activated by tyrosine phosphorylation. Frequently, these relays are GEFs which then further activate kinases or lipases (215).
Insulin
ROS production in response to insulin application to adipocytes has been one of the first demonstrations that nonphagocytic cells are able to produce ROS involving an NADPH-dependent oxidase activity (175).
Subsequently, activation of an NADPH oxidase activity by insulin was shown to involve dimerization of the heterotetrameric insulin receptor and autophosphorylation resulting in the recruitment and activation of insulin receptor substrate protein (IRS) and Shc. The activation of the PI3K pathway and the MAPK pathway by insulin (210) has been accompanied by increased ROS production due to an NADPH oxidase activity (129 –131, 175). In addition, the insulin receptor can interact with Gα-proteins, and Gαi-proteins have been associated with insulin stimulation of ROS production (110, 115, 129, 134, 213).
Insulin-dependent ROS generation has been implicated to inhibit several phosphatases such as PTEN, which antagonizes PI3K (79, 138, 220), or PTP1B, which counteracts AKT (154, 155) thus being critically involved in kinase signaling.
Although insulin has long been recognized as an activator of ROS production and has been associated with NADPH oxidase activity, only limited data are available regarding the specific NOXes involved. Whereas insulin increased NOX4 expression in adipocytes, thus explaining enhanced ROS levels after prolonged exposure to insulin (217), depletion of NOX4 or expression of a NOX4 mutant lacking the C-terminal FAD/NADPH-binding domain also prevented short term elevation of ROS after insulin treatment of adipocytes (2, 158). This fast NOX4-mediated response appears to be rather surprising, considering that NOX4 has been suggested to be constitutively active and not activated by any regulatory subunits.
Recently, an involvement of NOX3, which is dependent on Rac and several other regulatory subunits, has been shown in insulin-induced ROS production in HepG2 cells (21). IGF-1-mediated ROS production has also been shown to act via NOX4 and Rac1, although Rac1 could not yet be linked to NOX4-mediated ROS production (163).
PDGF
PDGF is a dimeric growth factor that exists as homo- or heterodimer of two isoforms termed PDGFA and PDGFB. PDGFAA binds to PDGF-receptor-α and PDGFBB and PDGFAB bind to PDGF-receptor-β (35).
PDGFRβ signaling has been initially described to activate NADPH oxidases (159) but subsequent studies also proved an involvement of PDGFRα (128, 130). The increase of ROS in response to PDGF is fast and transient showing maximum effects after 10 to 30 min (229) and has been reported for various cell types, such as VSMC, endothelial cells, hepatic and pancreatic stellate cells, HepG2 cells, and fibroblasts. PDGF-mediated ROS production has been associated with Rac1 (5), p22phox (82), and NOX1 (143). Furthermore, PDGF induced translocation of p47phox and p67phox to the plasma membrane (11, 22, 23, 225) whereas the PDGF response was not observable in pancreatic stellate cells derived from p47phox-/- mice (106). In endothelial cells, PKC-dependent phosphorylation of p47phox has been documented in response to PDGF (223). Other kinases involved in the induction of ROS in response to PDGF are PI3K, Src, and c-Abl (5, 11, 17, 22, 23, 161). PI3K has been shown to be particularly important for the recruitment of p47phox and Rac1 to the plasma membrane (11). The role of c-Abl is still unclear, although it may be activated by PDGF-activated c-Src (17, 22, 23). Since c-Src by itself seems to be redox-sensitive, this pathway can be part of a positive feedback loop amplifying PDGF signaling. Recently, an involvement of NOX5 in PDGF-induced ROS production and proliferation of human aortic smooth muscle cells has been shown (111). PDGF-mediated ROS production in endothelial cells seems to be Ca2+ dependent (140) and calcium sensitivity of NOX5 has been shown to be positively modulated by c-Abl (59) although a direct involvement of NOX5 in the response to PDGF in endothelial cells has not been shown, yet.
bFGF
bFGF also known as FGF2, is part of the large FGF superfamily. Four different receptors for bFGF are known (FGFR1 to FGFR4), in addition to several low affinity receptors (63). bFGF is involved in embryonic growth and differentiation processes and plays a role in angiogenesis (63).
Binding of bFGF to its receptor can activate PI3K which is important for the recruitment of PLC, which by itself is phosphorylated and activated by the FGFR. The main pathway leading to activation of the Ras/MAP kinases is performed by recruitment of Grb2/SOS-1 (63). Activation of PLC and subsequent activation of PKC can lead to Rac activation after bFGF stimulation (216). A role of PI3K, PKC, and Rac in the activation of NOX1 in response to bFGF has also been shown in smooth muscle cells (216). In addition, activation of FGFR leads to phosphorylation and activation of βPIX via a Ras/ERK dependent pathway. βPIX can bind to NOX1 directly thus activating NOX1 via Rac in the neurite outgrowth response to bFGF (222).
VEGF
The vascular endothelial growth factor is one of the most important endothelial growth factors inducing proliferation, migration, and angiogenesis of endothelial cells (20). VEGF triggers cellular responses mainly through receptor tyrosine kinase VEGF receptor-2 (VEGFR2) (247) and it was demonstrated that NADPH oxidase-derived ROS play a role in VEGF signaling (249, 251). VEGF induces membrane ruffles at the leading edge of migrating endothelial cells (262, 265). Within these ruffles, VEGFR2 was shown to interact with the actin binding scaffold protein IQGAP1 (265). Rac was recruited in response to VEGF to these ruffles and interacted with IQGAP1, thus mediating VEGF-induced ROS production. In migrating endothelial cells, NOX2 was found to interact with IQGAP1 in the leading edges of these cells (107). In addition, p47phox and PAK1 were found in the membrane ruffles associated in a complex with Rac1 and the WASP-family verprolin homologous protein-1 WAVE1 (262). Interaction of the NADPH oxidase subunits with IQGAP1 seems to be necessary for VEGF-induced ROS generation, as depletion of IQGAP1 not only decreased ROS generation, but also NOX2 translocation and endothelial cell migration (107). VEGF-induced ROS generation also appeared to depend on p47phox phosphorylation by PAK1, whereas VEGF-induced membrane ruffle formation seemed to be dependent on the interaction of p47phox with WAVE1 (262).
Angiopoietin
Angiopoietin-1 and −2 can induce angiogenesis and endothelial cell proliferation, migration, and vessel formation (137). They work mostly through binding to the receptor tyrosine kinase tie2 (97). Upon angiopoietin-1 stimulation, the tie2 receptor was found in lipid rafts of human umbilical vein endothelial cells (115). Angiopoietin-induced ROS generation was dependent on NOX2, p47phox, and Rac (26, 95, 122). Activation of Rac-dependent NADPH oxidases by angiopoietin is consistent with the fact that the activated tie2 receptor recruits and activates PI3K (126) and that several GEFs such as Vav, SOS-1, and βPIX are activated by PI4,5P2 and PI3,4,5P3 (19, 147, 204), although in these studies no link to the NADPH oxidase was shown.
The tumor necrosis factor receptor family
Tumor necrosis factor receptor (TNFR) is a member of the death receptor family. It is activated by trimerization upon ligand binding. This trimerization leads to a conformational change and subsequent dissociation of its inhibitor silencer of death domains (SODD) (24, 255). After dissociation, SODD is able to bind to TNFR associated death domain protein (TRADD). Via TRADD, three pathways can be activated. First, by association with TNF receptor-associated factor 2 (TRAF2) and receptor-interacting serine/threonine-protein kinase 1 (RIPK1), it can bind and activate IKK which leads to phosphorylation and inactivation of NFκB inhibitor (IκB) and activation of NFκB. Second, also via TRAF2, it can activate the MAP kinase pathway via apoptosis signal-regulating kinase 1 (ASK-1), MAP kinase kinase 7 (MKK7), and finally activation of c-Jun N-terminal kinase (JNK). As a third pathway, it can directly bind to Fas-associated protein with death domain (FADD) and subsequently activate caspase 8, leading to apoptosis (24, 257).
TNFα mediates the proinflammatory response of neutrophils (179), but has also been shown to regulate directly or indirectly the activation of NFκB and the expression of adhesion molecules like ICAM-1 in endothelial cells (65, 72).
TNFα can increase ROS production not only in neutrophils, but also in a variety of other cells including endothelial cells, human aortic smooth muscle cells, embryonic kidney cells, and mouse lung epithelial cells (71, 146, 170, 188). In neutrophils, TNFα is not only able to modulate the expression of NOX2, but also to activate the NOX2-containing NADPH oxidase directly via different mechanisms (40, 44, 71, 266). Activation of TNFR1 resulted in activation of p38MAPK which subsequently phosphorylated p47phox at S345, thus leading to NADPH oxidase activation (40). An earlier report showed the involvement of a tyrosine kinase in this process but which tyrosine kinase is still unknown (44). In endothelial cells, a similar mechanism involving PKCζ was shown to activate NOX2 by TNFα (71). In HeLa cells, TNFR1 stimulated NOX2 as well as NOX1 involving activation of the riboflavin kinase RFK. RFK bound to the TNFR1 death domain and to p22phox, thereby physically and functionally coupling TNFR1 to the NADPH oxidase (266). RFK was further found to catalyze the formation of the coenzyme FAD which is required for full NADPH oxidase activity (266). However, whether RFK is needed for the on site synthesis of FAD or if it serves as a scaffolding protein remains to be elucidated.
In mouse fibroblasts, NOX1 can be found in a complex formed by RIPK1, TRADD, and NOXO1 after TNFα stimulation (123). RIPK1 may lead to recruitment of TRADD which may stabilize the interaction between NOX1 and NOXO1, thereby leading to increased NOX1 activity upon Rac activation.
The NADPH oxidase cofactors including p47phox, p67phox, NOXO1, or NOXA1 are required to get the enzyme FAD in close proximity to NADPH in order to facilitate electron transfer. This rather fast refolding is followed by the rather slow intraenzyme rearrangement of the cytochrome b558. This second refolding is believed to be the reason for the lag phase in ROS production observed in neutrophils after applying TNFα (143, 235, 270). Interestingly, a second application of TNFα is usually much faster followed by ROS production compared to the first one, a phenomenon called priming. Therefore, it is tempting to speculate that TRADD or RFK are locking the cytochrome b558 in an active form just waiting for the recruitment of Rac in order to allow electron transfer from NADPH to FAD. Interestingly, in fibroblasts NOX2-derived H2O2 generation was necessary for recruiting TRAF2 to the TNFR1/TRADD complex in endosomes in order to activate NFκB (148). TNFα further induced the expression of NOX2 mRNA in a PU.1-dependent manner in monocytes (58, 179) and increased the expression of NOXO1 in colon epithelial cells (135).
Conclusion
The family of NADPH oxidases has been recognized to play an essential role in innate immunity and related host defense programs where the toxic levels of ROS need to be released in response to specific stimulation within a defined area. Thus, the activation of NADPH oxidases due to receptor-mediated signaling cascades allows the timely generation of ROS where they are needed. In recent years, substantial progress has been made to understand the signaling cascades initiated by receptor activation that help to organize and activate the many components of the NADPH oxidases in a proper way to allow efficient electron transfer and ROS generation.
In addition, recent findings that nonphagocyte NADPH oxidases generate ROS at levels suited for signal transduction processes in response to different stimuli resulted in an increasing interest in elucidating the mechanisms linking NADPH oxidase activation and ROS signaling to receptor-mediated signal transduction pathways. In contrast to the phagocyte enzyme, which does not produce ROS when not activated, nonphagocytic NADPH oxidases do not seem to underlie these strict regulations, but seem to be able to generate ROS at various levels, depending on the stimulus or pathway involved. Thus, understanding how specific receptor-initiated signalling cascades are able to stimulate complex formation, electron transfer, or even complex disassembly is of major importance.
This task is not only complicated by the many different types of receptors and receptor signaling pathways present in a cell or an organ, but also by the differences in cofactor requirement and complex formation, as well as in intracellular localization of the nonphagocytic NADPH oxidases. In fact, in addition to homologous forms of the catalytic subunit NOX2 there is increasing evidence that homologous proteins of the regulatory subunits exist which may act similarly or differently than the classical regulators. In addition to heterodimerization partners such as p22phox or possibly DUOXA1/2 for the DUOX proteins, organizing or scaffolding proteins such as p47phox, NOXO1, or Tks4/5, as well as activating proteins such as p67phox or NOXA1 or the GTPase Rac can be recruited to form an NADPH oxidase complex dependent not only on the cell type but also on the stimulus. To date, however, only limited data are available demonstrating the signaling mechanisms responsible to selectively activate those regulatory proteins in response to receptor activation. This picture becomes even more complicated by recent findings that additional proteins are either interacting with complex members or are associated in order to keep the complex functional or to direct it to a specific intracellular localization. These associated factors which seem to be required for full functionality of the enzymes, are presumably part of the regulatory network initiated by receptor activation.
To date, these complex regulatory networks directing the signal from the membrane to various intracellular localizations are only at the beginning to be fully understood. However, this knowledge will be mandatory for using NADPH oxidase-mediated ROS generation as a therapeutic target in various disorders.
Footnotes
Acknowledgments
This work was supported by DFG grant GO709/4-4, Metoxia (HEALTH-F2-2009-222741) under the seventh research framework program of the European Union and Fondation Leducq, to AG.