
Abstract
The discovery of ischemic postconditioning (IPost) has rejuvenated the field of cardioprotection. As an interventional strategy to be applied at the onset of myocardial reperfusion, the transition of IPost from a bench-side curiosity to potential clinical therapy has been impressively rapid. Its existence also confirms the existence of lethal myocardial reperfusion injury in man, suggesting that 40%–50% of the final reperfused myocardial infarct may actually be due to myocardial reperfusion injury. Intensive analysis of the signal transduction pathways underlying IPost has identified similarities with the signaling pathways underlying its preischemic counterpart, ischemic preconditioning. In this article, the reperfusion injury salvage kinase pathway and the more recently described survivor activating factor enhancement pathway, two apparently distinct signaling pathways that actually interact to convey the IPost stimulus from the cell surface to the mitochondria, where many of the prosurvival and death signals appear to converge. The elucidation of the reperfusion signaling pathways underlying IPost may result in the identification of novel pharmacological targets for cardioprotection. Antioxid. Redox Signal. 14, 893–907.
Introduction
In common with its preischemic counterpart, ischemic preconditioning (IPC), intensive investigation of the signaling pathways underlying IPost have identified a number of different signal transduction pathways conveying the cardioprotective signal from the sarcolemma to the mitochondria, some of which overlap with IPC. This article reviews two of these reperfusion signaling pathways, the reperfusion injury salvage kinase (RISK) pathway and the more recently described survivor activating factor enhancement (SAFE) pathway, two apparently distinct signal transduction pathways that may actually interact to convey IPost cardioprotection.
The RISK Pathway: Origins, Applications, and Controversies
The actual term RISK pathway was first coined by our research group in 2002, in a study investigating the signal transduction pathway underlying the infarct-limiting effects of urocortin administered at reperfusion (110). In that initial study, the mitogen extraregulated kinase 1/2 (MEK1/2)–extraregulated kinase 1/2 (Erk1/2) prosurvival kinase pathway was implicated as the mediator of urocortin-induced cardioprotection and the first member of the RISK pathway (110). Crucially, the administration of urocortin at the onset of myocardial reperfusion reduced infarct size and increased Erk1/2 activation over and above that achieved by reperfusion alone—importantly, the pharmacological inhibition of urocortin-induced Erk1/2 activation abolished the infarct-limiting effects of urocortin (110).
However, the concept of a cardioprotective antiapoptotic reperfusion pathway was actually first proposed in the late 1990s with the discovery that a variety of growth factors were capable of limiting myocardial infarct size when administered at the immediate onset of myocardial reperfusion (133). However, it must be appreciated that although these growth factors were first investigated for their potential antiapoptotic effects, it has in fact been their dramatic effects on necrosis and reductions in myocardial infarct size, which has been their most important contribution. Of course, the idea of protecting the heart by intervening at the time of reperfusion had been previously explored using a variety of approaches, some of which have been less successful, including antioxidant therapy, calcium channel blockers, anti-inflammatory agents, and so forth, with mixed results (134). However, initial results from more novel approaches such as cyclosporine A (105) and atrial natriuretic peptide (65) have been more promising. The novel aspect, therefore, was the possibility of recruiting prosurvival signaling pathways to protect the heart against lethal myocardial reperfusion injury. In this review article, the focus will be on the involvement of the RISK pathway in IPost signaling. For a more comprehensive account of the RISK pathway, the reader is directed to several recent reviews (17, 45, 48).
Reperfusion Signaling in IPost: The RISK Pathway
In the original description of IPost in 2003 (137), the infarct-limiting effects were attributed to the prevention of myocardial reperfusion injury-induced calcium overload, less oxidative stress, preserved endothelial function, attenuated apoptotic cell death, less myocardial inflammation, and edema. However, the first study to implicate an actual signal transduction pathway as a mediator of IPost was by our research group in 2004 (127). In that study, we demonstrated that six 10-s cycles of reperfusion and ischemia applied at the immediate onset of myocardial reperfusion phosphorylated Akt, endothelial nitric oxide synthase (eNOS), and p70S6K to a greater extent than uninterrupted reperfusion in the isolated perfused rat heart (Fig. 1). Crucially, the pharmacological inhibition of phosphatidyl inositol 3 kinase (PI3K) during the IPost protocol using either LY294002 or Wortmannin abolished the infarct-limiting effects of IPost and abrogated the phosphorylation of Akt, eNOS, and p70S6K (Fig. 1). Interestingly, the recruitment of the RISK pathway is shared with the phenomenon of IPC (43), suggesting that the RISK may be a common pathway for cardioprotection, which can be activated either prior to ischemia or at the onset of myocardial reperfusion (44, 47).
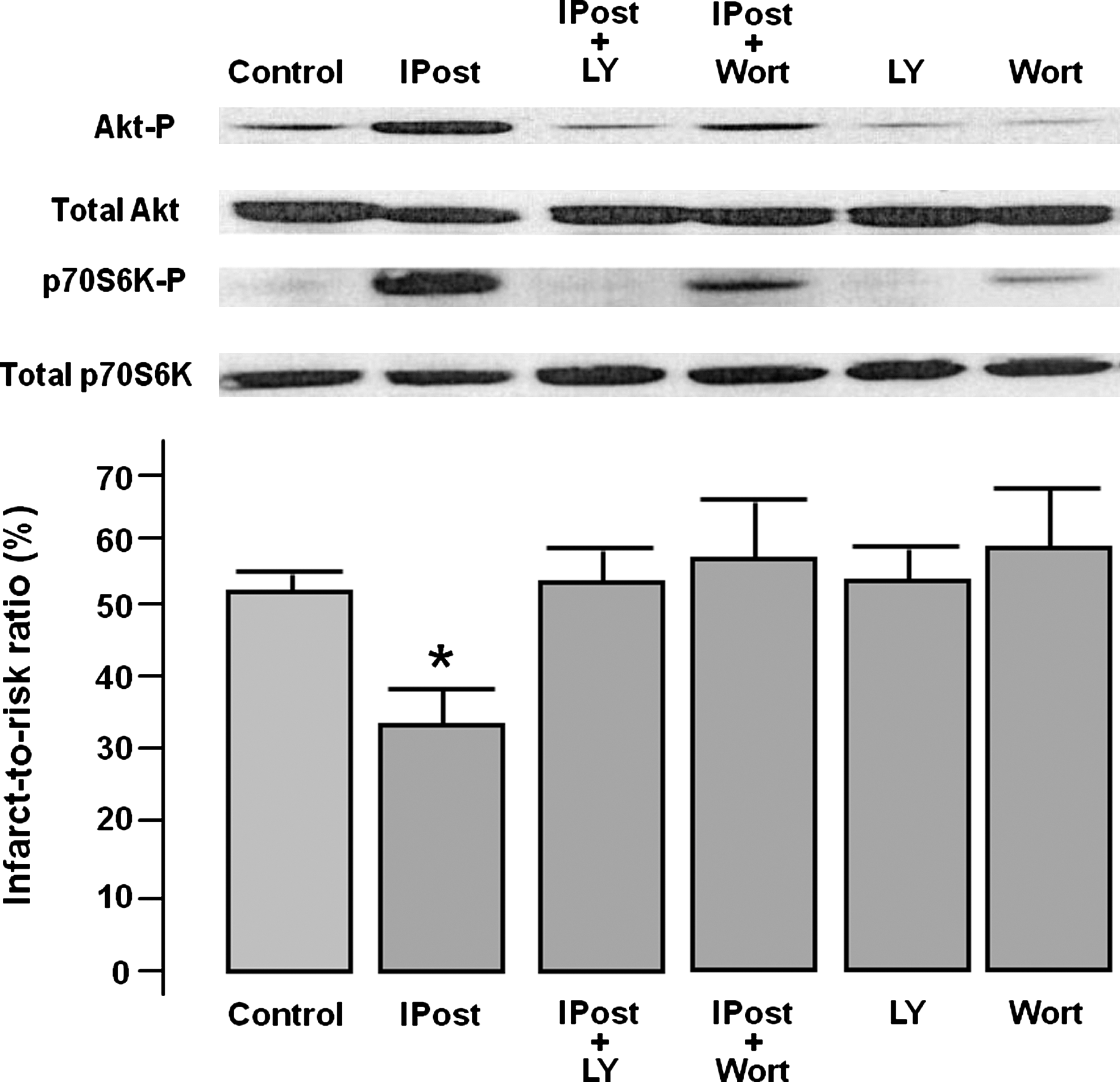
The first study to implicate the MEK1/2-Erk1/2 component of the RISK pathway was that by Yang et al. (132), who demonstrated that pharmacological inhibition of MEK/12 during IPost abrogated cardioprotetion in the isolated rabbit heart. A number of experimental studies have subsequently confirmed the involvement of the RISK pathway in IPost signaling and these are briefly summarized in Table 1. There are several other protein kinases that have been implicated in IPost signaling, which could also be considered components of the RISK pathway, such as protein kinase C (PKC), protein kinase G, p38 mitogen-activated protein kinase (p38MAPK), and Jun N-terminal kinase MAPK.
Erk, extraregulated kinase; eNOS, endothelial nitric oxide synthase; GSK3β, glycogen synthase kinase 3 beta; IS, infarct size; MEK, mitogen extraregulated kinase; PI3K, phosphatidyl inositol 3 kinase; RI, reperfusion ischemia; RISK, reperfusion injury salvage kinase.
The mechanism responsible for activating components of the RISK pathway in IPost is not clear, but has been attributed to cell-surface receptor binding of autacoids such as adenosine.
Upstream Activators of the RISK Pathway
The actual mechanism through which components of the RISK pathway are activated at the onset of myocardial reperfusion are not clear. Most evidence supports the activation of cell-surface receptors during the IPost stimulus, which recruit components of the RISK pathway. It is clear from the literature that recruiting components of the RISK pathway at the onset of myocardial reperfusion to confer cardioprotection can be initiated via the pharmacological activation of a variety of cell-surface receptors including G-protein coupled receptors, cytokine receptors, tyrosine kinase receptors, and serine threonine receptors (48, 49). The realization that the signaling pathways underlying IPost included components of the RISK pathway resulted in a number of different pharmacological postconditioning agents being investigated, including inhalational anesthetics, which have been extensively studied as pharmacological postconditioning agents capable of recruiting the RISK pathway and its downstream components (17). Interestingly, some of these receptors that convey cardioprotection when activated by pharmacological agents appear to be involved in IPost cardioprotection. Two experimental studies have demonstrated directly the involvement of cell-surface receptors in the activation of the RISK pathway. Morrison et al. (90) reported the failure of IPost to activate the RISK pathway in mice lacking the adenosine A2A receptor, suggesting that, in this case, the A2A receptor may be responsible for the endogenous activation of the RISK pathway in the postconditioned heart. There has been some disagreement on the adenosine receptor subtype involved in IPost signaling. Other experimental studies have implicated the adenosine A2B receptor and the A3 receptor as potential triggers of cardioprotection. However, why IPost should be selective for a specific adenosine receptor subtype is not clear, given that at the onset of myocardial reperfusion the myocardium would be awash with adenosine. A more recent study has linked the binding to the sphingosine kinase-1 receptor as a potential upstream activator of the RISK pathway (56). Mice lacking the sphingosine kinase-1 receptor were found to be resistant to IPost and did not phosphorylate Akt and Erk1/2 in response to a standard IPost stimulus. It is probable that many of the other cell-surface receptors implicated as triggers of IPost, such as the bradykinin (104) and opioid (136) receptors, also activate the RISK pathway but this remains to be demonstrated directly. IPost has also been reported to protect the heart by delaying the restoration of physiological pH (20, 31), which then prevents the activation of calpain (55). The actual interplay between these beneficial effects of IPost against myocardial reperfusion injury and the activation of the RISK pathway at this time is unclear, although preliminary evidence suggests that delaying pH restoration may facilitate RISK pathway activation through an unknown mechanism (31).
Downstream Effectors of the RISK Pathway
There are a number of downstream effectors of the RISK pathway that could be responsible for the cardioprotection elicited by IPost. Many of these terminate on the mitochondria, an organelle that occupies an essential role in cardiomyocyte survival and death signaling.
The mitochondrial permeability transition pore
The opening of the mitochondrial permeability transition pore (mPTP) at the onset of myocardial reperfusion is a critical determinant of lethal myocardial reperfusion injury, such that pharmacologically inhibiting its opening at this time can reduce myocardial infarct size by 40%–50% in both the laboratory (3, 40, 41) and clinical setting (105). Although the actual identity of the pore-forming units of the mPTP is unknown, several studies have demonstrated mitochondrial cyclophilin-D to be a major regulatory component of the mPTP, such that mice lacking cyclophilin-D appear resistant to mPTP opening and sustain greatly reduced myocardial infarct sizes (5, 77, 94). Studies from our laboratory and others have demonstrated that mPTP inhibition at the time of reperfusion is the end-effector in both IPC (41) and IPost (4). Experiments are ongoing to determine the mechanism through which mPTP is inhibited in these settings, but potential mechanisms include the prosurvival kinase pathways [RISK (12, 22, 59) and SAFE] and less oxidative stress (19). We and others have demonstrated that a variety of pharmacological agents capable of limiting myocardial infarct size when administered at the onset of myocardial reperfusion do this through the activation of the RISK pathway and the subsequent inhibition of mPTP (8, 22, 59, 78). The actual mechanism through which the Akt and Erk1/2 components of the RISK pathway mediate mPTP inhibition is unclear, although potential explanations include (i) the generation of nitric oxide by eNOS, a downstream target of the RISK pathway, can inhibit mPTP opening (62); (ii) Akt may modulate mitochondrial morphology, thereby rendering mitochondria more resistant to mPTP opening (42, 101); (iii) Akt may modulate intracellular calcium handling by increasing sarcoplasmic reticulum calcium uptake and thus may prevent mPTP opening (1); and (iv) glycogen synthase kinase 3 beta (GSK3β), a downstream target of both Akt and Erk1/2, may act as a point of convergence for a variety of prosurvival signaling pathways resulting in mPTP inhibition (59, 60).
Antiapoptotic signaling pathways
The possibility of recruiting antiapoptotic signaling pathways had been one of the original reasons for proposing the RISK pathway as a prosurvival signaling pathway, particularly given the close association of apoptotic cell death with the reperfusion phase (133). Interestingly, although a large number of potential antiapoptotic pathways exist downstream of the RISK pathway, relatively few have actually been investigated in the context of cardioprotection, yet alone IPost. These antiapoptotic mechanisms include the phosphorylation and inhibition of proapoptotic proteins such as Bcl2-associated death factor (57) and Bcl2-associated X protein, the activation of antiapoptotic proteins such as PIM-1 kinase (13), the effect of which is preservation of mitochondrial integrity, and a favorable increase in the antiapoptotic proteins such as B-cell lymphoma 2 and B-cell lymphoma 2 extra large. There are a number of other potential antiapoptotic mechanistic pathways recruited by the RISK pathway—these are reviewed elsewhere (46, 48).
Modification of the RISK Pathway in Comorbidities
Many of the comorbidities that exist with ischemic heart disease, such as diabetes, hypertension, age, hyperlipidemia, and so on, may impact on the cardioprotection elicited by IPost (26). The actual interplay between these confounding factors and IPost cardioprotection need to be taken into account when designing appropriate experimental animal studies. The effect of these comorbidities is particularly important with respect to signal transduction, as conditions such as diabetes and age may downregulate certain signaling pathways. For example, downregulation of the Erk1/2-GSK3β signaling pathway rendered the hearts excised from the metabolic syndrome rat resistant to the infarct-limiting effects of IPost (129). A similar loss of IPost cardioprotection was observed in relation to downregulation of PI3K-Akt and MEK1/2-Erk1/2 signaling in the presence of a coronary stenosis (98) and obesity (14). This effect of comorbidities on cardioprotection has been also observed in pharmacological postconditioning using inhaled anesthetics. In the presence of hyperglycemia, the in vivo rabbit heart was resistant to the infarct-limiting effects of isoflurane postconditioning and Akt-eNOS activation was absent (107).
Controversial Issues Surrounding RISK Pathway Signaling in IPost
The involvement of the RISK pathway in IPost signaling has sparked several controversial issues. The first of these was by Darling et al. (21), who failed to implicate the PI3K-Akt component of the RISK pathway in the ex vivo postconditioned rabbit heart. However, other studies have provided evidence for the role of the PI3K-Akt pathway in IPost signaling in the intact rabbit heart (17) (Table 1).
In the porcine heart, the disparate findings concerning the RISK pathway have been even more interesting. First, Schwartz and Granha (112) reported dissociation between RISK pathway activation and the infarct-limiting effects of IPost in the in vivo porcine heart. Using three 30-s cycles of reperfusion and ischemia, these authors were able to demonstrate significant Akt and Erk1/2 phosphorylation in the absence of cardioprotection. One could argue that although the IPost stimulus may have been adequate to activate the RISK pathway it was not strong enough to elicit infarct limitation. Curiously, a subsequent study by Skyschally et al. (115) reported almost the opposite findings—these authors managed to demonstrate infarct limitation with IPost (six 20-s cycles of reperfusion and ischemia), but in the absence of RISK pathway activation. Of note, inhalational anesthetics, which are known to activate components of the RISK pathway, were used in this study, which may explain why no difference in RISK pathway activation was observed in IPost-treated hearts. The failure of kinase inhibitors to block IPost cardioprotection may be due to the fact that they were administered via the intracoronary route as opposed to being given peripherally prior to the onset of myocardial reperfusion.
A recent study has questioned the role of GSK3β, a downstream component of the RISK pathway, as a target for IPost. A wealth of data, much of it pharmacological, suggests that the phosphorylation and inhibition of GSK3β constitutes a common mechanism of cardioprotection that underlies IPost (59). However, Nishino et al. (97) reported that mice containing a form of GSK3β resistant to phosphorylation and inhibition could still be cardioprotected by IPost. This finding contrasts with that of Gomez et al. (32), who reported that mice expressing a different form of GSK3β, which was also resistant to phosphorylation and inhibition, were resistant to IPost.
The controversial issues surrounding RISK pathway activation in IPost signaling have been used to argue for the existence of RISK-independent pathways underlying IPost. One of these alternative signaling pathways is the SAFE pathway, which is the next topic of this review article.
Reperfusion Signaling in IPost: The SAFE Pathway
In addition to the inconsistencies, noted in the previous section, surrounding RISK signaling in IPost, one particular postconditioning mimetic does not appear to limit myocardial infarct size at the time of reperfusion through the RISK pathway. Tumor necrosis factor (TNF) can mimic IPost in isolated murine hearts in the absence of Akt activation, and its protective effect is not affected by the PI3K inhibitor, Wortmannin (69). Taken together, these data support the existence of an alternative prosurvival signal transduction pathway for protecting the ischemic myocardium against lethal myocardial reperfusion injury. In this respect, Lecour has recently described this novel prosurvival pathway, which involves the activation of TNF and the transcription factor, signal transducer and activator of transcription 3 (STAT3), as the SAFE pathway (70, 71). The SAFE pathway was first discovered in the setting of IPC, but its role in IPost has been only recently confirmed.
TNF Is Part of the SAFE Pathway
TNF and the heart
TNF is a proinflammatory cytokine discovered in macrophages as a cytotoxic factor that can induce necrosis in certain murine tumors (16). First produced as a 212-amino-acid-long type II transmembrane protein, it is cleaved by TNF-converting enzyme (also called ADAM 17) (9) to release the homotrimeric soluble TNF (56 kDa). Expressed in all nucleated cell types of the myocardium, including cardiomyocytes (84), TNF exerts its major effects after binding onto its cell-surface receptors, TNF receptor 1 (TNFR1 or p55) and TNF receptor 2 (TNFR2 or p75). Both receptors are present in the heart (126), and when activated, they form homotrimers, thereby facilitating signal transduction (81). The existence of a mitochondrial binding protein to allow receptor-independent delivery of TNF from the cell surface to the mitochondria has been also proposed (15).
The two TNFRs differ in their signal pathways and are differentially expressed and regulated in the human heart (2). The concentration of both receptors is increased in coronary artery disease (108) and the signaling pathways coupled to TNFR1 include both apoptotic and protective signaling, whereas the signaling pathways coupled to TNFR2, although poorly studied, seem to convey protective signaling only (Fig. 2). Apoptosis is induced by the adaptor protein, Fas-associated death domain protein, which recruits and activates the apoptosis initiator caspase-8 (19). The protective effects mediated by the receptors may occur via the activation of nuclear factor kappa-B, Jun N-terminal kinase (81). TNFR binding also leads to diacylglycerol formation via phospholipase C (111), which in turn activates the sphingolipid signaling pathway and PKC (111). Another prosurvival factor activated in response to TNFRs binding is the Janus kinase (JAK)/STAT3 pathway (described later).
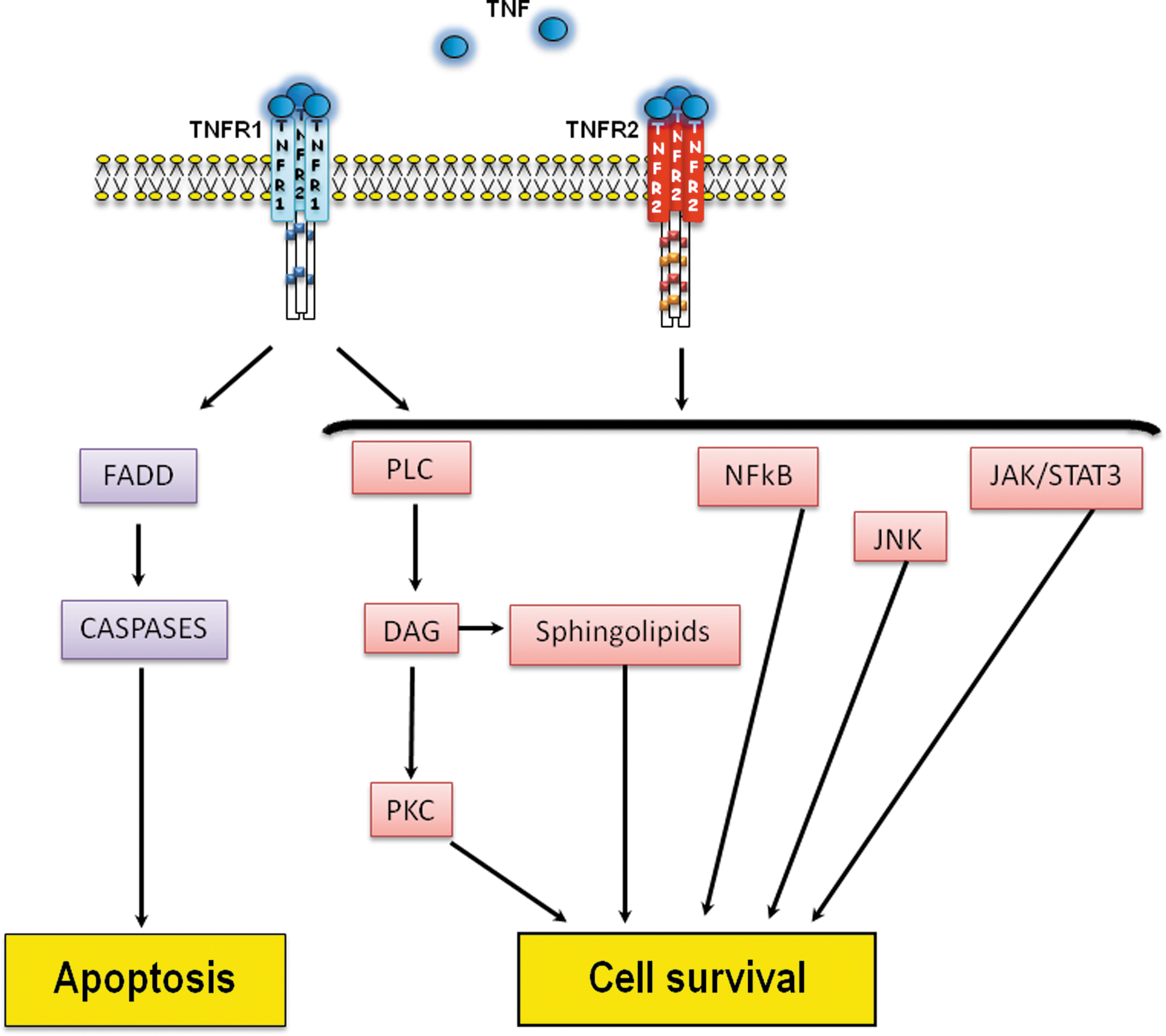
TNF and myocardial infarction
Although low physiological levels of TNF do not seem to alter the function of the heart (85), an increase in TNF, as observed in myocardial infarction (24, 50, 109) or heart failure (76), exerts a negative inotropic effect and leads to contractile dysfunction in a concentration-dependent manner (114, 135). This effect is most likely due to an alteration in intracellular calcium homeostasis (135) and the formation of free radicals (25). However, animal studies exploring the exact role of TNF in ischemia-reperfusion injury (IRI) have led to conflicting results. Mice lacking TNF are protected from IRI (82) and the presence of TNF antibodies limits the infarct size in animal studies (6, 38), therefore supporting the concept of a deleterious effect of TNF in myocardial infarction. In contrast, exogenous TNF protects cardiomyocytes against hypoxic injury (95). Similarly, knockdown of both TNFRs in mice subjected to coronary artery ligation results in an increase in infarct size, whereas the infarct size in TNFR1 knockout mice was reduced or unchanged (27, 69, 88). Apart from the concentration of TNF, the type of receptor activated in the setting of IRI is another critical factor that will influence the deleterious or beneficial effect of TNF.
TNF in pre- and post-postconditioning
The requirement of TNF release for both classic and delayed IPC to confer protection is now strongly supported in the literature. In rats, mice, or rabbits, classic IPC is associated with an increased level of preischemic TNF (7, 117), but a decreased level of endogenous TNF released during IRI (7, 63). TNF antibodies can reduce the infarct-sparing effect of delayed IPC in a rat model (131) or preconditioning with microembolization in a pig model (114). Similarly, TNF knockout mice fail to be preconditioned (54, 67, 117) and exogenous TNF given as a preconditioning mimetic can reduce the infarct size to a similar extent to IPC both in vivo or in vitro (72 –74, 124). Interestingly, the protection with exogenous TNF can only be achieved with a washout phase prior to the sustained ischemia, therefore suggesting that the time when TNF is produced is another criteria, together with the concentration and the type of receptors activated, to consider for the deleterious versus protective effect of TNF (70, 73).
Similar to IPC, IPost reduces levels of plasma TNF during the reperfusion phase in anesthetized rats subjected to coronary artery ligation (64). In a Langendorff system, TNFR1 knockout mice hearts but not TNF or TNFR2 knockout mice hearts are protected with an IPost stimulus (69) (Fig. 3). Similarly, the addition of TNFR2 antibodies abolishes the infarct-sparing effect of IPost, whereas TNFR1 antibodies have no effect (69). All together, these data strongly support the requirement of TNF release in a dose- and time-dependent manner in both IPC and IPost and the protective effect mediated by TNF is acting via its binding to the TNFR2.

JAK/STAT3 Is Part of the SAFE Pathway
JAK/STAT3 pathway in the heart
Traditionally believed to act as a transcription factor after activation and translocation of STAT3 to the nucleus, the JAK/STAT3 pathway plays a critical role in the myocardial response of the heart in various physiopathological conditions such as myocardial infarction, myocarditis, or hypertrophy. The recent discovery that activated STAT3 can also translocate to the mitochondria suggests a control of STAT3 in the energy production of the cells (130).
JAK2 proteins are a family of tyrosine kinases constitutively associated with the cytoplasmic domain of cytokine and growth factor receptors. Upon activation of the receptors, JAK proteins phosphorylate and create a docking site for a STAT3 protein, which in turn is activated by phosphorylation. Tyrosine phosphorylation of STAT3 enables STAT3 to dimerize and translocate to the nucleus. Serine phosphorylation of STAT3 is required for its translocation to the mitochondria, where it interacts with GRIM-19–containing complexes I and II to regulate the electron transport chain (92).
JAK/STAT3 and myocardial infarction
Following coronary artery ligation in vivo, phosphorylation of STAT3 occurs, which is maintained for up to 24 h (96). Reperfusion after 25 min of ligation further increases its activation (87). Although acute ischemia followed by up to 2 h of reperfusion did not affect the infarct size of cardiomyocyte-specific STAT3-depleted mouse hearts compared with their littermate controls (10, 69, 116), a 10-fold cardiac overexpression of STAT3 in mice reduced the infarct size following IRI (102, 103). In contrast, the addition of the JAK tyrosine kinase inhibitor, AG490, in an isolated rat heart undergoing IRI protected the heart (53, 86).
JAK/STAT3 in pre- and postconditioning
The role of STAT3 in early preconditioning was first suggested in an isolated rat heart model whereby STAT3 phosphorylation was increased following the preconditioning stimulus, and the addition of AG490, the JAK/STAT3 inhibitor, during the preconditioning stimulus ablated the protection against infarction (39). Further, hearts from mice in which STAT3 was ablated by MLC2v promoter–driven activation of the Cre-recombinase failed to be protected by either an IPC stimulus or a pharmacological preconditioning stimulus such as TNF and diazoxide (116). Additionally, the presence of AG490 at the onset of reperfusion also abolished the protective effect of IPC, thereby suggesting that STAT3 phosphorylation is also required during the reperfusion period and not limited to its activation during the preconditioning stimulus (74, 122). Interestingly, the coronary effluent from a preconditioned heart activates the JAK/STAT3 pathway in a donor heart, conferring cardioprotection by limiting apoptosis (52).
In 2006, the role of STAT3 in IPost was also suggested in an isolated rat heart model whereby the addition of the JAK/STAT3 inhibitor AG490 at the onset of reperfusion abolished the cardioprotective effect of IPost (121) (Table 2). Both ischemic and TNF postconditioning with TNF failed to confer an infarct-sparing effect in isolated STAT3-deficient murine hearts subjected to IRI (10, 69). Further, the absence of protection observed in TNF or TNFR 2 knockout mice was associated with the absence of STAT3 phosphorylation (69). Similarly, postconditioning the heart with sphingosine-1 phosphate (S1P) is associated with an increase in STAT3 phosphorylation and its cardioprotective effect is abolished in STAT3-deficient mice (119). In both IPC and IPost, activation of STAT3 occurs within minutes and its cardioprotective effect can be seen very rapidly, therefore suggesting that its effect is mediated by its translocation to the mitochondria (10, 130) rather than its translocation to the nucleus where it will regulate the transcription of various genes.
MI, myocardial infarction; SAFE, survivor activating factor enhancement; STAT3, signal transducer and activator of transcription 3; TNFR, tumor necrosis factor receptor.
Upstream Targets of the SAFE Pathway
The upstream activators of the SAFE pathway have been poorly studied. Nevertheless, many pharmacological agents capable of mimicking IPC or IPost may confer their cardioprotective effect via the SAFE pathway. Although not always demonstrated in the heart, angiotensin II (28, 100), bradykinin (58, 106), adrenoreceptors (33), leptin (37), opioids, and cannabinoids (18, 89) can activate both TNF and STAT-3. Similarly, insulin given at the time of reperfusion confers cardioprotection via the activation of the JAK/STAT-3 pathway (29), and preliminary data from the Hatter Institute in Cape Town demonstrate that insulin, given at the onset of reperfusion in isolated TNF knockout mice hearts, fails to protect the heart against IRI. Interestingly, adenosine can mimic IPC in wild-type mice and its protective effect is unaffected in TNF knockout mice (117) or in cardiomyocyte-specific STAT3-deficient mice (116). These data strongly suggest that the cardioprotective effect of adenosine is independent of the SAFE pathway. Another postconditioning mimetic is S1P (128), also known as a downstream target of TNFRs (Fig. 2) (73). Surprisingly, exogenous S1P requires the activation of the SAFE pathway as it fails to induce cardioprotection in both TNF knockout mice and cardiomyocyte-specific STAT3-deficient mice (61, 119), therefore suggesting a complex regulatory cross-talk mechanism between TNF and S1P.
Dowstream Targets of the SAFE Pathway
All these data clearly indicate that the activation of TNF, TNFR2, and STAT3, major components of the SAFE pathway, are required for IPost to confer protection, but very little is known about the signaling pathways activated downstream of STAT3. TNF-induced protection requires the activation of PKC, the mitochondrial ATP-dependent potassium channel, and NFκ-B (73, 118). In addition, pharmacological preconditioning with TNF is associated with an increase in mitochondrial free radicals and an addition of free radical scavengers such as mercaptoprpionyl glycine abolished its cardioprotective effect (68, 72). In contrast, neither p38MAPK, Akt, Erk, nor GSK3β seems to be involved in TNF-induced cardioprotection, therefore suggesting that they do not act as the downstream target of the SAFE pathway (69, 74, 124). STAT3 mediates cardioprotection via the phosphorylation and inactivation of the proapoptotic factors Bcl2-associated death factor (23, 74) and Bcl2-associated X protein (52). The mPTP is described as an end-effector of IPost and the RISK pathway; our initial data suggest that the SAFE pathway may also target the mPTP (unpublished data, 2010).
Modification of the SAFE Pathway in Comorbidities
Interestingly, IPost signaling through the SAFE pathway may also be influenced by comorbidities. Boengler et al. (10) noted that the infarct-limiting effects of IPost were lost in aged mice, and this inability to postcondition the aged mice was related to inadequate activation of the JAK-STAT3 pathway, a component of the SAFE pathway. Whether other comorbidities are able to impact on signaling through the SAFE pathway remains to be determined.
Two Sides of the Same Coin: Interplay Between the SAFE and RISK Pathways
It is clear that IPost can protect the ischemic heart from myocardial reperfusion injury through the activation of either the RISK or SAFE pathway (Fig. 4). Interestingly, the pharmacological inhibition of either pathway appears to abrogate IPost cardioprotection completely, which suggests that either there is an interaction between the two pathways, or the resolution of our animal models cannot distinguish between partial and complete inhibition of cardioprotection. Studies are ongoing to investigate the actual interplay between the RISK and SAFE pathways. All things considered, it appears that the mitochondria and specifically the mPTP may act as a point of convergence for the two IPost signaling pathways. The study by Goodman et al. (34) suggested the possibility of an interaction between the RISK and SAFE pathways in IPost signaling. These authors found that pharmacological inhibition of STAT3 with Stattic (a specific STAT3 inhibitor) abrogated both the IPost-induced phosphorylation of STAT3 and Akt, suggesting that Akt may be downstream of STAT3. Interestingly, the pharmacological inhibition of PI3K blocked the IPost-induced phosphorylation of Akt but not STAT3, suggesting that STAT3 is not downstream of PI3K-Akt. In the setting of morphine-induced postconditioning, Gross et al. (36) found that morphine-induced Akt phosphorylation could be blocked by AG-490, a JAK2 inhibitor, and that morphine-induced STAT3 phosphorylation could be blocked by Wortmannin, a PI3K inhibitor, suggesting an interaction between the two pathways.

Conclusions
The elucidation of the signal transduction pathways underlying IPost has identified a number of signaling pathways, which include the RISK and the SAFE pathways. The identification of the major components of these two pathways has identified several novel pharmacological targets for protecting the ischemic heart at the time of myocardial reperfusion. Further work is needed to elucidate the relationship between these two reperfusion signaling pathways.