
Abstract
Several signal transduction pathways are activated by cardioprotective stimuli, including ischemic or pharmacological postconditioning. These pathways converge on a common target, the mitochondria, and cardioprotection by postconditioning is associated with preserved mitochondrial function after ischemia/reperfusion. The present review discusses the role of mitochondria in cardioprotection, especially the involvement of ATP-dependent potassium channels, reactive oxygen species, and the mitochondrial permeability transition pore, and focuses on the effects of postconditioning on mitochondrial function (i.e., their oxygen consumption and calcium retention capacity). The contribution of mitochondria to loss of protection by postconditioning in diseased or aged myocardium is also addressed. Antioxid. Redox Signal. 14, 863–880.
Cardioprotection and Its Signaling: Background and Conceptual Framework
The signal transduction of ischemic preconditioning has traditionally been classified into initiating triggers, intracellular mediators, and end-effectors. Triggers were considered active during the preconditioning brief episode(s) of ischemia/reperfusion and antagonized by inhibitors bracketing the preconditioning ischemia/reperfusion, whereas mediators and effectors were active during the prolonged ischemia. However, with the recognition of the postconditioning phenomenon, it became apparent that pre- and postconditioning share a number of signaling elements and that preconditioning, like postconditioning, induces activation of signaling elements during the early reperfusion following the prolonged ischemia (37). Thus, an easy temporal distinction of triggers, mediators, and end-effectors was no longer possible. Mitochondria are involved in three different ways: a) as triggers or signal amplifiers (in terms of activation of mitochondrial KATP channels and the resulting formation of small amounts of reactive oxygen species (ROS), b) as end-effectors (in terms of inhibition of mitochondrial permeability transition pore (MPTP) opening and of release of pro-apoptotic factors into the cytosol), and c) as targets of damage and protection from it (in terms of their functional and morphological integrity). Also, ROS are involved in two different ways: a) as signaling elements when formed in small amounts, and b) as damaging elements when formed in excessive amounts.
The present review attempts to characterize in detail the multiple roles of mitochondria in cardioprotection, notably in postconditioning.
Role of Mitochondria in Cardiomyocyte Function After Ischemia/Reperfusion Injury
The most important role of mitochondria is the generation of adenosine triphosphate (ATP). Mitochondria also contribute to the generation and scavenging of ROS, are involved in cellular ion homeostasis, notably calcium homeostasis, and contribute to cardiomyocyte apoptosis and necrosis. Mitochondria consume large amounts of oxygen during oxidative phosphorylation. During ischemia, the lack of oxygen inhibits electron flow, and myocardial ATP utilization becomes inefficient. The F0F1ATP synthase, which normally produces ATP, switches into reverse mode and consumes ATP to pump protons from the matrix into the intermembrane space.
Depending on the species, 50%–80% of ATP is consumed by the F0F1ATPase during ischemia (49). The ATP, which is otherwise required for ion pumps to maintain the cellular ion homeostasis, is used to maintain the mitochondrial membrane potential (90). At reperfusion, an increased ATP concentration via restored oxidative phosphorylation may contribute to the reestablishment of cellular ion homeostasis; however, increased ATP content paradoxically contributes to reperfusion injury; in rapidly re-energized cells, Ca2+ oscillations between the sarcoplasmic reticulum, and the cytosol contribute to hypercontracture of cardiomyocytes, membrane disruption, and subsequent necrosis (114).
Two populations of mitochondria exist within cardiomyocytes: subsarcolemmal mitochondria (SSM) are located directly beneath the plasma membrane, and interfibrillar mitochondria (IFM) are located between the myofibrils (100). SSM and IFM differ in morphology, and IFM have a higher respiratory and calcium handling capacity than SSM (100, 119). Following ischemia/reperfusion mitochondrial oxygen consumption is reduced (17, 101, 110, 111), and this reduction in oxygen consumption is accompanied by decreased activities of protein complexes of the electron transport chain [e.g., complex I (101), complex II (24), and complex III (110, 111)]. Oxidative phosphorylation after 45 min ischemia is specifically reduced in SSM and not in IFM (81).
ROS are generated in different cellular compartments and by several enzymes, including NADPH oxidases at the plasma membrane (3, 80) and cytosolic xanthine oxidases (14). However, clearly mitochondria are the most important cellular source of ROS (11, 38, 91). Within mitochondria, the largest amount of oxygen is reduced to water at respiratory complex IV. However, at respiratory complexes I or III, electrons from the electron transport chain can be transferred to oxygen, resulting in the generation of partially reduced oxygen, especially superoxide anions. Superoxide dismutases then rapidly convert superoxide anions to hydrogen peroxide (43). Apart from the electron transport chain, ROS can also be produced by monoamine oxidases in the outer mitochondrial membrane, which transfer electrons from amine compounds to oxygen and thereby generate hydrogen peroxide. Within mitochondria, p66Shc oxidizes reduced cytochrome c, which induces the partial reduction of oxygen to peroxide (45). p66Shc is present in the cytosol under physiological conditions, becomes phosphorylated by protein kinase C β under stress conditions, and subsequently translocates into the mitochondrial intermembrane space (97, 112).
Detrimental effects of excessive ROS formation include protein and lipid damage, which leads to cellular dysfunction and cell death. The proximity of mitochondrial DNA to the production site of ROS, the lack of protection of mitochondrial DNA by histones, and the limited capacity of repair mechanisms render the mitochondrial DNA highly susceptible to increased oxidative stress (27).
Excessive oxidative stress contributes to reversible and irreversible myocardial injury after ischemia/reperfusion (2, 13, 77, 120, 139, 141). The reversible contractile dysfunction following myocardial ischemia and reperfusion (“stunning”) is clearly a manifestation of excessive oxidative stress (22, 50). ROS scavenging by administration of phenanthroline and MPG (mercaptopropionylglycine) at reperfusion reduces cell death of embryonic chick cardiomyocytes, confirming that the burst of ROS at the onset of reperfusion is causal for cell death (140). In contrast, ROS scavenging by a combination of superoxide dismutase and catalase during reperfusion did not reduce infarct size in dog hearts in vivo; however, microvascular injury and the low-reflow phenomenon were attenuated (115).
The burst of ROS at the onset of reperfusion facilitates opening of the MPTP, a large conductance pore in the inner mitochondrial membrane. MPTP opening at reperfusion enhances inner mitochondrial membrane permeability to solutes with molecular weights up to 1.5 kDa and leads to mitochondrial depolarization, followed by ATP depletion (35). Mitochondrial matrix volume increases and induces rupture of the outer mitochondrial membrane, leading to loss of pyridine nucleotides and release of pro-apoptotic factors such as cytochrome c and thus also inhibiting electron flow via the electron transport chain. Not only ROS, but also increased concentrations of calcium ions (Ca2+), inorganic phosphate and mitochondrial depolarization—conditions occurring during ischemia and reperfusion—favor MPTP opening (34, 57). These factors are counteracted by physiological MPTP antagonists, such as increased mitochondrial membrane potential, high concentrations of protons, magnesium ions, adenine nucleotides, notably ADP, as well as nitric oxide (34, 142). Whereas the pathophysiological conditions favoring MPTP opening and consequences resulting from MPTP opening are well established, the molecular identity of the protein(s) forming this pore is still unknown. Originally, it had been suggested that the MPTP is formed by the voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide transporter (ANT) in the inner membrane, and cyclophilin D in the matrix. However, experiments with VDAC and ANT knockout mice demonstrated efficient permeability transition upon Ca2+-stimulation, indicating that VDAC and ANT are dispensable for permeability transition (10, 74). Whereas in fibroblasts and in liver mitochondria of cyclophilin D knockout mice Ca2+-induced MPTP opening is clearly delayed (9, 12), the sensitivity of MPTP opening in response to adenine nucleotides or oxidative stress is similar in cyclophilin D-deficient and in wild-type mitochondria, indicating that cyclophilin D is important for MPTP opening but that permeability transition can also occur in the absence of cyclophilin D (12).
Functional mitochondria participate in fusion and fission, whereas nonfunctional and damaged mitochondria are removed by autophagy. Therefore, removal of mitochondria, which trigger apoptosis or necrosis, contributes to cardioprotection (for review, see (47)).
Taken together, mitochondria are damaged during myocardial ischemia and reperfusion. Mitochondria not only suffer from but also contribute to myocardial injury in that MPTP opening induces cell death. Thus, preservation of mitochondrial integrity appears an attractive target to reduce damage, notably infarct size after ischemia/reperfusion injury (56, 57).
Mitochondria in the Cardioprotection by Ischemic and Pharmacological Preconditioning
The activation of endogenous cardioprotective mechanisms attenuates irreversible myocardial injury following ischemia/reperfusion. One of these mechanisms is ischemic preconditioning (IP), that is, the infarct size reduction by brief, nonlethal episodes of ischemia/reperfusion preceding a period of sustained ischemia/reperfusion (92). The major signal transduction pathways involved in IP cardioprotection seem to converge at the level of mitochondria and impact on mitochondrial function (for review see (56, 88, 89)). Activated protein kinase C and protein kinase G, which are part of the intracellular mediator signaling program (56), project onto mitochondrial ATP-dependent potassium channels and facilitate their opening (29, 30, 144). Opening of ATP-dependent potassium channels (mitoKATP channels) in the inner mitochondrial membrane is essential for cardioprotection (5). Evidence for the involvement of mitoKATP channels in the inner mitochondrial membrane in IP is mainly derived from pharmacological modification of mitoKATP channel opening by substances such as diazoxide that opens mitoKATP channels, and 5-hydroxydecanoate (5-HD) that closes mitoKATP channels. Recently, increased potassium fluxes in response to diazoxide have been measured in intact mitochondria using the potassium sensitive dye PBFI (potassium-binding benzofuran isophthalate) or directly by patch-clamping of mitochondria devoid of the outer membrane (Fig. 1) (86, 122). The exact molecular structure of the mitoKATP channels has not been identified up to now, although immunoreactivity for the sarcolemmal KATP channel subunits Kir6.1 and Kir6.2 has been detected in heart mitochondria (79). Pharmacological opening of mitoKATP channels by diazoxide contributes to the formation of small amounts of ROS (53), mainly superoxide anions derived from complex I of the electron transport chain (4). Also, mitochondrial uncoupling (i.e., proton influx into the mitochondrial matrix without phosphorylation of ADP) contributes to ROS formation (123).

Small amounts of ROS function as trigger molecules of IP, presumably by activating protein kinases such as protein kinase C (8) or the mitogen-activated protein kinase (MAP kinase) p38 (31). Accordingly, scavenging of ROS with ascorbic acid attenuates infarct size reduction by IP in pig myocardium (127) and by pharmacological preconditioning with diazoxide in isolated rabbit hearts (99).
Within cardiomyocytes, connexin 43 (Cx43) is predominantly localized at gap junctions; however, the protein is also present at the inner membrane of cardiomyocyte subsarcolemmal mitochondria (16, 21, 121) where it regulates mitochondrial potassium fluxes (86, 122). Cx43 is a target of several protein kinases, and mitochondrial Cx43 is highly phosphorylated under physiological conditions (137). A decrease of the mitochondrial Cx43 content is sufficient to abolish the cardioprotection by pharmacological preconditioning with diazoxide (53, 121). Recent studies demonstrated that pharmacological inhibition or genetic ablation of mitochondrial Cx43 confers resistance to mitoKATP channel opening in response to diazoxide in patch-clamped mitoplasts (mitochondria devoid of the outer membrane). However, since the open-probability of these channels was not affected under baseline conditions, Cx43 rather regulates mitoKATP channel activity than constituting the pore forming unit of the mitoKATP channel (122).
In isolated cardiomyocytes, hypoxic preconditioning attenuates oxidative stress at the beginning of reoxygenation (140). Also, in mitochondria isolated from rat hearts in vitro, IP reduces the burst of ROS production at reperfusion (102). Large amounts of ROS favor MPTP opening, and IP and pharmacological preconditioning confer cardioprotection by inhibiting MPTP opening at the onset of reperfusion (52, 65). Vice versa, the preservation of mitochondrial function by IP results in an attenuation of the ischemia/reperfusion-induced decrease in ADP-stimulated respiration (150). In preconditioned hearts, tissue levels of ATP decline more slowly than in control hearts, resulting in diminished stimulation of anaerobic glycolysis, decreased reduction of intracellular pH, and attenuation of the ionic alterations occurring during ischemia (131, 132). Following an ischemic preconditioning stimulus, F0F1ATP synthase activity is reduced via binding of the inhibitor protein IF1 (36, 106). The inhibition of the F0F1ATP synthase may contribute to preservation of ATP content during ischemia, when the F0F1ATP synthase works in the reverse mode. Details on the role of the F0F1ATP synthase in cardioprotection are reviewed in detail elsewhere (84). As in preconditioned hearts, ATP hydrolysis during ischemia is also reduced by overexpression of bcl-2 (62). In addition, inhibition of glycogen synthase kinase 3 β (GSK3β) may slow adenine nucleotide transport through the outer mitochondrial membrane via dephosphorylation of the voltage-dependent anion channel (32, 131). The adenine nucleotide transporter (ANT) in the inner mitochondrial membrane exchanges ATP/ADP by an antiport mechanism. The ischemia-induced dephosphorylation of ANT is prevented by pharmacological preconditioning, and the preserved ANT phosphorylation may enhance ATP production during ischemia (40).
Taken together, the common target of the signal transduction pathways activated by ischemic and pharmacological preconditioning are the mitochondria. Within mitochondria, the generation of small amounts of ROS during the preconditioning ischemia/reperfusion phase through activation of mitoKATP channels reduces the excessive oxidative stress resulting from the subsequent prolonged phase of ischemia/reperfusion. The inhibition of MPTP opening contributes to cell survival.
Mitochondria in the Cardioprotection by Ischemic and Pharmacological Postconditioning
In contrast to IP, where nonlethal periods of ischemia/reperfusion before the sustained ischemia/reperfusion reduce infarct size, ischemic postconditioning induces cardioprotection by short cycles of ischemia and reperfusion at the immediate onset of reperfusion (155). Obviously, ischemic postconditioning specifically reduces reperfusion injury (54). Delaying the first re-occlusion of the postconditioning maneuver for several minutes abolishes cardioprotection (73, 151). Ischemic postconditioning can also be mimicked pharmacologically [e.g., by various anaesthetics (26, 118)]. Postconditioning activates signaling cascades [e.g., the RISK (reperfusion injury salvage kinase) and the SAFE (survivor activating factor enhancement) pathway, which are reviewed in detail elsewhere (53a). However, while established in small animals, mainly rodents, the causal involvement of the RISK pathway in ischemic postconditioning was not confirmed in pigs (126, 129).
Cx43 is clearly essential for the cardioprotection by ischemic preconditioning (125); however, data on the role of Cx43 in ischemic postconditioning are limited. Although the mitochondrial Cx43 content decreases with ischemic postconditioning (107), such reduction of Cx43 does not interfere with protection. In fact, infarct size in heterozygous Cx43-deficient mice is reduced by ischemic postconditioning to a similar extent as in wild-type mice (58). Whether a further decrease of the Cx43 protein content, which can be achieved in conditional knockout mice, abolishes the cardioprotection by ischemic postconditioning, remains to be established. Despite striking similarities between the signal transduction pathways of ischemic pre- and postconditioning, some elements such as mitochondrial Cx43 might be specific for either pre- or postconditioning.
STAT3 (signal transducer and activator of transcription 3) is part of the janus kinase (JAK)/STAT signal transduction pathway and of the SAFE pathway (78), and transduces stress signals from the plasma membrane to the nucleus, leading to changes in gene transcription. Apart from its role in cardiomyocyte hypertrophy and apoptosis, STAT3 is involved in the infarct size development following ischemia/reperfusion (for review, see (18)). In mice with a cardiomyocyte-specific deletion of STAT3, infarct size reduction by a postconditioning stimulus of 3 cycles 10 s ischemia and reperfusion each was abolished (15). Also, postconditioning with exogenous tumor necrosis factor α did not protect isolated hearts from cardiomyocyte-specific STAT3 knockout mice (78). Recently, STAT3 has been identified by Western blot analysis in cardiomyocyte mitochondria (145). We confirmed the presence of STAT3 in isolated mitochondria by confocal laser scan microscopy (Fig. 2). However, whether specifically mitochondrial STAT3 contributes to the cardioprotection by postconditioning must be addressed in further studies.

Similar to IP, the signal transduction pathways of postconditioning converge at the level of mitochondria. The subsequent review will focus on the ischemic or pharmacological postconditioning maneuvers that confer cardioprotection by interaction with mitochondrial function.
Role of mitochondrial ATP-dependent potassium channels in the cardioprotection by ischemic and pharmacological postconditioning
As in ischemic preconditioning, activated protein kinase C and G facilitate mitoKATP channel opening also in ischemic postconditioning (30, 108). In isolated rat hearts subjected to 30 min ischemia and 2 h reperfusion, ischemic postconditioning by 5 cycles of 10 s ischemia/reperfusion each reduced infarct size. Inhibition of mitoKATP channel opening by 5-HD during the entire reperfusion or during the postconditioning maneuver only abolished the protection by ischemic postconditioning (105); protection by postconditioning was also lost by 5-HD administered following the postconditioning maneuver for the remaining 117 min of reperfusion (105). These data suggest that mitoKATP channel activation persists during reperfusion. The loss of protection after mitoKATP channel inhibition by 5-HD was confirmed in isolated guinea pig hearts (39) and in rabbit and dog hearts in vivo (94, 151).
The mitoKATP channel opener pinacidil (10 μM) for the first hour of reperfusion reduced cell death after 1 h ischemia and 3 h reperfusion in embryonic chick cardiomyocytes (140). Intermittent mitoKATP channel opening in isolated rat hearts with 5 cycles 10 s oxygenated buffer and 10 s oxygenated buffer supplemented with 30 μM diazoxide at reperfusion reduced infarct size following 30 min ischemia and 2 h reperfusion (105).
The calcium sensitizer levosimendan impacts on both sarcolemmal (154) and mitochondrial (75) KATP channels. In isolated guinea pig hearts, pharmacological postconditioning with 3 cycles of 30 s perfusion with 0.1 μM levosimendan reduced infarct size (39); addition of 5-HD during the postconditioning maneuver abolished the protection by levosimendan. Protection by levosimendan postconditioning was confirmed in rat hearts in vivo, where an intravenous bolus of 24 μg/kg levosimendan 5 min before reperfusion reduced infarct size reduction following 30 min ischemia and 30 min reperfusion (59). Again, the beneficial effect of levosimendan was abolished by 5-HD. These data support a role for opening of mitoKATP channels in pharmacological postconditioning by levosimendan.
Morphine at 3 μM for the first 10 min of reperfusion reduced infarct size in isolated rat hearts, and the protection was abrogated by simultaneous 5-HD (25). Also, protection by isoflurane (1 minimum alveolar concentration (MAC)) in rabbit hearts in vivo for 5 min starting 3 min before reperfusion was abrogated by 5-HD (76).
Taken together, ischemic pre- and postconditioning share mitoKATP channels as an important signaling element for cardioprotection.
The studies on the role of mitoKATP channels in postconditioning have been summarized in Table 1.
5-HD, 5-hydroxydecanoate; isch, ischemia; iv, intravenously; MAC, minimum alveolar concentration; rep, reperfusion; ↓, reduction; ↔, no effect.
Role of reactive oxygen species for the cardioprotection by ischemic and pharmacological postconditioning
Ischemic postconditioning by 3 cycles of 5 min hypoxia and reoxygenation reduced superoxide anion formation in neonatal rat cardiomyocytes subjected to 3 h hypoxia and 30 min reoxygenation, demonstrating that postconditioning indeed impacts on ROS formation (133). The reduced superoxide production was paralleled by decreased cytosolic and mitochondrial calcium concentrations (133). Postconditioning also decreased superoxide anion formation in neonatal rat cardiomyocytes after longer periods (3 and 6 h, respectively) of hypoxia and reoxygenation (134), and in rat hearts undergoing 30 min ischemia and 3 h reperfusion in vivo (71).
Isolated rat hearts were subjected to 30 min ischemia and 2 h reperfusion, and postconditioning by 5 cycles of 10 s ischemia and reperfusion each significantly reduced infarct size. Whereas the protection by ischemic postconditioning was lost in hearts perfused with NAC (N-acetylcysteine, 4 mM) for the entire 2 h reperfusion period, infarct size was still reduced when the perfusion with NAC was initiated after the first 3 min of reperfusion, demonstrating an essential role of ROS formation during
Reperfusion of isolated rabbit hearts with acidic buffer for the first 2 min of reperfusion significantly reduces infarct size. This cardioprotection is blocked by MPG applied for 20 min starting 5 min before reperfusion (28), suggesting the involvement of ROS signaling.
In mice, pharmacological postconditioning by either 1.4 % isoflurane or 10 mg/kg of the delta-opioid receptor agonist SNC-121 was also abolished by MPG when applied 10 min before, but not 10 min after reperfusion (138). NAC for the first 15 min of reperfusion also abolished postconditioning by sevoflurane in isolated rat hearts (152). These data support a central role for ROS signaling during early reperfusion in the protection by ischemic postconditioning.
ROS signaling is downstream of mitoKATP channel opening, as evidenced from isolated rat hearts subjected to ischemia/reperfusion with an intermittent infusion of diazoxide or diazoxide + MPG at the onset of reperfusion, since MPG attenuated diazoxide-induced protection.
While ROS scavenging attenuates the infarct size reduction by postconditioning, increasing ROS formation at the onset of reperfusion does not confer protection. Increasing ROS by reperfusion with purine/xanthine oxidase for the first 3 min or with 5 cycles of 10 s oxygenated buffer and 10 s oxygenated buffer + purine/xanthine oxidase failed to reduce infarct size (105).
In summary, whereas excessive ROS formation during reperfusion enhances cell death, ROS signaling during
Isch, ischemia; MPG, mercaptopropionylglycine; NAC, N-acetylcysteine; rep, reperfusion; SNC-121, 4-[(aR)-a-((2S,5R)-4-propyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide; SOD, superoxide dismutase; ↓, decrease; ↔, no effect.
Ischemic and pharmacological inhibition of MPTP opening at reperfusion
In ischemic preconditioning, the program of activated mediator kinases converges on glycogen synthase kinase-3β (GSK-3β) which, when phosphorylated, inhibits MPTP opening (68). In ischemic postconditioning, the obligatory role of GSK-3β for protection is still controversial and possibly species specific (46, 55, 95, 129). Despite the unresolved molecular identity of the MPTP, mitochondrial permeability transition can be modified pharmacologically [e.g., by cyclosporine A (CsA), which delays MPTP opening by preventing binding of cyclophilin D to the inner mitochondrial membrane, or by atractyloside, which opens the MPTP via inhibition of adenine nucleotide transport]. Both agents have been used to study the contribution of the MPTP to protection by ischemic or pharmacological postconditioning in isolated mitochondria, isolated cardiomyocytes, mammalian hearts in vitro and in vivo, notably including patients with acute myocardial infarction.
Calcium retention capacity (i.e., the amount of calcium uptake into the mitochondrial matrix in response to pulses of exogenous calcium before MPTP opens) has been determined in isolated mitochondria from hearts following different durations of ischemia and reperfusion without and with postconditioning maneuvers as a measure of their MPTP function (57). In mitochondria isolated from rabbit hearts undergoing 30 min ischemia and 1 h reperfusion with a postconditioning maneuver of 4 cycles of 1 min ischemia and reperfusion each calcium retention capacity was increased (6). Similar results were obtained when the reperfusion period was shortened to 10 min (98) or prolonged to 4 h (7). CsA (5 mg/kg) 1 min before reperfusion enhanced calcium retention capacity of rabbit mitochondria following 30 min ischemia and 10 min reperfusion (98). The nonimmunosuppressive CsA derivative NIM811 5 mg/kg 1 min before reperfusion also increased calcium retention capacity of isolated rabbit mitochondria (7). Still at 24 h reperfusion following 1 h ischemia in mouse hearts in vivo, isolated mitochondria had increased calcium retention capacity when hearts were postconditioning by 3 cycles of 1 min ischemia and reperfusion each (46); again, calcium retention capacity was also increased by CsA 5 min before reperfusion.
Pharmacological postconditioning with different—chemically unrelated—substances also increases mitochondrial calcium retention capacity. In isolated mouse hearts undergoing 30 min ischemia and 30 min reperfusion and anaesthetic postconditioning by 1 MAC isoflurane from 5 min before to 3 min after reperfusion, mitochondrial calcium retention capacity was increased (44). Likewise, postconditioning by 0.3 mg/kg morphine for 3 min starting 5 min before reperfusion in rat hearts subjected to 35 min ischemia and 10 min or 2 h reperfusion increased mitochondrial calcium retention capacity (96).
Postconditioning by activation of adenosine receptors reduced infarct size (66, 72), and the adenosine A1/A2 receptor agonist NECA (5′-(N-ethylcarboxamido) adenosine) reduced swelling of mitochondria isolated from rat hearts undergoing 30 min ischemia and 10 min reperfusion in vitro (148), suggesting delayed/reduced MPTP opening.
Resveratrol, a polyphenole present in grapes and red wines, reduced infarct size of isolated rat hearts following 30 min ischemia and 2 h reperfusion when administered for 35 min starting 5 min before reperfusion, associated with reduced mitochondrial swelling (149). The phytoestrogen genistein administered 5 min before reperfusion reduced infarct size of rabbit hearts following 20 min ischemia and 4 or 72 h reperfusion, respectively; the protection by genistein was associated with reduced calcium-induced mitochondrial swelling, suggesting that genistein reduces MPTP opening (136).
In H9C2 cells undergoing 8 h hypoxia and 3 h reoxygenation, postconditiong by 3 cycles of 5 min hypoxia and reoxygenation each preserved mitochondrial membrane potential, as did CsA at reperfusion, suggesting delayed MPTP opening (94).
The protection by ischemic postconditioning was abrogated in rat hearts in vivo, which were treated with 5 mg/kg of the MPTP opener atractyloside 5 min before the postconditioning maneuver (64). Also, the infarct size reduction by 1.5 MAC isoflurane for the first 15 min of reperfusion was abolished, when isolated rat hearts simultaneously received 10 μM atractyloside (41). The infarct size reduction by 1 μM morphine, when given for 20 min starting 5 min before reperfusion to isolated rat hearts, was also abolished by 60 μM atractyloside (64). Atractyloside also attenuated infarct size reduction by postconditioning with the adenosine A3 receptor agonist IB-MECA (N6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide) (103) and with bradykinin (104) in isolated rat hearts.
NIM811 (5 mg/kg, 1 min before reperfusion) reduced infarct size in rabbit hearts in vivo undergoing 30 min ischemia and 4 h reperfusion (7). When the reperfusion period was extended to 24 h, CsA (10 mg/kg, 5 min before reperfusion) still reduced infarct size in mouse hearts in vivo (46). In rabbit hearts in vivo, infarct size reduction by CsA for 2 min starting 5 min before reperfusion was dependent on dose, whereas 5 mg/kg CsA did not reduce infarct size: significant protection was achieved by 10 mg/kg CsA (76). Inhibition of MPTP opening by CsA was not only cardioprotective in small animals, but also reduced infarct size in pig hearts in vivo equally well as ischemic postconditioning (Fig. 3) (128).

In patients with acute myocardial infarction undergoing percutaneous coronary interventional reperfusion, postconditioning by 4 cycles of 1 min inflation and 1 min deflation of the angioplasty balloon reduced infarct size (130). The protective effect of postconditioning improved contractile function up to 1 year after myocardial infarction (135). In a recent study, an intravenous bolus of 2.5 mg/kg CsA immediately before reperfusion also provided protection (113). Creatine-kinase release and the area of hyperenhancement on MRI imaging were reduced in CsA-treated patients, reflecting reduced infarct size and suggesting that inhibition of MPTP opening is also beneficial in patients with myocardial infarction.
Studies on the contribution of MPTP opening to postconditioning are summarized in Table 3.
CsA, cyclosporine A; IB-MECA, N6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide; isch, ischemia; iv, intravenously; NAC, N-acetylcysteine; NAD+, nicotineamide adenine dinucleotide; NECA, 5'-(N-ethylcarboxamido) adenosine; NIM811, N-methyl-4-isoleucine cyclosporin; MAC, minimum alveolar concentration; rep, reperfusion;,↑, increase; ↓, decrease; ↔, no effect.
In conclusion, reduced MPTP opening at reperfusion is achieved by ischemic and pharmacological postconditioning stimuli and associated with protection; the cause–effect relationship between reduced MPTP opening and protection is suggestive but not proven (57). Nevertheless, MPTP is certainly an attractive target to achieve protection, notably also in patients with acute myocardial infarction.
Mitochondrial function and morphology in response to ischemic and pharmacological postconditioning
In mitochondria isolated from rabbit hearts subjected to 30 min ischemia and 1 h reperfusion, neither ischemic postconditioning nor the MPTP inhibitor NIM811 affected basal state 4 or ADP-stimulated state 3 respiration, excluding uncoupling or inhibition of the respiratory chain as a mechanism of MPTP inhibition (6). Postconditioning did also not influence mitochondrial respiration, neither in SSM nor in IFM from rabbit hearts subjected to 30 min ischemia and 10 min reperfusion (98). In contrast, ADP-stimulated respiration was increased after pharmacological postconditioning with morphine in mitochondria isolated from rat hearts subjected to 35 min ischemia and 10 min or 2 h reperfusion whereas basal respiration was not affected (96). Ultrastructural alterations in mitochondria isolated from rat hearts undergoing 30 min ischemia and 2 h reperfusion were characterized by disruption of membranes and broken christae, and the mitochondrial damage was reduced by ischemic postconditioning (107).
Studies on the effect of postconditioning on mitochondrial integrity and respiration are summarized in Table 4.
CsA, cyclosporine A; IFM, interfibrillar mitochondria; isch, ischemia; iv, intravenously; NIM811, N-methyl-4-isoleucine cyclosporin; rep, reperfusion; SSM, subsarcolemmal mitochondria; ↑, increase; ↔, no effect.
Taken together, ischemic and pharmacological postconditioning clearly prevent the loss of mitochondrial membrane potential, inhibit MPTP opening, and preserve mitochondrial morphology; it is less established whether postconditioning also results in alterations of mitochondrial oxygen consumption. Methodologically, the measurement of mitochondrial respiration is dependent on the quality of the isolation procedure, since the normalization of oxygen consumption to total protein content does not account for different amounts of functionally active or inactive mitochondria in the respective preparations.
Reduction of apoptosis by ischemic and pharmacological postconditioning
Cardiomyoctye death after ischemia/reperfusion is primarily necrotic, but apoptosis also contributes. Anti-apoptotic substances when given during early reperfusion reduce infarct size (87, 156), and ischemic postconditioning is associated with reduced apoptosis (107, 134, 143).
Pro-apoptotic signaling pathways also involve mitochondria, for example, the release of cytochrome c or AIF (apoptosis-inducing factor) from the mitochondrial intermembrane space into the cytosol. In neonatal rat cardiomyocytes subjected to 3 h hypoxia and reoxygenation each, ischemic postconditioning reduces the release of cytochrome c from the mitochondrial intermembrane space to the cytosol (83).
Cytochrome c release is also decreased by ischemic postconditioning in isolated rat hearts (107) and by morphine postconditioning in rabbit hearts in vivo (146). The release of AIF from the mitochondria to the cytosol is also reduced by ischemic postconditioning in rat hearts in vivo (143).
Role of mitochondria in remote ischemic postconditioning
The heart is not only protected by short cycles of ischemia and reperfusion at the onset of reperfusion in the heart itself, but also by ischemia and reperfusion in remote organs or tissues, a phenomenon termed remote ischemic postconditioning (70). Remote ischemic postconditioning elicits cardioprotection originating from the kidney (85), skeletal muscle (82), or brain (48). The signal transduction of remote ischemic postconditioning may involve the mitoKATP channel (124). Reduced irreversible injury involves mitochondrial anti-apoptotic pathways; decreased apoptosis by renal postconditioning in rabbit hearts is associated with increased expression of the anti-apoptotic protein bcl-2 (69).
Potential Loss of Protection in Diseased and/or Aged Myocardium
Most studies analyzing ischemic or pharmacological postconditioning have been performed in young and healthy animals. However, myocardial infarction is a disease of older age and affects patients with co-morbidities and under pharmacological treatment that may result in loss of cardioprotection (42).
With aging, the myocardial tolerance for ischemia/reperfusion injury is reduced (1) and the expression of proteins important for cardioprotection is altered (19, 20). Aging also interferes with the protection by ischemic postconditioning. In fact, postconditioning by either 3 or 6 cycles of 10 s ischemia and reperfusion each failed to reduce infarct size in 20–24-month-old mouse hearts (116). In middle-aged mouse hearts (>13 months), postconditioning by 5 cycles of 5 s ischemia and reperfusion each reduced infarct size, whereas 3 cycles of 10 s ischemia and reperfusion did not (15). Four cycles of 10 s ischemia and reperfusion were sufficient to protect aged (16–18 months) rat hearts (153). Apparently, stimulus strength is critical for ischemic postconditioning in the aged heart.
The loss of protection in aged hearts could relate to impaired mitochondrial function. ROS are considered as causal for the process of aging (51) their amount increases with aging in the cardiovascular system (67, 93, 147), and they may favor MPTP opening. Calcium retention capacity was decreased in aged mitochondria of different origin (brain, liver, and lymphocytes) (33). Calcium retention capacity was decreased in mitochondria isolated from 24 months as compared to 6-month-old rat hearts (63). We also studied MPTP opening in aged mitochondria and quantified the calcium-induced loss of mitochondrial membrane potential (rhodamine 123 fluorescence); however, mitochondria from young (4 months) and aged (18 months) mice tolerated similar amounts of calcium until loss of mitochondrial membrane potential and MPTP opening occurred (Fig. 4).
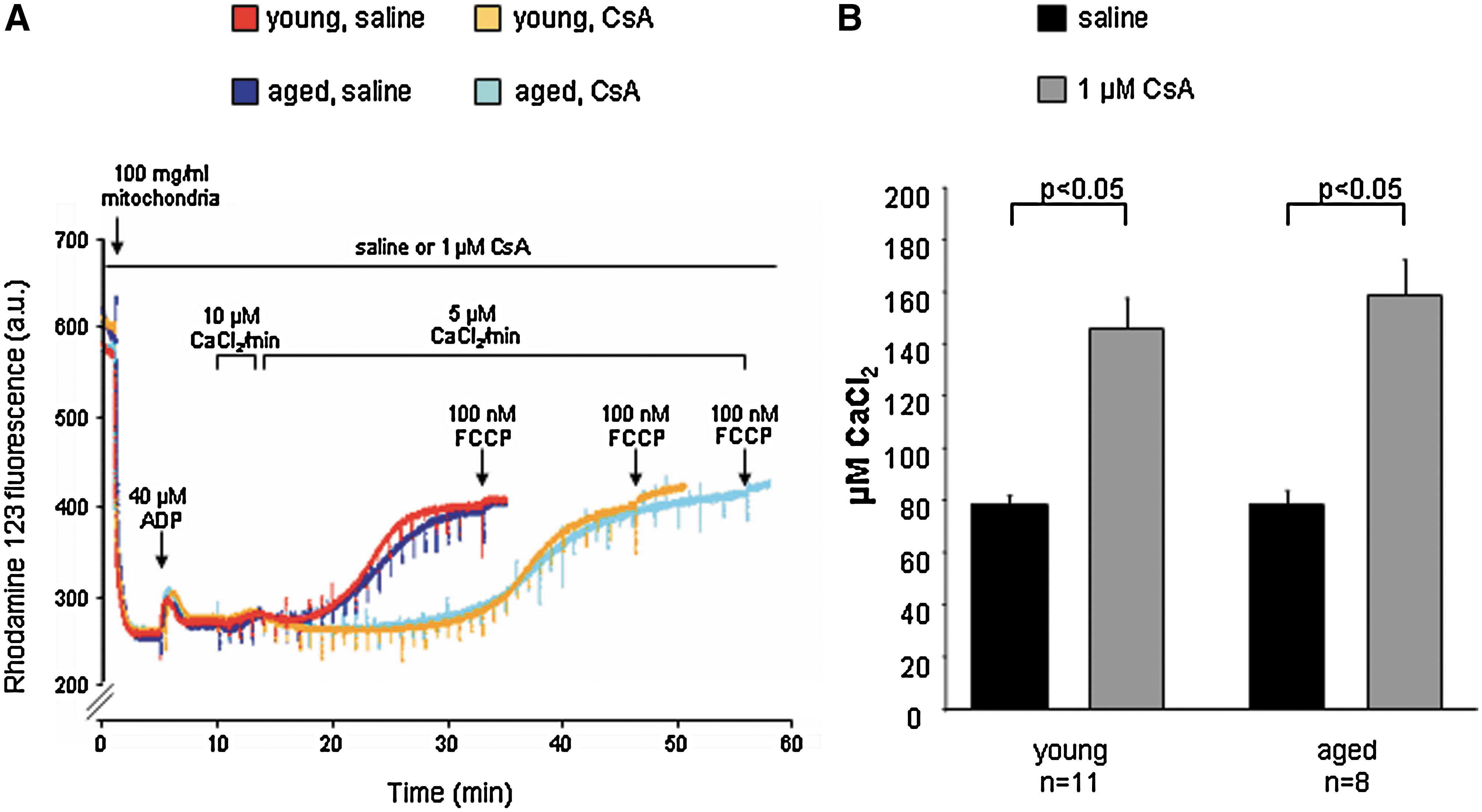
In isolated hearts from spontaneously hypertensive rats, ischemic postconditioning elicited only a slight but nonsignificant infarct size reduction, whereas infarct size was significantly reduced in normotensive rat hearts (109). Likewise, isoflurane reduced infarct size in rabbit hearts undergoing 40 min myocardial ischemia and 3 h reperfusion, but the protection was abrogated by hyperglycemia (117). In obese Zucker rats in vivo, the infarct size reduction by postconditioning with 70% helium for the first 15 min of reperfusion (61) or with sevoflurane (60) were abolished. CsA 5 min before reperfusion did not restore protection, suggesting that cyclophilin D-mediated MPTP inhibition is impaired in these prediabetic obese Zucker rats.
Further studies are needed to demonstrate the specific contribution of mitochondria to loss of protection by ischemic and pharmacological postconditioning in aged and/or diseased hearts.
Conclusion
Mitochondria and ROS are essential signaling elements of cardioprotection, including protection by postconditioning. Mitochondria serve a signal amplifier function insofar activation of mitoKATP channels induces the formation of small amounts of ROS. Mitochondria also serve as end-effectors of protection since inhibition of MPTP opening and of release of apoptotic signals into the cytosol contributes to cardiomyocyte survival. Finally, mitochondria are targets of functional and structural damage by ischemia and reperfusion, notably by excessive amounts of ROS. Clearly, mitochondria and ROS are attractive mechanistic targets for pharmacological cardioprotection. Indeed, a first proof-of-concept study demonstrated a beneficial effect of the MPTP inhibitor CsA during early reperfusion in patients with acute myocardial infarction. However, experimental studies suggest that the cardioprotection by postconditioning may be lost in aged and/or diseased myocardium.
Footnotes
Acknowledgments
KB, GH, and RS were recipients of grants from the Deutsche Forschungsgemeinschaft (Bo 2955/1-1, He 1320/18-1, Schu 843/7-2, respectively).