
Abstract
Ischemic postconditioning (PoCo) has been proven to be a feasible approach to attenuate reperfusion injury and enhance myocardial salvage in patients with acute myocardial infarction, but its mechanisms have not been completely elucidated yet. Recent studies demonstrate that PoCo may delay the recovery of intracellular pH during initial reperfusion, and that its ability to limit infarct size critically depends on this effect. Prolongation of postischemic intracellular acidosis inhibits hypercontracture, mitochondrial permeability transition, calpain-mediated proteolysis, and gap junction-mediated spread of injury during the first minutes of reflow. This role of prolonged acidosis does not exclude the participation of other pathways in PoCo-induced cardioprotection. On the contrary, it may allow these pathways to act by preventing immediate reperfusion-induced cell death. Moreover, the existence of interactions between intracellular acidosis and endogenous protection signaling cannot be excluded and needs to be investigated. The role of prolonged acidosis in PoCo cardioprotection has important implications in the design of optimal PoCo protocols and in the translation of cardioprotective strategies to patients with on-going myocardial infarction receiving coronary reperfusion. Antioxid. Redox Signal. 14, 923–939.
Reperfusion Injury and Ischemic Postconditioning
Experimental and clinical studies (30, 39, 117) demonstrate that the duration of ischemia is the main determinant of the outcome of reperfusion in terms of myocardial salvage. The time window for reperfusion consistently resulting in significant myocardial salvage varies between 30 and 90 min, depending on species and conditions. Unfortunately, most patients receive these treatments after this window has passed, and when reperfusion has little effect on myocardial cell death (30). Even patients receiving reperfusion very early usually end up with significant areas of myocardial necrosis (149). Since reducing the time interval between symptoms onset and coronary reperfusion is very difficult, strategies aimed to amplify reperfusion-induced myocardial salvage are strongly needed. The feasibility of this approach is based on the concept of lethal reperfusion injury (113). In contrast to the initially prevalent view of cell death secondary to transient ischemia as a mere consequence of cumulative cell injury caused by energy deprivation during the ischemic period, the new paradigm of reperfusion injury predicts that part of myocardial cell death induced by transient ischemia occurs at the time of reperfusion, and can be thus prevented by interventions applied at the time of reflow (60), as illustrated in Figure 1.
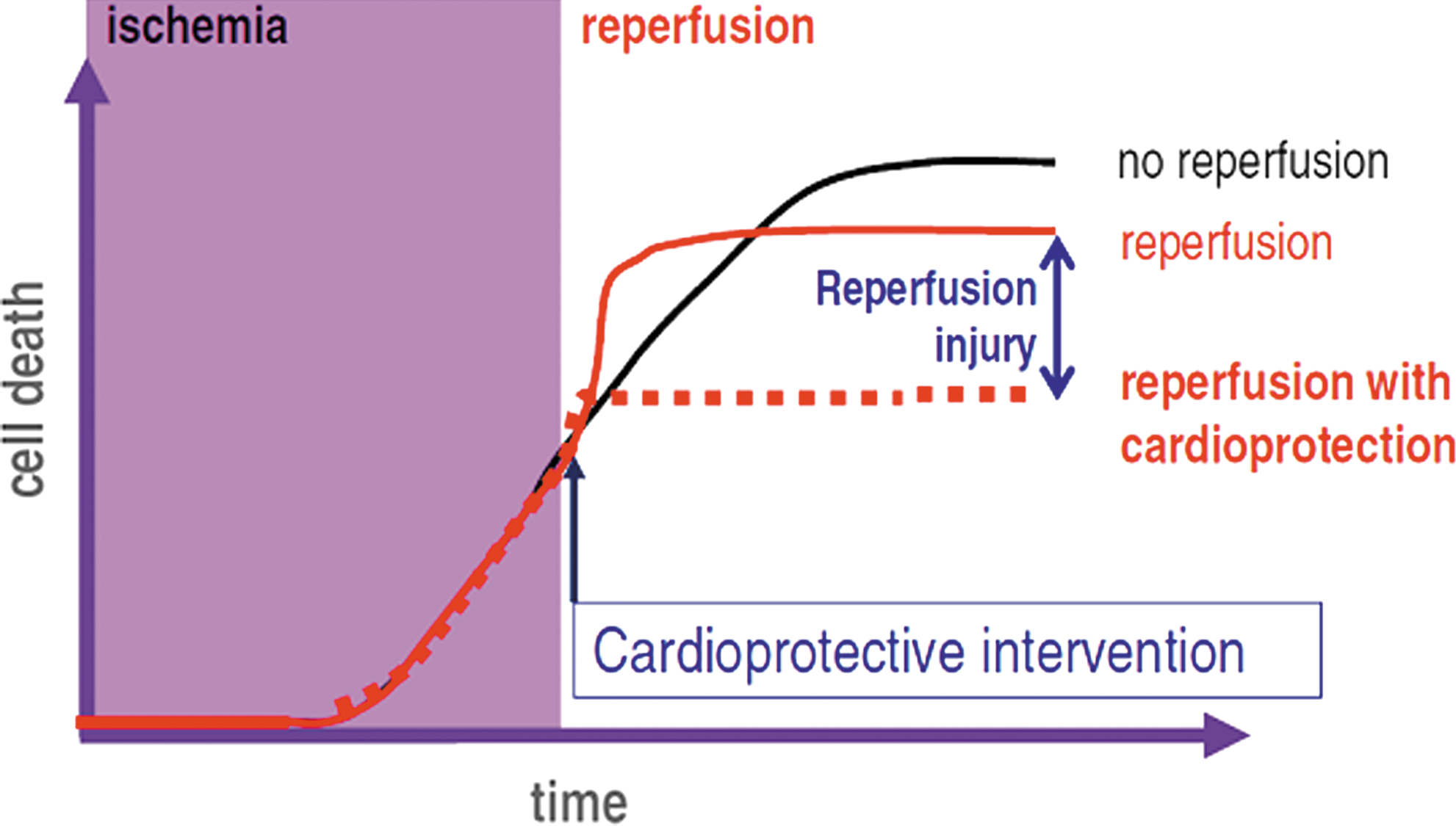
The existence of lethal reperfusion injury, defined as cell death preventable by treatments applied during reperfusion, is proven by extensive experimental data, but its demonstration has failed to result in the development of treatments applicable to patients (23). Recently, however, several strategies against reperfusion injury have shown a clear-cut infarct sparing effects in patients with acute myocardial infarction, including ischemic postconditioning (PoCo) (145), administration of cyclosporine (110) or atrial natriuretic peptide (77), and remote conditioning (13). Of these, the only one applied so far in different studies by different groups, although in a small number of patients, is PoCo (97). In fact, the translation of PoCo to patients has played a leading role in the acceptance of the paradigm of reperfusion injury in the arena of clinical cardiology (37).
PoCo was relatively recently described by the Vinten–Johansen group as the infarct size-limiting effect of brief episodes of ischemia applied during the initial minutes of coronary reperfusion (164). The effectiveness of PoCo limiting infarct size, as compared to other interventions and in particular to ischemic preconditioning, its dependence of conditions and protocols, and its mechanisms have been extensively investigated but have not been completely elucidated (56, 102). The present article reviews the hypothesis that the effects of PoCo on intracellular pH (pHi) during the initial minutes of reperfusion are a key element in the mechanism of its protective effect against myocardial necrosis.
pH as Modulator of Reperfusion-Induced Cell Death
Emerging data show that ischemic PoCo activates multiple and interacting active and passive components that protect against myocardial injury. The results of many laboratories have provided support to the notion that the protective effects of PoCo are mainly due to endogenous cardioprotective mechanisms activated with the intermittent reperfusion. However, PoCo cardioprotection is also due, at least in part, to the prevention of an abrupt reperfusion. In this regard, it has been proposed that the reduction of Starling forces during the PoCo maneuvers may play a role in determining the protective effects (101). The importance of passive components is clearly manifested by the strong dependency of the magnitude of protection on the PoCo protocol. The time interval between the onset of reperfusion and the PoCo maneuver, the duration of each cycle of ischemia/reperfusion, and the total duration of the protocol are determinant for the successful protection against infarction. Recent studies have compiled from the literature models and algorithms of ischemic PoCo in an attempt to yield clues to define the conditions for a protocol to be optimally effective (56, 102, 137). These extensive analyses of available data show that the conditions vary with the species and the experimental model. Duration of cycles of ischemia/reperfusion must be shorter in smaller animals (from 5 to 15 sec in mice and rats) (59, 68, 70) and longer in larger species (from 30 sec in dogs to 60–180 seconds in humans) (82, 145, 164). Even for a given species, the cycles must be shorter in models perfused with crystalloid buffer than in in situ hearts, probably due to the higher coronary flow in perfused hearts (156, 157). Globally, these results suggest that a gradual washout of metabolites is determinant for the cardioprotective effects of PoCo.
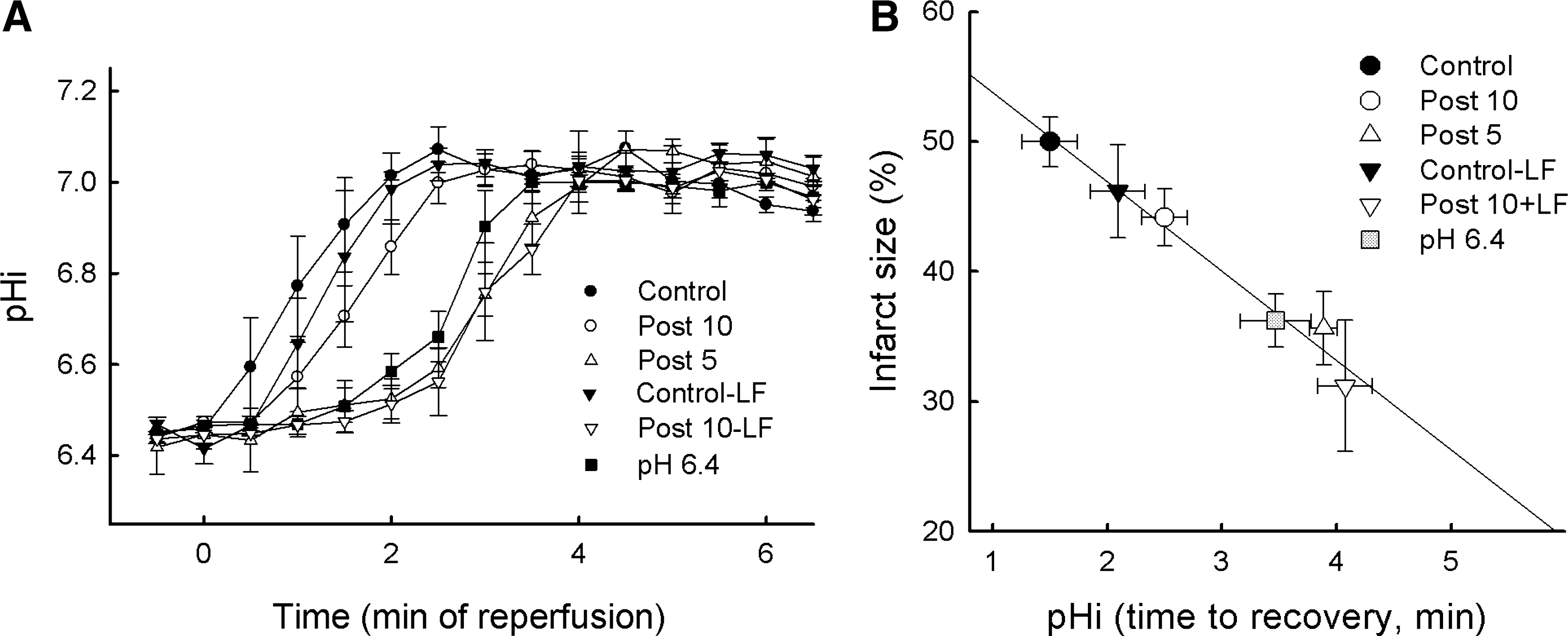
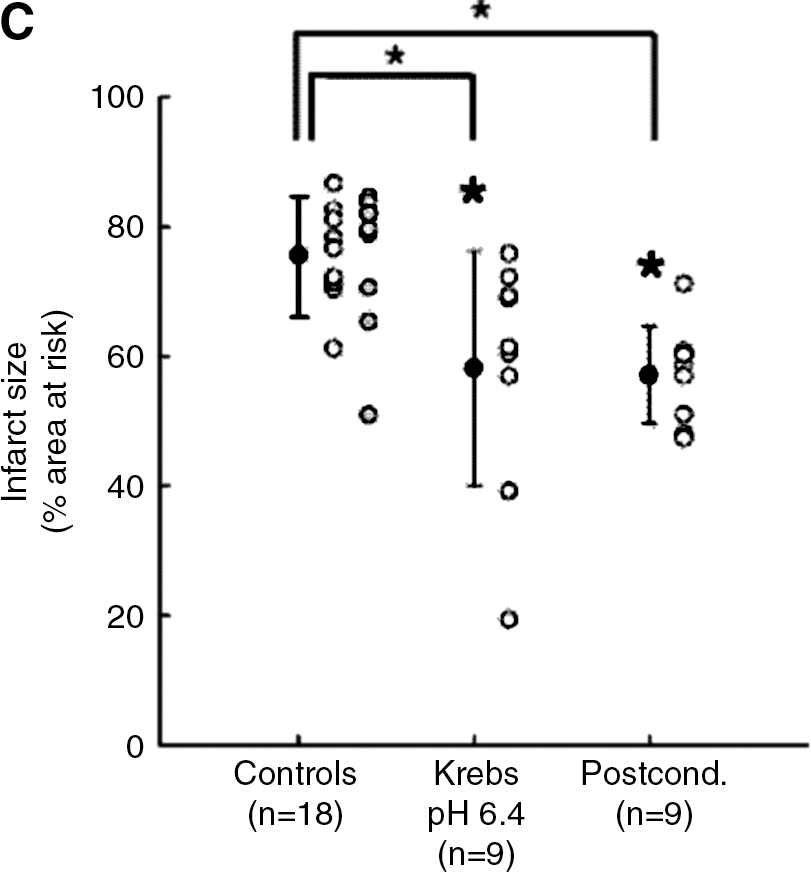
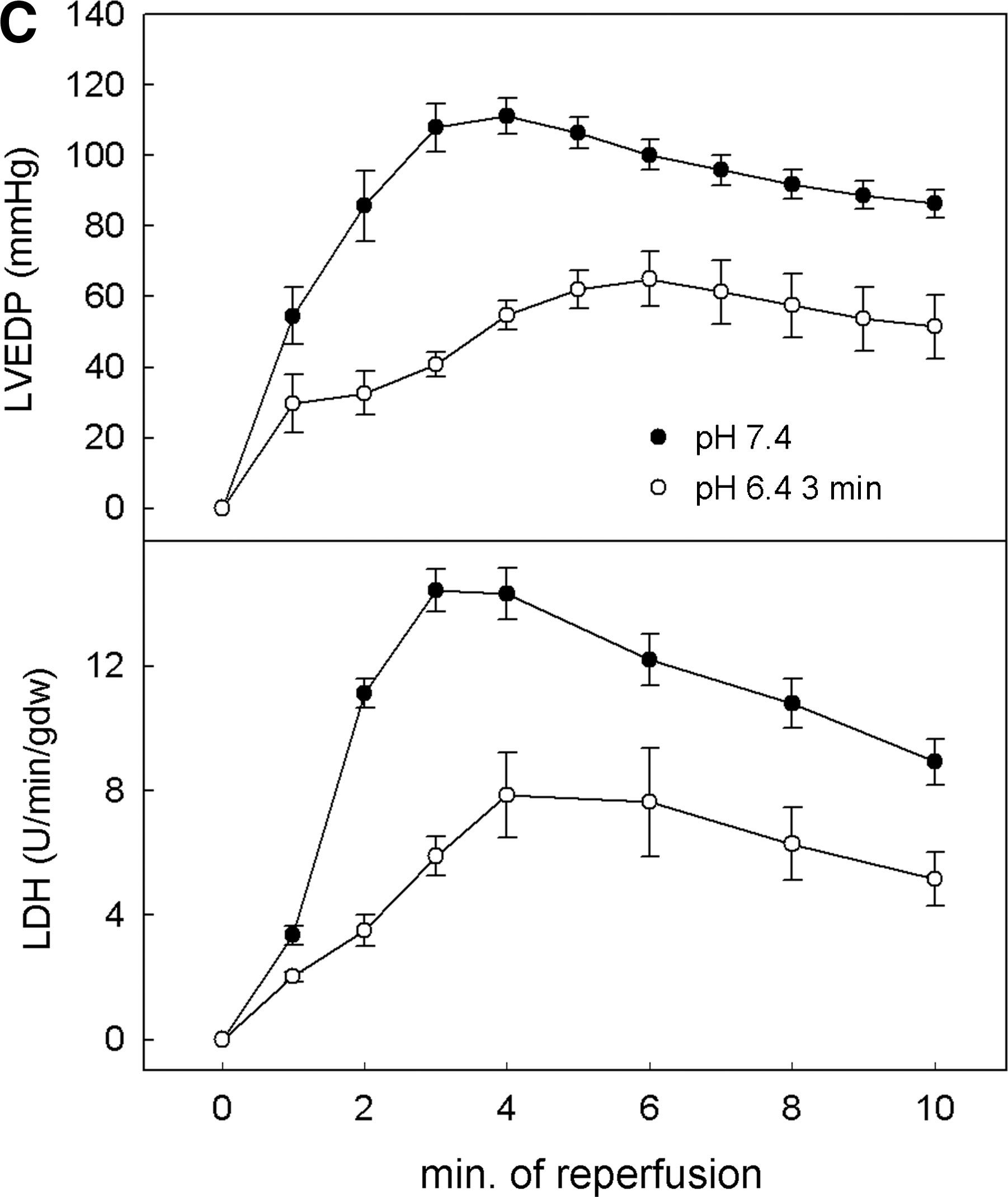
One recent hypothesis derived from this concept and from a previous study showing in dogs subjected to 15 min of coronary occlusion that staged reperfusion prolongs acidosis and attenuates stunning (53) is that PoCo protects because it slows the normalization of correction of intracellular acidosis occurring during the initial minutes of reperfusion. Initial experimental evidence for this hypothesis was provided by Cohen et al. (18). They found that using acidic buffer for the first 2 minutes of reperfusion was as protective as a PoCo protocol in isolated rabbit hearts. If either the duration of acidosis or the PoCo protocol were reduced to only 1 min, the protection was lost. By modifying the pH during initial reperfusion and using 31P-NMR in perfused rat hearts, we demonstrated that the pH value and duration of acidic perfusion that afforded optimal protection are 6.4 and 3 min, respectively (58). Prolongation of acidic infusion beyond 3 min failed to further delay pHi recovery, probably due to the activity of Na+/H+-exchanger (NHE) and Na+/bicarbonate-cotransporter (NBC), and was associated to loss of benefit (Fig. 2). In a more recent NMR spectroscopy study in rat hearts, we showed that PoCo delays pHi recovery for up to 3 min, depending on the protocol used (59). In fact, this went further to demonstrate that only PoCo protocols able to induce a significant delay in pHi recovery were able to afford cardioprotection. Moreover, a close correlation between the delay in pHi recovery and the magnitude of myocardial salvage was documented (Fig. 3) (59). These studies give us insight into the determinants of success of PoCo protocols. Independently of the species or experimental model, the cycles should not be long enough to allow pH normalization during a reperfusion cycle, and the overall duration of the PoCo protocol should maintain intracellular acidosis for at least the first 3 minutes of reperfusion.

Intracellular pH during Ischemia/Reperfusion
An excellent review outlining the mechanisms that regulate pHi in cardiomyocytes under physiological conditions has been recently published by Vaughan–Jones et al. (151). The pHi regulatory response of the myocardium is an important factor contributing to myocardial injury associated with ischemia/reperfusion. Myocardial ischemia causes a progressive fall in intra- and extracellular pH due to the increased production caused by an initial upregulation of anaerobic metabolism (15) and reduced washout of protons. Reperfusion removes extracellular protons and corrects intracellular acidosis, mainly through the lactate-H+ cotransporter (48), the activity of NHE and NBC (151). The low pHi overdrives these latter transporters, resulting in intracellular Na+ accumulation (14, 49). Reduction of transarcolemmal Na+ gradient favors the reverse mode activity of the sarcolemmal Na+/Ca2+ exchanger (NCE) and causes cytosolic Ca2+ overload (12). Studies in isolated cardiomyocytes identified the rapid correction of intracellular acidosis as an important determinant of reperfusion injury. Correction of acidosis has two main adverse effects: first, it favors Na+ and Ca2+ overload (112); second, it allows the activation of systems that remain otherwise inhibited during low pH (see below). The correlation between the time course of correction of the intracellular Ca2+ concentration and pHi during the first minutes of reperfusion decides between cell death (recovery of pH occurs first) and survival (recovery of Ca2+ occurs first) (37). The rapid normalization of the intracellular Ca2+ concentration is critically dependent on the rapid recovery of Na+ which depends on the ability of the cell to resume the activity of the sarcolemmal Na+/K+-ATPase upon re-energization (57).
During PoCo, controlled washout of extracellular metabolites prevents the rapid formation of a transarcolemmal gradient of H+ and the reactivation of NHE leading to a gradual restoration of pHi. An additional mechanism that could contribute to prevent the fast pHi normalization is related to the proposed activation of the cGMP/PKG pathway during postconditioning (101, 157). Several studies have shown that PKG-dependent phosphorylation of NHE modulates negatively the activity of this transporter affecting pHi recovery (66, 131). Conversely, it has been described that acidosis increases the activity of Ca2+/calmodulin-dependent kinase II (CaMKII) (81, 93) and more recently that CaMKII contributes to pHi recovery from acidosis via activation of NHE (152). Whether modulation of NHE activity by phosphorylation contributes to the delayed normalization of pHi during reperfusion remains to be demonstrated.
Effects of Reperfusion Injury Modulated by pH
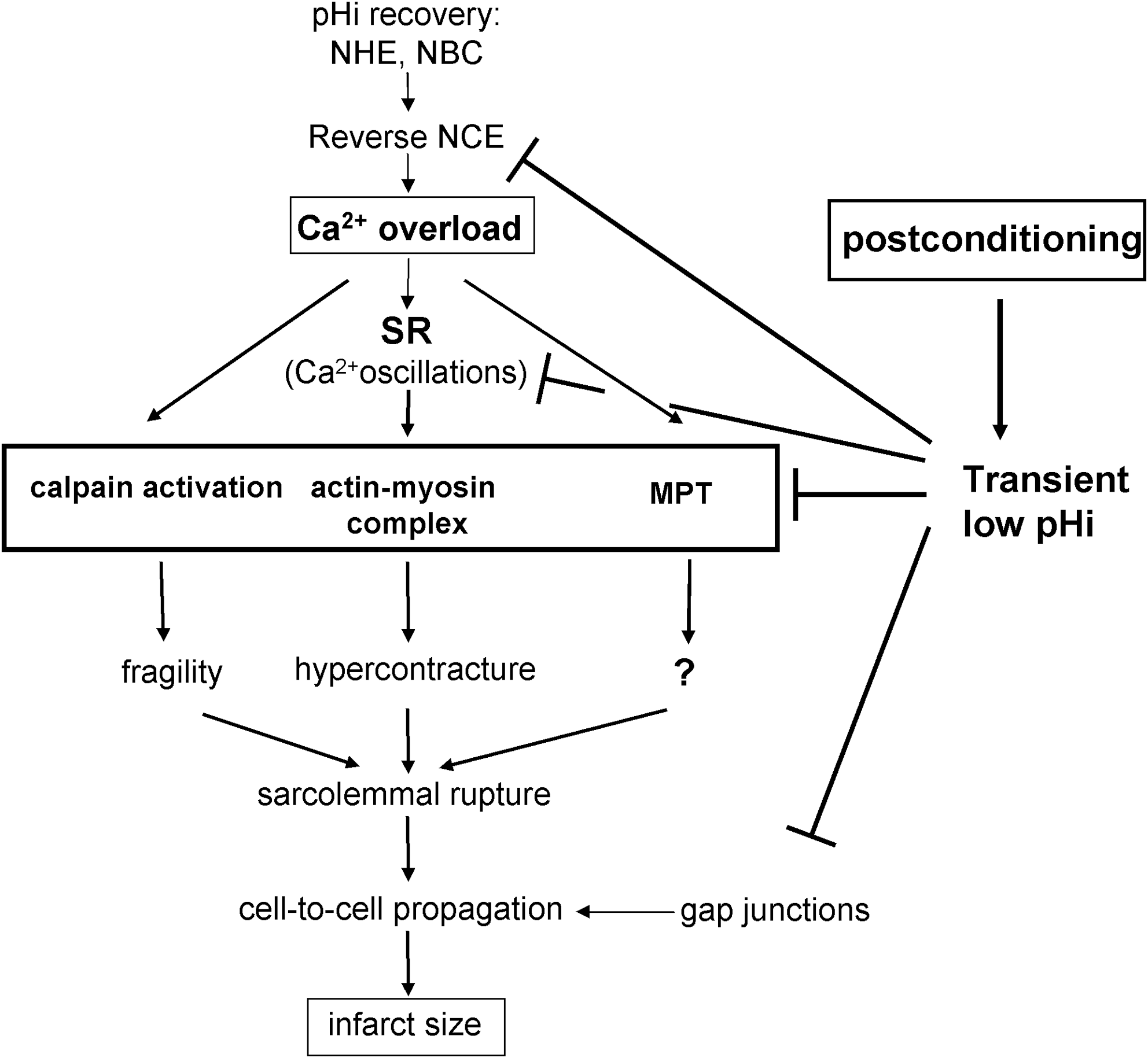
The protective effect of delayed pHi recovery could be predicted in view of the effects that acidosis has on many molecular mechanisms implicated in reperfusion injury (Fig. 4). Prolongation of pHi at reperfusion may be protective by reducing Ca2+ overload and myofibrillar contractility that causes the Ca2+-dependent hypercontracture in reperfused cardiomyocytes (71, 115, 130). Low pHi also inhibits other mechanisms involved in reperfusion injury, such as mitochondrial permeability transition (MPT) pore opening (47) and activation of calpains (62, 63). Finally, acidosis maintains gap junctions in a closed state, impairing the propagation of cell death (29). However, the relative importance of each mechanism to the cardioprotective effect of prolongation of acidosis has not been established.
Hypercontracture and Ca2+ homeostasis
The histological analysis of post reperfused hearts has shown that infarcts are almost exclusively composed by areas of contraction band necrosis generated by cardiomyocytes with the sarcolemma disrupted as consequence of the mechanical stress caused by extreme cell shortening (10, 88). This excessive activation of the contractile machinery, known as hypercontracture, is induced when cardiomyocytes recover ATP production in the presence of a high cytosolic Ca2+ concentration (150). Ca2+ overload is largely a consequence of its influx through a reverse mode of NCE during ischemia and the first minutes of reperfusion (64, 129). The reverse mode operation of NCE is caused by a reduction in the transarcolemmal Na+ concentration as a consequence of the Na+ entry coupled to the mechanisms of pHi correction and lasts until Na+/K+-ATPase activity normalizes Na+ concentration and membrane potential recovers. At reperfusion, recovery of mitochondrial energy production in reoxygenated myocytes induces oscillatory changes in the concentration of Ca2+ induced by cyclic uptake and release of Ca2+ into the sarcoplasmic reticulum. These fast Ca2+ transient cytosolic peaks are responsible of the uncontrolled activation of the contractile machinery that results in hypercontracture (111, 113).
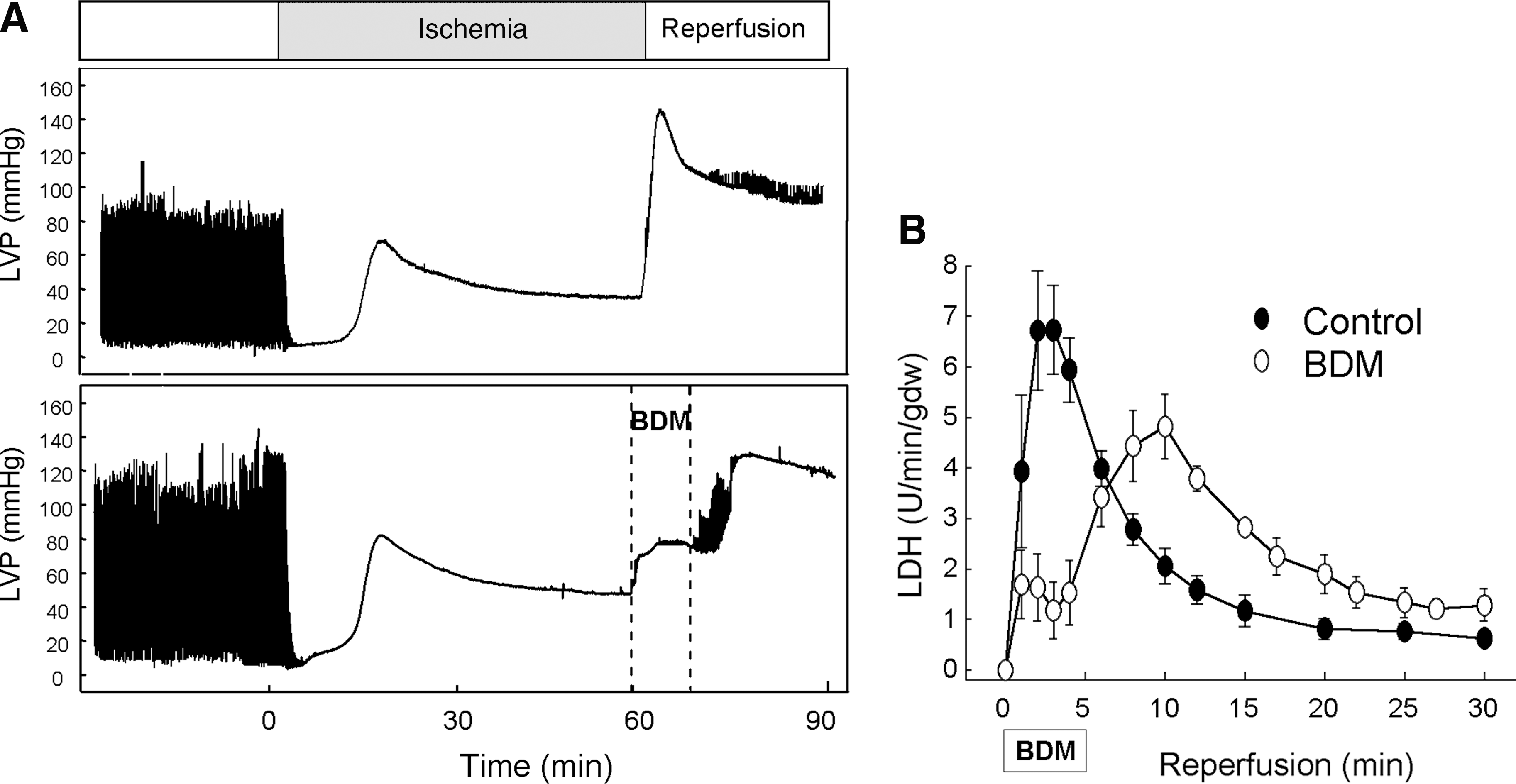
Prolongation of acidosis during reperfusion causes a pronounced attenuation in the development of hypercontracture (58, 71, 115, 130) (Fig. 5). This effect can be explained by different mechanisms. Acidosis inhibits myofibrillar machinery by reducing the sensitivity of myofibrils to Ca2+ (96) and, therefore, prevents the development of hypercontracture during the initial minutes of reperfusion when Ca2+ homeostasis has not been yet restored. This effect is similar to that observed in several studies with the use of 2,3-butanedione monoxime (BDM), a reversible inhibitor of actomyosin-ATPase, during initial reperfusion. BDM reduces cross-bridge force production and allows a complete blockade of contractile activity resulting in transient inhibition of enzyme release, and is rapidly followed by a rise in LVEDP and cell death upon its withdrawal when used for a short period of time (Fig. 5) (62). However, prolongation of contractile inhibition with BDM until cardiomyocytes recover Ca2+ control results in a pronounced infarct limitation in a variety of models (38, 142). The fact that the optimal protection achieved by delaying pHi recovery limits the prolongation of acidosis to the first 3 min of reperfusion (58), too short for contractile inhibition with BDM to be effective, indicates that additional mechanisms contribute to the cardioprotective effects of acidosis.
Acidosis may also modulate Ca2+ homeostasis during reperfusion and therefore the substrate for hypercontracture (Fig. 5D). By preventing the rapid washout of extracellular H+ upon reperfusion of ischemic myocardium and the formation of a transmembrane H+ gradient, acidosis attenuates the increase in intracellular Na+ concentration as a consequence of Na+ influx coupled to NHE and NBC and, in turn, the Ca2+ entry through reverse mode of NCE (151). In addition, several studies propose that acidosis could reduce Ca2+ overload by inhibiting NCE (58, 109) and the Ca2+ inward current (65), and the development of Ca2+ oscillations through its inhibitory effect on SERCA and RyR (27, 54). Supporting this effect on Ca2+ homeostasis, it has been demonstrated that brief prolongation of extracellular acidosis during the early phase of reoxygenation reduces intracellular Na+ and Ca2+ overload (100).
Mitochondrial permeability transition
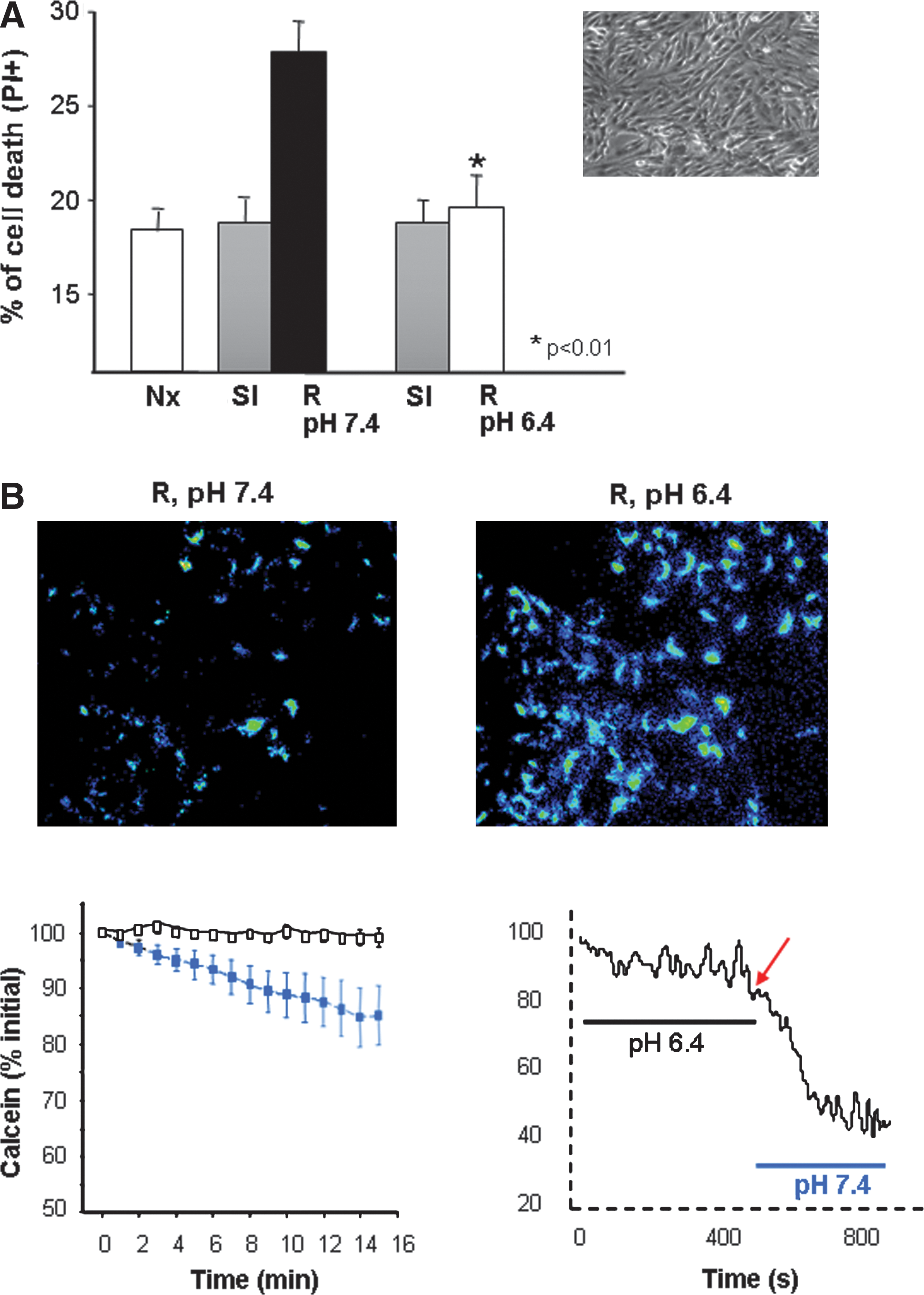
Mitochondrial permeability transition (MPT) is a well-established key end effector of cell death during myocardial reperfusion (8, 41). Low pH exerts a powerful inhibitory effect on MPT (46). Although the molecular mechanism responsible for this inhibitory effect has not been fully elucidated, it has been proposed that acidification reduces mitochondrial Ca2+ load (45) and that H+ may antagonize Ca2+ binding to the adenine nucleotide translocase (46). The rapid and profound fall in pHi that takes place during ischemia maintains MPT in its closed state, despite the concurrence of high Ca2+, low ATP concentration, oxidative stress, and accumulation of arachidonic acid and amphipatic molecules, conditions all that have been shown to act as MPT inducers (43). Indeed, correction of intracellular acidosis during reperfusion is necessary for MPT, as demonstrated by the close correlation between pH normalization and mitochondrial permeabilization observed in a number of studies involving several cell types (22, 42, 74, 116). The protective effect of acidic reoxygenation or reperfusion observed in different models could be therefore explained by its inhibitory effect on MPT. In HL1 cardiomyocytes, in which the role of hypercontracture in reperfusion injury is negligible due to the existence of a very rudimentary contractile apparatus, reperfusion after simulated ischemia (hypoxia and acidosis at pH 6.4) caused cell death within 20 min (propidium iodide labeling and LDH release) that was associated with the release of mitochondrial calcein, indicative of MPT (126) (Fig. 6). Preservation of extracellular acidosis during reperfusion prevented both mitochondrial calcein release and cell death that took place only upon pH normalization. These results, in a cell model in which contribution of hypercontracture and gap junction-mediated communication on sarcolemmal rupture is excluded, indicate that MPT inhibition is an important molecular end-effector of the protection afforded by intracellular acidosis during initial reperfusion, and strongly support the hypothesis that delayed normalization of pHi is a prominent mechanism of PoCo protection.
Intracellular acidosis may have other effects on mitochondrial function during ischemia-reperfusion injury, such as modification of ionic transport systems, substrate availability, and ATP synthesis, among others. A high H+ concentration in the mitochondrial intermembrane space might slow ATP hydrolysis under low energetic conditions, by the reversal of the F0-F1 ATPase, and delay progression of cell injury, increasing cell survival upon reperfusion by a mechanisms involving the preservation of the transmembrane electrochemical gradient (125). Low pH also reduces mitochondrial calcium uptake and favors calcium extrusion from mitochondrial matrix, due to the activation of mitochondrial NHE and subsequent NCE (24). High H+ concentration may activate uncoupling proteins (UCPs), inner mitochondrial membrane H+ carrier that uncouples ATP synthesis from oxygen consumption (25). Mild uncoupling secondary to activation of UCPs has been described to confer cardioprotection under several conditions, including myocardial reperfusion, likely by decreasing reactive oxygen species production (52). On the other hand, acidosis inhibits glycolysis and pyruvate production, ultimately resulting in a slower feeding of the respiratory complex chain. Interestingly, several lines of evidence indicate that transient complex I inhibition during reperfusion is cardioprotective, possibly by attenuating ROS production (4, 135). Nevertheless, experimental evidence supporting the involvement of these changes in PoCo protection has yet to be provided.
Calpains
Calpains constitute a wide family of nonlysosomal Ca2+-dependent thiol proteases with an important role in basic cellular functions, including apoptosis, proliferation, cell migration, and protein homeostasis (32, 139). The binding to its specific endogenous inhibitor calpastatin in a substrate competitive manner and the cellular control of Ca2+ homeostasis regulate calpain activation (40). However, under pathologic conditions in which Ca2+ homeostasis is lost, as occurs during ischemia–reperfusion, the control of the calpain system is altered, causing inappropriate activation (160).
Calpain proteolysis of known substrates and the use of calpain inhibitors in different models of ischemia–reperfusion have been consistently demonstrated by several groups and in different experimental models that calpain activation occurs prior to membrane damage and plays an important role in reperfusion-induced contractile dysfunction and cell death due to the proteolysis of a wide variety of proteins (17, 62, 63). However, the regulation and time-course of calpain activation secondary to transient ischemia was not defined until recently (51). In a study performed by our laboratory in perfused rat hearts, intracellular acidosis inhibited calpain activation during ischemia despite Ca2+ overload, while pHi normalization allowed its activation upon reperfusion. Furthermore, a delay in pHi recovery during reperfusion by either acidic perfusion or by PoCo protocol attenuated calpain activation (59) while the pharmacological inhibition of calpain exclusively during the first min of reperfusion reduced infarct size in an in vivo and in situ models (51, 91). Taking them together, these studies confirm previous in vitro results showing that calpain activity is highly dependent on pH (163) and support the contribution of its inhibition to the cardioprotective effects of PoCo.
Several mechanisms have been proposed by which calpains may contribute to reperfusion injury (Fig. 7). A first mechanism is related to sarcolemmal cell fragility. During reperfusion calpains hydrolyse proteins from the sarcolemma and the cytoskeleton including α-fodrin and ankyrin. Alpha-fodrin forms the backbone of the membrane cytoskeleton (11) and its degradation correlates with increased fragility of the membrane, reducing the tolerance of the sarcolemma to the mechanical stress induced by acute cell swelling and contractile activation during reperfusion (6, 63).

A second mechanism is related to the degradation of ankyrin. Ankyrin has a central domain that binds to α-fodrin and an N-terminal domain that interacts with several membrane receptors and channels, including the α subunit of Na+/K+-ATPase (69). The interaction of ankyrin with fodrin mediates the linkage of Na+/K+-ATPase to the fodrin-based membrane cytoskeleton and determines its specific localization in the membrane and its correct function (122). During reperfusion, calpain degradation of both α-fodrin and ankyrin causes detachment of the Na+-pump from its anchorage to the fodrin-based membrane skeleton inducing its dysfunction which results in impaired normalization of cytosolic Na+ concentration (62). The kinetics of Na+ recovery during reperfusion determines the influx of Ca2+ through the reverse mode of NCE and, consequently, the degree of injury (57). Degradation of ankyrin could result in the loss of ankyrin binding proteins other than Na+K+-ATPase, with potential effects on intracellular Ca2+. In fact, reduction in the levels of NCE in cardiomyocytes with a null mutation for ankyrin-B (89) and the cleavage of NCE3 by calpain in homogenates from brains subjected to ischemia (9) have been demonstrated. Reduced NCE extrusion of Ca2+ during reperfusion could impair the recovery of Ca2+ homeostasis and be detrimental (64).
Finally, calpain may also participate in mitochondria-driven cell death. Activated calpain has been described to induce the release of cytochrome c and other proapoptotic factors by cleaving Bid into an active form (17), and calpain but not caspase inhibitors appear to preserve Bid from cleavage and reduce cell death (16). A more recent study in forebrain and liver mitochondria (114) proposes that calpain-activated Bid induces Bak oligomerization and mitochondrial outer membrane permeabilization, granting calpain access to the intermembrane space and apoptosis-inducing factor (AIF) cleavage and release. These results indicate that calpain activation could initiate apoptosis by a pathway independent of caspases.
Cx43-channel gating
Previous studies have demonstrated that spreading of necrosis through gap junctions plays an important role in the pathophysiology of myocardial infarction following ischemia-reperfusion (36). Gap junction-mediated propagation of hypercontracture, sarcolemmal rupture, and cell death occur during reperfusion in connected pairs of freshly isolated cardiomyocytes, and in intact myocardial tissue (34). Propagation of injury involves passage of Na+ from dying cells to adjacent myocytes, and subsequent influx of Ca2+ through the reverse mode of the NCE (124).
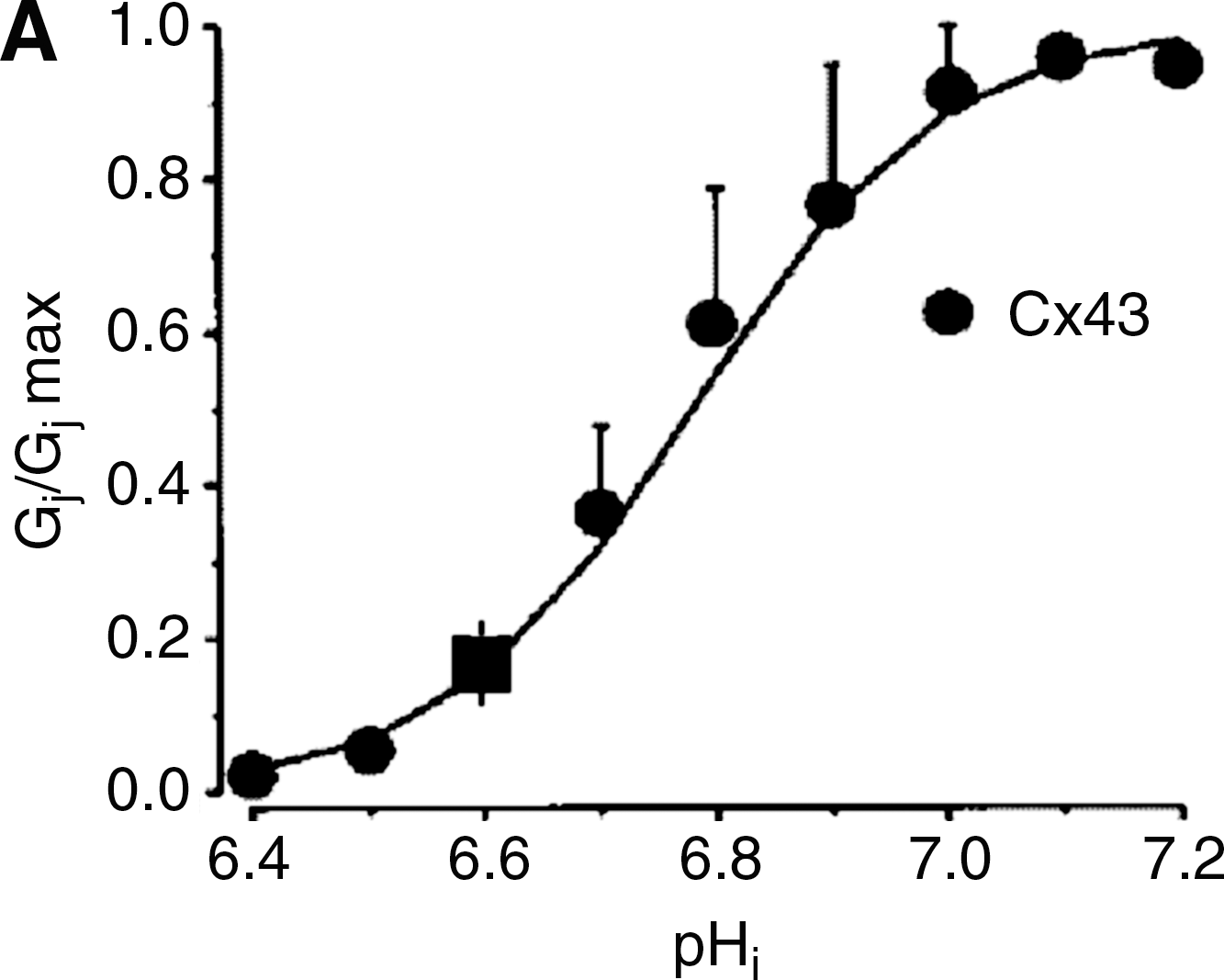
Furthermore, closure of gap junctions by several, chemically unrelated, gap junction uncouplers reduced infarct size in several animal models when given specifically during reperfusion (34, 121). Connexin channels can be regulated by a variety of factors, many of which are modified during acute myocardial ischemia, including calcium, calmodulin, or kinase activation (106, 138). Among these factors, intracellular acidosis is thought to play an essential role (106) (Fig. 8). The first evidence supporting that pH is an important regulator of gap junction channel permeability came from studies in Xenopus embryos (146). In this study, cells were reversibly uncoupled by lowering pHi to 6.4–6.85 using buffer solutions gassed with 100% CO2. Moreover, a close sigmoid relationship was shown between current flow between neighboring cells and pHi (146, 147). Importantly, studies in ventricular myocytes have suggested the existence of synergism between intracellular Ca2+ levels and pHi (154). However, other authors do not support the existence of this synergy and propose that Ca2+ mediates, in fact, the effects of H+ on chemical gating (83), probably through an intermediary molecule, possibly calmodulin (107). Cx43, as all other connexins isoforms, is markedly regulated by intracellular pHi, as demonstrated by studies in Xenopus oocytes, and in pairs of rat and guinea pig cardiomyocytes (26, 90, 92, 141, 154). Xenopus oocytes expressing Cx43 showed a pKa of 6.7, and macroscopic gap junctional conductance reached almost zero at pHi values of about 6.4 (26, 90, 141). Both the carboxyl terminal of Cx43 (85, 141), and a separate cytosolic sequence related to the pore-forming region (90) are essential for chemical gating of gap junctional channels by acidosis.
The delay in pHi recovery during initial reperfusion after PoCo (18, 59) may, thus, keep Cx43 gap junctional channels closed during initial reperfusion limiting spreading of necrosis between neighboring cells.
However, PoCo may also interfere with gap junctional gating by additional mechanisms. Cx43 is a phosphoprotein containing several consensus sites for regulation by a variety of serine, threonine, and tyrosine kinases (138). The modulation of gap junctional coupling by different kinases implicated in the cardioprotective effects of PoCo has been demonstrated in several studies (21, 134, 162). Recently, phosphorylation of Cx43 at specific serine residues has been linked to a cardiac-injury resistant state (140).
Moreover, PoCo may also modify gap junction-independent functions of connexins. Unopposed Cx43 hemichannels or connexons have been suggested to play a role in several pathological conditions, including ischemia-reperfusion injury (120), where they can play a key role in volume regulation. Hemichannel gating is closely regulated by several factors, including acidosis and phosphorylation (127). The delay in pHi recovery induced by PoCo during initial reperfusion, in addition to changes in phosphorylation status, may help to keep hemichannels closed, thus avoiding excessive cardiomyocyte swelling and cell injury (120).
Mitochondrial Cx43 has been described to be essential for preconditioning protection (84, 118, 132), but a recent study in mice heterozygous for Cx43 (Cx43+/-) indicates that it does not play a significant role in PoCo protection.
Sustained Protection
The existence of a potential link between delayed acidosis and other pathways reported to be involved in the cardioprotective effects of PoCo, specially pathways associated with survival kinases and inhibition of MPT formation, has received attention from several groups (18, 19, 31). It has been suggested that these links could explain the perpetuation of cardioprotection once the pHi has been normalized.
Although the signal pathways of PoCo are still under investigation, it has been proposed that they share many similarities with those described for preconditioning. PoCo allows accumulation of classical autacoids [adenosine (75, 76, 108), bradykinin (103), opioids (159)] that trigger the activation of the major components of the RISK pathway (44, 50, 102). A study from Fujita et al. has suggested an interrelation between RISK and acidosis (31). In open-chest anesthetized dogs, PoCo induced the phosphorylation of Akt and ERK in the ischemic myocardium. Administration of NaHCO3 reverted transient acidosis and blunted the infarct size reduction induced by PoCo and these effects correlated with attenuated phosphorylation of Akt and ERK in the ischemic region. However, it is worth noting that a recent study performed in an in vivo pig model observed phosphorylation of Akt and ERK1/2 during reperfusion, but failed to demonstrate their contribution to the infarct size reduction of PoCo (136).
ROS production is also included among the triggers of PoCo (104). In fact, the ROS scavenger N-2-mercaptopropionyl glycyne prevents the protective effects of PoCo (105), and the same ROS scavenger has been shown to block the protection afforded by acidic reperfusion (18). It has been proposed that reintroduction of oxygen after transient ischemia induces ROS production, but it does not protect against reperfusion injury because MPT opens before the activation of endogenous survival pathways. Prolongation of cellular acidosis during reperfusion by acidic perfusion or PoCo could thus inhibit MPT during the first minutes of reflow and allow for endogenous protective signaling pathways to be activated by ROS formation. The delivery of oxygen during acidic perfusion or the brief reperfusions of PoCo would promote mitochondrial ROS formation which has been proposed to activate PKC through redox signaling (19). PKC appears as a critical kinase in the signaling cascade leading to a reduced probability of MPTP opening after pH normalization (105, 108, 158).
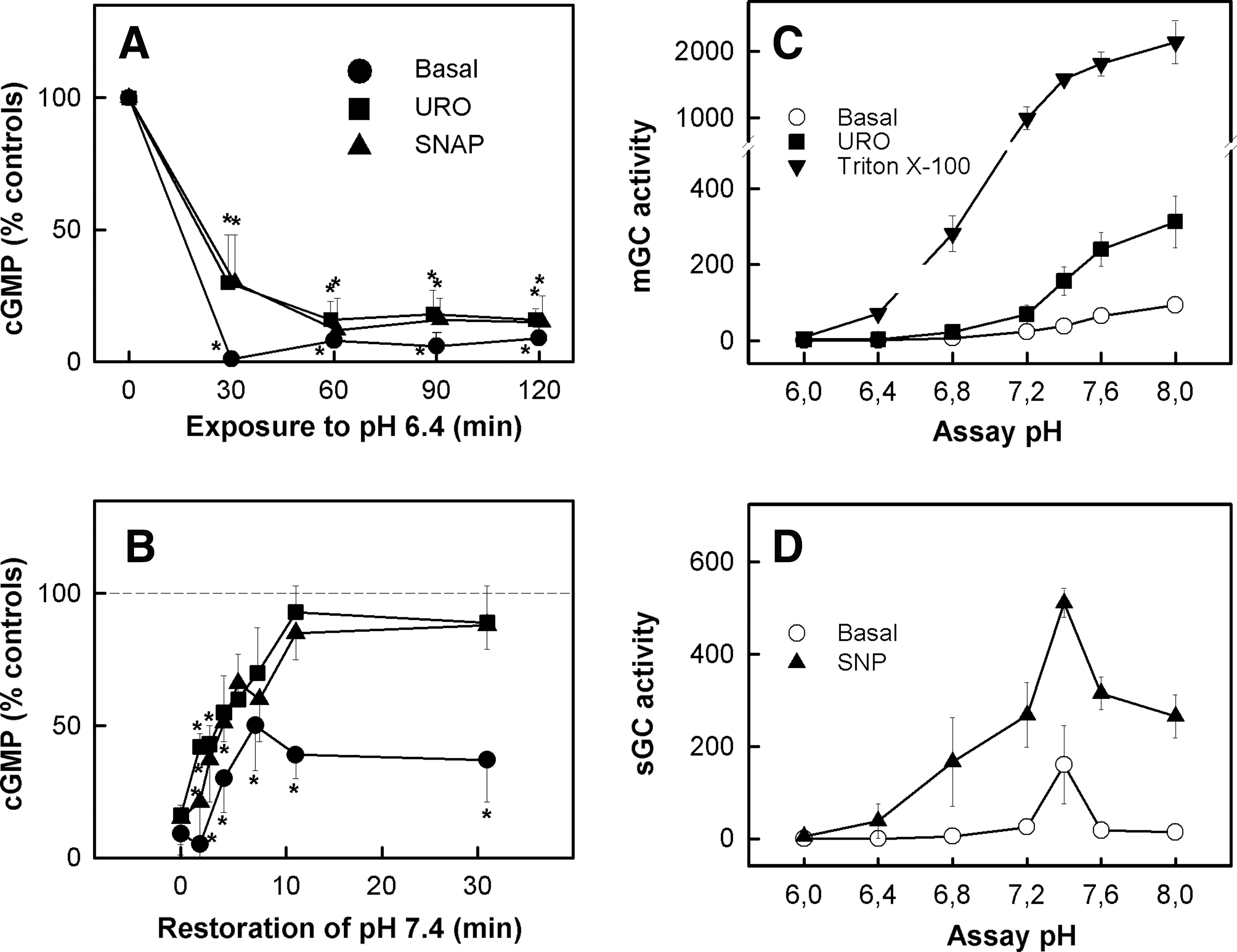
The cGMP/PKG pathway is severely depressed in cardiomyocytes and endothelial cells after prolonged ischemia (2, 3). Preservation of this pathway with sodium nitroprusside, natriuretic peptides, and cGMP analogues has been solidly demonstrated to protect against reperfusion injury (1, 98, 99). More recently, the use of either the NOS inhibitor L-NAME or ODQ blocked the infarct-sparing effect of PoCo (101, 157), suggesting that, as it occurs in preconditioning, cGMP-mediated signaling is involved in the pathways responsible for PoCo-induced cardioprotection against ischemia-reperfusion injury. cGMP, through the activation of PKG, has been suggested to mediate the transfer of the signals triggering protection from the cytosol to the mitochondria (20). PKG-dependent phosphorylation has been described to induce the opening of ATP-sensitive potassium channels in the mitochondrial inner membrane (mKATP) evoking K+ entry into mitochondria and generation of ROS (95). Although not clearly established, PKCɛ has been proposed to be the intermediate step between the cytosol and the mitochondrial inner membrane (67). Apart from mKATP, PKG could modulate other targets implicated in reperfusion injury. These include the phosphorylation of phospholamban, enhancing SERCA activity, and accelerating normalization of cytosolic Ca2+ concentration and attenuating SR-dependent Ca2+ waves (1), and the attenuation of contractility during reperfusion through PKG-dependent effects on the sensitivity of myofibrils to Ca2+ (61, 133) This effect could be the consequence of a direct phosphorylation of troponin I and/or related to direct or to indirect effects of PKG on myosin light chain phosphatase (128, 133).
Both the basal content of cGMP and the ability of myocardial cell to respond to cGMP activators are decreased in hearts subjected to ischemia (61). It has been shown that in cell models, pHi during ischemia is critical for cGMP synthesis. Guanylyl cyclase (GC) (soluble and particulate) activity in cell homogenates shows a typical bell-shaped curve with respect to pH, resulting in negligible cGMP synthesis at pH values close to 6.4 (Fig. 9) (2, 3). In experiments performed by our group in isolated rat hearts and in agreement with previous studies (101, 157), the use of the sGC inhibitor ODQ blocked the cardioprotective effects of PoCo. However, ODQ was ineffective in preventing the cardioprotective effects obtained after transient acidic reperfusion. 31P-RMN data obtained from these hearts could explain these differences since they showed that ODQ accelerates pHi recovery in hearts subjected to PoCo but not in those perfused with acidic buffer (unpublished data). Since NHE has been shown to be negatively modulated by PKG (66, 131), it is tempting to speculate that activation of PKG by PoCo could contribute to the delay in pHi recovery and, by this mechanism, to the cardioprotective effects of PoCo.
Despite the solid evidence supporting that a delay in pHi recovery is critical for the cardioprotective effects of ischemic PoCo, the effect of remote pre- or post- conditioning on the kinetics of correction of intracellular acidosis during reperfusion is not known. However, there is no reason to expect that these strategies should have a significant effect on pHi. Remote ischemic PoCo was recently reported to protect the heart against acute myocardial infarction with cycles of few minutes of ischemia/reperfusion applied to the distal artery territory (femoral or renal artery) either immediately after the beginning of reperfusion (5) or just few minutes before heart reperfusion (73). Loukogeorgakis et al. demonstrated that remote PoCo could be produced in both human volunteers and patients with CAD in a manner which was sensitive to inhibition by glibenclamide (a KATP channel blocker), using endothelial function, but not infarct size, to assess protection (86). In a recent proof-of-concept clinical study, Botker and co-workers (13) have demonstrated that remote ischemic perconditioning (four 5 min cycles of cuff inflation/deflation to the arm during the ongoing ischemia) administered in the ambulance, improved the myocardial salvage index in ST-elevation myocardial infarction (STEMI) patients receiving primary PCI. Although the efficacy of remote PoCo as compared to local PoCo and its mechanisms of action have not been established, its cardioprotective effects suggest that endogenous cardioprotection by mechanisms independent of delayed pHi recovery may be important and have relevant clinical implications.
Translation to Patients
While experimental evidence demonstrated that transient reoxygenation with acidic solutions could protect isolated myocytes, papillary muscle, and perfused hearts against cell death (18, 58, 71, 94, 126), only few studies have analyzed the effect of transient acidosis during reperfusion in models closer to the clinical setting (78, 115). In an early study, a protective action of 60 min of HCl intracoronary infusion or respiratory acidosis against infarction was described in in situ dog hearts submitted to 30–60 min of LAD occlusion (78). However, neither of these interventions appears as a practical therapeutics in patients submitted to primary percutaneous interventions. In a more recent study, intracoronary administration of an acidic crystalloid solution during the first 3 min of reperfusion was as effective as PoCo in reducing the infarct size in an in situ pig model subjected to 60 min of regional ischemia, but was accompanied by an increased incidence of early reperfusion malignant ventricular arrhythmias (119). Although the coincident reduction in infarct size by both interventions support the hypothesis of a common mechanism of action, the increased risk of ventricular fibrillation precludes the translation of acidic reperfusion to patients undergoing primary percutaneous interventions. In contrast to these results, others studies have shown that limiting the rate of normalization of pH by brief acidic reperfusion (2 min) reduces the incidence of ventricular fibrillation in an isolated rat heart model of regional ischemia (7, 55) by a mechanism that involved enhanced recovery of Na+/K+-ATPase activity while prolonged intracellular acidosis can cause electrical disturbance, partly through the activation of H+ extruders, which indirectly raises Ca2+ in the sarcoplasmic reticulum. Sarcoplasmic reticulum can spontaneously initiate a propagating Ca2+ wave of release that provides the substrate for delayed after-depolarizations, ectopic beats, and triggered arrhythmias. In addition, acidosis influences the action potential, partly through effects on ICa and K+ conductance pathways (65, 80), and partly through its ability to activate an outward current via electrogenic NBC (153, 155).
Pharmacological strategies expected to delay pHi recovery during reperfusion have been tested in clinical studies with largely disappointing results.
Extensive preclinical work had soundly demonstrated that inhibition of NHE prior to ischemia markedly reduces infarct size (33, 72). Available evidence indicated that NHE inhibitors delay the progression of myocardial injury during ischemia by attenuating intracellular Na+ accumulation which in turn reduces the Ca2+ entry through reverse mode of NCE (79) and by delaying ATP depletion through inhibition of mitochondrial NHE (125). In contrast, many studies indicate that the efficacy of NHE inhibitors administered just prior to reperfusion is very small (33, 79, 143). NHE enhances its activity associated to pHi correction during the first minutes of reperfusion and its inhibition was expected to delay pHi recovery and reduce Na+ entry. However, it has been reported that inhibition of NHE at reperfusion results in only a small effect on kinetics of pHi and Na+ recovery due to the compensatory action of bicarbonate transporters (143). In contrast, the simultaneous inhibition of both NHE and NBC significantly delayed recovery of pHi and attenuated reperfusion-induced cell death (33, 58, 130).
Despite this preclinical evidence, the therapeutic potential of NHE inhibitors used as an adjunct to reperfusion in patients with acute myocardial infarction was tested in clinical trials. In the ESCAMI trial, the NHE inhibitor eniporide given before reperfusion therapy in patients with ST elevation AMI did not limit infarct size or improve clinical outcome (161). In contrast, when the NHE inhibitor cariporide was administered before coronary artery bypass graft surgery, a reduction in perioperative infarction was observed by retrospective analysis of data from a subgroup of the GUARDIAN trial (144). This apparent benefit in the setting of CABG surgery led to the initiation of the EXPEDITION trial (87). However, in this study using a dose higher than in previous trials, treatment with cariporide was associated with higher rates of short-term mortality and cerebrovascular events precluding its clinical use.
Conclusion
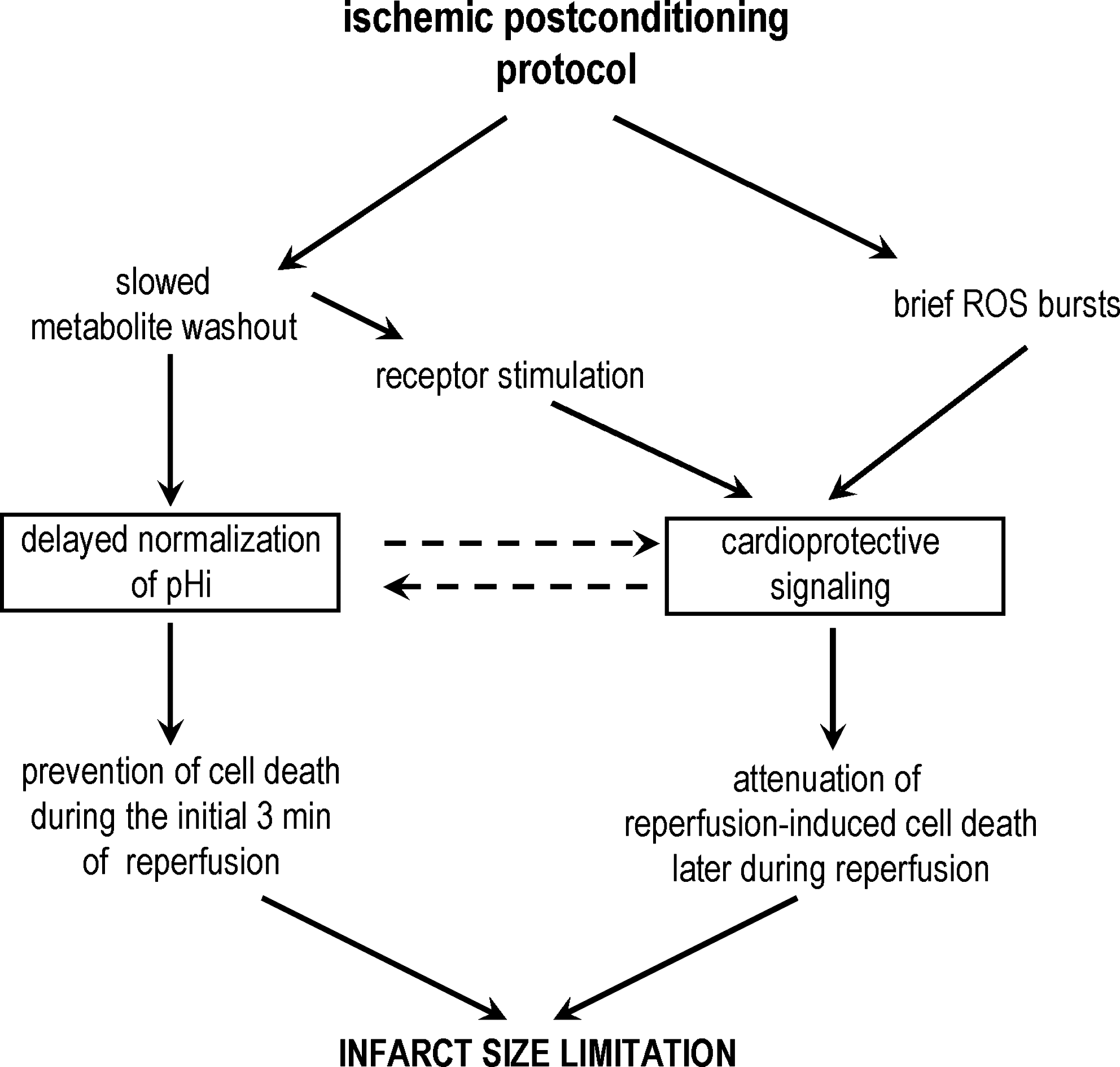
There is evidence that delayed normalization of pHi during reperfusion plays a relevant role in the cardioprotective effects of PoCo. The relation between this and other previously proposed mechanisms of PoCo protection needs to be established (Fig. 10). PoCo could allow other mechanisms to act by preventing immediate cell death upon re-flow. Further research is needed to investigate this relationship and to identify potential ways to translate this knowledge to the treatment of patients with acute myocardial infarction.
Footnotes
Acknowledgments
This work was supported by Redes Temáticas de Investigación Cooperativa Sanitaria (RETICS-RECAVA RD06/0014/0025); Comisión Interministerial en Ciencia y Tecnología (CICYT SAF/2008-03736); Fondo Investigación Sanitaria (FIS-PI080238 and FIS-PS09/02034).