
Abstract
Redox modulation by antioxidants, such as selenium (Se), has emerged as an important regulator of erythropoiesis. Using Se-deficient (0.04 ppm), Se-adequate (0.1 ppm), and Se-supplemented (0.4 ppm) C57/BL6 mice, we show that Se deficiency caused anemia, when compared to the Se-supplemented and Se-adequate groups. Increased denaturation of hemoglobin, methemoglobin, protein carbonyls, lipid peroxidation, Heinz bodies, and osmotic fragility of erythrocytes were observed in Se-deficient mice. Increased oxidative stress upregulated forkhead transcription factor (FoxO3a) and hypoxia-inducible factor-(HIF)1α in the spleen and kidney of Se-deficient murine as well as in the proerythroblast G1E cells cultured in Se-deficient media. A significant increase in the expression of erythropoietin, a downstream target of HIF1α, and expansion of stress erythroid progenitors (burst forming units-erythroid) were seen in the Se-deficient mice. Despite the increase in erythroid progenitors, lowered reticulocytes suggest a defective erythroid differentiation pathway. While Se deficiency led to increased nuclear levels of active FoxO3a, Se-adequate conditions reversed this effect and increased nuclear export by its binding partner, 14-3-3βζ, that is under the redox control of selenoproteins. In summary, these results provide insight into the importance of adequate Se nutrition in regulating red cell homeostasis by mitigating oxidative stress-dependent modulation of FoxO3a and HIF1α to effect differentiation of erythroid progenitors. Antioxid. Redox Signal. 14, 1403–1412.
Introduction
Hematopoiesis is under the tight control of expansion, differentiation, and survival of progenitors (4, 28). This characteristic of erythroid cells exposes them to highest order of oxidative stress (28), which makes hemoglobin (HGB) highly prone to oxidative damage (42). Erythroid cells have developed stress–response systems, involving transcription factors such as Nrf-2 and FoxO3, to counter oxidative stress (28). Oxidative stress also activates hypoxia-inducible factor-1α (HIF-1α), a key transcription factor that upregulates the expression of erythropoietin (Epo), a critical growth factor, which is expressed by the peritubular capillary endothelial cells of the kidney that controls the steady-state production of erythrocytes (41). The specific erythroblastic progenitors burst forming units-erythroid (BFU-E)/CFU-E (burst and colony-forming unit-erythroid) are Epo-sensitive and critical in the amplification of the differentiation process in response to erythropoietic stress mechanisms (34).
Forkhead transcription factor (FoxO3a), a member of Forkhead transcription regulators, is one of the most abundantly expressed proteins in erythroid cells and is essential for the maintenance of hematopoietic stem cell pool (29). Previous studies have shown reciprocal relationship between FoxO3a and Se status by demonstrating that Se affected the activity and expression of FoxO3a in cancer cells (25, 28). FoxO3a has also been found to mitigate the oxidative threat posed during erythropoiesis by upregulating many antioxidant genes, including GSH-Px1 (13, 23, 28, 31). The activity of FoxO3a depends upon its cellular localization and phosphorylation status, which is controlled by 14-3-3 family of highly conserved regulatory proteins involved in a vast array of processes such as the response to stress, cell-cycle control, and apoptosis, serving as adapters, activators, and repressors (2, 14, 46). Phospho-FoxO3a, which is transcriptionally inactive, binds to 14-3-3 proteins to be exported out of the nucleus into the cytosol (6). Conversely, stress stimuli increase the nuclear localization and activation of unphosphorylated FoxO3a (15). Recently, selenoprotein W, a selenoprotein that modulates control of cell cycle entry in breast and prostate cancer cells, was shown to modulate the activity of 14-3-3 proteins in a redox-dependent manner (11). Taken together, this reinforces the possibility that Se-variation-induced regulatory mechanisms involving 14-3-3βζ, a highly expressed 14-3-3 isoform in erythroid cells, and FoxO3a are critical for maintaining erythropoietic homeostasis. However, the contribution of these factors in the physiological control of oxidative stress during variation in Se status is not well understood. Here we show that cellular Se status plays a crucial role in red cell homeostasis by modulating FoxO3a localization, which is pivotal for mitigating oxidative stress in erythroid cells.
Materials and Methods
Experimental design
C57/BL6 mice were fed semipurified diets purchased from Harlan Teklad (Madison, WI) containing different concentrations of Se (added as sodium selenite), namely, 0.04 ppm (Se-deficient), 0.1 ppm (Se-adequate), or 0.4 ppm (Se-supplemented) for ∼8 weeks. There were no significant differences in the diet consumption or gross weight changes in any of the groups. After the completion of diet feeding schedule, blood was obtained by puncture of the retro-orbital plexus and used for various hematological parameters and biochemical experiments as described below. All experiments were preapproved by the Institutional Animal Care and Use Committee.
Cell culture
For mechanistic studies murine G1E cells, derived from a targeted disruption of GATA-1 in embryonic stem cells, were used. G1E cells recapitulate erythropoiesis from the proerythroblast through the basophilic erythroblast stage as seen from a detailed transcriptome analysis (47). These cells were cultured in Se-deficient (7 nM) Iscove's media containing 5% defined fetal bovine serum that was quantitated for Se by atomic absorption spectroscopy. G1E cells were supplemented with graded levels (25–500 nM) of Se, as sodium selenite, for 4 days to create differential Se status. Such cells were harvested and nuclear and cytoplasmic extracts were prepared from these cells as described earlier from our laboratory (45). The nuclear extracts were tested for cytoplasmic contamination using glyceraldehyde 3-phosphate dehydrogenase as a marker.
Hematological parameters
Blood (200 μl) was collected from mice on different treatment groups (n = 5 per group) and analyzed for hematocrit, reticulocytes, leukocytes, and platelets, using the Advia 120 Blood Analyzer with veterinary software program.
Lipid peroxidation assay
The levels of lipid peroxidation (LPO) were assayed in the postmitochondrial fraction of liver samples of different treatment groups by the method of Wills (48). Since malondialdehyde (MDA) is a degradation product of peroxidized lipids, the development of 2-thiobarbituric acid-MDA chromophore with the absorption maxima at 532 nm was taken as an index of LPO. 1,1′,3,3′-tetraethoxypropane was used (2–10 nmol) to create a standard calibration curve against a Milli-Q water background. MDA levels were expressed as n mol MDA per mg protein.
Determination of protein carbonyl content
Protein carbonyl content was measured using 2,4-dinitrophenylhydrazine (DNPH) as described by Levine et al. (24). In each experiment fresh blood was centrifuged and the cell pellet was resuspended in 5 mM phosphate buffer (pH 7.5) containing the protease inhibitors leupeptin (0.5 μg/ml), aprotinin (0.5 μg/ml), and pepstatin (0.7 μg/ml), and 0.1% Triton X-100. From the resulting supernatant, ∼2.0 mg of protein was treated with 300 μl of 10 mM DNPH solution in 2 M HCl. Samples were then incubated for 1 h at room temperature, stirred every 10 min, precipitated with 10% trichloroacetic acid, and centrifuged for 3 min. The pellet was washed three times with 1 ml of ethanol/ethyl acetate, 1:1 (vol/vol), and redissolved in 1 ml of 6 M guanidine-HCl in 10 mM phosphate buffer/trifluoroacetic acid, pH 2.3. Any trace insoluble material was removed by centrifugation. The differences in absorbance at 366 nm between the DNPH-treated and the HCl-treated control samples were determined and the results are expressed as nmol of carbonyl groups per mg of protein, using the extinction coefficient of 22.0 mM −1 cm−1 for aliphatic hydrazones.
Analysis of globin precipitates
For globin chain analysis in membrane skeleton, 100 μl of freshly drawn blood was lysed and membranes were washed extensively in 0.05% phosphate buffered saline. Membrane lipids were removed by extraction with 56 mM sodium borate (pH 8.0). Precipitated globins were dissolved in 8 M urea containing 10% acetic acid, 10% β-mercaptoethanol, and 0.04% pyronin. The samples were electrophoresed on triton-acetate-urea (TAU) gels and stained with Coomassie brilliant blue as described (44).
Levels of MetHGB
MetHGB content was determined by the method of Sakata et al. (38). Sodium cyanide solution (10%) was added to heparinized blood (100 μl) and the disappearance of the characteristic absorption at 635 nm, signifying the formation of cyanmethemoglobin, was monitored spectrophotometrically.
Osmotic fragility
Heparinized venous blood (10 μl) was added into tubes with increasing concentration of buffered salt solution (pH 7.4; NaCl [%] 0, 0.30, 0.35, 0.40, 0.45, 0.50, 0.55, 0.60, 0.70, and 0.90). The tubes were gently mixed and incubated at 25°C for 30 min. Samples were centrifuged at 1500 g for 10 min, and hemolysis was evaluated spectrophotometrically at 540 nm on a Beckman UV-VIS DU700 diode array spectrophotometer.
Heinz body staining
Blood smears were placed on microscopic slides, and stained with rhodaline blue solution (Sigma) for 1 min at room temperature. The slides were washed in Milli-Q water, dried, and mounted using Paramount mounting solution. Photographs were taken on an Olympus BX6 epi microscope.
Expression of Epo
To examine the effect of Se status on the expression of Epo, mRNA was isolated from snap-frozen kidneys from Se-deficient and Se-adequate mice using the Trizol reagent (Invitrogen). cDNA was synthesized using the cDNA Archive Kit (Applied Biosystems) and used in semiquantitative polymerase chain reaction (PCR) using primers specific for murine Epo and β-actin. The PCR products were analyzed on a 1.2% agarose gel and the band intensities were analyzed using Image J software program from the National Institutes of Health.
Expression of FoxO3a, 14-3-3βζ, and HIF1α
Protein expression studies were carried out in G1E cells that were cultured in Iscove's media (5% fetal bovine serum) with varying concentrations of Se (0, 25, 50, 100, 250, and 500 nM) as described earlier. Cells were harvested and nuclear and cytoplasmic extracts were prepared as described earlier (45). To correlate these findings in an in vivo system, the expression of HIF-1α in the total lysates from kidneys and FoxO3a, phosporylated FoxO3a (pFoxO3a) in the nuclear fractions isolated from the spleen of Se-deficient and Se-adequate mice, were examined by Western blot analyses (10). Protein estimation was performed using the bicinchoninic acid protein assay (Thermo Pierce) and equal amounts of protein were used for sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blotting with antibodies specific to HIF1α, FoxO3a, pFoxO3a, and 14-3-3 βζ antibodies (Abcam). The density of protein bands was quantified and normalized to appropriate housekeeping genes.
Assay for BFU-E
Spleen and bone marrow cells were isolated from mice fed Se-deficient, Se-adequate, or Se-supplemented diet. Splenocytes or bone marrow cells were washed in phosphate-buffered saline following lysis of red blood corpuscles in ammonium chloride buffer and resuspended in Dulbecco's modified Eagle's medium. About 1 × 106 total cells were plated in methylcellulose (Methocult; StemCell Technologies) semisolid media with Epo (3 U/ml), whereas bone marrow cells were cultured with Epo and IL-3 (10 ng/ml) for 5 days. BFU-Es were counted on day 5 using benzidine stain as reported earlier (34).
Statistical analysis
All experiments were repeated at least four times and values are expressed as means ± standard error of the mean. Statistical analyses were carried out using SigmaPlot, and Student's t-test was applied. The overall significance level was set to p < 0.05.
Results
Se deficiency leads to alterations in the hematological parameters
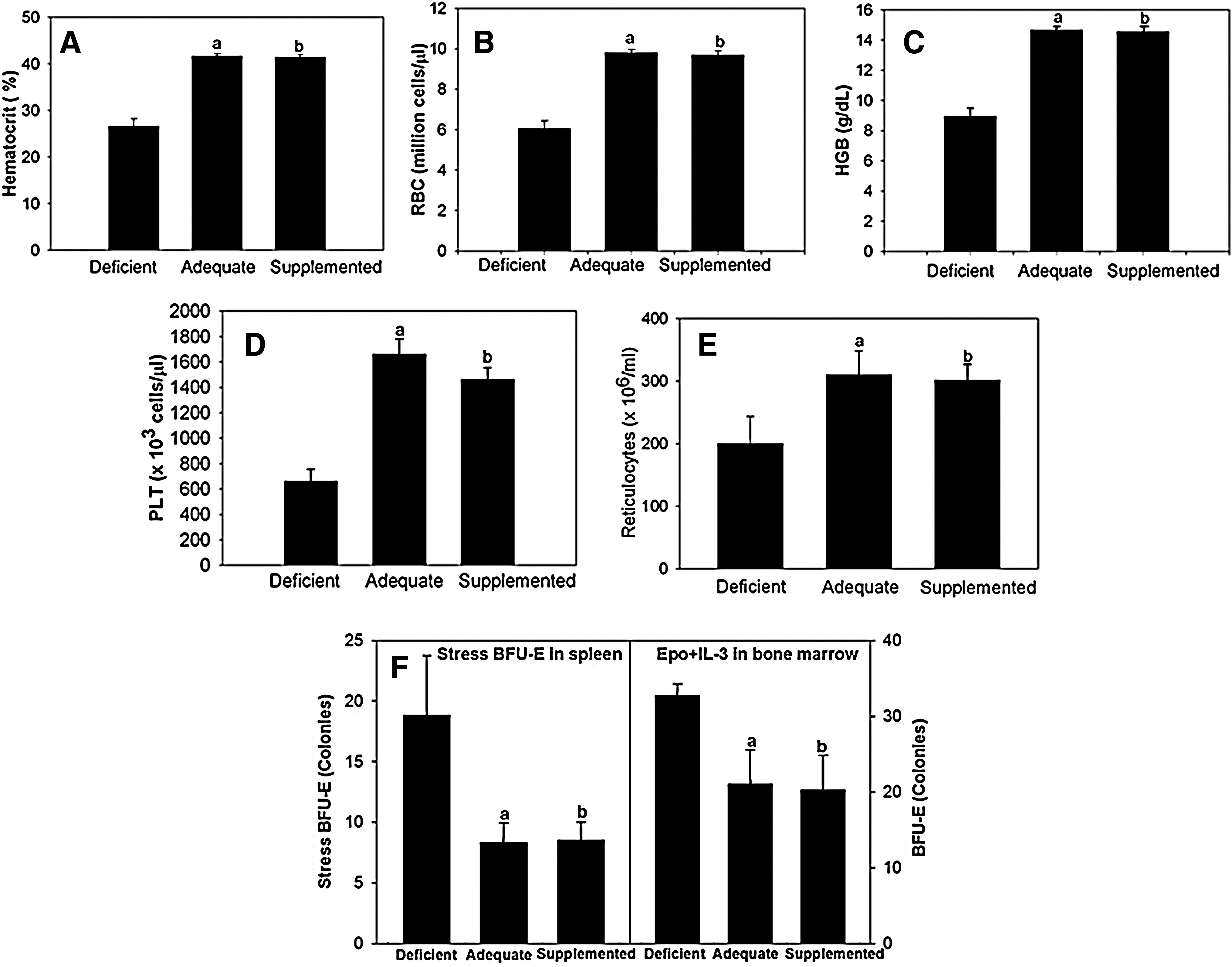
C57/BL6 mice exhibited phenotypic changes in erythroid and platelet parameters as a function of Se in the diet. As shown in Figure 1, hematocrit, red blood corpuscle counts, HGB, and platelets were significantly reduced in mice maintained on a diet deficient in Se for 8 weeks when compared to those fed on either Se-adequate or Se-supplemented diets (Fig. 1A–D). However, despite anemia, Se-deficient mice did not exhibit an increase in reticulocytes (Fig. 1E). These data suggest that compensatory stress erythropoiesis is defective in these mice. We also analyzed early erythroid progenitors in the bone marrow and specialized stress erythroid progenitors (stress BFU-E) in the spleen. The data in Figure 1F show that BFU-E in the bone marrow and spleen increased in Se-deficient mice, which further suggests that Se-deficient mice exhibit inefficient erythropoiesis, which impedes terminal differentiation of erythrocytes. Since no defects in the hematological parameters were observed between Se-adequate and Se-supplemented-diet-fed groups, further studies compared only Se-deficient and Se-adequate diet groups.
Se deficiency increases the LPO, protein carbonyls, and MetHGB in erythrocytes
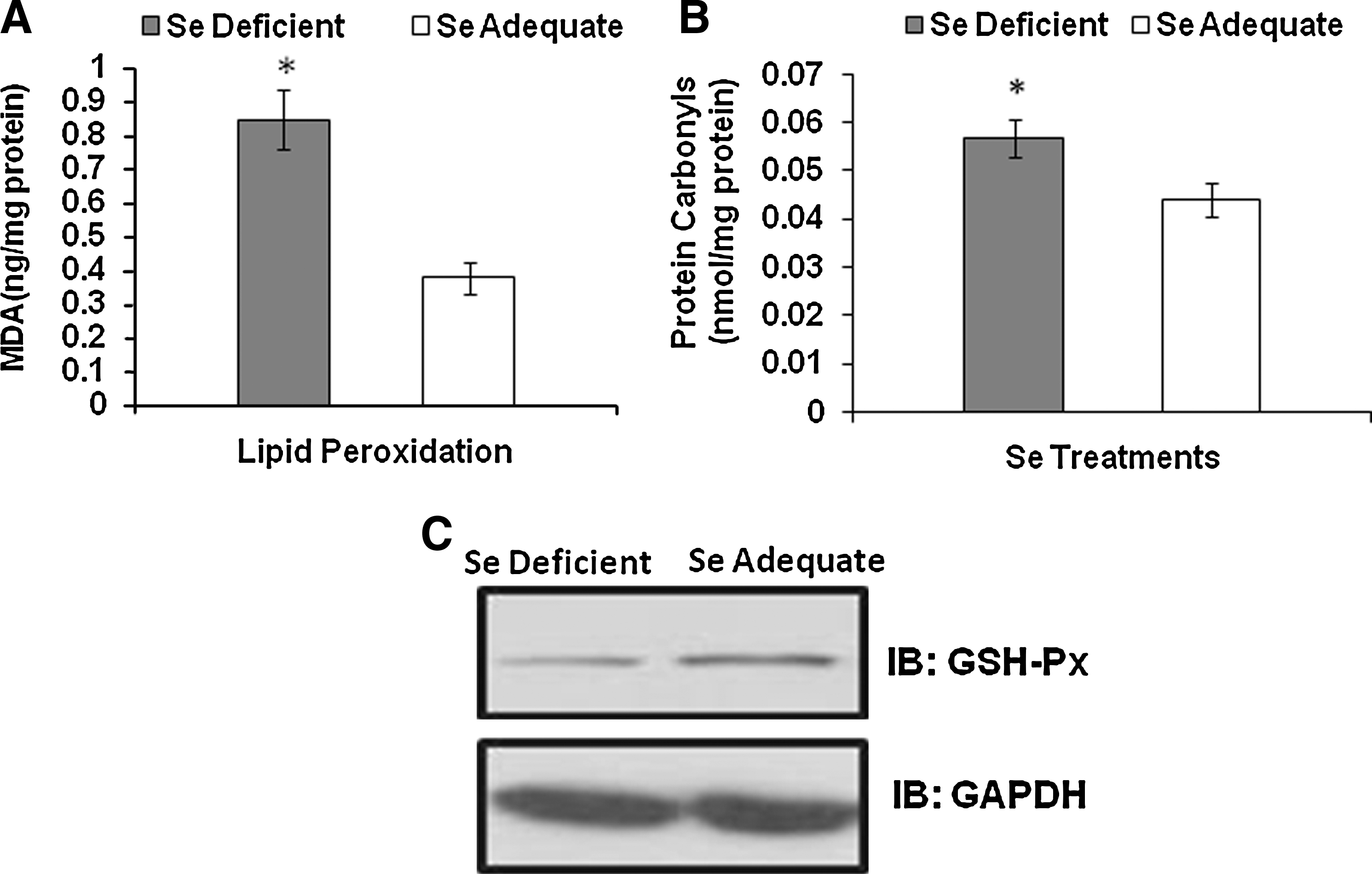
Significant increase in hepatic LPO was observed in the Se-deficient group when compared to the Se-adequate group (Fig. 2A). Thus, given that Se status was a critical determinant of cellular oxidative tone, we analyzed if Se modulated the levels of protein carbonyls in erythrocytes. Carbonyl moieties, formed by the oxidation of amino acid residues in proteins, represent the abundance of free radicals and ROS in cells. As expected, the erythrocytes isolated from Se-deficient mice demonstrated a significantly increased level of protein carbonyls compared to those from Se-adequate mice (Fig. 2B). Analysis of the expression of GSH-Px1 in erythrocytes indicated a dose-dependent effect with low expression in the Se-deficient group (Fig. 2C). A similar pattern of increase in GSH-Px1 with Se supplementation has been reported in lymphoid tissues (spleen and lymph nodes) as well (18, 33). Taken together, these results clearly demonstrate the importance of cellular Se status in modulating erythrocyte oxidative stress.
Se deficiency leads to denaturation of HGB
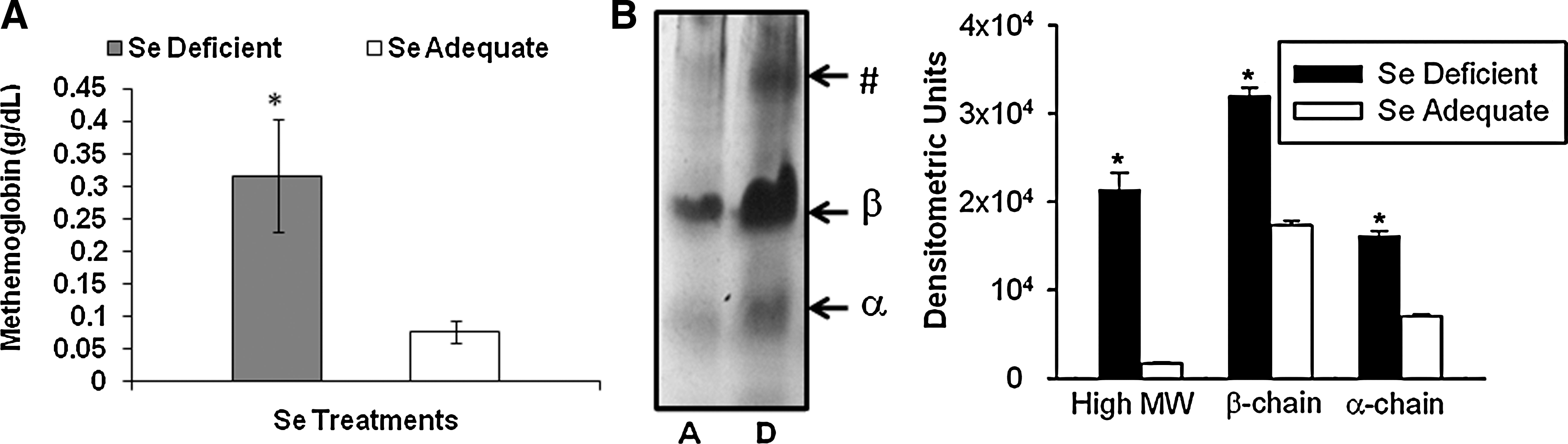
To further extend our observation on the modulation of erythroid oxidative stress Se, the formation of MetHGB and oxidative denaturation of the α and β chains of HGB were analyzed. As shown in Figure 3A, an increase in the levels of MetHGB in the erythrocytes was seen in Se-deficient group, which signifies increased oxidation of ferrous ions in HGB. Further, analysis of the membrane ghosts for precipitated globin chains by TAU gel electrophoresis demonstrated a higher intensity of bands corresponding to α and β chains in the Se-deficient group, whereas those in the Se-adequate were substantially reduced. We consistently observed an additional high-molecular-mass band of denatured β-globin chain only in the Se-deficient group (Fig. 3B; indicated by asterisk). These data suggest that Se deficiency exacerbates oxidative-stress-mediated denaturation of globin subunits of HGB.
Heinz bodies in Se-deficient mice
To further explore the relationship of Se status to HGB oxidation, blood smears from two treatment groups were stained with rhodaline blue for the presence of Heinz bodies. Heinz bodies represent small round inclusions within the red cell body indicative of the denaturation of HGB and formation of MetHGB. Increased number (∼2.5-fold) of granulated spots in the Se-deficient group clearly indicated the prevalence of oxidative stress-mediated damage to the erythrocytes to a much greater extent when compared to the Se-adequate diet groups (Fig. 4 and Table 1). In addition, there was a 18-fold increase in echinocytes (erythrocytes with spicules) in blood smears of Se-deficient mice when compared to the Se-adequate-diet-fed group. These results strongly suggest that oxidative stress, a consequence of Se deficiency, may contribute to a decrease in mature erythrocytes.

The prevalence of Heinz bodies and echinocytes in the blood smears from Se-deficient and Se-adequate mice is shown. Average of n = 3 per group.
Se, selenium.
Se deficiency makes the erythrocytes osmotically fragile
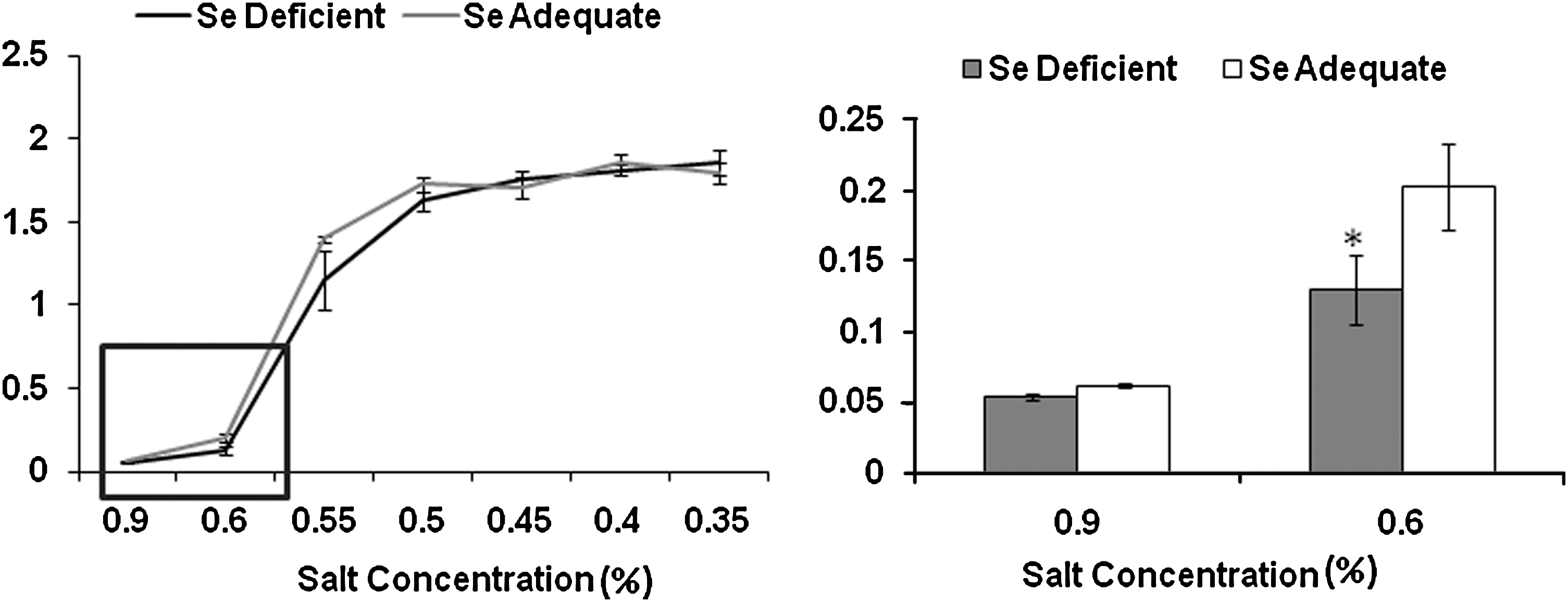
Based on our studies above, we tested the possibility that increased oxidative modification of proteins, including membrane components, could affect the osmotic tolerance of erythrocytes. Standard osmotic fragility tests with fresh erythrocytes from the two groups revealed that the erythrocytes from Se-deficient group were more prone to hemolysis compared to those in Se-adequate group when subjected to hypotonic stress (Fig. 5), which could be due to oxidative stress-mediated changes in cells.
Differential Se status modulates the activation of FoxO3a
To elucidate the underlying mechanism as to how differential Se-deficiency-induced oxidative stress affected hematopoiesis, we examined the regulation of FoxO3a. We utilized G1E cells, a widely used in vitro murine cell line developed to study gene regulation during erythropoiesis from the proerythroblasts through the basophilic erythroblast stage. G1E cells cultured in Se-deficient media or that supplemented with graded amounts of selenite were used. Culturing of G1E cells in Se-deficient media significantly decreased the GPX1 levels, as seen by Western blot (data not shown). Modulation of FoxO3a, pFoxO3a, and 14-3-3βζ in the nuclear and cytoplasmic extracts was examined as a function of Se concentration. The ratio of total:pFoxO3a was taken as an index of active (unphosphorylated) FoxO3a. As shown in Figure 6A and B, Western immunoblot analysis indicated increased nuclear localization of active form of FoxO3a in cells cultured in Se-deficient media compared to those cells cultured in the presence of adequate (100 nM) levels of Se. Exogenous addition of Se-deficient cells with Se (50–100 nM) decreased the levels of active FoxO3a. Further addition of supra-physiological levels of Se (250–500 nM) caused an increase in active FoxO3a in the nucleus. An increase in 14-3-3βζ in the nuclear fractions of cells supplemented with Se at adequate (50–100 nM) concentrations was seen, supporting the data with increased levels of phosphorylated FoxO3a in the cytoplasm. Supraphysiological (>250 nM) concentrations of Se also increased the activity of FoxO3a in G1E cells, but with no obvious hematological changes in Se-supplemented mice (0.4 ppm). These changes in FoxO3a expression were also similar in vivo where increased expression of active FoxO3a (measured as TotalFoxO3a/pFoxO3a) was seen in Se-deficient animals as compared to adequate Se group (Fig. 6C). These data demonstrate that Se-deficient erythroid progenitors activate FoxO3a-dependent response to adapt to increased oxidative stress.
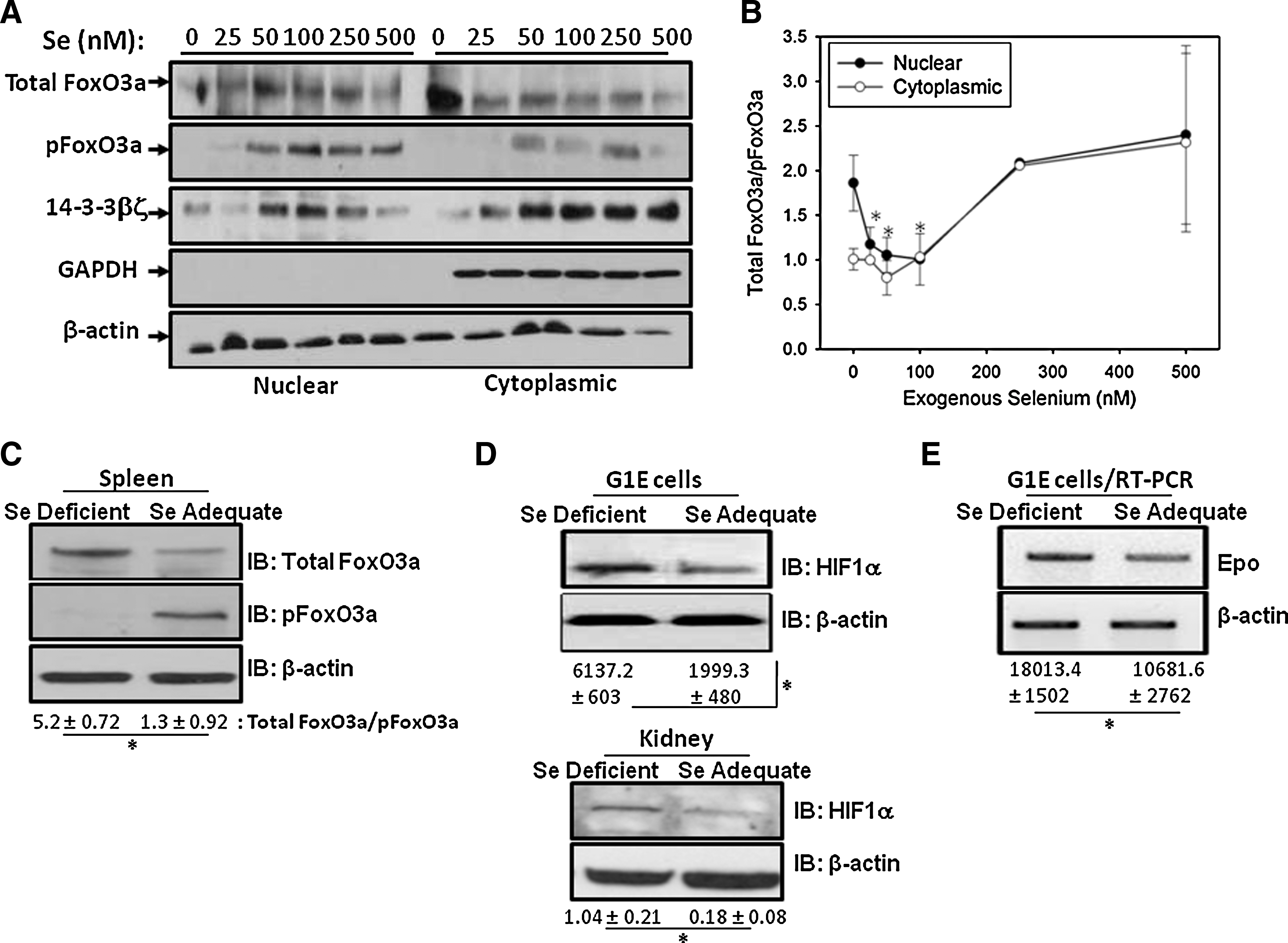
Se deficiency increases Epo levels
Given the fact that oxidative stress accompanying Se deficiency led to reduced hematological parameters that culminated in increased activation of FoxO3a, we speculated that oxidative stress also activated HIF1α and downstream genes in Se deficiency. Consistent with our hypothesis, Western blot analysis of HIF1α expression showed a significant increase in Se-deficient G1E cells as well as Se-deficient kidney extracts when compared to their Se-adequate counterparts (Fig. 6D). Further, the regulation of Epo by HIF1α was examined as a function of Se status. Semiquantitative reverse transcription-PCR analysis showed a significant increase in the levels of Epo in the kidney of mice fed on an Se-deficient diet when compared to the Se-adequate-diet-fed group (Fig. 6E). Taken together with the hematological data, such an increase in Epo is indicative of disrupted erythropoiesis in Se-deficient animals. HIF1α regulates Epo in response to hypoxia; therefore, this increase in Epo expression in Se deficiency could be caused by anemia in those mice.
Discussion
The critical role of Se in the etiology of a number of physiological and pathological states has been studied extensively in the past and still continues to be investigated from a mechanistic standpoint. Literature is replete with reports demonstrating the existence of an inverse causal relationship between the levels of Se and free radical-mediated oxidative stress (12, 19, 35). Rotruck et al. (36) demonstrated that dietary Se, which functions as an antioxidant through selenoproteins, prevented the erythrocyte lysis (autohemolysis) and formation of MetHGB in vitro. However, there has been no follow-up of these studies to address the underlying molecular mechanisms. We report here the importance of adequate Se nutrition in regulating red cell homeostasis by mitigating oxidative stress-dependent modulation of two important transcription factors, FoxO3a and HIF1α, in erythropoiesis.
Protein carbonyls, formed by oxidation of proteins, are considered to be one of the reliable markers of oxidative stress (30). Along these lines, increased protein carbonyls and LPO in the Se-deficient group demonstrated uncontrolled production of ROS and free-radical-mediated oxidative stress. Such changes in the Se-deficient group can be explained on the basis of decreased levels of selenoproteins, such as GSH-Px1 (21, 22), leading to peroxide accumulation and altered redox environment in cells, where biomolecules become more susceptible to oxidative damage (26).
Selenium-deficiency-induced oxidative stress explains some of the changes observed in the hematological parameters and enhanced levels of MetHGB. Previous reports also suggest an inverse relationship between dietary Se, hemolysis, and oxidation of HGB to MetHGB (36). Studies in the past have suggested the accumulation of denatured globins as possible mechanism of variety of hematological disorders (1, 20, 43). This prompted us to investigate the possibility that Se-mediated oxidative stress may denature the globin subunits. TAU gel electrophoresis indicated an increase in the membrane association of both α and β subunits of HGB in Se-deficient erythrocytes, which is suggestive of an ROS-mediated damage. In fact, monomeric α-HGB is known to generate ROS that damages cellular biomolecules that further aids in the propagation of free radical production (39, 40, 42). This explains increased susceptibility of the Se-deficient group to these changes. Similarly, enhanced levels of MetHGB, Heinz bodies, and echinocytes were seen in the blood smears of Se-deficient group than in the mice maintained on an Se-adequate diet (Table 1). Taken together, these data corroborate well with the increased levels of globin denaturation that is possibly a result of oxidation of ferrous ions in HGB leading to abnormalities in the structure and function of this protein.
The regulation of Epo by Se status is an intriguing finding. Hypoxia and oxidative stress regulate Epo, via HIF1α (41), which is critical for red blood cell survival and erythropoiesis. The increased expression of Epo in Se-deficient mice indicates that increased hypoxic stress possibly stimulates stress erythropoiesis. However, these erythrocytes do not mature causing a net decrease in the hematocrit due to an apparent lack of selenoproteins. Such an increase in Epo could serve as a compensatory mechanism in Se-deficient mice. Increased expression of HIF1α both in vitro and in vivo models of Se deficiency further lends credence to the idea that selenoproteins are perhaps indispensable for red cell homeostasis. The role of Epo in the protection against oxidative stress has been reviewed recently (27). Such an increase in cellular oxidative stress may have several ramifications not only on the gene expression, but also on the integrity of the red cell. In this regard, the “myelin figures” arising in the form of spicules in echinocytes suggest potential fragmentation and spherocytosis, signifying enhanced susceptibility to lysis during circulation (5). Consistent with this idea, the Se-deficient erythrocytes were found to be more prone to hemolysis when subjected to osmotic fragility test. Increased LPO leading to changes in membrane fluidity, membrane potential, and permeability (16), as well as increased globin denaturation, could collectively contribute to decreased erythrocyte life span and the development of anemia.
Despite anemia, Se-deficient mice do not exhibit increased reticulocytes, which would be indicative of compensatory stress erythropoiesis. When we analyzed erythroid progenitors in the bone marrow and stress erythroid progenitors in the spleen, Se-deficient mice had elevated numbers of progenitors. These data suggest that Se deficiency leads to inefficient erythropoiesis. Further, this observation suggests that erythroid progenitors and, in particular, stress BFU-E expand, but do not readily differentiate in response to Se-deficiency-induced anemia. One might speculate that stress erythropoiesis requires an Se-dependent function, in addition to FoxO3a and other stress-response genes, to rapidly generate new red cells at times of acute need.
FoxO3a is one of the pivotal transcription factors critical to repress oxidative stress, particularly during the differentiation and maturation phase of erythrocytes (28). Most importantly, activation of FoxO3a mitigates oxidative stress via the upregulation of many ROS scavenging enzymes, including GSH-Px1 (23, 31). Our results are in accordance with these recent findings that suggest the activation of FoxO3a in response to oxidative stress, particularly under Se-deficient conditions as well as in cells cultured under supraphysiological (>250 nM) Se levels. The variation in the levels of phosphorylated FoxO3a and unphosphorylated FoxO3a explains the differences in the response of erythrocytes when exposed to Se-induced oxidative stress under deficient and excess (250–500 nM) conditions. Earlier studies have shown that FoxO3a serves as an important sensor of cellular stress that helps in cell-fate decisions, particularly during conditions of oxidative stress (7). FoxO3a is regulated by 14-3-3 family of proteins. 14-3-3 dephosphorylates FoxO3a and directs it to the nucleus under oxidative stress conditions, which then leads to upregulation of a battery of antioxidant enzymes, including GSH-Px1. Further, given the Se-dependent redox regulation of 14-3-3 via direct binding through the Cys-rich motif (11), such a binding of SelW to 14-3-3 family members perhaps culminates in differential responses based on the levels of oxidative stress and selenoproteins present. The Se-dependent changes in the expression of 14-3-3βζ further supports the possible role of these proteins in the modulation of FoxO3a under conditions of Se deficiency. Even though FoxO3a are increased and 14-3-3 expression and nuclear levels are decreased, there is still a defect in the erythropoiesis pointing to the pivotal role of selenoproteins in erythropoiesis. However, under conditions of excess Se levels (>100 nM) it appears that there is some level of protection afforded by the complete expression of the selenoproteome along with all other FoxO3a-target genes that are likely to be expressed to alleviate oxidative stress-related effects without any apparent changes in the hematological parameters.
Based on these data, we believe that during Se deficiency, which is more common than Se toxicity, erythroid progenitors are under the influence of highest degree of free radical-mediated oxidative stress, leading to the activation of FoxO3a by 14-3-3 regulatory proteins. Consequently, together with the changes in downstream gene expression pathways, these events ultimately lead to loss of mature erythrocytes as observed in the Se-deficient mice. In the Se-adequate group, the high level of cellular pool of antioxidant selenoproteins promotes the FoxO3a pathway toward DNA repair and activation of detoxification genes, leading to efficient erythropoiesis. In conclusion, we have shown that cellular Se status plays a pivotal role in erythropoiesis by differentially regulating two major transcription factors, HIF1α and FoxO3a, to mitigate oxidative stress-dependent effects on the hematological parameters exacerbated by Se deficiency.
Footnotes
Acknowledgments
The authors thank Drs. Ross Hardison (Penn State University) and Mitchell Weiss (Children's Hospital of Philadelphia) for providing the G1E cells. These studies were supported by funds from the National Institute of Health PHS grant DK 077152 to K.S.P.
Author Disclosure Statement
No competing financial interests exist.