
Abstract
Reactive oxygen species (ROS) and cellular oxidative stress are involved in many physiological and pathophysiological processes, including cellular and organismal aging, migration, proliferation, senescence or death of normal and cancer cells, and stress resistance of stem cells. The forkhead homeobox type O (FOXO) transcription factors FOXO1, FOXO3a, and FOXO4 are critical mediators of the cellular responses to oxidative stress and have been implicated in many of the above ROS-regulated processes. In cancer cells they converge oxidative stress signaling to cell cycle arrest and cell death or promote a motile phenotype. Dependent on their posttranslational modifications FOXOs can also actively regulate the detoxification of cells from ROS and promote stress resistance. Thus, FOXO transcription factors are of vital importance in processes regulating tumor survival or progression, stem cell maintenance, age-related pathological processes, and lifespan extension. Antioxid. Redox Signal. 14, 593–605.
Introduction
Generation of cellular oxidative stress


Regulation of cellular oxidative stress
Intracellular ROS homeostasis is steadily maintained by cellular detoxification systems to prevent cells from damage. Detoxification of cells from oxidative stress is mediated through antioxidant enzymes that specifically scavenge different kinds of ROS and by nonenzymatic molecules (Fig. 3) [reviewed in (75)]. The dismutation of O2 •− anions to oxygen and H2O2 is mainly mediated by superoxide dismutases (SODs). SOD enzymes are located in different compartments within the cell, such as MnSOD in the mitochondrial matrix and Cu/ZnSOD in the cytosol (25).
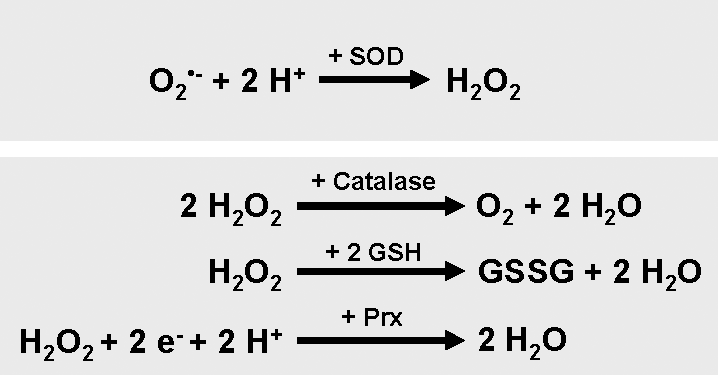
Decomposition of H2O2 to water and oxygen is mediated by various enzymes such as catalase, located at the cytosol (8, 45, 76) and several peroxiredoxins (Prxs) located at peroxisomes, mitochondria, and the cytosol (46, 101, 133). Further, glutathione (GSH) peroxidases, localized in cytosol and mitochondria, catalyze the breakdown of H2O2 and organic hydroperoxides (13, 124). Thioredoxins (Trx) act as electron donors to peroxidases and also facilitate the reduction of proteins by cysteine thiol disulfide exchange (3). Nonenzymatic molecules that detoxify cells from ROS are for example vitamins A, C, and E, and GSH. GSH protects cells from oxidative stress by reducing disulfide bonds of cytoplasmic proteins to cysteines. During this process, GSH is oxidized to GSH disulfide. The regeneration of GSH pools is mediated by GSH reductase, an enzyme that reduces GSH disulfide (10, 22).
Cellular sensing of oxidative stress
Within recent years it became apparent that ROS-sensing signaling molecules exist that are activated at the location of ROS generation and translate increased oxidative stress into induction of protective or apoptotic signaling pathways [discussed in (112)]. Such cellular ROS sensors can be activated by direct oxidation through O2 •− or H2O2. O2 •− regulates the activity of proteins that contain iron sulfur centers (37). H2O2 reversibly oxidizes the sulfhydryl group in the active site cysteine residues of several phosphatases, and similar regulation was shown for kinases and transcription factors (84, 91, 117, 134). Whereas the importance of oxidative stress in inducing necrosis and apoptosis is focus of intensive investigation, only little is known about ROS-sensing signaling molecules that mediate cell survival or protection from ROS under moderate levels of oxidative stress through induction of antioxidant nuclear genes. One example, for ROS-induced protective signaling is an Src-initiated signaling pathway that promotes cancer cell survival through activation of NF-κB (112), and Src has a cystein residue motif that allows activation of the kinase through direct oxidation (91). Forkhead homeobox type O (FOXO) transcription factors can also be viewed as sensors for oxidative stress since their activity is regulated by H2O2 and dependent on the cellular context they relay these stresses to induce apoptosis, stress resistance, or senescence (17, 34). Recently, it was reported that FOXO4 can be directly regulated by the cellular redox state through either monoubiquitination (126) or the induction of cysteine-thiol disulfide-dependent complexes of FOXO with the acetyltransferase p300/cAMP-response element binding protein (CREB)-binding protein (CBP) (27). Binding of p300/CBP to FOXO is essential for FOXO-mediated transcription, since both CBP and p300 act as FOXO cofactors by weakening histone/DNA interactions (125). The modulation of FOXO biological activity by p300/CBP-mediated acetylation is completely dependent on the formation of the redox-regulated complexes, suggesting that FOXO transcription factors are bona fide sensors for the redox status of cells. The modulation of such ROS-sensing signaling molecules and the development of inhibitors or mimetics may reveal strategies to extend lifespan in humans or to prevent ROS-caused diseases.
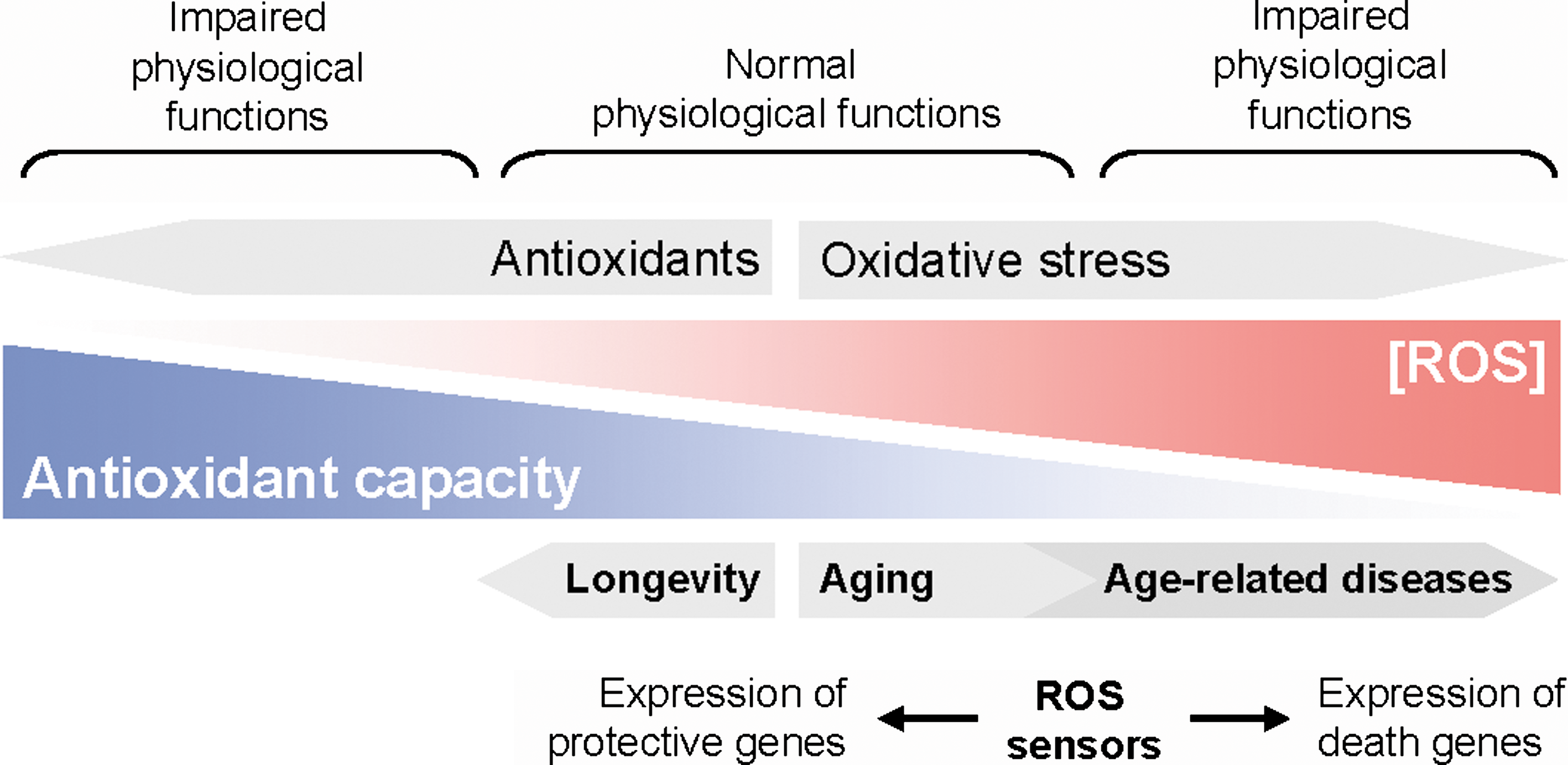
Oxidative stress in aging and age-related diseases
A cellular shift toward oxidative stress conditions can lead to macromolecule damage such as lipid peroxidation, protein oxidation and aggregation, or DNA modification. Over time, ROS-mediated damage is accumulated and this may facilitate aging processes within cells and overall in the organism as it was formulated in the free radical theory of aging (Fig. 4) (44). This hypothesis is supported by the observation that the lifespan of a wide spectrum of animals is proportional to their metabolic rate and therefore to the rate at which the organisms generate ROS [summarized in (111)]. Dietary and chemical antioxidants as well as caloric restriction can lower cellular ROS production and this translates to an extension of lifespan [summarized in (36)]. Consequently, increased life span correlates with increased stress resistance and expression of nuclear stress response genes encoding antioxidant and stress-regulating proteins. Increases in intracellular oxidants are believed to be responsible for the onset and progression of >40 human diseases, including ischemic hard disease, neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease, diabetes, and cancer [summarized in (35, 105)]. Therefore, aging as well as the development of age-related disease are dependent on the rates of ROS generation or detoxification, whereby ROS levels are impacted by dietary and cellular antioxidants as well as the ability of cells to replace oxidative-damaged macromolecules over time (66).
FOXO Transcription Factors in the Responses to Oxidative Stress
Regulation of FOXO transcription factor activity by oxidative stress
FOXO transcription factors are regulated by a multitude of kinases of which some mediate activating and others inhibiting phosphorylations (in detail reviewed by Van den Berg et al. in this issue). An increase in intracellular ROS facilitates the localization of FOXO to the nucleus where it is transcriptionally active (34). Mimicking such oxidative stress conditions by treating cells with H2O2 revealed that in response to ROS, c-Jun-N-terminal kinase (JNK) is a major inducer of FOXO activity. However, additional posttranslational modifications are needed to exert the full spectrum of FOXO-mediated responses to ROS (Fig. 5) [reviewed in (126)].
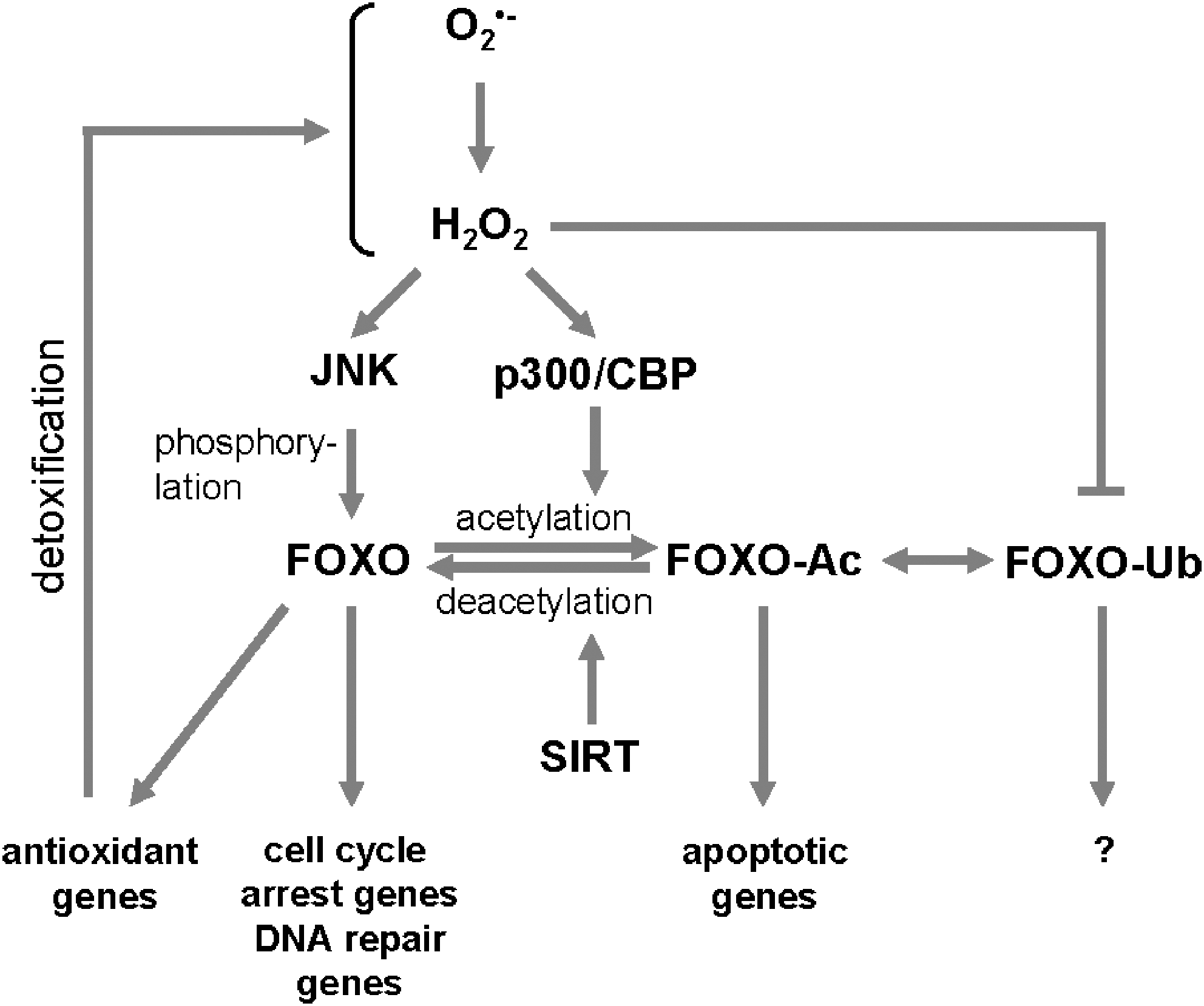
Activation of FOXO transcription factors through ROS-regulated kinases
Particularly two kinases, JNK and mammalian ortholog of the Ste20-like protein kinase (MST-1), have been implicated in H2O2-mediated activating phosphorylations of FOXOs. Upon treatment with H2O2 or after activation of the small GTPase Ral, JNK interacts with FOXO and phosphorylates FOXO directly at two threonine residues (Thr447 and Thr451 in FOXO4), leading to its nuclear translocation and activation (34, 64). JNK previously has been described to be activated by mitochondrially produced H2O2. Once activated at these organelles in cancer cells JNK induces apoptosis by negative regulation of Bcl-2 function and activation of Bax [summarized in (112)]. Other functions of ROS-JNK-FOXO signaling is a contribution to insulin resistance (32) and to an increased lifespan in worm, fly, and rodents (97, 131).
Like JNK, MST-1 is a kinase involved in ROS-induced signaling. MST-1 mediates oxidative stress-induced and growth factor withdrawal-induced neuronal apoptosis through phosphorylation and activation of FOXO (i.e., phosphorylation of FOXO3a at S207) (139). This phosphorylation induces nuclear accumulation of FOXO by disrupting its association with 14-3-3 proteins (55, 70). MST-1 is also a physiological substrate for Akt and phosphorylation of MST-1 at Thr 387 by Akt blocks the activation of FOXO3a by this kinase (55).
Role of ROS in signaling pathways that negatively regulate FOXO
FOXO transcription factors are negatively regulated by insulin/insulin-like growth factor-1 (IGF-1) signaling pathways, mainly by PI3K-mediated activation of the kinases serum- and glucocorticoid-inducible kinase (SGK) (16) and Akt (14). Phosphorylation of FOXO by Akt or SGK leads to binding of 14-3-3 proteins and subsequent nuclear export (15). Other negative regulators of FOXO-mediated gene induction are inhibitor of nuclear factor-κB kinase (IKK) and Pin1. All these proteins have been shown to be activated by oxidative stress (14, 53), adding an additional level of complexity to the regulation of FOXO. Pin1, for example, in response to oxidative stress interacts with FOXO4 and attenuates its monoubiquitination to inhibit transcriptional activity (12). Dependent on the cellular context, oxidative stress can increase Akt activity. However, in some cell systems, it was shown that oxidative stress actually downregulates IGF-1-mediated Akt signaling. Others have shown that oxidative stress allows FOXO to accumulate in the nuclei of cells in response to stresses though active growth factor signaling and the presence of Akt/SGK-mediated phosphorylations (17, 38, 62). Moreover, increases in oxidative stress under conditions where Akt is inactive lead to increased activity of FOXO transcription factors. This may occur in starved cancer cells in which starvation-mediated increase in cellular oxidative stress leads to the activation of FOXO3a (113). Other examples are quiescent cells in which FOXO3a activity is increased by H2O2-induced oxidative stress (64).
Regulation of FOXO by nutrients and dietary restriction
Glucose and glucose metabolism can induce oxidative stress and activate JNK, suggesting a ROS-JNK-mediated regulation of FOXO at high glucose levels. Additionally, O-linked beta-N-acetylglucosamine modification regulates FOXO1 in response to glucose and results in increased expression of gluconeogenic and of ROS detoxifying genes. This provides a mechanism for direct nutrient control of transcription to regulate metabolism and stress response through FOXOs (51). Overall, FOXO activity induces a metabolic switch that is similar to low glucose and fasting conditions. This is mediated by FOXO-induced suppression of genes involved in glycolysis and lipogenesis (140) and expression of genes essential for the regulation of gluconeogenesis (7, 90).
Dietary restriction is the most potent intervention to slow aging in a variety of species (132). Possible mediators of caloric restriction are the sirtuins, which possess nicotinamide adenine dinucleotide-dependent protein deacetylase activities (54). SIRT1 protein levels are upregulated by caloric restriction or fasting (24, 103) and deacetylate histones and multiple transcription factors such as p53, NF-κB, and FOXO (17, 78, 88, 127, 136). For example, upon peroxide stress deacetylation by SIRT1 prolongs FOXO4-dependent transcription of stress-regulating genes (127). Similarly, SIRT1 increases FOXO3a's ability to induce cell cycle arrest and resistance to oxidative stress but inhibited its signaling to induce cell death (17). The SIRT1 homolog SIRT2 deacetylates α-tubulin, but also enters the nucleus and deactylates histones and FOXO transcription factors (shown for FOXO3a) in response to both oxidative stress (as induced by H2O2) and caloric restriction (130). After its induction by oxidative stress, SIRT2 activity induces cytostatic, pro-apoptotic, or antioxidant factors such as p27kip1, Bcl-2-interacting mediator of cell death (Bim), and MnSOD probably through its effects on FOXO.
Targets for FOXO in Response to Oxidative Stress
The activation of FOXO transcription factors due to absence of insulin/IGF-1 signaling or increased oxidative stress can induce a wide range of genes that regulate cellular responses such as cell cycle arrest and apoptosis or stress responses, including resistance to oxidative stress (Fig. 6). These various functions can be mediated by posttranslational modifications and/or different FOXO interaction partners that modulate FOXO targeting [reviewed in (21)].

FOXO targets genes involved in cellular detoxification of ROS
Management of intracellular ROS to prevent macromolecule damage is achieved through cellular enzymes or small molecules that collectively form the cellular antioxidant system (49). Important ROS within cells are O2 •−, H2O2, or the DNA-damaging hydroxyl ions (111). One role of FOXO in the stress response is the upregulation of antioxidant proteins that mediate detoxification of ROS and stress resistance that is tightly linked to increased lifespan. Moreover, FOXO-mediated redox regulation has been implicated in tumor suppression (21). The activation of FOXO transcription factors leads to the induction of a variety of genes encoding antioxidant proteins, including MnSOD, which detoxifies cells from mitochondrially generated O2 •− (64). In Caenorhabditis elegans mRNA levels of MnSOD are negatively regulated by the insulin/IGF-1 signaling pathways (48). The exposure of quiescent cells to H2O2 activates FOXO3a, leading to expression of MnSOD via regulation of the SOD2 gene (64). However, it was also shown that in tumor cells under normal growth conditions FOXO transcription factors may be dispensable from mROS-mediated induction of SOD2 (114).
H2O2 is hydrolyzed by a variety of antioxidant proteins of which several are also regulated in their expression by FOXO, indicating that activation of FOXO can indeed lead to a complete detoxification of ROS. For example, the forkhead-type transcription factor DAF-16 in nematodes induces expression of catalase after its activation by hyperosmotic stress, high fever stress, oxidative stress, and nutrient starvation (52, 67, 96). In human cells FOXO3a also induces expression of catalase (40, 93). Moreover, the JNK-FOXO signaling pathway upregulates expression of Prx II a protein that is mainly expressed in the adult brain (69). Another FOXO target in human cardiac fibroblasts is the gene promoter of the mitochondria-located Prx III (23). Depletion of Prx III in cardiac fibroblasts was associated with decreased resistance to oxidative stress (23). This is important since after myocardial injury, human cardiac fibroblasts are protected from oxidative stress (72) by expressing FOXO-regulated antioxidant molecules such as SODs, catalases, and Prxs.
Selenoprotein P, the major selenoprotein in human plasma, can act as an antioxidant and its expression in hepatoma cells is regulated at the promoter level through the interaction of FOXO1, peroxisomal proliferator-activated receptor-γ coactivator 1α, and hepatocyte nuclear factor-4α (108, 129). Moreover, the sestrin 3 gene is a FOXO3a target that acts as a ROS scavenger (94). Sestrins accumulate in cells exposed to oxidative stress and loss of seatrain in drosophila results in age-associated pathologies, including mitochondrial dysfunction and cardiac malfunction (68).
FOXO1 also directly represses expression of Trx-interacting protein in human cells. Trx-interacting protein hinders the cellular response to oxidative stress and affects life span (28) by inhibiting the reducing activity of Trx, an antioxidant protein that acts as electron donor to peroxidases and also facilitates the reduction of proteins by cysteine thiol disulfide exchange (3). Additionally, FOXO upregulates the fatty acyl-CoA carriers sterol carrier protein X and sterol carrier protein 2, which both protect unsaturated fatty acids from oxidative damage (26). Finally, FOXO3a directly activates the human MstA promoter in human cells and in C. elegans, contributing to maintain the methionine sulfoxide reductase system that protects against oxidative stress and was implicated in the regulation of aging (86).
Regulation of ROS-induced cell cycle arrest and apoptosis by FOXO
The mechanisms by which FOXO transcription factors switch from antioxidant signaling to apoptotic signaling are not well defined. FOXO transcription factors integrate stress stimuli via phosphorylation, acetylation, and mono-ubiquitination events [summarized in (43)], and this as well as alternate binding partners may define the cellular program activated. FOXO functions in ROS signaling may also be dependent of the differentiation state of cells. For example, it was shown that in undifferentiated 3T3-L1 cells FOXO3a mediates ROS-induced apoptosis, whereas in differentiated 3T3-L1 adipocytes FOXO3a suppressed ROS-induced apoptosis via a switch to increased expression of ROS scavenging enzymes (63).
The therapeutic potential of FOXO transcription factors in cancer is due to their ability to induce cell cycle arrest and apoptosis [for a review on the therapeutic value of FOXOs see (80)]. In cancer cells, FOXO is activated by anticancer drugs, radioisotope irradiation, or serum starvation, all inducers of intracellular oxidative stress. All these events can cause cell cycle arrest and/or apoptosis through FOXO. In response to oxidative stress FOXO induces expression of genes that regulate DNA repair and cell cycle arrest, including growth arrest- and DNA damage-inducible gene 45α (GADD45α), p130, and cyclin D2 and the cell cycle inhibitors p21 and p27kip1 (18, 39, 82, 83, 120). For example, in thyroid cancer, FOXO is activated by H2O2 through JNK to induce the genes encoding p27kip1 and GADD45α (59). Serum starvation-mediated nuclear localization of FOXO (here FOXO1) was also observed in medulloblastoma cells where it induces growth suppression (109). In C. elegans the homolog of human host cell factor-1 binds to DAF-16 and acts as a negative regulator to modulate stress response and longevity (71). In mammals host cell factor-1 is required for appropriate transition from G1 to S phase and progression of M phase and cytokinesis (56, 123), and the binding and inactivation of FOXOs by this factor can be one of the mechanisms of how it exerts its functions (71).
Some anticancer drugs activate apoptosis through FOXO. For example, adriamycin regulates cell death through activation of FOXO1 and tumor necrosis factor receptor-associated death domain (104). Pro-apoptotic targets for FOXO are for example Bim, Bcl-6 (B-cell/lymphoma 6), and the Fas ligand (30). In addition, other signaling proteins such as p66Shc and p53 that interact with forkhead transcription factor-mediated pathways have been implicated in the induction of cell cycle arrest and apoptosis in response to ROS (14, 138). For example, p53 is stabilized by stresses and, once activated, induces expression of cell cycle arrest genes, including p21 (95). In response to DNA damage p53 activation can lead to FOXO3a inhibition (137). Moreover, coexpression of p53 with FOXO3a under oxidative stress conditions leads to decreased expression of the FOXO3a-regulated apoptotic genes Bim and Bcl-6, but had no effect on p27 and CyclinG2 expression (87). Therefore, a crosstalk between FOXO and p53 may be a threshold for apoptotic signaling (87). FOXO and p53 interact in response to cellular stresses that increase ROS levels such as serum deprivation, drug treatment, and treatment of cells with H2O2 (87). However, it is not known if direct interaction is the mechanism of how p53 regulates FOXO3a-mediated transcription.
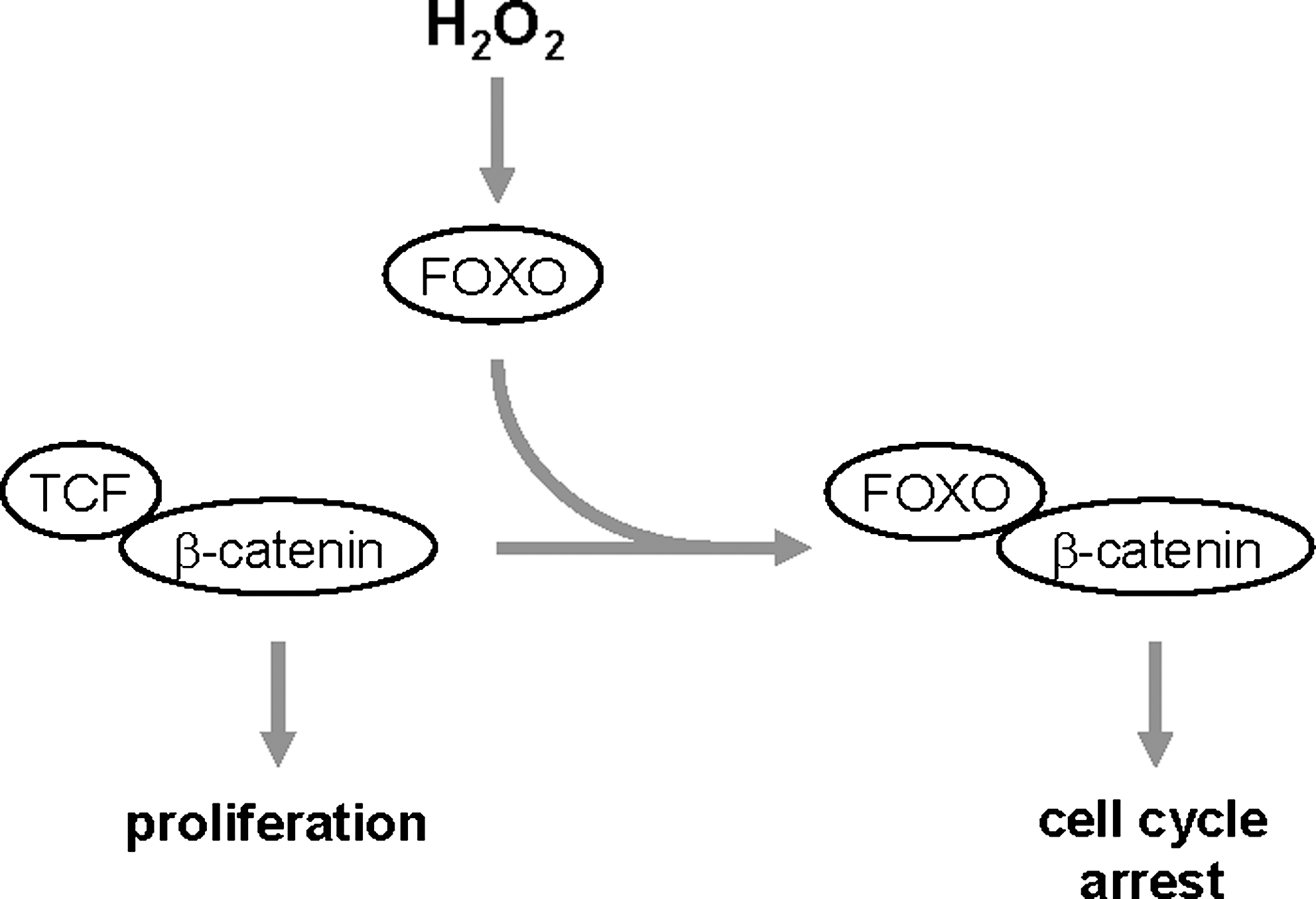
The WNT signaling pathway as a target for ROS-activated FOXO
The WNT (wingless) signaling pathway is involved in many physiological and pathophysiological activities and regulates lipid metabolism and glucose homeostasis. A major effector of canonical WNT signaling is a transcription complex formed by free β-catenin and TCF (T cell factor) that drives gene transcription. Overexpression of FOXO led to a reduced binding between TCF and β-catenin and to FOXO/β-catenin complex formation (50). Additionally, oxidative stress as induced by H2O2 strengthened the binding between FOXO and β-catenin (49, 50) and led to enhanced FOXO transcriptional activity (33). FOXO competes with TCF for the same binding site at β-catenin and suppresses β-catenin/TCF signaling toward proliferation, thus attenuating WNT-mediated signaling activities (Fig. 7) (65). This is supported by findings showing that colon carcinoma cells with activated β-catenin/TCF are arrested in their cell cycle in G1 when active FOXO is ectopically expressed (33). The antagonism of WNT signaling by oxidative stress and FOXO could be a common molecular mechanism that contributes to several pathologies that become more prevalent with increasing age, including insulin resistance, arteriosclerosis, and osteoporosis (81).
Functions of FOXO in the Stress Response
Aging is associated with an increased onset of age-related diseases, including neurodegenerative diseases and cancer. However, the molecular mechanisms of how increased age sensitizes organisms to such diseases are not well understood (35, 111). FOXO transcription factors were implicated in regulating aging and multiple age-related diseases (Fig. 8). One potential mechanism of how FOXOs impact these processes is by increasing the antioxidant capacity of cells (48, 57, 64).
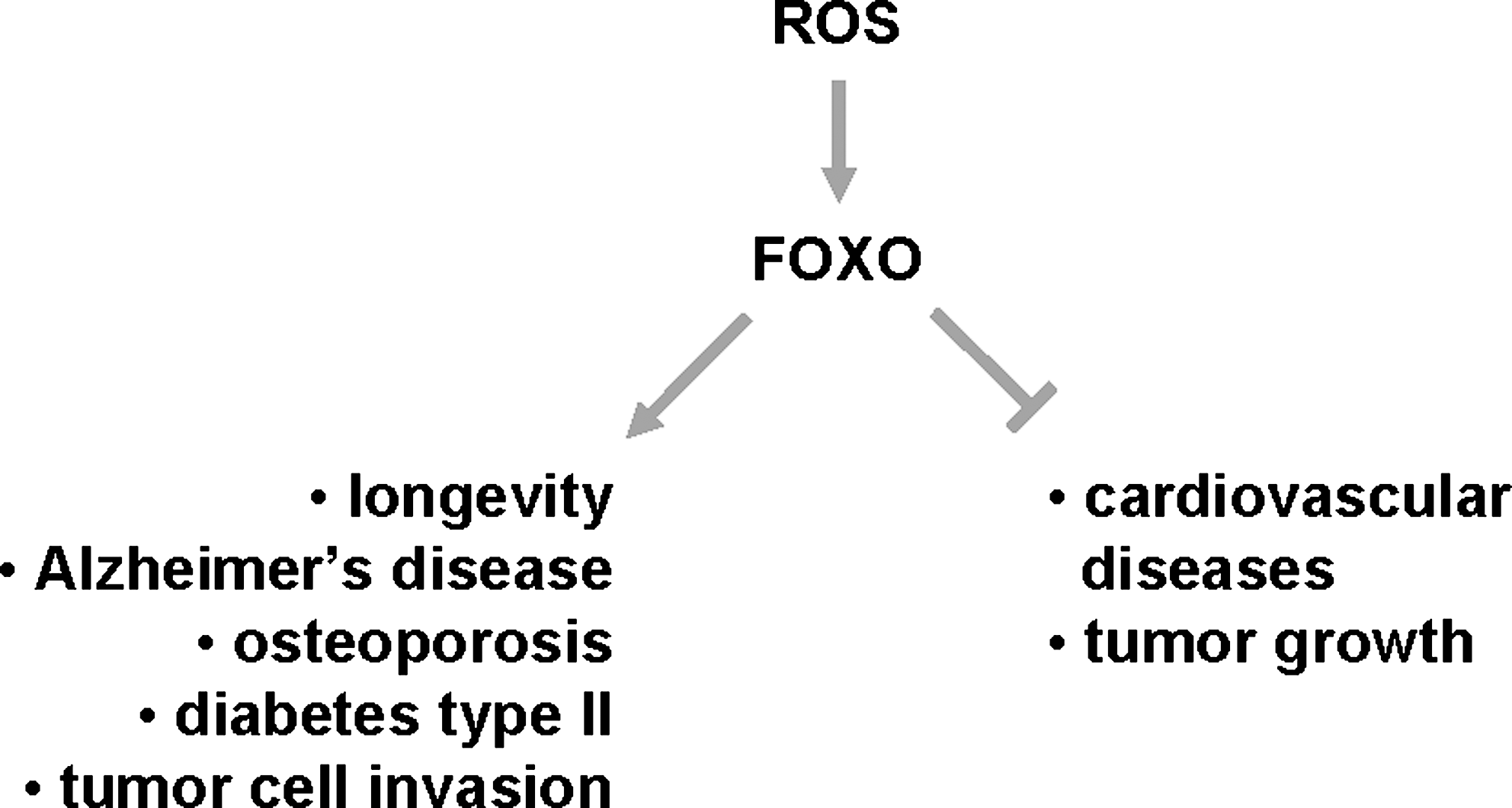
Role of ROS-FOXO signaling in the regulation of lifespan
Oxidative stress is one of the key factors that limit the lifespan of organisms, since aging as well as age-related diseases can result from the accumulation of damage caused by oxidative stress (44). Consequently, increases in oxidative stress resistance in invertebrates are associated with an increase in their lifespan (74, 85). For example, in C. elegans an increase in MnSOD or catalase expression was linked to increased lifespan (48, 98, 99, 116). In C. elegans longevity is regulated by the DAF-2 signaling network, which induces the expression of antioxidant defense enzymes such as MnSOD (48). The daf-2 gene encodes a member of the insulin receptor family and mutations in this gene confer the longevity and the dauer stage phenotype, a growth-arrested larval form specialized for dispersal. Similarly to worm, the attenuation or disruption of the insulin/IGF-1 signaling pathways in mice or rats resulted in increased longevity (11, 47, 106).
The adapter protein p66Shc of which a fraction of the intracellular pool localizes to the mitochondria accelerates aging (9). Mice with a homozygous deletion p66Shc have an extended life span and cells derived from these mice exhibit lower levels of ROS (92). It was suggested that p66Shc may impact life span by repartitioning metabolic energy conversion away from oxidative and toward glycolytic pathways. Such a metabolic switch was demonstrated with p66Shc negative cells that showed a defect in their mitochondrial oxidative capacity and reduced mitochondrial NADH metabolism, but also an increase in lactate production and a stricter requirement for extracellular glucose to maintain intracellular ATP levels. FOXO mediates oxidative stress resistance in p66shc negative cells (93).
In contrast to humans and rodents, which have four functional foxo genes (encoding FOXO1, FOXO3a, FOXO4, and FOXO6), D. melanogaster and C. elegans have only one foxo gene encoding dFOXO in fly and DAF-16 in worm, respectively (121). This makes them ideal systems to investigate the role of FOXO transcription factors in aging. In C. elegans insulin/IGF-1 signaling decreases stress resistance and longevity by inhibiting the FOXO protein DAF-16 (122). On the other hand, altered expression or regulation of FOXO in both D. melanogaster and C. elegans resulted in increased longevity (41, 73, 96). The effects on DAF16/FOXO on lifespan have been linked to the antioxidant capacity of FOXO. Additionally, insulin/IGF-1 signaling inhibits SKN-1, a C. elegans transcription factor that also has a function in the defense against oxidative stress that mobilizes the conserved phase 2 detoxification response. Like in FOXO, this inhibition is mediated by the insulin effector kinases Akt1/2 and SGK-1, which both phosphorylate SKN-1. However, although SGK-1 and FOXO both contribute to increased stress tolerance and longevity, they are most likely independent downstream targets for the insulin signaling pathway (122).
ROS-FOXO signaling in age-related diseases
In AD the accumulation of amyloid beta-peptide (Abeta) plays a critical role in synapse and neuronal loss. One of the mechanisms by which Abeta is believed to mediate neuronal cell death is through increased oxidative stress. The treatment of primary neuronal cultures with Abeta induced p66Shc phosphorylation through JNK and caused apoptotic death (107). Abeta also induced the phosphorylation and inactivation of forkhead transcription factors, overriding the activating effects of JNK on FOXO. On the other hand, ectopic expression of a p66Shc mutant that cannot be phosphorylated by JNK or antioxidant treatment protected cells against Abeta-induced death and also reduced FOXO inactivating phosphorylations. This suggests that the redox regulation of forkhead proteins through JNK/Shc signaling underlies Abeta toxicity, a finding that could be potentially useful to develop therapeutic targets for AD.
Loss of bone mass with advancing age or during osteoporosis is associated with decreased canonical WNT signaling correlating with decreased osteoblast numbers (2). WNT is an essential stimulus for osteoblastogenesis and oxidative stress is a pathogenic factor of bone loss in mice, leading to a decrease in osteoblast number and bone formation [reviewed in (81)]. In osteoblasts H2O2 promotes a diversion of the limited pool of β-catenin from TCF- to FOXO-mediated transcription. This leads to an attenuation of osteoblastogenesis, osteoblast differentiation, and bone formation as observed in mice after the age-dependent increase in oxidative stress. However, among the different FOXO transcription factors, FOXO1 was shown to be a positive regulator of bone formation, since it is required for proliferation, protein synthesis, and resistance to oxidative stress in osteoblasts (2a, 100).
Increased production of ROS was also implicated in the development of age-related cardiovascular diseases. It was shown that moderate overexpression of SIRT1 can protect the heart from oxidative stress. This is due to increased expression of FOXO-regulated genes, including catalase. High levels of SIRT1, however, can also increase basal levels of oxidative stress (1).
Some observations suggest that FOXO1 can promote type II diabetes when constitutively active. Increased FOXO activity contributes to insulin resistance in several animal models for insulin resistance and diet-induced diabetes (89). FOXO factors can act at multiple levels to increase systemic glucose. For example, in hepatocytes sirtuin activation or H2O2 cause nuclear translocation of FOXO1, leading to activation of gluconeogenesis and glycogenolysis (38). Increased glucose concentrations and oxidative stress can also result in JNK-dependent nuclear localization of FOXO1 in pancreatic β-cells, where active FOXO1 reduces the net insulin production through decreasing insulin secretion or attenuating β-cell division [reviewed in (42, 60)]. Pancreatic β-cells are located in the islets of Langerhans and regulate glucose homeostasis through insulin production. FOXO1 expression in islets is increased in diabetic patients (61). Pancreatic β-cell failure is commonly seen in type II diabetes and is caused by chronic exposure of cells to elevated glucose concentrations (glucose toxicity). This glucose toxicity is induced by hyperglycemia-generated oxidative stress. When the glycolytic capacity of β-cells is exceeded by high glucose concentrations, glucose is shunted to other pathways leading to O2 •− production (102). In pancreatic β-cells expression of a constitutively active FOXO1 represses glycolysis, increases free fatty acid oxidation, and also reduced insulin secretion (20). Additionally, expression of constitutively active FOXO1 in β-cells of transgenic mice prevents the compensatory proliferation of β-cells that occurs during insulin resistance (60). On the other hand, FOXO1 haploinsufficiency protects against β-cell failure, probably through inhibition of the pancreatic duodenal homeobox-1 gene promoter activity, a gene that is a key regulator of β-cell development (61). In contrast to the above results obtained by introducing constitutive-active alleles of FOXO1, others have shown that FOXO1 can regulate β-cell proliferation and protect against ROS-induced β-cell failure through induction of NeuroD and MafA, two Insulin2 (Ins2) gene transcription factors (19). Such regulation requires acetylation of FOXO1 by p300/CBP to allow the formation a complex with the promyelocytic leukemia protein and the deactylase SIRT1 (62).
FOXOs' antitumor functions have been discussed above and are mainly mediated through the regulation of genes that control cell cycle arrest and apoptosis. However, with recent work it becomes evident that FOXO may not be a bona fide tumor suppressor. For example, the redox-sensitive transcription factors NF-κB and FOXO3a have been described as regulators of matrix metalloproteinase (MMP) expression (110, 114). In response to starvation conditions that previously have been shown to induce oxidative stress, FOXO3a also contributes to increased cell invasion through regulation of the genes encoding MMP-9 and MMP-13 (113). Thus, FOXO transcription factors may contribute to tumor expansion and metastasis. This is interesting since tumor cell metastasis can be viewed as an integrated escape program that is triggered by changes in the redox state and serves to avoid oxidative stress-mediated damage of cells within the primary tumor. Therefore, oxidative stress-sensitive transcription factors such as FOXO may either induce the stress resistance of cancer cells or induce the migration of cells out of stress areas. Taken together, FOXOs may act as a first line of defense and are tumor suppressors for as long as cells have not become tumorigenic. However, once a tumor is formed many tumor suppressors do not function properly. Similar as known for the multiple roles of transforming growth factor β in tumorigenesis, FOXOs may become pro-tumorigenic or pro-metastatic when reactivated.
FOXO-induced stress resistance could also be important for tumor recurrence. Cancer stem cells (CSC) express stem cell markers, are highly drug resistant, and after radio- or chemotherapy can initiate recurrence. Keeping endogenous and induced oxidative stress at moderate levels mediates drug resistance and allows these cells to survive during treatment (6, 128). For example, human mammary epithelial cancer stem cells contain lower concentrations of ROS, specifically O2 •−, than normal epithelial cells and the more mature progeny of cancer cells (29). Moreover, these differences in ROS levels are critical for maintaining stem cell function. It is likely that FOXO transcription factors also regulate these important aspects of tumor biology.
Other FOXO-regulated stress responses
FOXO signaling is required for the response of hematopoietic stem cells (HSCs) to physiological oxidative stress and the long-term regenerative potential of the hematopoietic stem cell compartment (118). This was implicated for a broad spectrum of tissues [for an additional review see (118)]. FOXO proteins induce stress resistance that led to quiescence and enhanced survival in the HSC compartment (119). Increased expression of FOXO1 was implicated in the regulation of other ROS scavengers such as SOD and catalase to confer resistance to oxidative stress in hematopoietic stem cells (119). Moreover, FOXO-deficient bone marrow showed defective repopulating activity correlating with increased apoptosis of HSC. Further, increases in ROS in FOXO-deficient cells correlated with decreased expression of antioxidant proteins. As an example, the loss of FOXO3a led to defects in HSC and these defects resulted from an accumulation of ROS. FOXO is essential for expression of ataxia telangiectasia mutated, which is critical for oxidative stress-mediated homeostasis of hematopoietic stem cells to preserve the hematopoietic stem cell pool (135).
Another function of ROS-mediated regulation of FOXO at the organismal level is the regulation of the sensitivity of the circadian clock (141). Oxidative stress is one regulator of circadian rhythms. When exposed to oxidative stress, drosophila shows attenuated clock gene cycling in peripheral tissues. Flies lacking FOXO expression in the fat body also lost behavioral rhythms driven by the central clock. It was suggested that FOXO has noncell-autonomous effects on central circadian clock function, but may also regulate age-associated rest and activity rhythms (141).
During development of disease cell can experience low oxygen (hypoxic) conditions. Hypoxia induces FOXO transcription via direct binding of hypoxia-inducible factor-1α (HIF-1α) to the FOXO3a promoter. Increased FOXO3a expression also resulted in enhanced activity (5). During hypoxia FOXO3a induces expression of CITED2, a transcriptional cofactor that functions as a negative feedback loop to control HIF-1 activity and to inhibit HIF-1-induced apoptosis. Such fine-tuning of HIF-1 activity by FOXO3a contributes to the survival response of normal and cancer cells in response to stress induced by hypoxia (5).
Summary
ROS are essential for a large number of cellular processes but are also highly reactive in nature and can result in considerable damage to cellular macromolecules, which contributes to cellular and organismal aging. Moreover, excessive or prolonged ROS formation leads to cellular oxidative stress and was implicated in the onset of a variety of human diseases. Management of ROS levels in cells, whether to prevent potential damage (i.e., in normal cells), or to increase damage (i.e., in cancer cells), can be effective strategies to prevent aging and disease or to cure disease, respectively. Due to their functions as modulators of cellular redox states and decision makers for cellular survival or apoptosis, FOXO transcription factors may be ideal targets for therapeutic approaches aiming to modulate intracellular ROS levels. As an example, nuclear FOXO4 can enhance doxorubicin-mediated cytotoxicity in cancer, and it was suggested that targeting Akt or FOXO4 may provide possibilities in sensitizing cancer cells to cytostatic drugs. This would allow the use of lower drug concentrations to minimize drug-induced adverse effects in patients (79). A potential inducer of FOXO activity is Kaempferol, a flavenoid that increases the lifespan of C. elegans probably by translocating DAF-16 to the nucleus (58). Flavenoids are natural dietary components that posses antioxidant activity. On the other hand, many anticancer drugs exuberantly increase intracellular ROS levels to kill cancer cells. It will be interesting to see if the additional inhibition of antioxidant systems will be effective in sensitizing cancer cells to such treatment.
Footnotes
Acknowledgments
The author would like to thank Heike Döppler for critical reading of the article. This work was supported by grants from the Mayo Clinic SPORE for Pancreatic Cancer (P50 CA102701), the AACR (08-20-25-STOR), and the NCI (R21 CA135102 and R01 CA140182)—all to P.S.