
Abstract
The current study was designed to evaluate the effects of silymarin (SM) on advanced glycation endproduct (AGE) formation and monocyte activation induced by S100b, a specific ligand of receptor for AGEs. The in vivo verification of antiglycation, antioxidant, and antiinflammatory capacities was examined by 12 weeks of SM administration in streptozotocin-diabetic rats. In vitro glycation assays demonstrated that SM exerted marked inhibition during the late stages of glycation and subsequent crosslinking. Dual action mechanisms, namely, antioxidant and reactive carbonyl trapping activities, may contribute to its antiglycation effect. SM produced a significant decrease in monocytic interleukin-1β and COX-2 levels and prevented oxidant formation caused by S100b, which appeared to be mediated by inhibition of p47phox membrane translocation. Chromatin immunoprecipitation demonstrated that S100b increased the recruitment of nuclear factor-kappaB transcription factor as well as cAMP response element-binding–binding protein and coactivator-associated arginine methyltransferase-1 cofactors to the interleukin-1β promoter, whereas these changes were inhibited with SM treatment. In vivo, SM reduced tissue AGE accumulation, tail collagen crosslinking, and concentrations of plasma glycated albumin. Levels of oxidative and inflammatory biomarkers were also significantly decreased in SM-treated groups compared with the diabetic group. These data suggest that SM supplementation may reduce the burden of AGEs in diabetics and may prevent resulting complications. Antioxid. Redox Signal. 14, 353–366.
Introduction
Acute exposure to hyperglycemia induces reversible oxidative stress and circulating monocyte activation. Natarajan and colleagues (12, 35, 36) have suggested that high glucose levels and AGEs can generate large amounts of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and chemokines, including MCP-1 and IP-10. These inflammatory responses were found to be related to the modulation of signaling molecules, such as protein kinase C, p47phox, and/or mitogen-activated protein kinases, through oxidant stress-dependent or independent pathways. These pathways, in turn, control the activation of the nuclear factor-kappaB (NF-κB) transcription factor, thereby influencing the synthesis and expression of downstream inflammatory mediators. Antioxidants, such as α-tocopherol (40) or natural polyphenols (45), inhibit the aforementioned formation of ROS and cytokines as well as postpone inflammation.
The administration of supplemental antioxidants in response to the inhibition of protein modifications is a theoretical strategy for preventing diabetic complications correlated with glycooxidative stress (1). This hypothesis has been supported by clinical results that the development of diabetes and accumulation of AGEs may be reduced by the intake of natural antioxidants in the diet (26). Fruits, vegetables, and beverages are important dietary sources of flavonoids. Flavonoids are of current interest in research due to their important biological and pharmacological properties, particularly their antioxidant components (22). The flavonoid blend, silymarin (SM), is an extract from milk thistle (Silybum marianum L.) and is composed mainly of the following components: silybin A and B; isosilibybin A and B; silychristin; and silydianin. These flavonolignans are well known for displaying a remarkable spectrum of biological processes, including antioxidant, cytoprotective, and anticarcinogenic activities. In addition to its free radical scavenging properties, SM has been clinically used for its beneficial effects on various hepatic diseases where the pathogenesis involves an inflammatory response (47).
To date, studies of protein glycation (Maillard reaction in vivo) contribution to diseases have primarily focused on its relationship to diabetes and diabetes-related complications (17). However, there is no relevant information determining if phytochemicals offer protective effects that guard against glycotoxin-induced damage. This study is the first to report SM as a promising inhibitor of protein glycation for the prevention of oxidative and inflammatory injury against AGEs in vitro and in vivo, providing evidence to support the health benefits of milk thistle for diabetic patients. Considering that SM behaves similarly to aminoguanidine (AG), which was the first inhibitor of AGEs investigated in clinical trials, it has great potential to be used as an agent to alleviate diabetic complications without adverse side effects.
Materials and Methods
Chemicals and reagents
AG, N-acetyl-glycyl-lysine methyl ester (G.K.) peptide, bovine serum albumin (BSA; fraction V, essentially fatty acid free), δ-gluconolactone (δ-Glu), methylglyoxal (MG), phenyl-tert-butyl-nitron, S100b (bovine brain), SM, and streptozotocin (STZ) were purchased from Sigma Chemical Co. An anti-AGE monoclonal antibody (6D12) was purchased from Trans Genic, Inc. Antibodies to p47phox and β-actin were obtained from Cell Signaling Technology. The antiepidermal growth factor receptor antibody was obtained from Santa Cruz Biotechnology. Anti-p65, anticoactivator-associated arginine methyltransferase-1 (CARM1), anti–CAMP response element-binding (CREB)-binding protein (CBP), antiacetyl-histone H3 (acetyl-histone 3 [HH3] that recognizes Lys9 and Lys14), antimethylated histone H3 at Arg17 (methyl-HH3R17), and anti-receptor for AGE (RAGE) antibodies were obtained from Upstate Biotechnology, Inc. The pharmacological inhibitor specific for NADPH oxidase (apocynin [APO]) was obtained from Biosource. Immunohistochemical reagents were obtained from DakoCytomation. All of the chemicals and solvents used were of analytical grade.
Assays for individual stages of protein glycation
Early stages of protein glycation were determined with a hemoglobin-δ-Glu assay (32). This method is specifically devised for the investigation of inhibitors of the formation of Amadori products, as evidenced by decreased glycated hemoglobin (HbA1C) levels. A BSA-glucose assay was used to evaluate the ability of SM to inhibit glucose-mediated protein glycation, and the development of AGE-related fluorescence was measured according to the method described by Wu and Yen (43). A G.K. peptide-ribose assay was used to evaluate the ability of SM to inhibit the crosslinking of G.K. peptides (late glycation products) in the presence of ribose using the method described by Nagaraj et al. (28) and Rahbar et al. (32). To evaluate the reactive carbonyl trapping activity, the inhibition of MG-mediated protein crosslinking and aggregation by SM was determined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) in a BSA-MG model (43). AG, an established anti-AGE agent (39), was used as a positive control.
Determination of free radicals generation by electron spin resonance
Free radical generation was measured according to the methods described by Finotti et al. (10). Electron spin resonance (ESR) spectra were measured on glycated samples (BSA-glucose assay) with a reaction mixture containing 0.2 mM phenyl-tert-butyl-nitron. The ESR spectra were recorded on a Bürker (EMX-10/12) spectrometer under previously described conditions (43). No ESR signals were detected in any of the reagents used in the ESR analysis. All spectra were recorded at room temperature.
Cell culture and treatments
The human acute monocyticleukemia cell line (THP-1) was obtained from the Bioresource Collection and Research Center (BCRC 60430; Food Industry Research and Development Institute) and was cultured in glucose-free RPMI 1640 medium (Gibco BRL, 11879-020) supplemented with 10% FBS, HEPES (10 mM), streptomycin (100 μg/ml), penicillin (100 U/ml), 50 μM β-mercaptoethanol, and 5.5 mM D-glucose (normal glucose, NG) in a 5% CO2 incubator at 37°C. For AGE challenge experiments, S100b protein (6.5 μg/ml), a specific RAGE ligand, was given to cells for 4 h with or without SM. The cell viability test was determined by a Trypan blue dye exclusion assay. With NG culture conditions, SM had no cytotoxicity on THP-1 monocytes at a concentration range of 5–25 μg/ml (cell viability >95%; data not shown).
RNA preparation and quantitative real-time polymerase chain reaction
Total RNA was prepared from THP-1 cells (1 × 106 cells/ml) by a Trizol RNA isolation kit, as described in the manufacturer's manual. From each sample, 1 μg of RNA was reverse transcribed to cDNA using SuperScript III First-Strand Synthesis SuperMix for qRT-PCR kit (Invitrogen). Polymerase chain reaction (PCR) analyses were performed to detect IL-1β and COX-2 gene expression in THP-1 cells using a PCR Master Mix 2 × kit (Fermentas, Glen Burnie, MD). Multiplex PCRs were performed for 29–35 cycles by using a P × 2 Thermal cycler (Thermal Electron Co.). The qRT-PCR primers were as follows: IL-1β, forward (5′-CTCTCTCACCTCTCCTACTCAC-3′) and reverse (5′-ACACTGCTACTTCTTGCCCC-3′); COX-2, forward (5′-ATCTACCCTCCTCAAGTCCC-3′) and reverse (5′-TACCAGAAGGGCAGGATACAG-3′); β-actin, forward (5′-ACAAAACCTAACTTGCGCAG-3′) and reverse (5′-TCCTGTAACAACGCATCTCA-3′). PCR products were fractionated on 1.8% agarose gels, and the signals were detected using SYBR DNA staining (Invitrogen). For quantification, the PCR bands on the gel photograph of the gel were scanned using a densitometer linked to a computer analysis system. After normalizing the gene signal relative to the corresponding β-actin signal from each sample, the results were expressed as fold stimulation over S100b.
Western blotting
The membrane protein fractions were extracted by a compartmental protein extraction kit. Western blotting was performed as previously described (45). Antibodies against p47phox and epidermal growth factor receptor were used. The relative protein expression was quantified densitometrically using LabWorks 4.5 software and was calculated in relation to the loading control reference band.
Measurement of monocytic IL-1β release and ROS production
THP-1 cells were incubated in NG medium with 0.2% BSA. The cells were challenged with S100b (6.5 μg/ml) and either with or without SM for 4 h. The supernatant-conditioned medium was then harvested and assayed for IL-1β secretion using a specific ELISA kit according to the manufacturer's instructions (Pierce Endogen). The pure medium (without cells) was incubated under the same conditions and was used as a blank control for the ELISA. Intracellular ROS generation was detected using a fluorescent probe, 5-(and-6)-carboxy-2′,7′-dichlorodihydro-fluorescein diacetate (DCFH-DA) (Molecular Probes, Eugene, OR), as previously described (45).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed with an EZ-ChIP™ chromatin immunoprecipitation kit (Millipore) according to the manufacturer's instructions. Briefly, THP-1 cells (1 × 107 cells/ml) were crosslinked with 1% formaldehyde and then sheared with 4–5 sets of 10 s pulses using a Misonix sonicator ultrasonic processor (model XL2020) set to 30% of maximal power. One-tenth of the total lysate was used for total genomic DNA as an Input DNA control. Immunoprecipitation was performed overnight with 5–10 μg each of specific antibodies. Precipitates were washed and extracted with 1% SDS containing 0.1 M NaHCO3. Elutes were pooled and heated to 65°C for 5 h to reverse protein/DNA crosslinked complexes to free the DNA. The DNA fragments were purified with spin columns. For PCR, 2 μl of the DNA sample was used. The PCR primers correspond to sequences within the promoter regions as follows: IL-1β, forward (5′-CACTCTTCCACTCCCTCC-3′) and reverse (5′-AGCCTCAAACCCTTCCTC-3′); TNF-α, forward (5′-CCCTCCAGTTCTAGTTCTATC-3′) and reverse (5′- GGGGAAAGAATCATTCAACCAG-3′); COX-2, forward (5′- CAAGGCGATCAGTCCAGAAC-3′) and reverse (5′- GGTAGGCTTTGCTGTCTGAG-3′).
Animals and experimental procedure
Experimental diabetes was induced in male Sprague Dawley rats (200 ± 10 g) by i.p. injection of STZ (55 mg/kg diluted in 0.1 mol/L citrate buffer at pH 4.5) after an overnight fast according to the methods described by Forbe et al. (11). Sham-injected control animals (0.1 mol/L citrate buffer at pH 4.5) were followed concurrently. Only animals with a plasma glucose concentration more than 15 mmol/L 1 week postinduction of diabetes were included in the study as being diabetic. The experimental animals were randomly divided into five groups where each group contained 8 to 10 rats. Group I was the nondiabetic control (NC), whereas group II was the diabetic control (DC). These two groups were only fed the basal diet. Group III consisted of DC rats that were treated with a diet containing 0.3% (w/w) AG. Groups IV and V consisted of the DC rats that were treated with a diet containing 0.1% SM (SM-L) and 0.3% SM (SM-H) SM, respectively. The doses of SM and AG administered to experimental animals used were estimated from food consumption of 5% of body weight per day. For example, the estimated dosage of SM-H groups for a 300 g rat was as follow: 15 g/day × 0.3% = 150 mg/kg body weight/day.
All animals were housed individually in an air-conditioned room maintained at 25°C ± 1°C with 55%–60% humidity and a 12-h light/dark cycle. Sterilized water was accessible ad libitum. Treatments commenced from the seventh day after STZ or control vehicle injection and continued for 12 weeks. Body weights and fluid intake were recorded daily, and blood glucose levels were monitored weekly throughout the study. At the end of the experiments, the rats were sacrificed after an overnight fasting and blood was withdrawn from the abdominal aorta under diethyl ether anesthesia. The blood samples were later centrifuged (1500 g for 10 min at 4°C) for plasma isolation. The aorta and kidney were immediately collected, rinsed in ice-cold PBS, and weighed. The specimens were then snap-frozen in liquid nitrogen or fixed in 10% (v/v) formalin for further immunohistochemical analyses. All experimental procedures involving animal studies were conducted in accordance with the National Institute of Health (NIH). This experiment was approved by the Institutional Animal Care and Use Committee of the National Chung Hsing University, Taichung, Taiwan.
Tissue extraction
The method used for tissue extraction was previously reported (42). Briefly, aorta and renal samples (100 mg) were homogenized with 1 ml of RIPA buffer (Cell Signaling Technology). Tissue proteins were prepared by the addition of 1 ml of 50% (w/v) trichloroacetic acid to the homogenated samples and were centrifuged at 20000 g. The resulting pellets were recovered and washed with diethyl ether before drying. The drying pellets were dissolved in sample buffer for subsequent identification analysis using SDS-PAGE. The aorta and renal AGEs were measured by immunoblot using an anti-AGE antibody as described above. The relative expression of protein was quantified densitometrically using LabWorks 4.5 software and was calculated in relation to the reference band of the loading control.
Immunohistochemical detection of AGEs
For immunohistochemical AGE staining, formalin-fixed, paraffin-embedded section cuts (2 μm thick) were mounted on slides coated with 2-aminopropyltriethoxy silane, baked for 3 h at 58°C, deparaffinized, rinsed with 3% H2O2, and incubated with proteinase K (0.5 mg/ml) for 5 min at room temperature as previously described (8). These sections were washed with rinse buffer and blocked with StartingBlock™ blocking buffers (Pierce) for 5 min and subsequently incubated with an anti-AGE monoclonal antibody (6D12) for 30 min. After washing with rinse buffer, the sections were incubated with EnVision + labeled polymer peroxidase-conjugated anti-IgG for 30 min at room temperature, followed by detection with a 3,3-diaminobenzidine tetrahydrochoride solution (chromogen) and hematoxylin (counterstain).
Measurement of glycative parameters in experimental rats
HbA1c and urinary albumin excretion were examined by the Union Clinical Laboratory (Taichung, Taiwan). Determination of AGE-related fluorescence was based on spectrofluorimetric detection according to Münch et al. (27) with slight modifications. Briefly, blood serum was diluted (1:50) with PBS (pH 7.4), and the fluorescence intensity was detected with a FLUOstar galaxy fluorescence plate reader (BMG Labtechnologies) with an excitation wavelength of 370 nm and emission wavelength of 440 nm. The fluorescence intensity was expressed in arbitrary units (AU). Plasma-glycated albumin levels were estimated with Glycalbumin ELISA kits (GAR-50; Exocell, Inc.) using a monoclonal antibody (A717) that specifically recognizes the glycated moieties in glycated albumin. Glycated albumin values were expressed relative to total albumin content after determination of total plasma albumin. Intraassay and interassay precision for samples within the useful range of the assay had a C.V. within 10% of the mean. Analysis of tail tendon breaking time was assessed according the method described by Yue et al. (48). Collagen fibers were cut in 5 cm lengths weighing 2–2.5 mg. The mean breaking time was taken as tendon breaking time.
Measurement of oxidative biomarkers in experimental rats
The levels of 8-isoprostane in the plasma were examined with a commercial ELISA kit (Cayman Chemical Corporation) following the manufacturer's protocol. Carbonyl proteins were analyzed by the determination of absorbance (14). The concentration of carbonyl groups was calculated using the absorbance of 1 nmol/ml of carbonyl at 365 = 0.021. The data were expressed as the amount in nanomoles of carbonyl protein formed per milligram of total protein. The determination of erythrocyte oxidative hemolysis was performed with [2,2-azobis(2-amidinopropane)dihydrochloride]-derived peroxyl radicals as previously described (37). DNA damage of peripheral blood lymphocytes in rats was determined by Comet assay according the methods previously described by Wu et al. (44). The slides were examined on a Nikon EFD-3 fluorescence microscope with excitation filter BP at 543/10 nm and a 590-nm emission barrier filter. Objective measurements of the distribution of DNA were performed for a sample of cells using Komet 3.1 software (Kinetic Imaging Ltd.). One hundred cells on each slide (scored at random) were classified according to the relative intensity of the fluorescence in the tail. The degree of DNA damage was scored by determining the percentage of DNA in the tail as follows: Tail DNA% = [Tail DNA/(Head DNA + Tail DNA)] × 100.
Measurement of inflammatory mediators in experimental rats
Plasma TNF-α levels were estimated with an ELISA kit according to the manufacturer's instructions (Pierce Endogen). Nitric oxide (NO) release was determined spectrophotometrically by measuring the accumulation of its stable degradation products, nitrite and nitrate, according to the methods previously described (15). Briefly, the fresh plasma was treated with a ZnSO4 and Cd suspension. After centrifugation, the supernatant was incubated with 1% sulfonamide and 0.1% naphtylethylenediamine for 10 min at 60°C. The absorbance at 546 nm was measured with a spectrophotometer, and the results were expressed in nanomolar units.
Statistical analysis
Each experiment was performed in triplicate. The results were expressed as the means ± SD. Statistical comparisons were made by one-way analysis of variance, followed by a Duncan multiple-comparison test. Differences were considered significant when the p-values were <0.05.
Results
Effects of SM on individual stages of protein glycation
The effects of SM on individual stages of protein glycation are demonstrated in Figure 1A and were compared with the effects of AG (Fig. 1B), an established AGE inhibitor. Three assay methods were adopted to evaluate the antiglycation effects of SM as follows: (a) δ-Glu assay, which is an assay based on inhibition of the Amadori product (HbA1c levels) generated during early stages of protein glycation (32); (b) BSA-glucose assay, which is an assay based on AGE-related fluorescence generated in the course of glycation (45); and (c) G.K. peptide-ribose assay, which is an assay based on the inhibition of protein-AGE crosslinking (28, 32). The results demonstrated that SM and AG had only a moderate inhibitory effect on the early stages of glycation (Fig. 1A, B, upper panel), whereas SM exerted marked inhibitory effects on late glycation, AGE formation (Fig. 1A, B, middle panel), and the subsequent protein crosslinking (Fig. 1A, B, bottom panel). As a result, SM is likely to behave in a similar fashion as AG in inhibiting protein glycation.

MG, a reactive carbonyl species (RCS), readily reacts with protein lysine and arginine protein residues to produce high-molecular-weight, cross-linked products (19, 28, 38). Time- and dose-dependent effects of MG-mediated protein glycation were determined by AGE-related fluorescence and SDS-PAGE protein maps. MG at a concentration of 2.5 mmol/L developed the most significant fluorescence after an incubation period of 9 days (Fig. 1C). There was a decrease in the detectable amount of BSA in its position at the bottom of the gel in addition to less resolution and spreading of bands compared with untreated protein (Fig. 1D). This result was similar to MG-treated BSA and ovalbumin as previously described (19). When SM (50 and 500 μg/ml) or AG (1 and 10 mmol/L) were present in the incubation mixture, losses of BSA and the formation of the high-molecular-weight protein were suppressed (Fig. 1E). This was an indication of SM- and AG-mediated in vitro protection against glycation. As mentioned above, AG has been demonstrated as an antioxidant and nucleophilic agent, possessing a potent scavenging effect on highly RCS (39). Thus, it is speculated that SM may exhibit an RCS trapping effect.
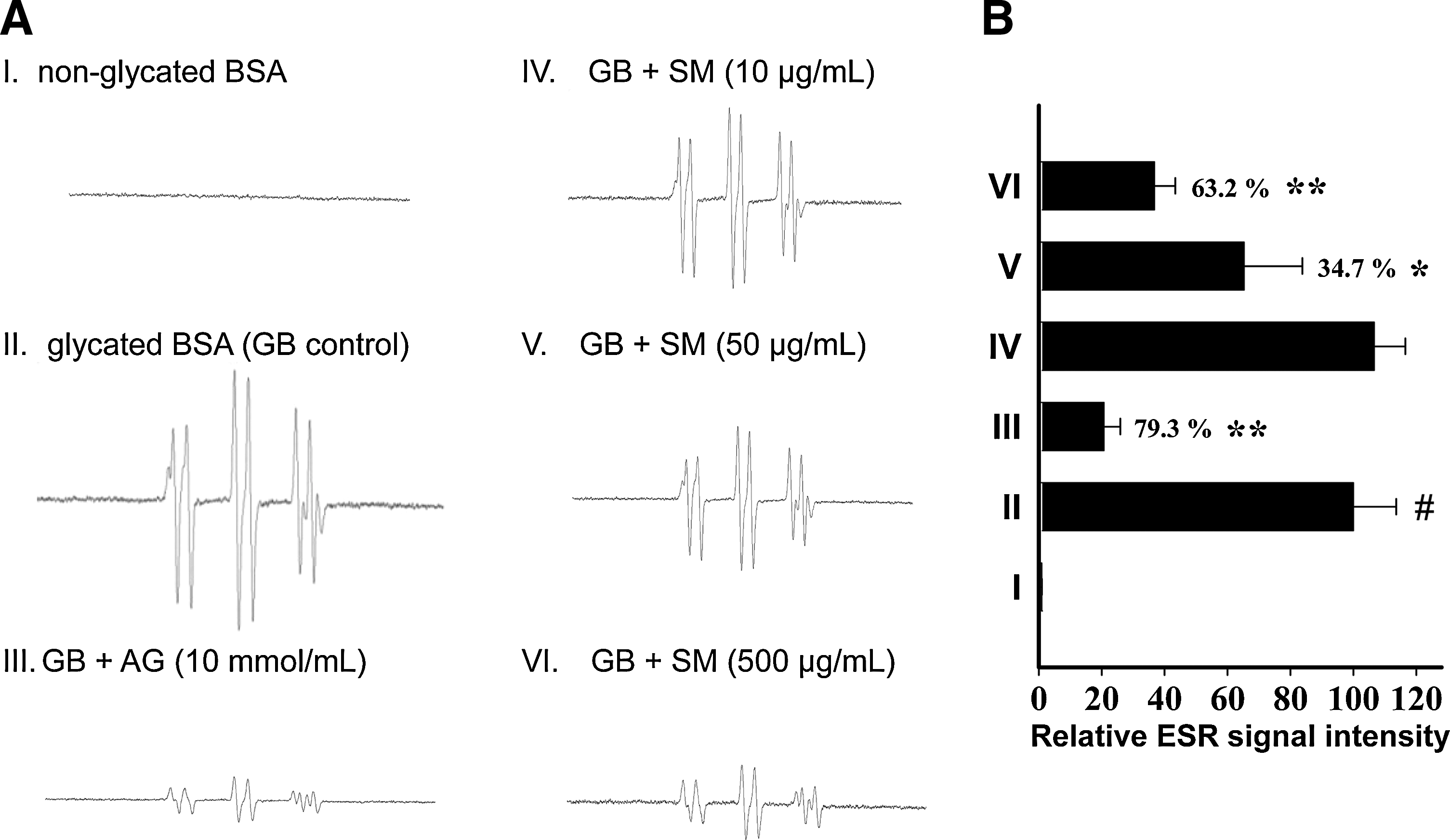
Scavenging activity of SM on glycation-derived free radicals
ROS are involved in the Maillard reaction, and that the formation of free radicals occurs during the glycation process (13, 33). On the basis of the significant inhibitory effect of SM against in vitro glycation (Fig. 1), ESR spectrometry was utilized to investigate the scavenging effect of SM on glycation-derived radicals. The ESR spectra clearly showed that protein glycation led to increased production of free radicals (Fig. 2A). The addition of SM to the reaction system caused a dose-dependent decrease in the ESR signal intensity (Fig. 2B), indicating that SM-mediated glycation inhibition may be related to its antioxidant properties.
SM inhibited expression of inflammatory mediators in S100b-stimulated monocytes
The ligation of S100b to RAGE is an important part of complex interactions of the oxidative stress and inflammatory responses (7). S100b serves as a valuable tool in the study of AGE-RAGE signaling and the diabetic inflammatory conditions (9, 35). As shown in Figure 3A, S100b (6.5 μg/ml) treatment in THP-1 cells for 4 h resulted in an approximate three- to fourfold increase in mRNA expression levels of IL-1β and COX-2. Moreover, SM dose dependently (p < 0.05) inhibited both the gene and protein levels of IL-1β in monocytes (Fig. 3).

Effect of SM on intracellular ROS production and p47phox translocation in monocytes
We next examined whether S100b induced a pro-oxidant status in cultured monocytes and estimated the protective effect of SM against intracellular ROS production. DCFH-DA staining showed that S100b-treated cells exhibited a striking increase in the basal levels of ROS compared with NG control cells (Fig. 4A, B; p < 0.05). A significant reduction of ROS was observed after the treatment with SM (5–25 μg/ml, p < 0.05). Further, the S100b-driven ROS production was completely blocked by pretreatment with APO, an NADPH oxidase inhibitor, indicating that the activation of NADPH oxidase may have a role in the elevated levels of oxidative stress caused by S100b.
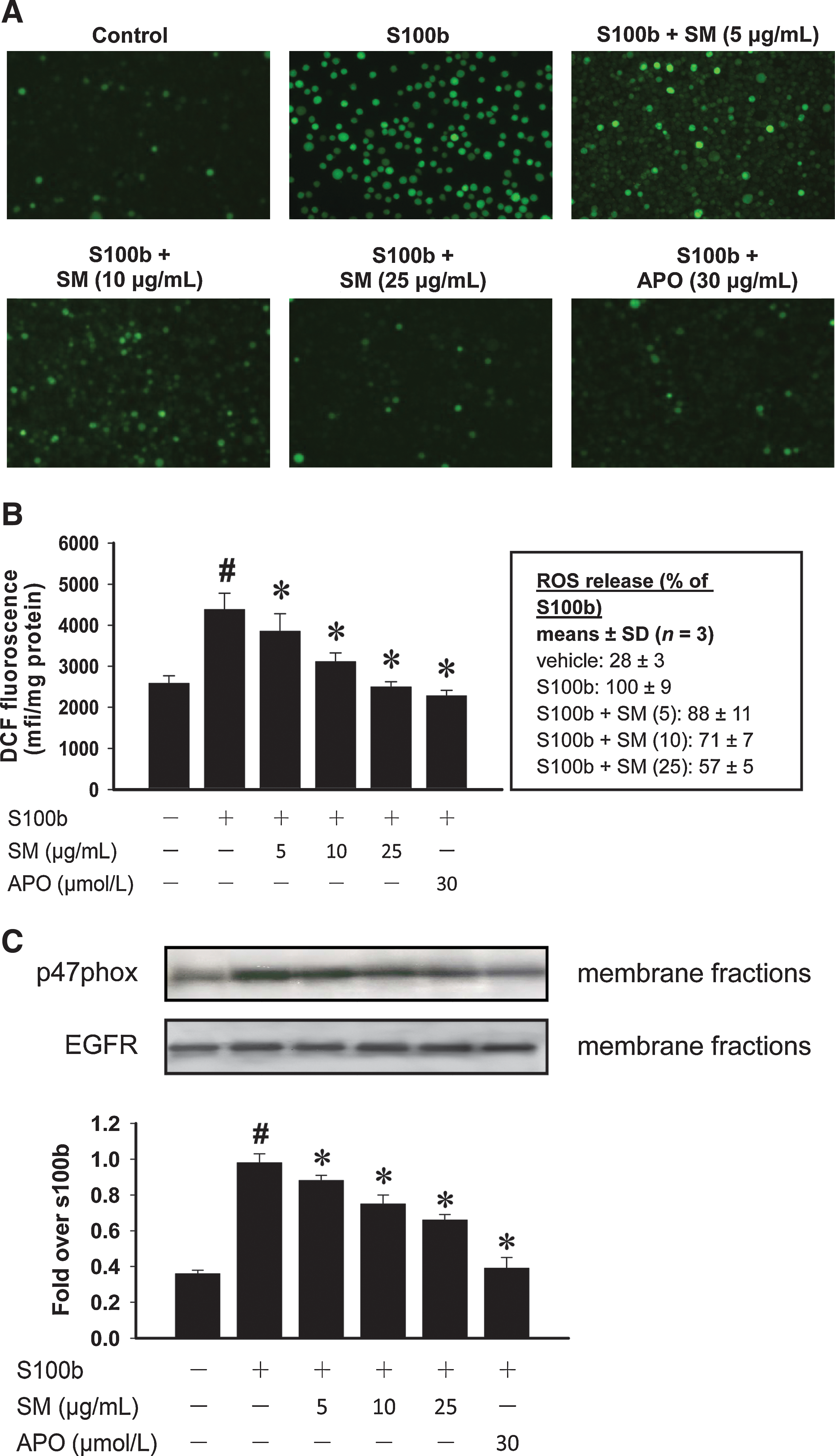
Therefore, the effect of S100b on p47phox protein expression, an NADPH oxidase subunit, was investigated. As shown in Figure 4C, membrane translocation of p47phox was significantly increased in S100b-treated cells as compared with untreated control cells (p < 0.05), whereas the addition of SM inhibited these translocation events. Moreover, the S100b-induced p47phox translocation was also blocked by APO treatment. These results suggest that SM has a potent impact on intracellular ROS formation, and may be partly due to downregulation of the NADPH oxidase activities.
S100b increased the recruitment of NF-κB p65 to the IL-1β gene promoter in monocytes
Diabetic stimuli such as hyperglycemia (6, 12) and/or AGEs (22) augment expression of inflammatory genes by transcriptional mechanisms. We demonstrated with ChIP assays that S100b increased the binding of NF-κB p65 subunit to the promoter of the IL-1β gene in monocytes, and these nuclear events were significantly enhanced when cells were challenged with S100b (Fig. 5A). Little or no binding behavior was found in NG control cells and NG cells without the addition of a p65 antibody (Fig. 5A, lanes 1 and 2), indicating the specificity of the S100b effects. There was no change in the amplification of input DNA (loading control) in all cases (Fig. 5A, lower panel). In addition, the recruitment of p65 to the promoters of COX-2 and TNF-α was also observed (Fig. 5B).
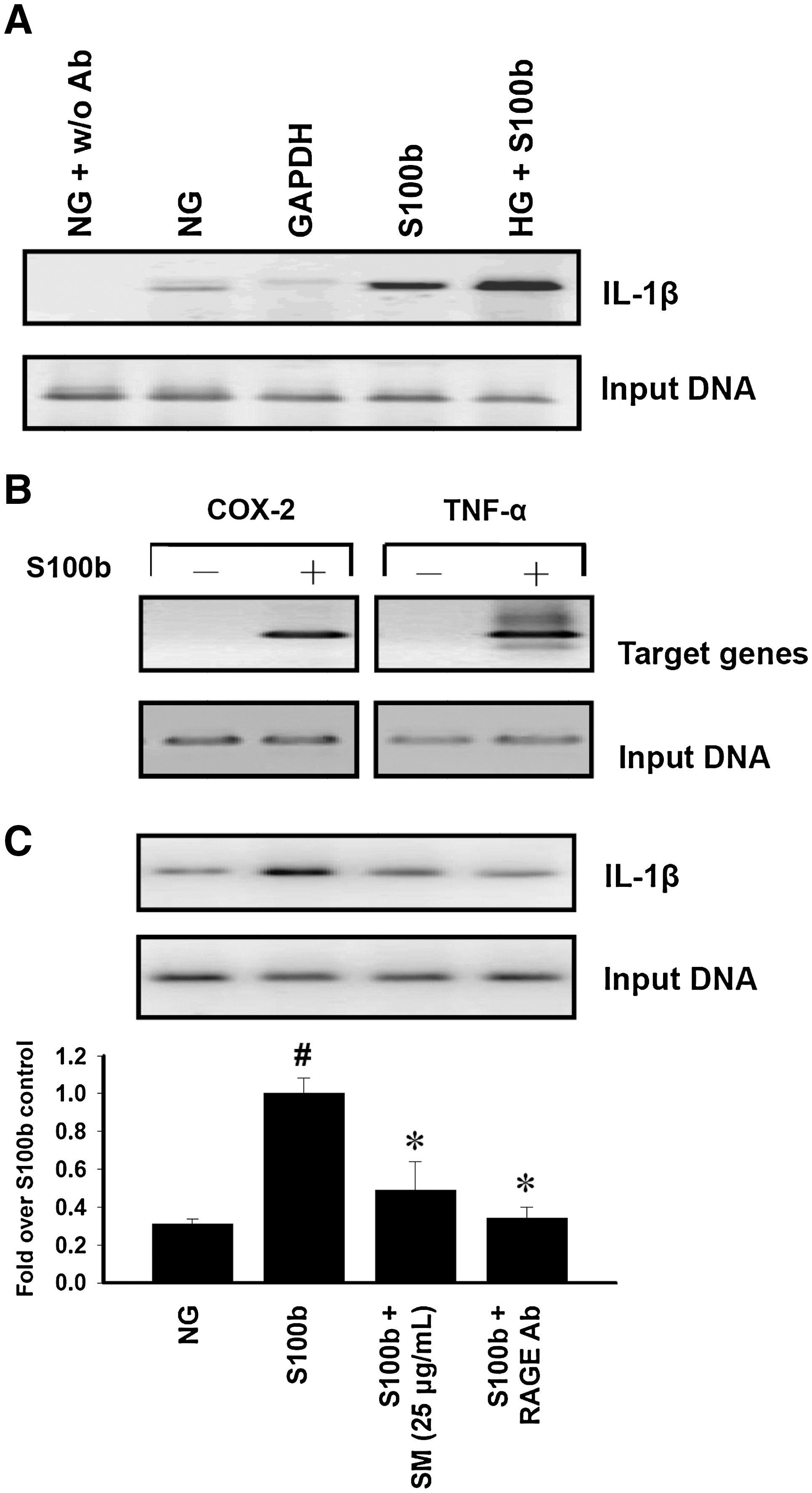
To clarify the molecular transcription mechanisms and nuclear chromatin remodeling events of antiinflammatory activities, SM was examined with ChIP assays. The RAGE antibody was also included as a reference drug. As the results show in Figure 5C, the recruitment of p65, which was involved in S100b-stimulated IL-1β promoter activation, was blocked by RAGE antibody treatment (lane 4). At a concentration of 25 μg/ml, SM had a marked inhibitory effect against S100b-mediated p65 recruitment in monocytes (lane 3).
ChIP assays of coactivator recruitment and histone modifications in response to S100b stimulation in monocytes
A time course of S100b-induced recruitment of chromatin remodeling factors to the IL-1β promoter was performed with antibodies to CBP, CARM1, acetyl-HH3, and methyl-HH3R17 with ChIP assays. Figure 6A shows that (a) S100b increased the recruitment of the CBP and CARM1 coactivators to the IL-1β promoter as early as 15 min after S100b treatment; (b) acetylation of HH3 occurred as early as 15 min after S100b treatment and reached a plateau at a 60 min incubation period and decreased after 2 h; and (c) similar results were also obtained with a methylation of HH3 at Arg17, which reached a plateau at a 30 min incubation period and decreased after 1 h. The input DNA was not altered by the S100b treatment (Fig. 6A; lowest panel). Additional experiments revealed that SM actively abrogated the amounts of acetylated Lys residues and methylated Arg residues of HH3 within the IL-1β promoter in response to S100b stimulation (Fig. 6B). Our results indicated that S100b stimuli induced histone acetylation and methylation at specific Lys and Arg residues within the IL-1β gene. These results provide novel evidence that a close relationship exists between proinflammatory gene activation and acetylated and/or methylated histone accumulation under high glycotoxin conditions.

Physical and biochemical parameters in experimental rats
At 12 weeks, the DC group had a significant increase in blood glucose and HbA1c levels compared with the levels in the NC group (p < 0.05 vs. DC group). However, no differences in glycemic levels were noted between treated and untreated diabetic groups (Table 1). The administration of SM-H (0.3% of diet) to rats showed only moderate hypoglycemic and urinary albumin excretion lowering effects that did not reach statistical significance (p > 0.05). Diabetic animals had decreased body weights but large increases in daily fluid intake indicating classic diabetes symptoms. No attenuation of these changes was demonstrated with AG or SM treatments (Table 1). In addition, normal rats treated with AG or SM did not show any differences from the NC group with respect to glycemic levels, body weights, or fluid intake (p > 0.05; data not shown).
Values are given as the means ± SD for 8–10 rats in each group.
p < 0.05 versus nondiabetic control.
p < 0.05 versus diabetic control.
AG, aminoguanidine; SM, silymarin.
Oxidative and inflammatory biomarkers in experimental rats
In vivo biomarkers of oxidative stress, such as 8-isoprostane, carbonyl groups, lymphocyte DNA damage, and erythrocyte hemolysis, were significantly increased in the DC group compared with the NC group (p < 0.05). These parameters were reduced in diabetic rats receiving SM-H (0.3% of diet) compared with untreated diabetic animals. No differences were observed in 8-isoprostane, lymphocyte DNA damage, and erythrocyte hemolysis in diabetic animals treated with SM-L. We also confirmed that treatment with AG resulted in a 26% reduction in lymphocyte DNA damage compared with the DC group (Table 1).
Levels of inflammatory mediators in plasma at the end of the 12-week experimental period are shown in Table 1. Diabetes found in the DC group was associated with a significant increase in plasma levels of TNF-α and NO compared with animals in the NC group. Treating the DC group with AG or SM-H reduced plasma TNF-α and NO levels, and to a lesser extent with animals in the SM-L group (Table 1).
Glycative biomarkers in experimental rats
The diabetic animals had significant increases in the tail tendon breaking times (Fig. 7; upper panel), development of AGE-related fluorescence (Fig. 7; middle panel), and levels of plasma albumin AGEs (Fig. 7; bottom panel) compared with NC group (p < 0.05). The inhibitory effects on either parameter were noted with all of the treatment regimens except the SM-L group, which had no significant effect on the inhibition of AGE-related fluorescence compared with the DC group (Fig. 7; p > 0.05).

AGE immunohistochemistry and immunoblotting
The monoclonal antibody raised against AGEs (6D12) was used for the immunohistochemical analysis and western blotting of the Maillard reaction products in the experimental rats. The specificity of the antibody was tested by preincubation with free antigens. As a result, no positive staining was observed after such preincubation (data not shown). Figure 8 shows immunohistochemical staining for AGEs in the aortas and renal glomeruli. With diabetes, AGEs were primarily localized in the extracellular matrix surrounding the smooth muscle cells of the aorta (Fig. 8A) and were also intensified in the glomeruli of the kidney (Fig. 8B). AG or SM-H treatment reduced the labeling of AGEs in the aortas and glomeruli of diabetic rats. Subsequent immunoblotting analysis the formation of AGEs showed similar results. The formation and accumulation of AGEs in the aortas (Fig. 8C, p < 0.05) and kidneys (Fig. 8D, p < 0.05) were increased in the DC group compared with the NC group. SM treatment was associated with reduced formation of AGEs in both of the diabetic treatment groups (SM-L and SM-H) as compared with their untreated counterparts. The AG-treated group had the most potent decrease in the formation of AGEs in the organs mentioned above (Fig. 8C, D).

Discussion
In this study, the phenomenon of protein glycation was demonstrated in the reaction mixtures of albumin with sugar by several model systems in vitro. Sugars, including glucose, δ-Glu, and ribose, in addition to a dicarbonyl compound, such as MG, were used as glycated agents, which are commonly adopted in Maillard reaction-associated studies (18, 29, 38). Human hemoglobin, BSA, and G.K. peptides, representing the amine sources serve as targets for glycated agents although G.K. peptides are not found in physiology or food systems (13, 32). These experimental methods uniquely differentiate between specific inhibitors of the early stage (Amadori products), middle stage (RCS), and the last stage of glycation (formation and crosslinking of AGEs) (18, 32). Our results revealed that SM inhibited AGE-related fluorescence, RCS-derived protein modifications, and the generation of ROS during glycation process (Figs. 1 and 2). A high correlation was proposed between antioxidant capacity and antiglycation action of SM. To the best of our knowledge, this is the first report showing SM as a potential anti-AGE agent.
There is positive feedback in the pathogenetic mechanism of glycation; ROS increase the formation of AGEs, the process being termed glycoxidation or autoxidative glycation (41). Owing to these adverse conditions, a supplement of antioxidants in response to protein glycation should be a theoretical strategy for preventing diabetic complications (1, 2). Having available clinical and experimental information, it may be of great interest to propose that administration of antioxidant flavonoids may be beneficial for the prevention of protein glycation. Daflon 500, a clinical drug that is composed of flavonoids, has attenuated effects on HbA1C levels and protein glycation in a group of 28 type 1 diabetic patients (21). Phytoestrogenic isoflavonoids, such as daidzein and genistein, have been shown to interfere with AGE-mediated oxidative DNA damage in hypertensive rats, which is attributed to direct scavenging action on AGE-derived radicals (25). Antiglycation candidates, such as green tea extract (30) and tomato paste fraction (18), are also related to their flavonoid contents in a similar system.
Increasing evidence points to the adverse effects of AGEs that are mediated by ligation of the RAGE. Aside from AGEs, several short peptides, such as S100b, can also upregulate RAGE expression, rendering the cells more susceptible to the effects of AGEs. Accordingly, S100b serves as a valuable tool in the study of AGEs and RAGE signaling in diabetic inflammatory conditions (9, 35). Because circulating monocytes are continuously exposed to conditions of high AGEs in diabetes, we examined if SM influenced S100b-driven oxidative stress and inflammatory responses in monocytes. The results may provide additional information to better understand the beneficial effects of SM in situations where either AGEs accumulate or NF-κB regulation of RAGE is activated.
In the present study, we have focused on S100b-stimulated monocytes in which a large panel of NF-κB-responsive genes, including IL-1β, was enhanced. Interestingly, we observed that SM was potent in inhibiting S100b-induced IL-1β activation (Fig. 3). Dasu et al. (4) reported that hyperglycemic conditions led to ROS and release of inflammatory mediators from monocytes that interfere with mitogen-activated protein kinases, protein kinase C, and NADPH oxidase. Further, the underlying mechanism was suggested to be an increase in the activation of an ROS-dependent NF-κB cascade. Similar results were obtained in the present study with S100b stimuli (Fig. 4). The SM-related decrease in IL-1β release from monocytes was likely through the abolishment of NADPH oxidase-associated p47phox expression (Fig. 4C). This process may mediate the ROS formation resulting in NF-κB-dependent genes expression. Indeed, the results indicated that S100b evoked IL-1β production, which was associated with increased recruitment of the CBP and CARM1 coactivators and p65 to the IL-1β promoter (Figs. 5 and 6). Nuclear acetylation controlled by histone acetylase and histone deacetylase (23) as well as methylation controlled by several site-specific methyltransferase, such as CARM1 (20, 24), are critical events during diabetes and inflammation conditions. Intrinsic histone acetylase, CBP, and CARM1 are able to acetylate and methylate histones and inflammation-responsive transcription factors (24). Among the histone modifications, we have observed that acetylation of HH3 at Lys9 and/or Lys14 and methylation of HH3 at Arg17 were associated with IL-1β gene activation in response to S100b stimulation (Fig. 6). However, despite S100b induction of nuclear NF-κB, CBP, and CARM1 in monocytic cells, the treatment of SM prevented the recruitment of these coactivators to their respective sites on the promoter. SM interfered with S100b-induced histone acetylation and methylation surrounding the IL-1β promoter, which provided an explanation for the abolished to recruit transcription factor NF-κB.
In this work, we further reported the influence of SM on the levels of AGEs in experimental diabetic rats. Previously, we have screened a series of polyphenol antioxidants, including 10 flavonoids (43, 45), 12 phenolic acids (46), curcumin, and SM to evaluate their effects on protein glycation, crosslinking, and subsequent formation of AGEs to find promising candidates as anti-AGE agents for further investigation. Two of these polyphenol compounds had consistent evidence of inhibitory activities toward the formation of AGEs in streptozotocin-induced insulin-deficient diabetic rats (Supplemental Material; see
In conclusion, SM has potent inhibitory effects on protein glycation and the subsequent formation of AGEs. SM also targets the chromatin remodeling machinery to prevent inflammatory mediators transcription. Further, this study confirms the significance of oxidative stress and inflammation in the accumulation of AGEs in diabetes, and it supports SM administration for the prevention of glycotoxin-associated complications of diabetes mellitus.
Footnotes
Acknowledgments
This research work was partially supported by the National Science Council, NSC94-2321-B005-001 and NSC96-2628-B005-004-MY3, Taiwan, Republic of China. The authors also would like to thank Dr. J. W. Liao of the Graduate Institute of Veterinary Pathobiology for his technical assistance on immunohistochemical staining.
Author Disclosure Statement
The authors declare no conflicts of interest in this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.