
Abstract
Hypoglycemia is the main complication for patients with type 1 diabetes mellitus receiving intensive insulin therapy. In addition to the obvious deleterious effects of acute hypoglycemia on brain function, recurrent episodes of hypoglycemia (RH) have an even more insidious effect. RH impairs the ability of the brain to detect and initiate an appropriate counterregulatory response (CRR) to restore euglycemia in response to subsequent hypoglycemia. Knowledge of mechanisms involved in hypoglycemia detection and counterregulation has significantly improved over the past 20 years. Glucose sensitive neurons (GSNs) in the ventromedial hypothalamus (VMH) may play a key role in the CRR. VMH nitric oxide (NO) production has recently been shown to be critical for both the CRR and glucose sensing by glucose-inhibited neurons. Interestingly, downstream effects of NO may also contribute to the impaired CRR after RH. In this review, we will discuss current literature regarding the molecular mechanisms by which VMH GSNs sense glucose. Putative roles of GSNs in the detection and initiation of the CRR will then be described. Finally, hypothetical mechanisms by which VMH NO production may both facilitate and subsequently impair the CRR will be discussed. Antioxid. Redox Signal. 14, 505–517.
Introduction
Hypoglycemia counterregulation and hypoglycemia-associated autonomic failure
It is clear that maintaining a satisfactory range of blood glucose is a fundamental concern of the brain since this organ cannot synthesize glucose. Rather, the brain relies on a constant supply of glucose coming from the bloodstream. Thus, hypoglycemia is a profound threat to the brain. As a result, hypoglycemia triggers a coordinated series of neuroendocrine events that restore euglycemia. These events known as the counterregulatory response (CRR) are initiated in sequence as blood glucose levels fall (Fig. 1). As the blood glucose level decreases to 80–85 mg/dL, insulin secretion is inhibited. When the blood glucose level reaches 65–70 mg/dL, glucagon secretion increases followed by increases in epinephrine and then cortisol secretion (22, 46). The net result of the CRR is an increase in liver and kidney glucose production and a decrease in muscle and fat glucose uptake, which ultimately restore euglycemia (Fig. 1).
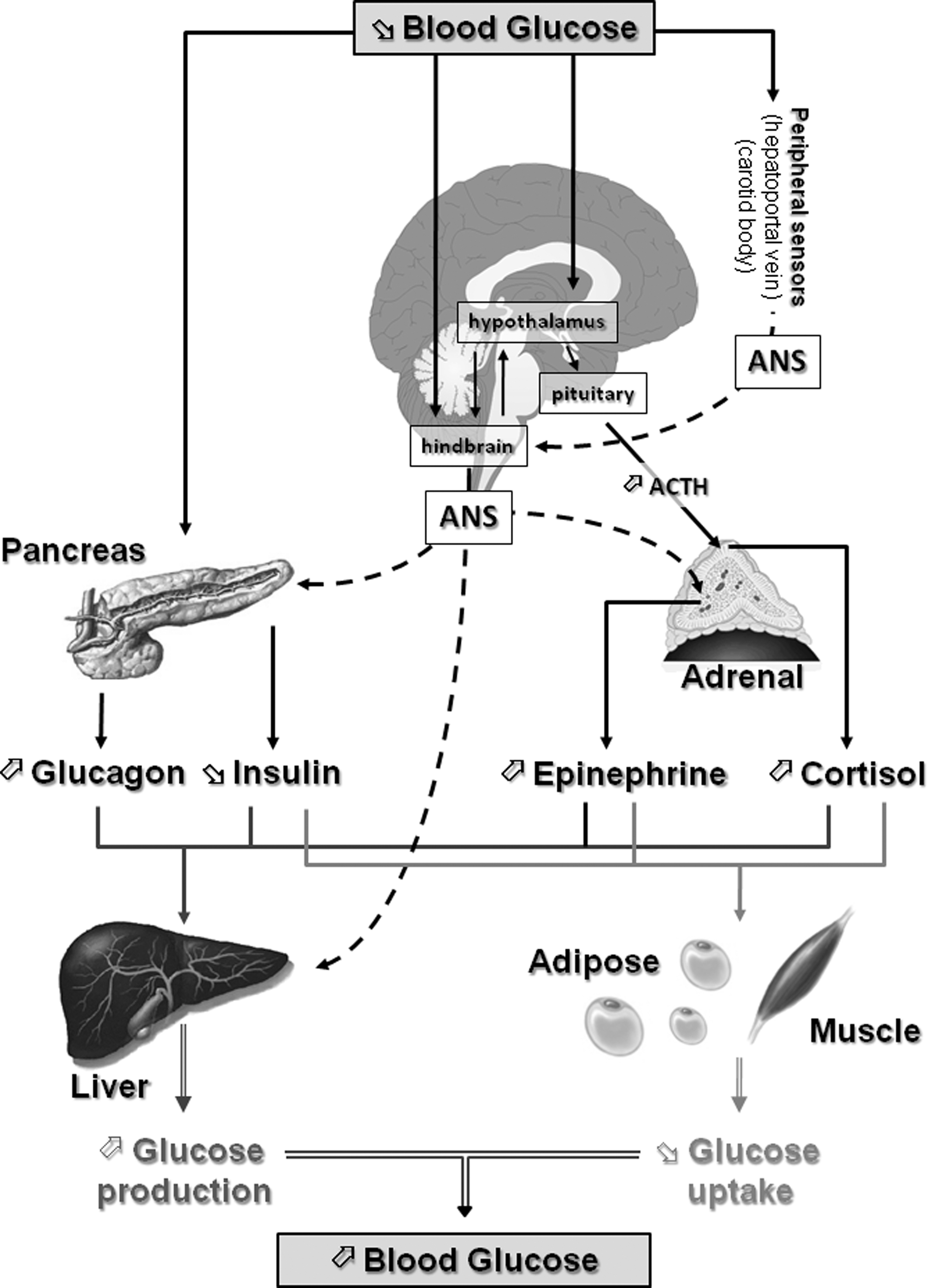
Unfortunately, CRR mechanisms become impaired as a consequence of recurrent hypoglycemic episodes (recurrent hypoglycemia, RH). This impairment is known as hypoglycemia-associated autonomic failure (HAAF). During HAAF, in addition to CRR impairment, hypoglycemia awareness is also altered. As a consequence, the glycemic threshold for CRR initiation and hypoglycemia awareness shifts to lower glucose levels. The major risk of HAAF is that blood glucose levels can fall to dangerously low or lethal levels without detection. Thus, the development of HAAF after RH is a major limiting factor in the management of patients with T1DM as well as patients with advanced type 2 diabetes mellitus who require intensive insulin therapy. Interestingly, HAAF can be reversed after 2–3 weeks of rigorous avoidance of insulin-induced hypoglycemia (22). Although HAAF is a serious health issue, the mechanisms by which it occurs, as well as reverses, are not well understood.
In this review, we will discuss what is known regarding central regulation of the CRR, and specifically the putative roles of glucose-sensing neurons (GSNs) in the ventromedial hypothalamus (VMH) in this response. We will focus on the novel role played by hypothalamic nitric oxide (NO) in CRR initiation and VMH glucose sensing. Finally, we will present possible mechanisms by which VMH NO production could also contribute to HAAF.
Central Nervous System Control of the CRR
Role of the VMH in the CRR
In animal studies, glucose sensors have been found both in peripheral tissues (pancreatic β-cells, intestine, carotid body, and hepatoportal vein) and in the central nervous system (amygdala, septum, striatum, motor-cortex, and hindbrain) (62), including the hypothalamus (15, 31, 68, 76). The respective roles of these peripheral and central glucose sensors in hypoglycemia detection and the control of the CRR will not be discussed here. However, it is noteworthy that the relative importance of the central versus peripheral glucose sensors in the initiation of the CRR depends on the rate of blood glucose level decline. In an elegant study, Donovan's group has recently shown that the hepatoportal glucose sensor dominates when hypoglycemia develops slowly and is responsible for over 90% of the sympathoadrenal response to hypoglycemia. When hypoglycemia develops faster, brain glucose sensors play a predominant role in the hypoglycemia detection and CRR initiation (64). While there are numerous studies that preferentially evaluate either peripheral or central glucose sensors during hypoglycemia, less is known about how these glucose sensors interact and coordinate the generation of the CRR. A recent review by Watts and Donovan begins to address this issue (77).
In the current review, we will focus our attention on the role played by the glucose sensors that exist in the VMH. It is generally well accepted that the VMH plays a key regulatory role in the control of the CRR. Electrical stimulation of the VMH in a rodent model activates the sympathoadrenal system in a manner similar to that seen during the CRR (72). Chemical destruction of the VMH with ibotenic acid inhibits the CRR to acute hypoglycemia (13). Local perfusion of the VMH with 2-deoxyglucose (2-DG, a nonmetabolizable form of glucose that mimics local hypoglycemia) stimulates the production of CRR hormones, whereas local VMH perfusion with glucose during systemic hypoglycemia inhibits the CRR (11, 14).
It is important to note the VMH actually consists of two different nuclei: the ventromedial (VMN) and the arcuate (ARC) nuclei. Their respective role in the control of the CRR is poorly understood. The VMN projects to autonomic outflow areas of the spinal cord via a brainstem projection to the periaqueductal gray and the reticular formation (24). Such projections are consistent with a stimulation of sympatho-adrenal responses seen during the initiation of the CRR. On the other hand, the ARC may be more involved in the control of food intake and energy expenditure [for review, see ref. (66)]. In vivo studies of the VMH lack the specificity to distinguish between the ARC and the VMN. Since VMN projections appear most likely to be involved in the CRR, we have chosen to use VMN glucose sensors in our in vitro studies as prototypes to understand the cellular basis of central regulation of the CRR. This, however, does not exclude a role for ARC or other central glucose sensors in CRR regulation.
The VMN glucose sensors
Hypothalamic neurons that modulate their electrical activity in response to changes in extracellular glucose level were first characterized in the 1960s (5, 57). These GSNs use glucose, not only as fuel, but also as a signaling molecule that regulates their electrical activity. In most cerebral areas protected by the blood–brain barrier (including the VMN), brain extracellular glucose level is about 30% of that found in blood. Thus, the extracellular brain glucose level ranges from 0.16 to 4.5 mM during peripheral hypoglycemia (2–3 mM) or hyperglycemia (≥15 mM), respectively. During euglycemia, brain extracellular glucose concentrations are approximately 2 mM (63). In the VMN, two types of GSNs have been found that respond directly to changes in extracellular glucose levels within this physiological range: the glucose-excited (GE) neurons and the glucose-inhibited (GI) neurons (68). GE neurons decrease whereas GI neurons increase their electrical activity as the glucose level decreases from 2.5 to 0.1 mM (68). Interestingly, VMN GE and GI neurons are exquisitely sensitive to decreases in glucose below 2.5 mM while remaining relatively insensitive to glucose increases above this level (68). This suggests that VMN GE and GI neurons are capable of playing a role in the detection of hypoglycemia and initiation of the CRR.
GE neurons
GE neurons exhibit a concentration-dependent increase in activity as extracellular glucose concentration rises (Fig. 2) (20, 68, 76). The mechanisms by which these neurons sense changes in glucose levels are fairly well understood. It has been proposed that GE neurons use similar glucose-sensing mechanisms as the pancreatic β-cell, which secretes insulin in response to increased glucose levels. In the β-cell, glucose is transported by the high-capacity glucose transporter 2 (GLUT2) and phosphorylated by the hexokinase IV isoform, glucokinase (GK). Glucose metabolism increases the intracellular ATP/ADP ratio, which consequently closes ATP-sensitive potassium channels (KATP channels). KATP channel closure depolarizes the β-cell and leads to Ca++ entrance, which in turn stimulates the exocytosis of insulin-containing vesicles. In the β-cell, GLUT2, GK, and KATP are considered the key glucosensors responsible for glucose-stimulated insulin secretion.

KATP channels, the pancreatic form of GK and GLUT2, are found throughout the brain, including hypothalamic regions involved in glucose sensing (6, 25, 27). Subunits of KATP channels and GK are expressed and functional in VMH GE neurons (41). Modulating the activity of KATP channels or GK alters glucose sensitivity of VMH GE neurons (7, 40, 68). Less is known regarding the involvement of GLUT2 in VMN GE neurons glucose sensing. GLUT2 has been mainly found in astrocytes but not in neurons (6). In addition, Kang et al. showed that GLUT2 is only expressed in ∼20% of VMH GE neurons (41). Other GLUTs such as GLUT3 and the insulin-sensitive transporter GLUT4 are expressed in hypothalamic neurons (4, 28, 41, 75). GLUT3 is expressed in 83% and GLUT4 in 57% of VMN GE neurons (41). Thus, whether a specific GLUT confers glucose sensitivity has yet to be determined.
The question that remains to be answered is whether VMH GE neurons play a role in the CRR to hypoglycemia. Alterations in the glucose-sensing machinery of VMN GE neurons are associated with CRR impairment, which is consistent with a role of GE neurons in the CRR. For example, glucagon production in response to insulin-induced hypoglycemia is impaired in Kir6.2-deficient mice lacking both functional KATP channels and VMH GE neurons (52). In addition, Sherwin's group showed that in vivo modulation of activity of KATP channels with sulphonylureas alters the CRR. VMH infusion of glibenclamide (KATP channel blocker) suppresses, whereas diazoxide (KATP channel opener) amplifies, the CRR during hypoglycemic clamp (29, 47). Levin and colleagues showed that modulation of GK activity altered both the glucose sensitivity of VMH GE neurons in vitro and the CRR in vivo (26, 40, 43). These data suggest that VMH GE neurons, through KATP channels and GK, play an important role in the control of the CRR. However, KATP channel expression is ubiquitous and is not specific to GE neurons. Thus, modulation of KATP channel activity would alter the electrical activity of other GSNs as well as non-GSNs. In contrast, GK expression seems relatively specific to VMH GSNs (41). However, GK has a very low glucose affinity (∼10 mM), which is well above the brain glucose concentration range. Thus, the role played by GK in hypoglycemia detection still remains controversial.
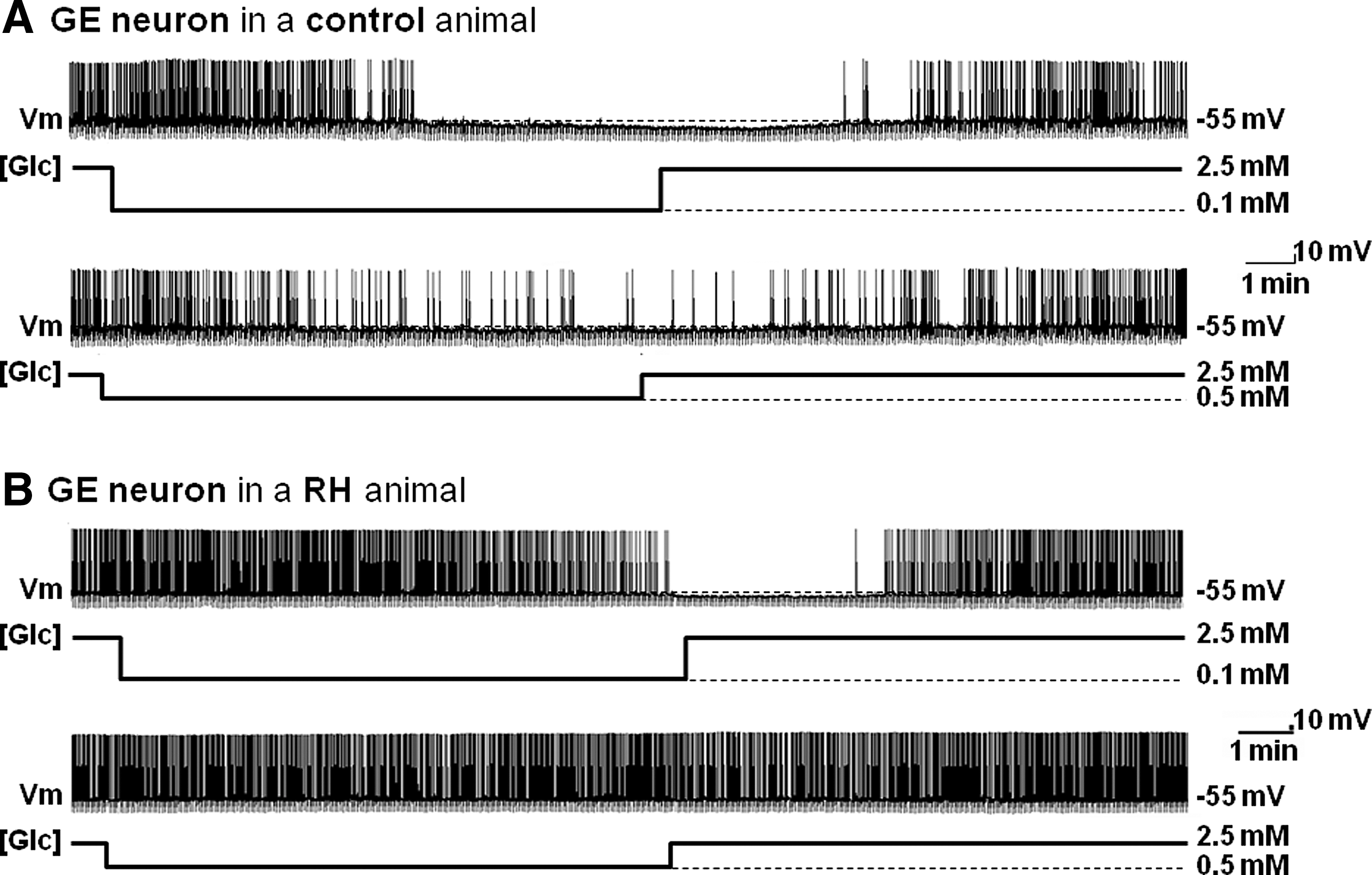
Further evidence from our laboratory suggests that GE neurons are involved in the CRR. We found that the response of VMN GE neurons to decreased glucose is attenuated under conditions where the CRR is impaired. Our laboratory and others have shown that the CRR is impaired after three consecutive daily episodes of insulin-induced hypoglycemia in rats (RH model) (23, 70). Using this model, we found that RH impairs the response of VMN GE neurons to decreased glucose (Fig. 3). In control animals, VMN GE neurons decreased their input resistance, membrane potential, and action potential frequency (APF) in response to glucose decreases from 2.5 to 0.5 or 0.1 mM. However, in RH animals, VMN GE neurons only decrease their input resistance, membrane potential, and APF when glucose is lowered to 0.1 but not 0.5 mM (Fig. 3). Thus, the threshold glucose level at which GE neurons become inhibited is lowered after RH. These results are consistent with a role for VMN GE neurons in the CRR. However, more work is necessary to further confirm a causal role between impaired VMN GE neuronal glucose sensing and impaired CRR.
GI neurons
GI neurons increase their electrical activity as extracellular glucose concentration decreases (Fig. 4) (68). Recently, significant progress has been made in the elucidation of the VMH GI neuronal glucose-sensing mechanisms. GK, AMP-activated kinase (AMPK), neuronal NO synthase (nNOS), and the cystic fibrosis transmembrane regulator (CFTR) are all critical for glucose sensing. The specific GLUT is still uncertain. Among VMN GI neurons, 22% express GLUT2, 96% express GLUT3, and 63% express GLUT4 (41). Thus, as for GE neurons, a specific GLUT has yet to be implicated. GK is expressed in about 50% of VMH GI neurons (41). Modulation of GK activity alters the glucose sensitivity of VMH GI neurons (26, 40). These data suggest that, as in GE neurons, GK is an important regulator of glucose sensing in at least some VMN GI populations.
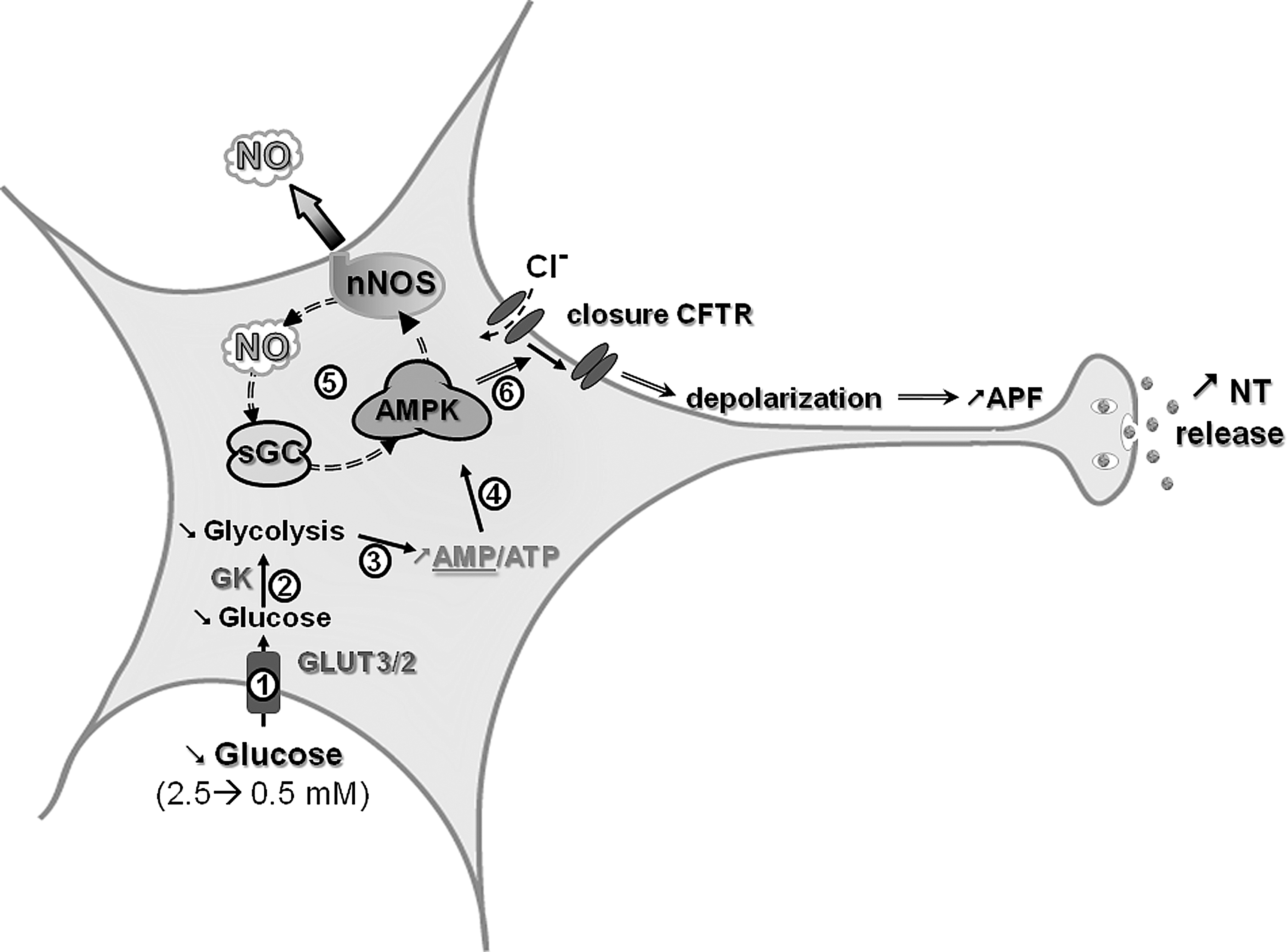
In contrast to GE neurons, AMPK is a key component of glucose sensing in GI neurons (16, 21, 56). Alterations in AMPK activity are associated with changes in glucose sensitivity of VMH GI neurons. Decreased glucose decreases the ATP/AMP ratio and increases VMH AMPK activity (56). Activation of AMPK activity mimics while inhibition of AMPK activity inhibits the response of VMH GI neurons to decreased glucose (54, 56). We have recently shown that nNOS is a downstream mediator of AMPK effects in GI neurons. It is well established that AMPK phosphorylates and activates nNOS in many tissues, including the brain (2, 44, 80). We have found this to be true in VMN GI neurons as well (16, 56). Further, VMH GI neurons produce NO in response to decreased glucose (16). NO production is a fundamental step in VMH GI neuronal glucose sensing. nNOS-deficient mice lack VMH GI neurons (32). In addition, nNOS inhibition impairs VMH GI neuronal responses to decreased glucose as well as AMPK activation (56).
The downstream effects of NO on the glucose response of VMN GI neurons are mediated through its receptor, soluble guanylyl cyclase (sGC), which is found in the majority of VMH neurons (16). When activated by NO, sGC increases cyclic GMP (cGMP) levels, leading to depolarization of VMN GI neurons. Interestingly, the NO-sGC-cGMP pathway is not only downstream of AMPK but also provides a necessary amplification of AMPK activity in VMN GI neurons. Our data indicate that AMPK inhibition prevents the cell permeable analog of cGMP, 8-Br-cGMP, from depolarizing GI neurons. Thus, as in skeletal muscle (44) and endothelial cells (80) an AMPK-NO-AMPK signaling loop links changes in intracellular energy availability to physiological responses.
KATP channels are expressed and functional in VMH GI as well as GE neurons (41, 69). However, in GI neurons increased electrical activity in response to decreased glucose is correlated with the closure of a chloride conductance (30, 68). The CFTR is believed to be the chloride channel responsible for the electrical activity changes in GI neurons. The CFTR is expressed in several brain regions, including the hypothalamus (55). We have recently shown that CFTR phosphorylation is regulated by AMPK in the VMH (56). Moreover, the CFTR modulator gemfibrozil inhibits VMH GI neurons response to decreased glucose (30). The CFTR channel is a member of ATP binding cassette and possesses sulphonylurea-binding subunits (SUR) as do KATP channels (60). Thus, it is important to note that in vivo studies using sulphonylureas do not target GE neurons specifically through modulation of KATP channel activity since GI neurons may also be affected as a result of interactions with the sulfonylurea subunit of either KATP or CFTR channels. The hypothetical mechanism for glucose sensing of VMN GI neurons as indicated by our data is shown in Figure 4.
Like GE neurons, the fact that VMN GI neurons are particularly sensitive to glucose deficit makes them well suited to play a role in hypoglycemia detection and counterregulation. This hypothesis is supported by that fact that components of VMN GI neuronal glucose-sensing machinery found to be activated by decreased glucose in vitro are also activated in vivo. That is, VMH AMPK activity is increased in response to insulin-induced hypoglycemia (34) and to cerebral glucopenia (3). VMH NOS activity is also increased in response to insulin-induced hypoglycemia. In addition, manipulating VMN GI neuronal glucose-sensing machinery in vivo alters the CRR. For example, modulation of VMH GK activity affects the CRR (43). Moreover, VMH AMPK inhibition attenuates the CRR to acute hypoglycemia (34, 50). In contrast, stimulating VMH AMPK activity amplifies the CRR to acute hypoglycemia and restores the impaired CRR after RH (48, 49). Interestingly, while the AMPK activator AICAR stimulates VMH GI neurons in vitro, VMH AICAR infusion during euglycemia does not initiate the CRR (48). This is consistent with the idea that multiple glucose sensors are necessary to coordinate the initiation of the CRR.
The hypothesis that VMN GI neurons play a role in the control of the CRR is further supported by numerous studies, indicating that VMN GI neuronal glucose sensitivity is altered under conditions where the CRR is impaired. First, the response of VMN GI neurons to decreased glucose is reduced after RH when the CRR is impaired (70). Second, alterations in VMH fuel source availability and/or utilization during hypoglycemia causes CRR impairment and reduces the response of VMN GI neurons to decreased glucose. Like glucose, other energy sources (e.g., lactate and ketone bodies) enter the brain and support energy metabolism during glucose deficiency. VMH lactate infusion inhibits the CRR during hypoglycemia (12). Similarly, we found that increased extracellular lactate levels attenuated the increase in APF of VMN GI neurons as glucose decreased from 2.5 to 0.1 mM (69). Thus, lactate supplementation reduces the response of VMN GI neurons to glucose deficit. Third, activation of the hypothalamic-pituitary-adrenal axis impairs the CRR to subsequent hypoglycemia and reduces VMN GI neuronal response to decreased glucose. Systemic perfusion of corticotrophin-releasing factor or VMH infusion of the endogenous corticotrophin-releasing factor receptor 2 agonist urocortin inhibits the CRR in vivo (51). Similarly, we found that urocortin decreases VMN GI neuronal response to decreased glucose in vitro (51). Finally, our recent data showing that VMH NO signaling is crucial for both the full generation of the CRR in vivo and the ability of VMN GI neurons to sense glucose in vitro provide a further support for the hypothesis that VMN GI neurons play a key role in hypoglycemia detection and the initiation of the CRR (32). For the remainder of this review we will focus on the putative role of NO signaling in central glucose sensing and the development of HAAF.
The Role of NO in the CRR
NO is critical for activation of the CRR
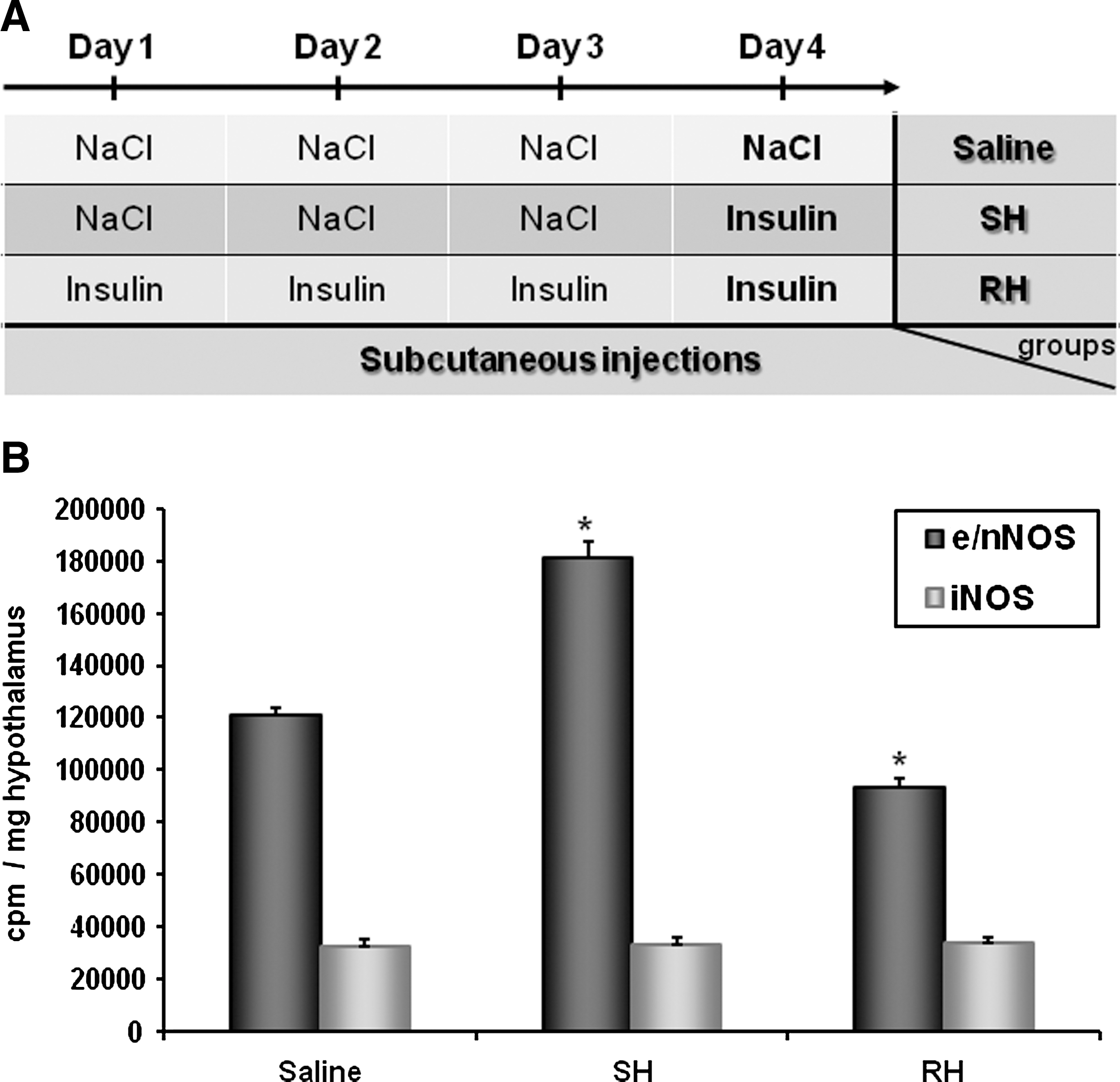
As mentioned previously, decreased glucose activates nNOS and increases NO production in VMH GI neurons (16). Using in vitro and in vivo strategies, we recently confirmed that nNOS-derived NO is produced in the VMH during hypoglycemia. Insulin-induced hypoglycemia increases VMH constitutive NOS (nNOS + endothelial [e] NOS) activity. Increased NOS activity during hypoglycemia is associated with phosphorylation of the nNOS but not endothelial NOS (eNOS) isoform. Moreover, we found that decreased glucose levels in vitro increase extracellular NO release using electrochemical detection of NO production. Next, we established that hypothalamic NO production is necessary for the full generation of the CRR. Plasma epinephrine production is impaired during a hyperinsulinemic/hypoglycemic clamp in animals injected in the VMH with a nonselective NOS inhibitor. Additionally, nNOS-deficient mice have an impaired CRR and lack VMN GI neurons (32). To strengthen our hypothesis that hypothalamic NO production is a critical step for the full generation of the CRR, we measured hypothalamic NOS activity in response to insulin-induced hypoglycemia after RH. Constitutive NOS activity is increased in response to insulin-induced hypoglycemia in naive animals injected with saline daily for 3 consecutive days. In contrast, no increase in constitutive NOS activity was detected in RH animals (injected with insulin daily for 3 consecutive days) in response to a subsequent episode of insulin-induced hypoglycemia (Fig. 5). This shows that increased NOS activity in response to insulin-induced hypoglycemia is impaired after RH. These data support the idea that VMH NO production is an important component of the central response to hypoglycemia and initiation of the CRR. Determination of the mechanisms leading to impaired NOS activity after RH may lead to the development of new therapeutic strategies to prevent the development of HAAF.
Potential functions of hypothalamic NO during hypoglycemia
NO, as a gas, diffuses freely across cell membranes, providing the opportunity to affect adjacent cells. The NO receptor sGC is distributed widely in the brain, and is present in virtually all VMH neurons (16, 33). Thus, NO produced by VMH GI neurons could modulate the activity of surrounding VMH glucose sensing and non-GSNs or even glia or neighboring capillaries. The consequences of such effects of NO are discussed in the following sections.
NO and blood flow
In peripheral tissues, one of the main physiological functions of NO is related to the vascular system. It is well established that NO produced by endothelial cells through the eNOS isoform induces the relaxation of neighboring smooth muscle cells. This leads to vasodilatation and increased blood flow (53). Human and animal studies showed that insulin-induced hypoglycemia is associated with increased cerebral blood flow in different brain areas, including the hypothalamus (42, 58). Hypoglycemia-induced increases in cerebral blood flow could increase nutrient availability to brain cells during energy deficit. Insulin-induced hypoglycemia does not increase eNOS activity (32). However, NO produced by VMH GI neurons via the nNOS isoform could diffuse to adjacent smooth muscle cells and increase blood flow locally. The role of NO on the control of brain blood flow during hypoglycemia remains controversial. Horinaka et al. showed that hypoglycemia-induced increased blood flow was not inhibited by peripheral administration of a nonselective NOS inhibitor (37). Nevertheless, concerns have been raised regarding the ability of NOS inhibitors administered peripherally to cross the blood–brain barrier and enter the brain in adult rats (9, 19). Further in vivo studies using local inhibition of NOS activity during hypoglycemia are required to determine whether hypothalamic NO production is involved in hypoglycemia-related vasorelaxation.
NO and nutrient supply
Studies performed by Bolanos' group suggest that NO increases energy supply during energy deficit (Fig. 6). NO increases GLUT1 and GLUT3 expression in astrocytes and neurons, respectively (17). In primary cortical cultures NO increases glycolytic flux in astrocytes (2, 10). Bolanos and colleagues proposed that NO impairs mitochondrial respiration via cytochrome c inhibition, which initially decreases the rate of ATP formation. The resulting increase in the AMP/ATP ratio increases AMPK activity. AMPK phosphorylates and activates 6-phosphofructo-2-kinase. 6-Phosphofructo-2-kinase increases the formation of fructose-2,6-bisphosphate from fructose-6-phosphate. Fructose-2-6-bisphosphate is the most potent activator of the rate limiting enzyme in glycolysis, phosphofructo-1-kinase. Thus, increased fructose-2-6-bisphosphate increases the rate of glycolysis, leading to ATP and pyruvate formation. Because mitochondrial respiration is inhibited, pyruvate is then transformed to lactate by lactate dehydrogenase rather than metabolized through the Krebs cycle. Increased glycolytic ATP level compensates for the energy deficit in astrocytes (9, 10) (Fig. 6). Lactate cannot be used by astrocytes as a source of ATP under anaerobic conditions but may be transported to adjacent neurons to fulfill their energy needs during hypoglycemia (see below). Astrocytes are also known to store energy as glycogen. Glycogen stores provide substrates for glycolysis, leading to a further increase in ATP and lactate in response to NO. Thus, NO produced by GI neurons may restore energy deficit in astrocytes during hypoglycemia by increasing glycolytic ATP.

In neurons, however, because of low expression of 6-phosphofructo-2-kinase, decreased ATP levels resulting from NO-dependent cytochrome c inhibition do not enhance the glycolytic production of ATP as seen in astrocytes (2, 9, 10). Thus, the ATP deficit could lead to energy crisis and neuronal death. However, Magistretti's group has proposed that astrocytes have the ability to fuel neurons through an astrocyte-neuron coupling pathway involving lactate transport. They called this model the “astrocyte-neuron lactate shuttle” (45, 59). The astrocyte-neuron lactate shuttle model states that during increased neuronal activity, the decrease in neuronal energy level would be compensated by astrocytic lactate production from glycogen stores and/or circulating glucose. Lactate produced by astrocyte could be transported to adjacent neurons via monocarboxylic acid transporters and fulfill their energy demands. Combining Bolanos' and Magistretti's models leads to the hypothesis that NO produced by VMH GI neurons during insulin-induced hypoglycemia increases lactate production by neighboring astrocytes to fuel surrounding glucose- or nonglucose-sensitive neurons via the astrocyte-neuron lactate shuttle (Fig. 6). In support of this hypothesis, monocarboxylic acid transporter 1 expression is increased during hypoglycemia (74). However, no one has been able to actually measure increased extracellular lactate during hypoglycemia. One possibility is that lactate is taken up by neurons as fast as it is released from astrocytes. Thus, increased extracellular lactate concentration during hypoglycemia would not be detectable. The other possibility, of course, is that the astrocyte-neuron lactate shuttle hypothesis is not valid in the hypothalamus during hypoglycemia. More work is required to determine whether NO-stimulated lactate production by astrocytes fuels neurons during insulin-induced hypoglycemia.
NO and GE neurons
We found that VMN GE neurons are fully functional in nNOS knockout mice. However, NO produced by GI neurons could diffuse to adjacent GE neurons and affect their glucose sensitivity. As discussed above, NO inhibits the mitochondrial cytochrome c and decreases mitochondrial respiration and ATP formation. Consequently, VMH GI neuronal NO production could further decrease ATP level in GE neurons during decreased extracellular glucose. Thus, the response of VMH GE neurons to decreased glucose would be greater in the presence of surrounding NO producing GI neurons. On the other hand, lactate produced by astrocytes in response to GI neuronal NO production could provide an energy source to GE neurons masking glucose deficit. In this case, NO produced by GI neurons could decrease the response of VMH GE neurons to subsequent glucose decrease. This hypothesis is discussed more in detail in the next section. Further investigation is needed to determine whether NO produced by GI neurons influences the glucose sensitivity of GSNs.
To summarize, there are several putative functions for NO produced by VMH GI neurons in the control of energy homeostasis in addition to its role in GI neurons glucose sensing. VMH NO production in GI neurons could participate in increased blood flow locally during hypoglycemia. NO production in GI neurons could also stimulate glycolysis in astrocytes and increase neuronal fuel supplies as a result of the astrocyte-neuron lactate shuttle. Finally, VMH NO production during insulin-induced hypoglycemia could modulate glucose sensitivity of VMH GE neurons.
Involvement of hypothalamic NO in the CRR impairment after RH
As mentioned in the Introduction, the CRR becomes impaired after recurrent episodes of insulin-induced hypoglycemia. While the mechanisms involved in the initiation of the CRR are fairly well understood, mechanisms involved in its impairment after RH are not. Our data show that VMH NO production is required for full generation of the CRR and glucose sensing in GI neurons. In the last part of this review we will introduce the hypothesis that VMH NO production also plays a role in the CRR impairment to subsequent hypoglycemia after RH.
NO and lactate
We hypothesized above that VMH NO production increases astrocytic lactate production and transport to surrounding neurons through an astrocyte-neuron lactate shuttle. VMH infusion of lactate impairs the CRR during hypoglycemic clamp (12). Moreover, increased extracellular lactate concentration impairs the ability of VMN GI and GE neurons to respond to decreased glucose (69). The glucose-sensing machinery of both GI and GE neurons involves changes in intracellular ATP level. In GI neurons, decreased ATP/AMP resulting from decreased extracellular glucose activates AMPK, leading to CFTR channel closure and depolarization. In GE neurons, decreased intracellular ATP directly opens KATP channels, leading to hyperpolarization. Increased extracellular lactate concentration may raise neuronal ATP levels. Thus, increased lactate concentration would mask the effect of decreased glucose on the intracellular ATP level (Fig. 6). Enhancing astrocytic production of lactate to GSNs is therefore one mechanism by which NO production during hypoglycemia could decrease the sensitivity of GSNs to subsequent hypoglycemia and impair the CRR. This phenomenon could be exacerbated by the fact that hypothalamic glycogen content might be increased after RH as described by Alquier and colleagues (3). These data are, however, controversial since Herzog and colleagues found no difference in hypothalamic glycogen level after acute or RH (35). The difference between these two studies may be due to the different RH models used (3 consecutive days of intracerebro-ventricular 2-Deoxy-
NO and reactive oxygen species
NO may also play a role in the development of HAAF through the production of reactive oxygen species (ROS) (Fig. 7). ROS production in response to hypoglycemia is believed to occur in many brain regions, including the hypothalamus. Insulin-induced hypoglycemia increases the level of lipid peroxidation and DNA fragmentation while decreasing the level of reduced glutathione in rats (67). In addition, Leloup and Penicaud (CSGA, Dijon, France) evaluated ROS production in the VMH of rats injected with saline or insulin (4 U/kg, subcutaneously) by measuring the fluorescence of the ROS-sensitive probe H2DCFDA (5,6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester). They found that H2DCFDA fluorescence was significantly increased by ∼50% in the VMH of insulin-treated animals compared with saline controls (personal communication). These results provide further support that ROS are produced in the VMH in response to insulin hypoglycemia.
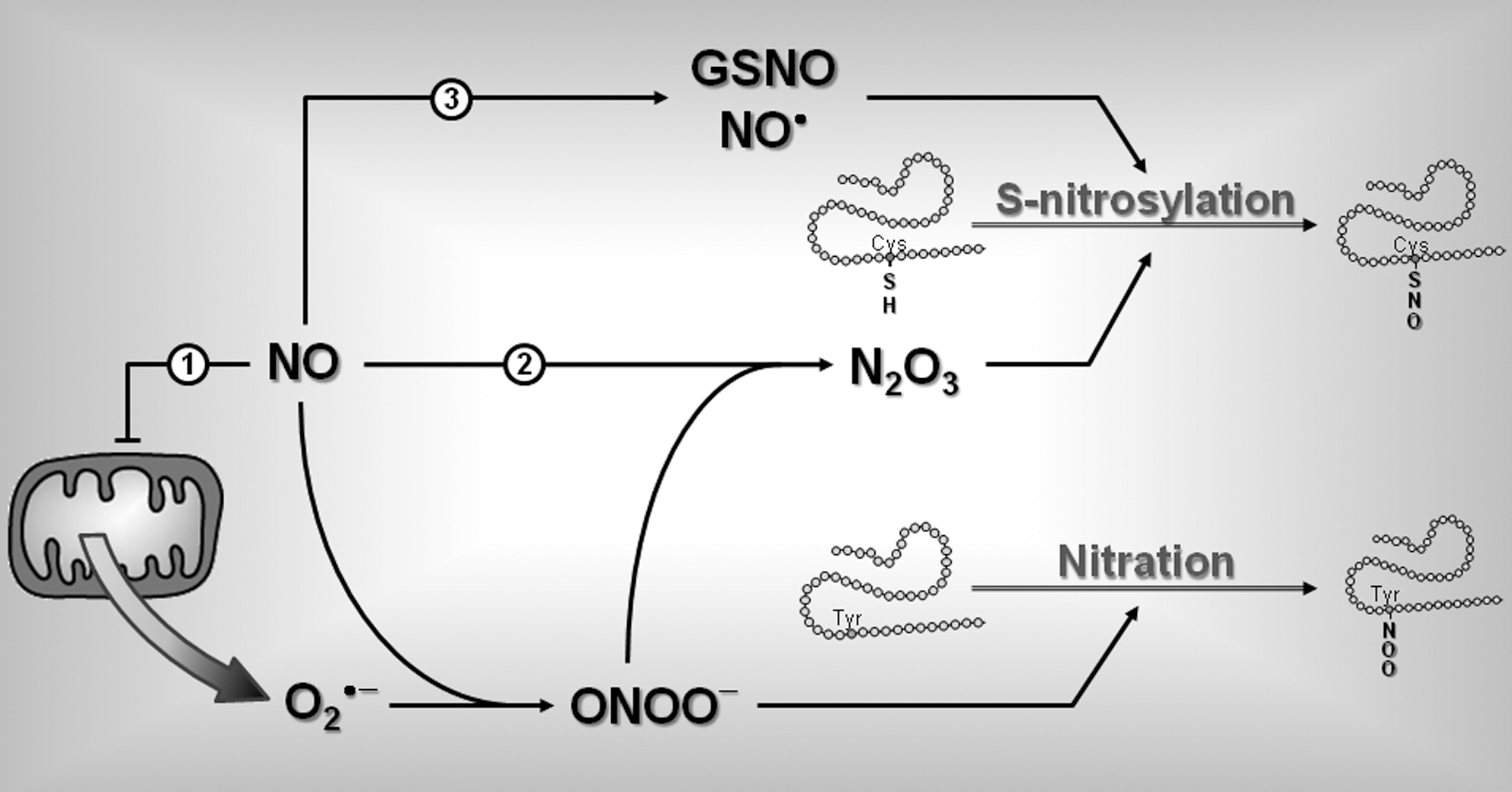
The mechanisms and cell types responsible for VMH ROS production during hypoglycemia are unknown. One hypothesis is that ROS formation is mediated by increased NO production. As mentioned earlier, NO inhibits cytochrome c and blocks mitochondrial respiration. A consequence of mitochondrial respiration inhibition is the production of the superoxide anion O2 •− (18) (Fig. 7). Since VMH GI neurons produce NO during insulin-induced hypoglycemia, one might hypothesize that these GI neurons are the source of ROS production. However, since NO can diffuse across cell membranes it is also possible that NO triggers ROS production in other cell types as well.
The effects of ROS on CRR impairment after RH are purely speculative at this time. However, increased NO and ROS production can alter protein function, which could lead to CRR impairment (Fig. 7). NO and superoxide readily combine to form peroxynitrite (ONOO−) (8). Peroxynitrite can modify protein structure in several ways, including the nonenzymatic reactions tyrosine nitration and S-nitrosylation. Tyrosine nitration consists in the addition of a nitro group (−NO2) to exposed tyrosine residue (38). In contrast, S-nitrosylation is a reaction that results in the addition of an NO moiety to the free thiol of a cysteine (Cys-SH) to form an S-nitroso-thiol (Cys-S-NO) (36, 71). Peroxynitrite can also react with another molecule of NO to form dinitrogen trioxide (N2O3), which increases the level of S-nitrosylation. In an oxidative environment, increased NO levels can also increase formation of nitroso-glutathione (GSNO) and/or reactive NO (nitrosium ion, NO•), both of which can react with free cysteine thiols on proteins, leading to increased S-nitrosylation (Fig. 7). A large number of peripheral and central proteins are targets of S-nitrosylation (39). Protein S-nitrosylation may be involved in the control of glucose homeostasis since proteins involved in glucose sensing are known targets of S-nitrosylation, including GK (61), nNOS (78), sGC (65), and CFTR (79). For instance, S-nitrosylation decreases NOS activity (78) and the sensitivity of sGC for NO (65). In both cases, S-nitrosylation of nNOS or sGC would decrease NO signaling. Since VMH NO-signaling is important for the generation of the CRR, inhibition of VMH NO-signaling by S-nitrosylation could lead to CRR impairment to subsequent hypoglycemia and development of HAAF. It is worth noting that this hypothesis is consistent with our results showing that RH reduces NOS activity and with the results of Leloup and Penicaud (personal communication) showing that hypoglycemia increases VMH ROS levels. However, the hypothetical role of protein nitration and/or S-nitrosylation in the development of HAAF remains to be determined.
Another hypothetical consequence of NO and ROS production is induction of cell apoptosis. Prolonged or repetitive concomitant NO and ROS production induces neuronal apoptosis. In support of this, one episode of severe hypoglycemia is sufficient to increase the number of apoptotic cells in the hypothalamus (73). The implication of apoptosis in the development of HAAF is yet another hypothesis that requires further evaluation
Conclusion
Our knowledge regarding mechanisms involved in the brain detection of hypoglycemia and the initiation the CRR has significantly evolved over the past 20 years (Fig. 8). A growing body of evidence supports a role for VMH GI and GE neurons in the regulation of the CRR. Further, the mechanisms by which they sense glucose are fairly well characterized with the notable exception of GLUT isoform involved. Work from our laboratory and others suggests that GK, and the KATP channel in GE neurons and AMPK in GI neurons are necessary for both glucose sensing and the CRR.
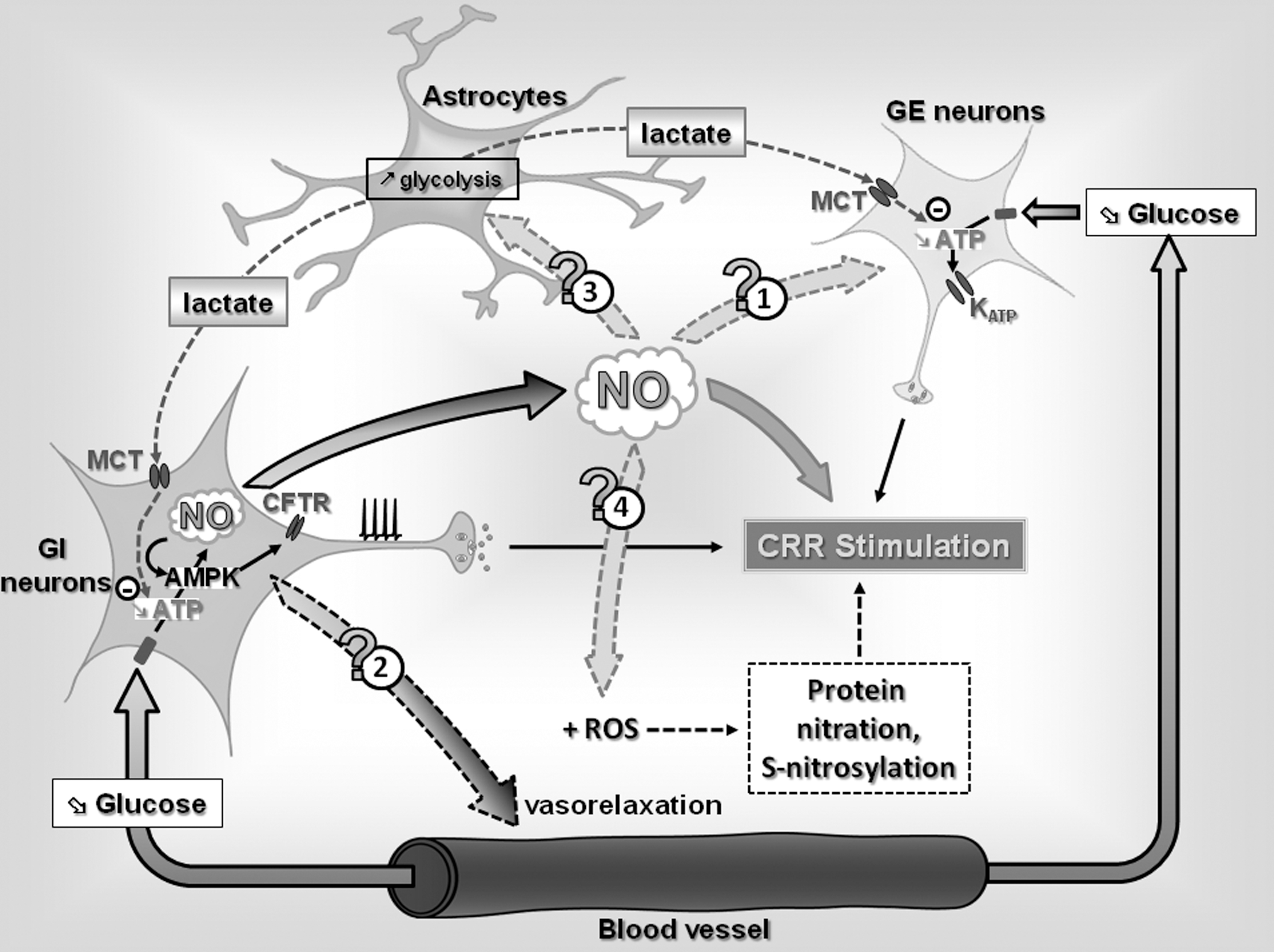
In the last few years, our laboratory has discovered the involvement of a new and important key determinant of glucose sensing in GI neurons and hypoglycemia counterregulation: NO. We found that (a) VMH GI neurons produce NO in response to decreased glucose; (b) production of NO by VMH GI neurons is required for glucose sensitivity in vitro, and (c) VMH NO production is important in the regulation of the CRR in vivo.
However, less is known regarding the mechanisms involved in CRR impairment after RH. As discussed in this review, we hypothesize that while NO is critical for central glucose sensing, NO may also participate in the development of HAAF. VMH NO production could (a) increase energy supply to GSNs and decrease their glucose sensitivity via increasing local blood flow or stimulating the astrocyte-neuron lactate shuttle or (b) increase ROS production during hypoglycemia and lead to alterations in glucose-sensing machinery and/or neuronal apoptosis (Fig. 8).
VMH NO production by GI neurons appears to be a necessary component of hypoglycemia detection and counterregulation. However, overproduction and/or a coproduction with ROS may impair the CRR and play a role in the development of HAAF. Determining whether and how VMH NO and ROS signaling are involved in the development of HAAF will enable us to safely wield the powerful two-edged sword: NO. In turn, such knowledge will provide new therapeutic targets to reduce the risks of hypoglycemia in patients with T1DM.
Footnotes
Acknowledgments
Authors would like to thank Dr. Thierry Alquier for helpful discussions and critical reading of the article. This work was supported in part by research awards from the Juvenile Diabetes Research Foundation (V.H.R.), the National Health Institute (2RO1DK55619 and 1RO1DK81358; V.H.R.), and a Juvenile Diabetes Research Foundation postdoctoral award (X.F.).