
Abstract
Hepatocyte growth factor (HGF) by stimulating the receptor tyrosine kinase c-Met induces angiogenesis and tissue regeneration. HGF has been shown to antagonize the angiotensin II-induced senescence of endothelial progenitor cells (EPCs), which is mediated by NADPH oxidase-dependent reactive oxygen species (ROS) formation. As growth factors, however, usually require ROS for their signaling, we hypothesized that the proangiogenic effects of HGF require NADPH oxidases and focused on the homolog Nox2, which is most abundantly expressed in EPCs and endothelial cells. Indeed, HGF increased the H2O2 formation in EPCs and human umbilical vein endothelial cells (HUVECs), and this effect was not observed in Nox2-deficient cells. HGF induced the mobilization of EPCs and vascular outgrowth from aortic explants in wild-type (WT) but not Nox2y/− mice. HGF also stimulated migration and tube formation in HUVECs, and antisense oligonucleotides against Nox2 prevented this effect. To identify the signal transduction underlying these effects, we focused on the kinases Jak2 and Jnk. In HUVECs, HGF increased the phosphorylation of these in a Nox2-dependent manner as demonstrated by antisense oligonucleotides. Also, the HGF-induced Jak2-dependent activation of a STAT3 reporter construct was attenuated after downregulation of Nox2. Accordingly, the HGF-stimulated tube formation of HUVEC was blocked by inhibitors of Jak2 and Jnk. In vivo treatment with the Jnk inhibitor SP600125 blocked the HGF-induced mobilization of EPCs. Ex vivo, SP600125 blocked HGF-induced migration and tube formation. We conclude that HGF-induced mobilization of EPCs and the proangiogenic effects of the growth factor require a Nox2-dependent ROS-mediated activation of Jak2 and Jnk. Antioxid. Redox Signal. 15, 915–923.
Introduction
The biological actions of HGF are mediated by the receptor tyrosine kinase c-Met, which dimerizes upon ligand binding and subsequently transautophosphorylates (7). Tyrosine phosphorylation of the receptor allows docking of Src and the adaptor proteins Gab1 and Gab2 (36). Via these pathways subsequently the focal adhesion kinase pathway, the PI3K-Akt pathway (4), and the Ras-MEK-Erk pathway are activated (36).
Interestingly, it recently has been suggested that HGF prevents the angiotensin II-induced dysfunction of endothelial progenitor cells (EPCs) and lowers the angiotensin II-induced reactive oxygen species (ROS) formation in these cells (30). This finding is unexpected given that signaling via tyrosine kinase receptors, like the VEGF (26) or the EGF receptor (6), usually requires ROS formation to achieve a transient inactivation of tyrosine phosphatases, which, as a consequence of their high specific activity, otherwise instantaneously dephosphorylate the active tyrosines (24, 38).
The main sources of ROS in endothelial cells are, besides mitochondria, the enzymes of the NADPH oxidase family (2). The family of NADPH oxidases consists of seven members (Nox1–Nox5 and DUOX 1 and 2) named by the large catalytically active membrane-bound subunit that is different among all members of the NADPH oxidase family. Endothelial cells express mainly two members of this family, Nox2 and Nox4, although recent findings support an expression of Nox5 in human microvascular endothelial cells. Most studies suggest that the classic NADPH oxidase Nox2 is involved in the signaling of tyrosine kinases in endothelial cells as demonstrated for tyrosine kinase receptors like the VEGF receptor (39) but also for receptors depending on intracellular tyrosine kinases like the erythropoietin receptor (34). Nox2, which is also highly expressed in leukocytes, comprises two membrane-bound (Nox2 and p22phox) and several cytosolic (p47phox, p67phox, and Rac) subunits. The activation of Nox2 occurs via the PI3-kinase pathway, which leads to the activation of Rac1 and the p21-activated protein kinase Pak-1, which phosphorylates and activates the cytosolic NADPH oxidase subunit p47phox. Active p47phox binds p67phox and transfers this activator protein to the membrane, where it interacts with Nox2. Active Rac1 also binds to the membrane through its geranylgeranyl side chain, and its interaction with p67phox and Nox2 is required for the activation of this NADPH oxidase (1).
In this study we determined whether NADPH oxidase-derived ROS formation is required for the signaling of HGF and the physiological responses of EPCs and endothelial cells. We provide evidence that HGF induces the mobilization of EPCs and a proangiogenic phenotype in endothelial cells via a process depending on the NADPH oxidase Nox2.
Materials and Methods
Animals and animal procedure
Animal experiments were approved by the local governmental authorities (approval number: F28/14) and were performed in accordance with the animal protection guidelines. C57Bl/6 mice were purchased from Charles Rivers. Nox2 knockout mice were obtained from Jackson Laboratories via Charles Rivers and bred at the local animal facility. HGF (5 μg/kg) or 0.9% NaCl (solvent) was injected in a series of 2 days followed by 1 day intermission before sacrifice. The Jnk inhibitor SP600125 (30 mg/kg/day) or solvent (40% PEG400-Glycol) was injected in a series of 5 days in a volume of 100 μl before treatment with HGF similar as reported previously (17).
FACS analyses of EPCs
Circulating EPCs were prepared from blood obtained by cardiac puncture in heparinized syringes as reported previously (34). The plasma was obtained by centrifugation at 800 g for 10 min. The remaining blood fraction was resuspended in 1 ml phosphate-buffered saline containing 0.5% bovine serum albumin and 2 mM EDTA, overlaid on top of 3 ml LSM 1077 lymphocyte separation medium (PAA Laboratories) and subjected to density gradient centrifugation (800 g, 10 min). Mononuclear cells were counted, and lineage-negative cells were enriched using a lineage cell-depletion kit (Miltenyi; Bergisch-Gladbach) according to the manufacturer's instruction. Lineage-negative cells were stained for Sca1- and Flk-1 with the aid of phycoerythrin and allophycocyanin-labeled antibodies or with the proper control IgGs in a dilution of 1:100 in the phosphate-buffered saline mentioned above. All antibodies were purchased from Becton-Dickinson. After washing, FACS analysis was performed with the aid of a FACS-Calibur (Becton-Dickinson).
Aortic ring outgrowth
Experiments were performed as reported previously (33). In brief, murine aortas were prepared and cleared from surrounding tissue and blood. Aortas were cut into rings of 1 mm length, placed on a Matrigel matrix, and stimulated with or without HGF as indicated. After 3 days of culture, images were taken and outgrowth areas were analyzed using the freely available ImageJ software.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords by dispase digestion or obtained from Lonza. HUVECs were used between passages 1–3 and cultured in endothelial basal medium (EBM) medium supplemented with 10% fetal calf serum (FCS), bovine brain supplement, and human recombinant EGF. EPCs were cultured in EBM medium containing 20% FCS, bovine brain supplement, and human recombinant EGF according to the instructions provided by Lonza.
Murine lung endothelial cells (LECs) were isolated from freshly prepared murine lungs as described before (34). The tissue was minced digested by dispase at 37°C. After several washing steps, LECs were separated magnetically using CD114-coated Dynabeads (MACS). LECs were used between passages 5 and 9.
Colony-forming units (CFU) were obtained after plating 1 × 105 cells per 12 well in methylcellulose (Methocult GF H4535, Stem Cell Technologies) including stem cell factor, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, interleukin 3, and interleukin 6 as indicated by the manufacturer. CFU were counted after 14 days of incubation by three independent investigators.
Tube formation assay
For inhibition of Jnk and Jak2 cells were pretreated with a solvent, SP600125 (30 μM) or AG490 (50 μM), for 30 min. Knockdown of Nox2 by antisense-oligonucleotides as described below was carried out over 3 days before the experiment. The assay was performed as described previously (27) with minor modifications. Briefly, 15-well μ-Slide Angiogenesis (ibidi) were coated with ice-cold Matrigel (10 μL/well), which was allowed to polymerize at 37°C for about 30 min. Thereafter, 50 μl of a suspension of HUVECs (1.5 × 105 cells/ml) were seeded onto the Matrigel and cultured in EBM medium (Lonza), supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2% (v/v) FCS, with or without 10 ng/ml HGF in the presence of solvent or the appropriate inhibitor. Tube formation was assessed after 2 h with a magnification power of 10×.
Scratch wound migration assay
HUVECs were transfected with sense- or antisense-oligonucleotides and cultured for 3 days until they reached confluence and subsequently serum reduced (0.5% FCS) overnight. The next day the monolayer was wounded with the aid of a sterile pipette tip and cells were allowed to migrate in the presence or absence of HGF (10 ng/ml) for 5 h.
Boyden chamber migration assay
Spleen-derived monocytic cells were separated by ficoll-gradient centrifugation and preincubated with or without the Jnk inhibitor SP600125 (30 μM) for 30 min and seeded as 2 × 106 cells per cm2 on Boyden chamber inlets (8 μm pore size, transwell; Becton-Dickinson). Migration was allowed for 4 h with the cells in RPMI in the upper compartment and EBM containing 0.1% FCS with or without HGF (10 ng/ml). Subsequently, cell in the lower compartment were counted by an automated cell counter (Casy).
Amplex red assay for H2O2 production
Cells were grown on 12-well dishes to 90% confluence. After starvation over night in serum-reduced medium (0.5% FCS) cells were incubated with or without HGF (10 ng/ml) in phenol red-free medium containing Amplex red (50 μM; Invitrogen), horse-radish peroxidase (2 U/ml), and 0.5% FCS. After 45 min the supernatant was transferred into 96-well plates and H2O2-dependent oxidation of Amplex red was measured in a microplate fluorimeter (excitation 540 nm, emission 580 nm). Experiments were performed in the presence and absence of catalase (50 U/ml). H2O2 formation was determined as the catalase-sensitive part of the Amplex red oxidation.
Transfections
Oligonucleotides as previously published (41) were synthesized with phosphothioate modifications indicated by asterisks (*) (Biospring). The following sequences were used: Scrambled Nox2 5′- C*A*T* T*GT GGA GTG ACA G*G*A *G*-3′ and antisense Nox2 5′- A*A*C* T*GG GCT GTG AAT G*A *G* G*-3′. For transfection, the oligonucleotides were diluted to 50 μM stock solution and given to the cells in a final concentration of 50 nM without transfection reagent. Oligonucleotides were replaced together with the medium every other day and experiments were performed at the third day after 1 day of serum-reduction. Efficient knockdown with this approach was demonstrated previously by us (34).
GAS-luc and pGL2P reporter gene plasmids were generous gifts from R. Jaster (University of Rostock, Germany) (12). Transfection of the plasmid was performed using the Transpass V HUVEC transfection reagent (New England Biolabs) according to the manufacturer's instructions.
Luciferase assay
Luciferase activity was assayed with a luciferase assay kit from Promega following the manufacturer's instructions and a Berthold LB9505 luminometer (Bad Wildbad).
Immunoblotting and immunoprecipitation
Western blot analyses were performed with an infrared-based detection system (Odyssey; Licor). All primary antibodies were purchased from Cell signaling and infrared-fluorescent-dye-conjugated secondary antibodies were obtained from Licor (Bad Homburg). The following lysis buffer was used (pH 7.4, concentrations in mM): Tris-HCl (50), NaCl (150), sodium pyrophosphate (10), sodium fluoride (20), nonidet P40 (1%), sodium desoxycholate (0.5%), proteinase inhibitor mix, phenylmethylsulfonyl fluoride (1), orthovanadate (2), and okadaic acid (0.00001).
Statistics
All values are mean ± standard error of the mean. Statistical analysis was performed by analysis of variance followed by LSD post hoc testing. Densitometry was performed with the odyssey software. A p-value of <0.05 was considered statistically significant.
Results
HGF induces mobilization of EPCs in wild-type but not Nox2y/− mice
In wild-type WT mice, injection of HGF but not saline control on three consecutive days increased the number of circulating EPCs by approximately fivefold in WT mice. In contrast, no mobilization in response to HGF was observed in Nox2y/− mice (Fig. 1A). Moreover, HGF increased the number of spleenocytes in WT mice but not in Nox2y/− animals (Fig. 1B). This effect is reflected in the ability of the spleenocytes to form CFU: the number of spleenocyte-derived CFUs from WT donor animals was significantly higher after HGF treatment compared to solvent control. In contrast, CFU number was not affected by HGF-treatment in Nox2y/− mice (Fig. 1C).

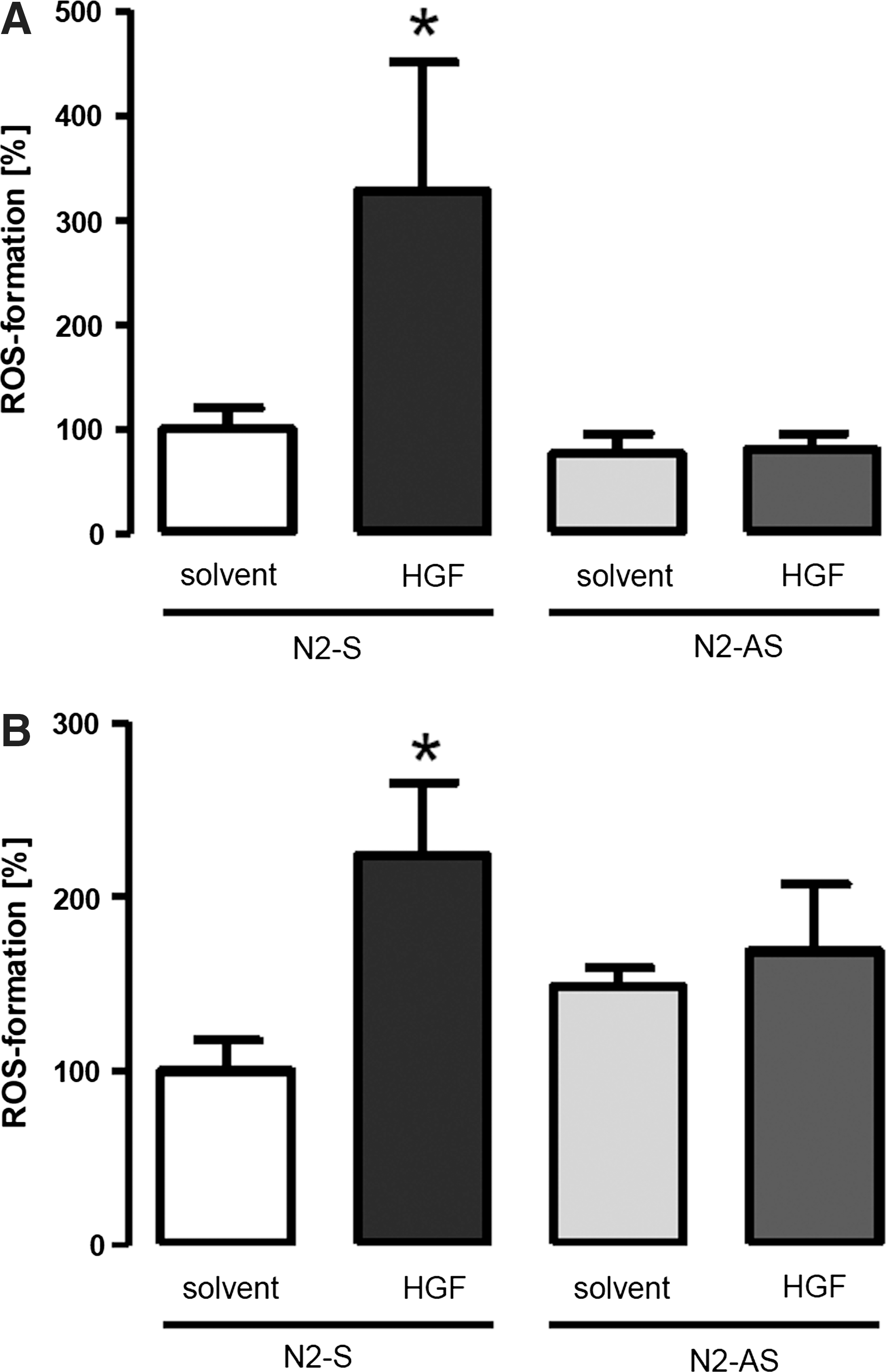
HGF induces ROS formation in a Nox2-dependent manner
As the previous data indicated that the action of HGF involves the NADPH oxidase Nox2, we determined whether or not HGF acutely stimulates a Nox2-dependent ROS formation. Amplex red measurements demonstrated that the H2O2 production of EPCs (Fig. 2A) as well as of HUVEC (Fig. 2B) more than doubles in response to HGF. If the cells were pretreated with Nox2 antisense oligonuclotides, this effect was not observed, demonstrating that HGF acutely activates Nox2 (Fig. 2).
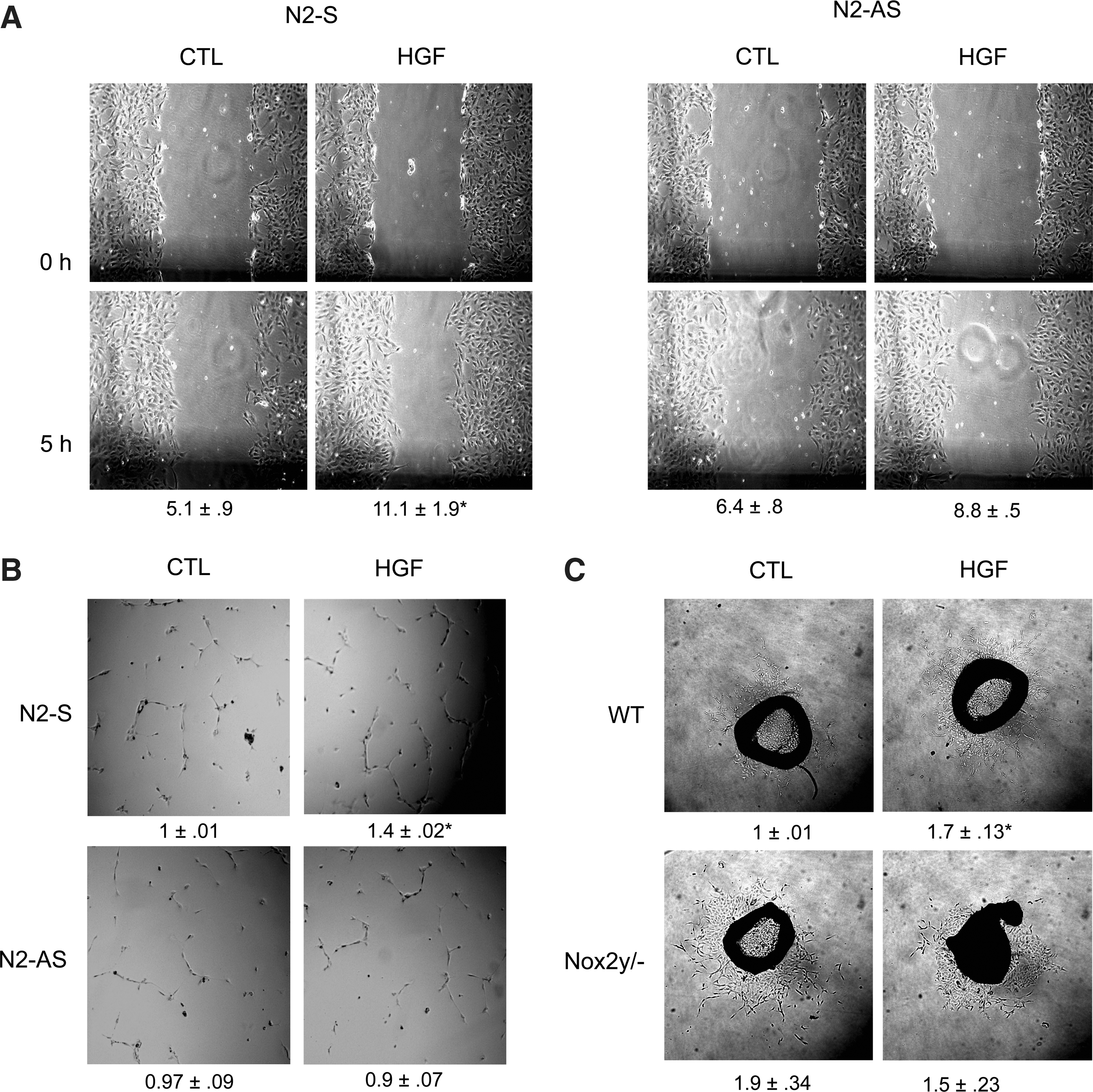
Nox2 mediates the proangiogenic effects of HGF
To determine whether the Nox2-derived ROS production is required for the proangiogenic effect of HGF, several ex vivo angiogenesis assays were performed. In the scratch wound assay, HGF approximately doubled the migration speed of HUVECs and this effect was not observed after downregulation of Nox2 (Fig. 3A). Moreover, HGF stimulated the tube formation of HUVECs in a Matrigel assay and also this effect was attenuated after downregulation of Nox2 (Fig. 3B). To get a closer insight into the process of new vessel formation, aortic outgrowth assays were performed. HGF stimulated outgrowth in aortic rings of WT mice but not of Nox2y/− mice. Interestingly, basal outgrowth in the absence of HGF was higher in aortic rings of Nox2y/− mice than in WT animals, suggesting that potentially these mice compensate the irresponsiveness to growth factors by a basally increased growth rate (Fig. 3C).
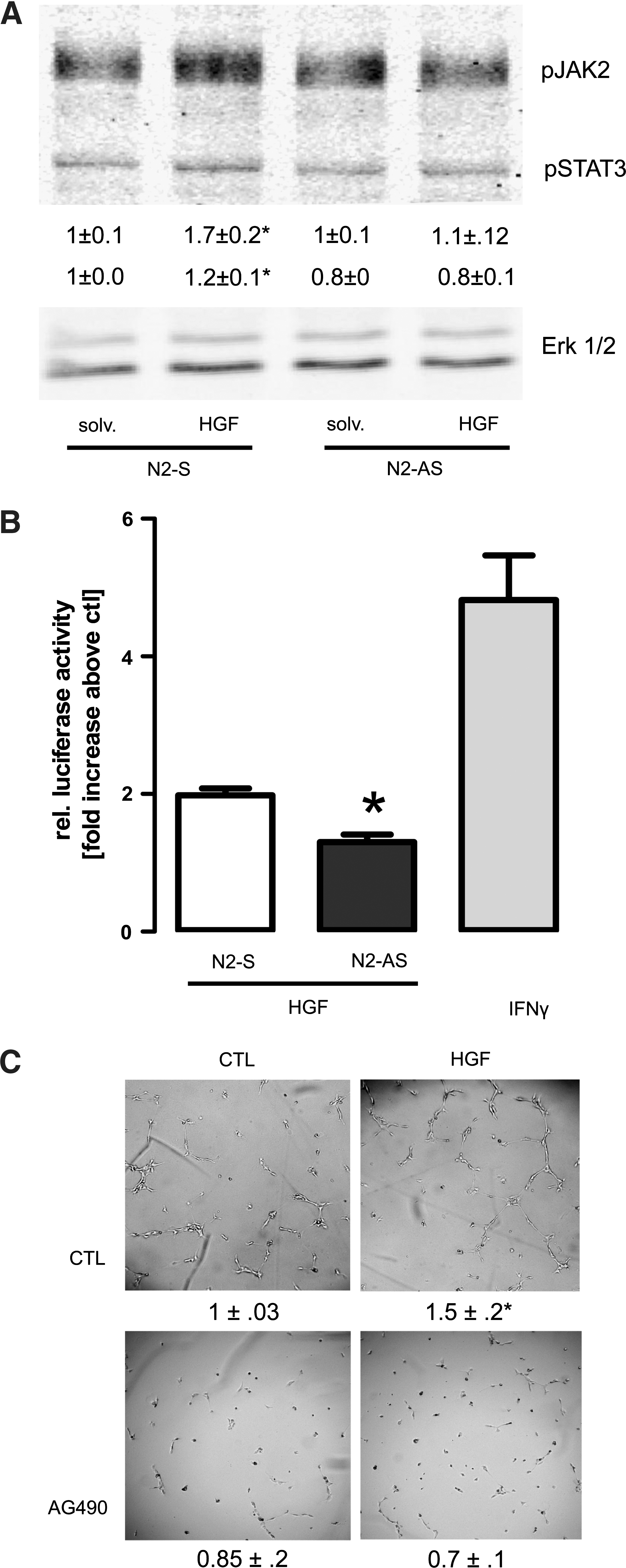
A Nox2-mediated activation of Jak2 contributes to the proangiogenic effect of HGF
A large aspect of the signaling of HGF depends on the transcription factor STAT3. Although the c-Met receptor tyrosine kinase should be able to directly increase STAT3 activity, for other growth factors, an additional involvement of the Jak2 kinase has been suggested. Jak2 activity has been shown to increase in response to ROS and the angiotensin II-induced activation of Jak2 in rat smooth muscle cells is NADPH oxidase dependent (32). In HUVECs, HGF slightly increased the phosphorylation of Jak2 and STAT3 and this effect was not observed in Nox2-deficient cells. As we, however, detected a substantial basal phosphorylation in these cells, the relative increase in response to HGF was minor (Fig. 4A). Thus, luciferase reporter gene assay for STAT3 activity was performed in HUVECs and demonstrated that HGF approximately doubled STAT3 activity and also this effect was attenuated by the downregulation of Nox2 by antisense oligonucleotides. Although a twofold increase in luciferase activity is also not considered strong, it should be realized that even the positive control interferon γ only increased the signal by fivefold (Fig. 4B). To study whether Jak2 contributes to the proangiogenic effect in HUVECs, we studied tube formation in the presence of the Jak2 inhibitor AG490. This compound prevented the HGF-induced increase in tube formation and, as expected from the basal phosphorylation of Jak2 and STAT3, also lowered basal tube formation (Fig. 4C). These data demonstrate that Jak2 is required for tube formation and suggest that the Nox2-dependent stimulation of Jak2 by HGF contributes to the proangiogenic effects of the compound.
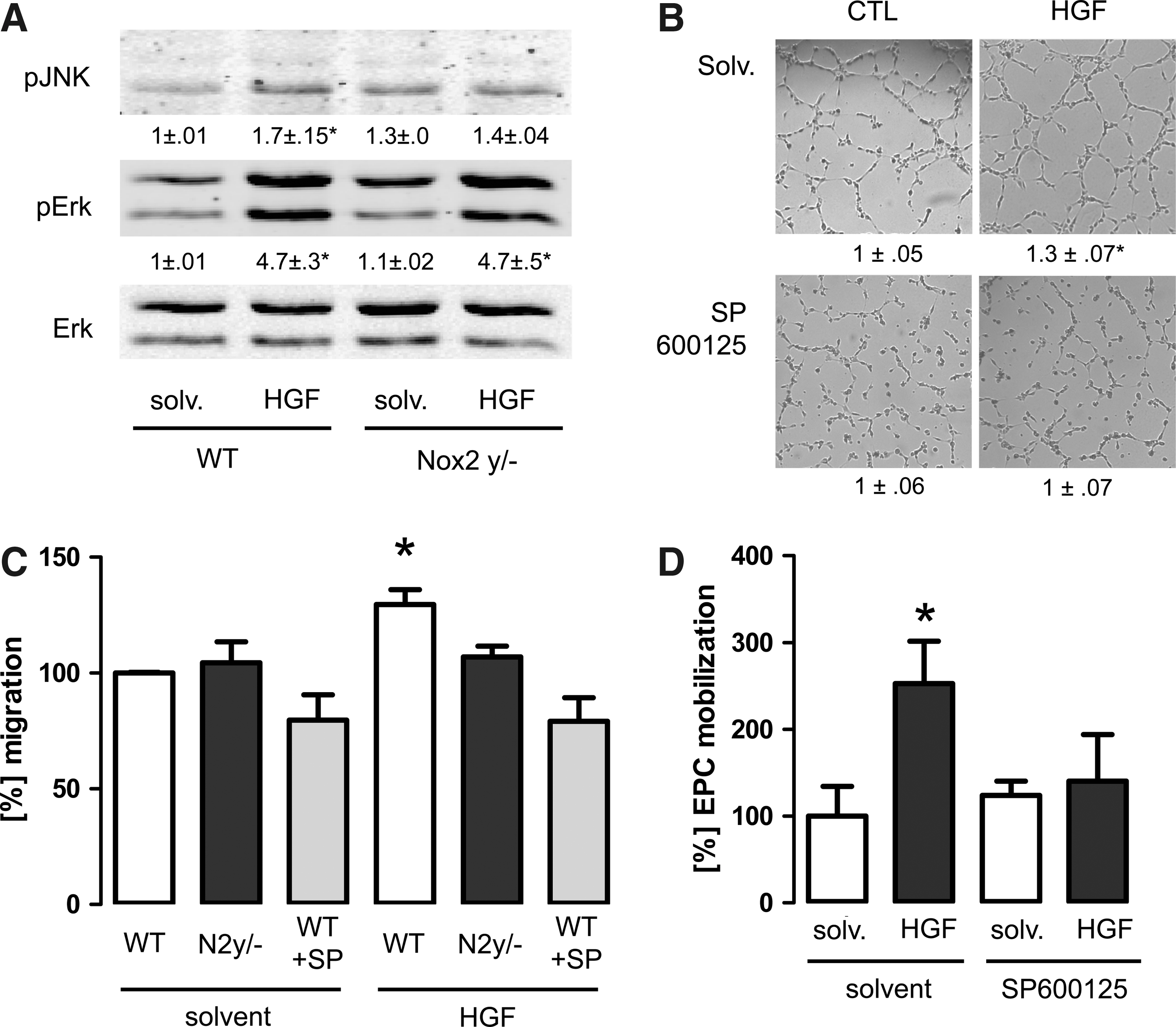
A Nox2-dependent stimulation of Jnk is required for EPC mobilization in vivo
MAP kinases are known effectors of NADPH oxidases. In particular, the activation of p38-MAP kinase and Jnk has been suggested to require Nox2. In LECs from WT mice, HGF increased the phosphorylation of Jnk and Erk1/2 but not of p38-MAP kinase. A different pattern was observed in cells derived from Nox2y/− mice as only the phosphorylation of Erk1/2 but not of Jnk was increased in response to HGF. Interestingly, the basal Jnk phosphorylation of Nox2-deficient cells was slightly higher than that of WT cells (Fig. 5A). To determine whether or not Jnk may contribute to the proangiogenic effects of HGF, the Jnk inhibitor SP600125 was used. In the tube formation assay, the compound blocked the HGF-induced increase in tube formation. Similar as the Jak2 inhibitor, however, SP600125 also lowered basal tube formation, although the effect was less pronounced (Fig. 5B). SP600125 also prevented the migration of spleenocytes in response to HGF as studied by a Boyden chamber assay (Fig. 5C). We although sought to determined the effect of SP600125 on the ex vivo proliferation of cultured EPCs in response to HGF. Under this condition, however, SP600125 inhibited the basal proliferation by >50% so that specific effect of HGF could not be uncovered (data not shown). As on such a basis, the in vivo relevance of the findings is difficult to judge, we determined the in vivo mobilization of EPCs in the presence of SP600125. Treatment of mice with solvent had only a minor effect on HGF-induced mobilization. In contrast, in mice treated with SP600125, no HGF-induced EPC mobilization was observed. Importantly, in vivo the compound had no effect on basal EPC level, demonstrating that Jnk is indeed essentially and specifically involved in the HGF-induced mobilization of EPCs (Fig. 5D).
Discussion
In this article, we demonstrate that HGF induces the mobilization of EPCs in a Nox2-dependent manner. The process of mobilization is accompanied by proliferation and subsequently migration of EPCs out of the bone marrow. Nox2 was also required for the HGF-induced migration of HUVECs and for in vitro angiogenesis from aortic rings. HGF acutely increased Nox2-dependent ROS formation in HUVECs as well as in EPCs and the acute HGF-induced activation of Jak2, STAT3, and Jnk, but not of Erk1/2, was dependent on Nox2. Importantly, ex vivo inhibition of Jak2 and Jnk prevents the proangiogenic effect of HGF, and in vivo inhibition of Jnk blocked HGF-induced EPC mobilization. These observations qualify Nox2-derived ROS as an essential intermediate in the beneficial vascular effects of HGF.
As a potential target of Nox2-dependent signaling, we identified Jak2. Nox2 deficiency attenuated the phosphorylation of Jak2 as well as of its downstream target STAT3. Moreover, pharmacological inhibition blocked the HGF-induced tube formation in HUVECs. These findings were unexpected given that the receptor tyrosine kinase c-Met on its own should sufficiently activate the STAT3 pathway. Interestingly, it was previously noted that also other tyrosine kinases like c-Src were required to elicit an effective response to HGF. Phospho-sites of the c-Met adaptor protein Gab1 were differentially phosphorylated by the two tyrosine kinases (5), which emphasizes that the complex signaling of a tyrosine kinase receptor occurs via several overlapping cascades and is not a simple linear mode.
Agonist-dependent redox signaling by NADPH oxidases is frequently mediated by a transient oxidation of thiols in proteins that either increases or reduces their activity (2). Several studies document a transient inhibition of phosphatases in response to NADPH oxidase activation. This event is required to perpetuate signaling by tyrosine kinases as the activities of the latter enzymes are usually several orders of magnitude lower than that of tyrosine phosphatases. We recently suggested that a transient inhibition of the phosphatase SHP-2 underlies Nox2-dependent signaling in response to erythropoietin (34). Although it has not yet been shown that c-Met stimulation results in Nox-dependent SHP-2 inactivation, a dominant negative mutant of this phosphatase enhanced c-Met signaling in Madin Darby canine kidney cells and promoted Vav2-dependent signaling, whereas overexpression of the phosphatase prevented HGF signaling (16).
The fundamental inhibitory role of phosphatases has previously been noted also for HGF signaling: in HUVECs tumor necrosis factor-α (TNF-α) blocks c-Met signaling by the induction of the SHP-2 homolog SHP-1 (37). Interestingly, SHP-1 is also a direct negative regulator of Nox-activation as shown for the AT2-receptor-mediated inhibition in response to angiotensin II (18). Accordingly, downregulation of SHP-1, different to the response observed by downregulation of SHP-2, did not restore agonist-induced signaling in Nox2-deficient cells (34). In hepatocytes c-Met also induces the receptor protein-tyrosine phosphatase leukocyte antigen-related protein (LAR) (22), which subsequently limits the responses to HGF. Interestingly, LAR itself is readily inactivated by H2O2 (40) and thus a target of Nox-derived ROS. Moreover, genetic deletion of LAR in vascular smooth muscle cells promoted the H2O2-induced phosphorylation of Jak2 and STAT3 (20).
In addition to Jak2 and STAT3, HGF-induced signaling also involves MAP-kinases (36). These enzymes are important mediators of NADPH oxidase-dependent redox signaling and activation. Depending on the stimulus, a differential activation of p38 MAP-kinase, Jnk, and Erk1/2 has been linked to individual Nox proteins (3). Although the underlying mechanisms of this central and specific redox-signaling is still obscure, also in the present study we observed that only the phosphorylation of Jnk but not Erk1/2 in response to HGF was Nox2 dependent. Importantly, in hepatic cells obtained from knockout mice of the phosphatase PTP1B, an increased phosphorylation of this MAP-kinase was observed in response to HGF (31). Interestingly, this phosphatase is a known target of Nox-derived ROS: agonist-stimulated inactivation of PTP1B by Nox1 is required for interleukin 4 signaling (35) and by Nox4 for insulin-induced signaling (23).
We previously reported that a Nox1-dependent activation of Jnk in smooth muscle cells is required for migration in response to the basic fibroblast growth factor (33) and inhibition of Jnk also blocked the migration of human brain microvascular endothelial cells in response to HGF (29). Accordingly, we observed that also the migration of murine spleenocytes and the proangiogenic effects of HGF in the tube formation assay in cell culture and, most importantly, the mobilization of EPCs in vivo were absent not only after loss of Nox2 but also after pharmacological blockade of Jnk. To our knowledge this is the first report on a role of Jnk for EPC mobilization. We have previously reported that Jnk is required for the induction of matrix metalloproteinases (33). It is attractive to speculate that this process is also required for HGF-induced mobilization as matrix metalloproteinases degrade the matrix that retains the EPCs in the bone marrow. Indeed, other mobilizing factors such as SFD-1α/CXCR4 also activate Jnk (44) although up to now the physiological significance of this finding was obscure.
It was previously noted that HGF has a protective effect against H2O2-induced oxidative stress and cell damage in the heart (14, 25). HGF also induced heme oxygenase 1 (HO-1), which is involved in the inhibitory effect of HGF on ROS-induced apoptosis (13, 28). Interestingly, HO-1 itself appears to be induced by NADPH oxidase activation (11, 19), providing some support for our present finding that HGF activates the NADPH oxidase. Induction of HO-1 also helps understanding why on the long run HGF prevents negative effects of NADPH oxidase overactivation as the enzyme indirectly through its product carbon monoxide is potently able to inhibit NADPH oxidase activity (8). Despite this, also HO-1 is thought to be involved in the mobilization of EPCs (21). As in vivo, however, HO-1 controls the plasma level of VEGF and SDF-1α, it is uncertain whether NADPH oxidase inhibition in vivo contributes to this process (21).
In conclusion, in the present study we provide evidence that the NADPH oxidase Nox2 is required for the expression of a proangiogenic phenotype in response to HGF. Nox2 contributes to the activation of STAT3 and the phosphorylation of Jnk and both pathways were observed to be essentially involved in the proangiogenic response of HGF.
Footnotes
Acknowledgments
This study was supported by grants from the Deutsche Forschungsgemeinschaft (SFB815/TP1 [K.S.], SFB834/TPA2 [R.P.B.]), the excellence cluster cardio-pulmonary system (ECCPS) (R.P.B.), the Hugelschaffner-Stiftung (K.S.), and the Goethe-University.
Author Disclosure Statement
No competing financial interests exist.