
Abstract
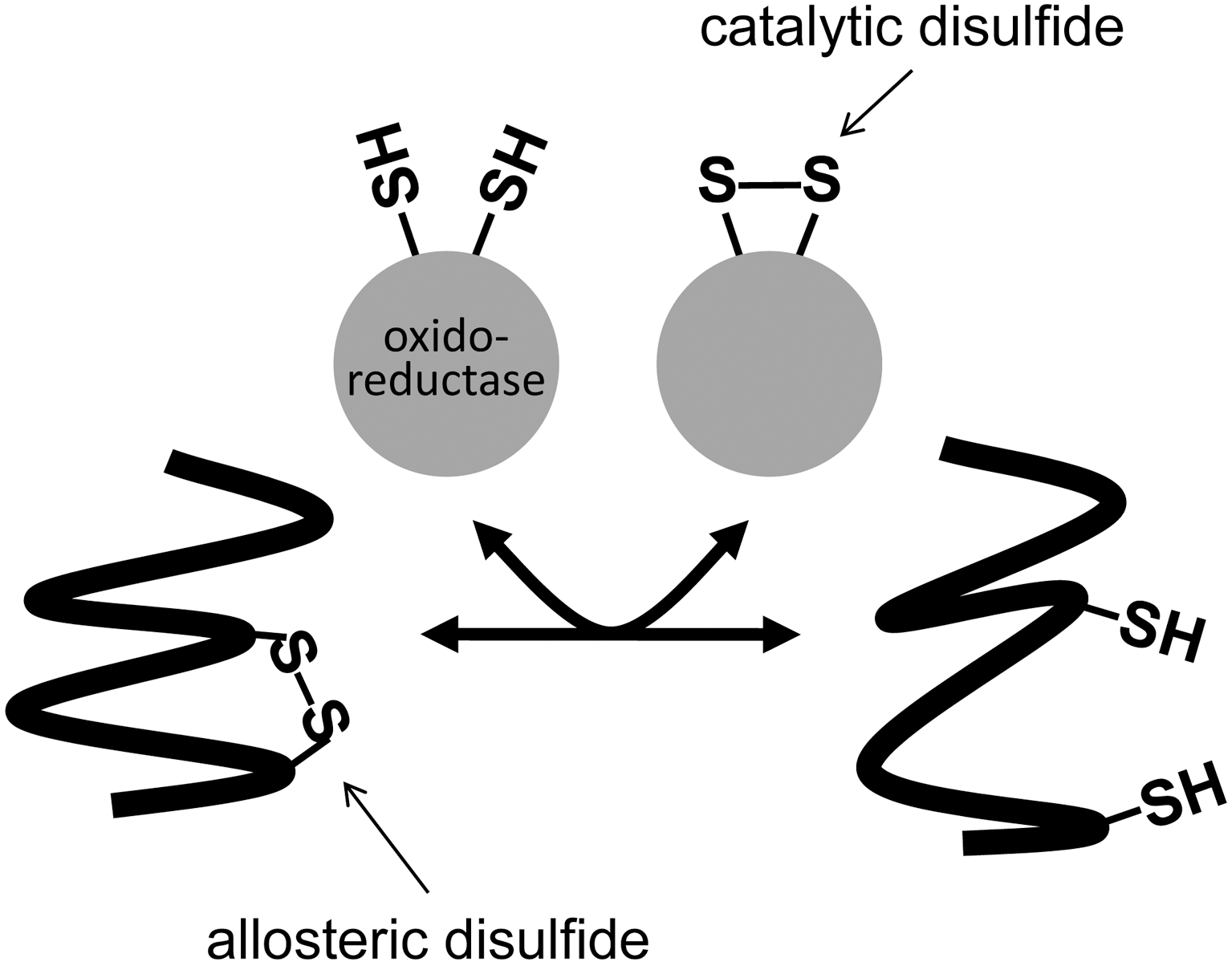
Protein disulfide bonds are the links between the sulfur atoms of two cysteine amino acids. All the known life forms appear to make this bond. Most disulfide bonds perform a structural role by stabilizing the tertiary and quaternary structures. Some perform a functional role and can be characterized as either catalytic or allosteric disulfides. Catalytic disulfides/dithiols transfer electrons between proteins, whereas the allosteric bonds control the function of the protein in which they reside when they undergo redox change. There are currently five clear examples of allosteric disulfide bonds and a number of potential allosteric disulfides at various stages of characterization. The features of these bonds and how they control the activity of the respective proteins are discussed. A common aspect of the allosteric disulfides identified to date is that they all link β-strands or β-loops. Antioxid. Redox Signal. 14, 113–126.
Introduction
Evolution of Cysteine and Cystine
From a physicochemical perspective, cysteine stands out from the other 20 common amino acids. The side chain thiol of cysteine affords the amino acid a wide range of functional roles including disulfide bond formation, metal binding, electron donation, and redox catalysis. Disulfide bond formation is unique for cysteines. Given the reactivity of cysteines, its acquisition is likely to have a significant role in protein evolution.
Using a conserved set of 65 proteins from 26 species across the three kingdoms, it was shown that the cysteine content in the set of proteins has more than doubled from 0.4% to 0.85% since the last universal ancestor (9). The expansion of this initially underrepresented amino acid continues in current protein evolution. Jordan et al. (46) compared the change in amino acid frequencies in 15 triplets of closely related genomes across all three domains of life. In all sets of genomes analyzed, cysteine was gained in the sister genomes when compared with the outgroup. In particular, cysteine was the strongest gainer in 11 of the 15 sets. The same trend is seen when the analysis was performed on human single-nucleotide polymorphisms. The finding suggests that, even today, cysteines are continuing to be positively selected.
Considering that cysteines are the second most conserved amino acid after tryptophan (45), it is commonly accepted that cystine residues or disulfide bonds are also highly conserved in proteins. We have analyzed the evolution of disulfide bonds across different species using a dataset of 5181 nonredundant cystines (96). We compared the conservation of disulfide-bonded cysteines, nondisulfide-bonded cysteines, tryptophan, and all amino acids combined. When conservation is expressed as a function of the time between humans and various last common ancestors through evolution, it is apparent that disulfide-bonded cysteines are significantly more conserved than cysteines not involved in disulfide bonds (96). Moreover, disulfide-bonded cysteines are even more conserved than tryptophans. The high conservation of disulfide-bonded cysteines is likely to reflect the requirement for the individual cysteines to be acquired or lost in pairs. Over all species, >90% of disulfides are conserved once acquired. This highlights the importance of disulfide bonds in protein evolution.
There is also a positive correlation between the rate of disulfide bond acquisition and organismal complexity as measured by distinct numbers of cell types (96). In comparison, the rate of unpaired cysteine acquisition is uncorrelated with organismal complexity. The faster rate of accrual of disulfide bonds in multicellular organisms is in accordance with the greater diversity of protein function in these species. Moreover, the relatively higher rate of acquisition of new disulfide bonds is probably a contributing factor to the sophistication of protein function in higher eukaryotes.
Disulfide Bond Structures
We are working on the hypothesis that the different types of disulfide bonds, structural versus functional, have in common particular configurations. To test this hypothesis, we examined the general features of all the disulfide bonds in X-ray and nuclear magnetic resonance spectroscopy (NMR) structures in the Protein Data Bank (86, 87).
We have characterized a disulfide bond as consisting of six atoms including the two α-carbons of the cysteine residues, Cα–Cβ–Sγ–Sγ′–Cβ′–Cα′ (Fig. 2A). These six atoms define five χ (chi) angles and each χ angle can be either positive or negative. This definition equates to 20 possible disulfide bond configurations (86). The three fundamental types of disulfide are defined by the angle of the central three bonds: the spirals, hooks, or staples (Fig. 2B). If the χ3 angle is positive the bond is right handed, and if the χ3 angle is negative it is left handed (80). A disulfide is a minus right-handed spiral (−RHSpiral), for example, if the χ1, χ2, χ3, χ2′, and χ1′ angles are negative, positive, positive, positive, and negative, respectively.
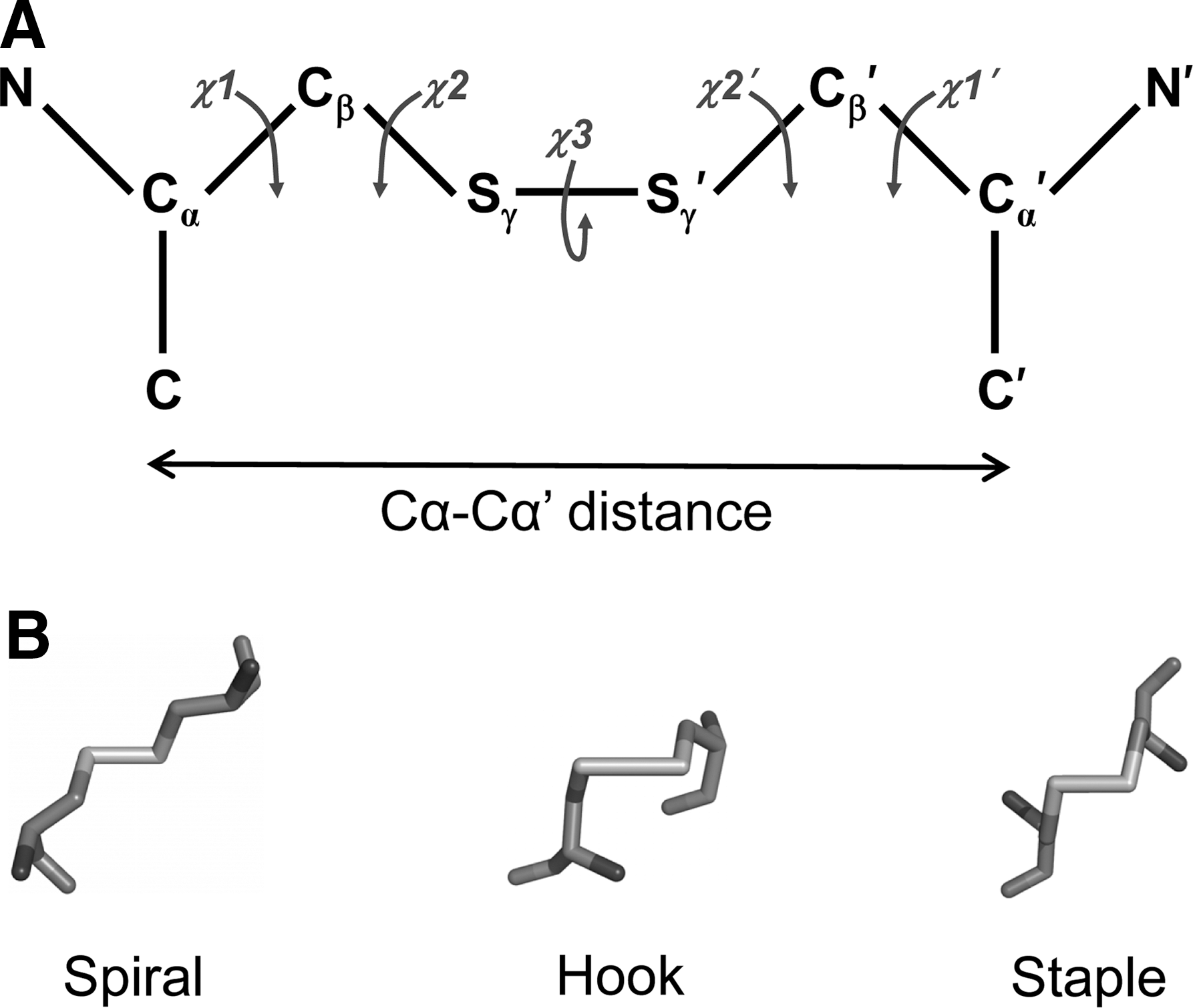
Structural disulfide bonds are most often spirals, in part because they are the most common disulfide. −LHSpiral bonds, for instance, account for nearly 30% of all disulfides in X-ray structures and 20% of the disulfides in NMR structures (87). Nearly all the catalytic disulfides are +/−RHHooks. This is because they share a common CysXXCys motif in a thioredoxin-fold domain structure. The −RHStaple is the most common allosteric configuration (86). A defining feature of −RHStaples is the close proximity of the α-carbon atoms of the two cysteine residues: 4.3 Å compared with a mean of 5.6 Å for all disulfides (86). The α-carbon atom distance is short because of the secondary structures that the cysteines link. About 60% of these bonds link adjacent strands in the same β-sheet. The strands are so close in the β-sheet that they need to pucker to accommodate the disulfide bond. The puckering can result in a significant strain on the bond (86, 97).
Analysis of the −RHStaple disulfides in NMR structures indicates that they can exist in the −LHStaple configuration. That is, the disulfides can switch between the two configurations in the solution structure. The −LHStaple disulfides have an even shorter mean Cα–Cα′ distance and higher mean torsional energy than the −RHStaple bonds (87). It is an interesting question which configuration might be functional.
Specific examples of allosteric disulfide bonds are now discussed. The examples are separated into two groups: clear examples of regulation of mature protein function by allosteric disulfides and possible examples of such bonds (Table 1). Criteria for inclusion in the table include the existence of one or more X-ray structures of the disulfide bond in question and the consequence of cleavage or formation of the bond for protein function is known. The oxidoreductase that controls the redox state of the disulfide has been determined in some cases. There are a number of other examples of possible allosteric disulfides, but their current characterization does not meet these criteria (11, 97).
The examples have been separated into two groups. The first are examples where it is clear that the disulfide bond regulates the function of the mature protein. The second are examples where there is currently no definitive proof that the bond is active in the mature protein. The oxidoreductase that controls the redox state of the allosteric disulfide is listed if known.
ERp5, endoplasmic reticulum protein 5; HIV, human immunodeficiency virus.
Examples of Allosteric Disulfide Bonds
gp120
The human immunodeficiency virus (HIV) is the causative agent of acquired immune deficiency syndrome. The HIV envelope glycoprotein (env) is translated as a single polypeptide chain (gp160) that is proteolytically cleaved by host cell subtilisins into two noncovalently associated fragments, the surface glycoprotein subunit (gp120) and the transmembrane (gp41) subunit, the latter is anchored in the viral membrane (19). env is a trimer of gp41/gp120 heterodimers on the viral surface, which is activated by binding to CD4 and chemokine receptor (usually CXCR4 or CCR5) on susceptible cells. gp120 dissociates from gp41, which allows the fusion peptide (a six-helix bundled gp41 ectodomain) to be inserted into the target membrane (28).
One (possibly two) of the nine disulfide bonds in gp120 are cleaved during HIV entry (2, 4, 27, 60, 82). The hypothesis is that reduction of the gp120 disulfide bond facilitates unmasking of the gp41 fusion peptide and its insertion into the target cell membrane. The thiol content of gp120 increases from 0.5–1 to 4 mol of thiol per mol of gp120 following interaction of gp120 with cells expressing CD4 and CXCR4 chemokine receptor (4). Cleavage of the gp120 disulfide bond is independent of the chemokine receptor usage of the virus (27). Reduction of the gp120 disulfide is critical for env-mediated cell–cell fusion and HIV entry and infection as these processes are blocked by both small thiol alkylators and protein inhibitors of cell-surface oxidoreductases (4, 27, 60, 82).
The gp120 disulfide(s) can be reduced by protein disulfide isomerase (PDI) (4, 27, 44), thioredoxin (71), or glutaredoxin-1 (2). This observation implies that the gp120 disulfide bond(s) must have a high redox potential, as PDI is a poor reductant with a standard redox potential of −175 mV (57). Thioredoxin has been reported to be a more efficient gp120-reducing agent than PDI (71).
We have mapped the disulfide cleaved in gp120 using a mechanism-based kinetic trapping approach (2a). A thioredoxin-trapping mutant cleaved a single disulfide bond in purified gp120 and cell surface gp120 and the reaction was enhanced when CD4 was bound to gp120. The labile gp120 disulfide was identified by mass spectrometry analysis of the gp120–thioredoxin complex and found to be the bond constraining the V3 domain β-loop structure, Cys296-Cys331. The Cys296-Cys331 bond in mature gp160 is intact, required for conversion of gp160 to gp120, and not cleaved by thioredoxin.
The Cys296-Cys331 V3 disulfide bond is a −RHStaple in all eight crystal structures of gp120 in the protein databank (87). It constrains a β-loop motif and has a very short α-carbon–α-carbon distance of 3.84 ± 0.04 Å (mean ± standard error for the eight structures), compared with a mean distance of 5.6 Å for all disulfides (87) (Fig. 3).
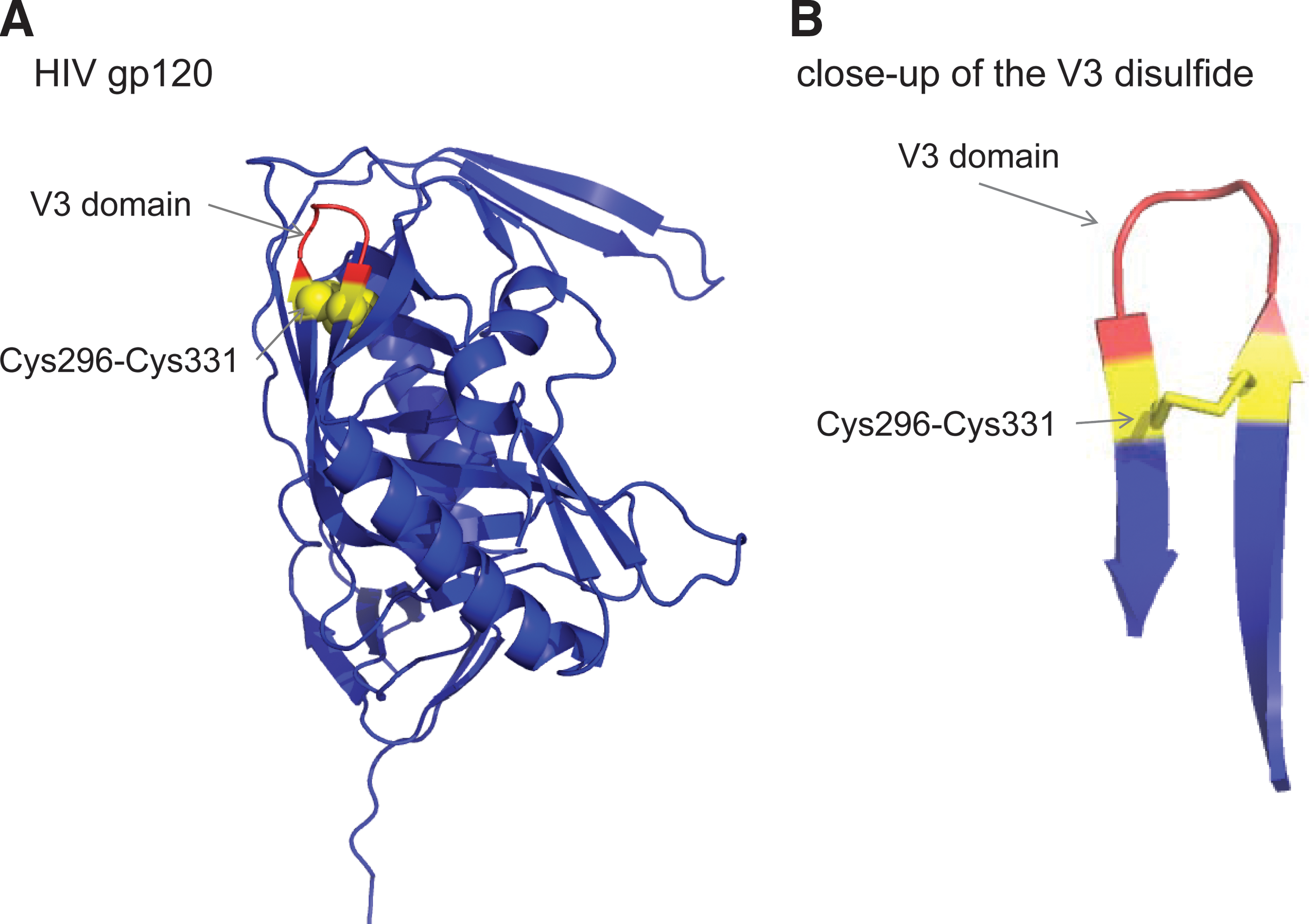
We suggest that binding of HIV gp120 to immune cell CD4 both brings gp120 close to a cell surface oxidoreductase and induces a conformational change in gp120 that facilitates reduction of the V3 domain disulfide bond by the oxidoreductase. Cleavage of the V3 disulfide bond then triggers a conformational change in the V3 domain, which results in dissociation of chemokine receptor from gp120 and allows refolding of gp41 and viral-membrane fusion. This mechanism is supported by the following observations: (i) CD4 binding results in a major reorganization of the env trimer, causing an outward rotation and displacement of each gp120 monomer, which is coupled with a rearrangement of the gp41 trimer, leading to closer contact between the viral and target cell membranes (55), and (ii) gp120 containing two unpaired cysteine thiols (one cleaved disulfide bond) bound normally to CD4 but did not interact with CXCR4 (4).
Botulinum neurotoxins
Clostridial botulinum neurotoxins are the causative agent of botulism and are the most poisonous proteins known. They block synaptic exocytosis in peripheral synapses, causing flaccid paralysis [reviewed by Montal (66)]. The neurotoxins are synthesized as a single polypeptide chain with a molecular mass of 150 kDa and the polypeptide is then cleaved by bacterial or host proteases into the active di-chain form consisting of a 100-kDa heavy chain and a 50-kDa light chain. The heavy and light chains are linked by a −RHStaple disulfide bond (Cys429-Cys453 in type A neurotoxins and Cys436-Cys445 in type B) (Fig. 4). The toxins consist of three modules. The N-terminal light chain is the catalytic domain and the heavy chain comprises the translocation domain (N-terminal half) and the receptor-binding domain (C-terminal half). The catalytic domain is an endopeptidase that shares structural similarity to the metalloprotease, thermolysin.
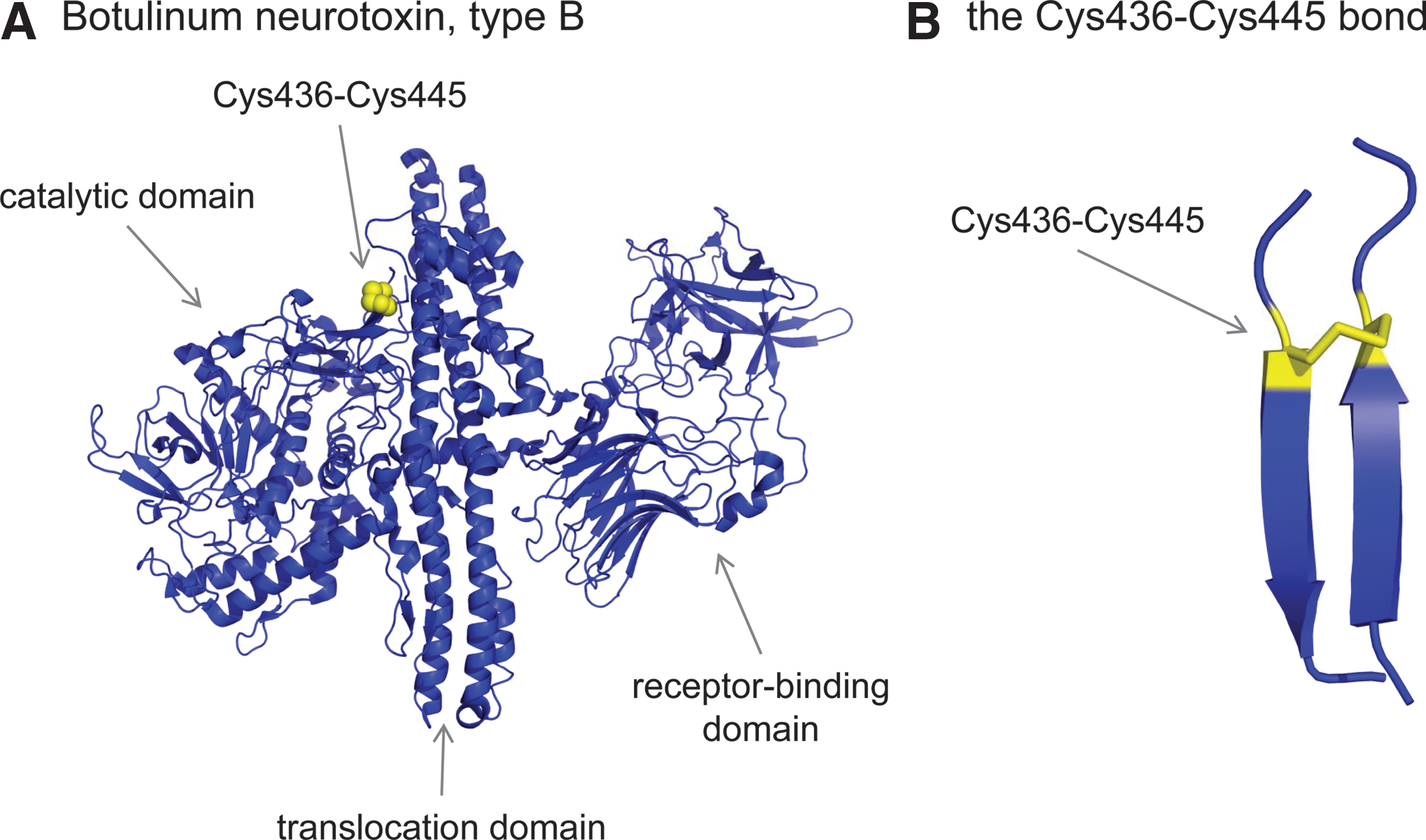
The neurotoxins enter cells by receptor-mediated endocytosis. The receptors involve a specific ganglioside, namely GT1b, together with other protein components that determine the neurotropism. The two-chain molecule integrates into the endosomal membrane where the −RHStaple is reduced, releasing the light chain into the cytosol. The sequence of events appears to be the following: pH-induced heavy chain insertion in the endosomal membrane, coupled light chain unfolding and its conduction through the heavy chain channel, release of the light chain from the heavy chain by reduction of the −RHStaple disulfide bond, and finally, light chain refolding in the cytosol (24). The light-chain endopeptidase cleaves unique components of the SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor) complex, the synaptic vesicle fusion complex required for membrane fusion.
The −RHStaple disulfide linkage is crucial for the chaperone function of the heavy chain and light chain translocation and release (24). The identity of the oxidoreductase that reduces the −RHStaple disulfide is not known.
CD4
CD4 is a type I integral membrane glycoprotein consisting of one V-type (domain 1) and three C2-type (domains 2, 3, and 4) immunoglobulin-like domains, a transmembrane domain, and a short cytoplasmic region. It is a coreceptor for binding of T cells to antigen-presenting cells and the primary receptor for HIV. The −RHStaple disulfide bond in the second extracellular domain of CD4, Cys130-Cys159, is reduced on the T-cell surface by thioredoxin, which leads to formation of disulfide-linked homodimers (61, 71, 89). A large conformational change must take place in domain 2 to allow formation of the disulfide-linked dimer. Domain swapping of domain 2 has been identified as the most likely mechanism for the conformational change leading to formation of two disulfide bonds between Cys130 in one monomer and Cys159 in the other one (58, 84) (Fig. 5).
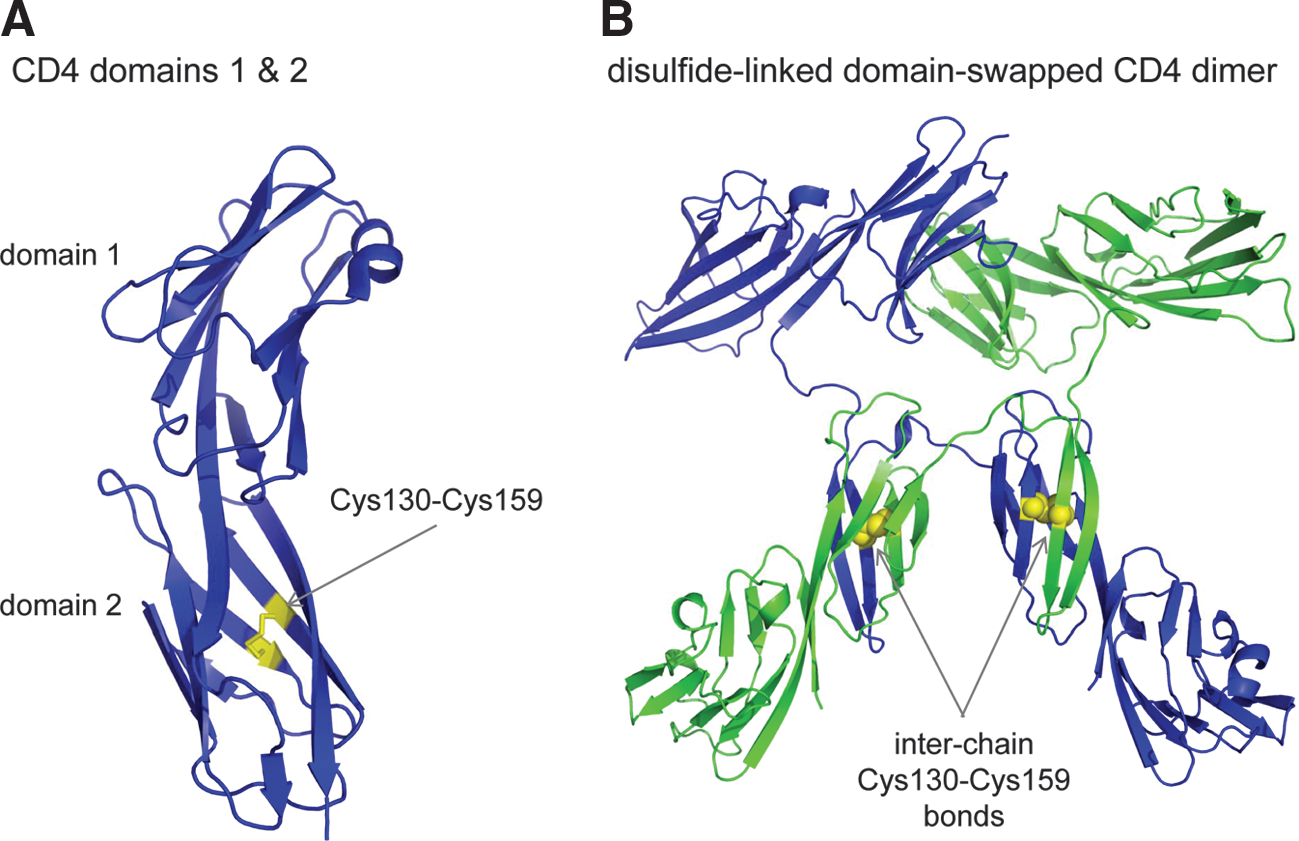
Ablating the domain 2 disulfide bond by mutagenesis markedly promotes HIV entry and env-mediated cell–cell fusion (60a) but impairs CD4's coreceptor function (58). These observations suggest that HIV enters susceptible cells preferably through monomeric reduced CD4, whereas dimeric CD4 is the preferred receptor for binding to antigen-presenting cells. This conclusion is supported by Bourgeois et al. (8), who found that disrupting CD4 dimerization increases HIV-1 entry into susceptible cells. Modeling of the complex of the T-cell receptor and domain-swapped CD4 dimer bound to class II major histocompatibility complex and antigen also supports the domain-swapped dimer as the immune coreceptor (58). The involvement of D4 residues Lys318 and Gln344 in dimer formation is accommodated by this model (65).
Considering the unusual nature of the domain 2 bond and its involvement in CD4 function, we examined all known C2-type immunoglobulin superfamily domains for an equivalent disulfide link. Of the 1275 domains analyzed, only one protein in addition to primate and rodent CD4 was found to contain a similar disulfide-bonded structure, CEACAM20 (J.W.H. Wong and P.J. Hogg, unpublished observations). This finding indicates that acquisition of the unusual disulfide bond in CD4 is a rare event in the evolution of immunoglobulin domains.
MICA
NKG2D is a C-type lectin-like activating immunoreceptor that is expressed on most natural killer (NK) cells, CD8 αβ T cells, γδ T cells, and macrophages (33). Among its ligands is the distant major histocompatibility complex class I (MHCI) homolog, MICA (33). Unlike MHCI proteins, which are ubiquitously expressed, the expression of MICA is restricted to intestinal epithelium and epithelium-derived tumors (37, 38). MICA has no function in antigen presentation and is induced by cellular stresses, such as infection, noxious conditions, or transformation.
The crystal structure of the NKG2D homodimer–MICA monomer complex is analogous to that of the T-cell receptor–MHCI complexes (51). The MICA and MHCI proteins comprise three extracellular domains (α1, α2, and α3), a transmembrane-spanning domain, and a small cytoplasmic domain (52). Although they have the same domain structure the sequence identity is <30% (52). The MHCI family proteins contain two highly conserved disulfide bonds, one each in the α2 (Cys101-Cys164) and α3 (Cys203-Cys259) domains. MICA contains these two disulfide bonds (Cys96-Cys164 and Cys202-Cys259), plus an extra one in the α1 domain (Cys36-Cys41).
NKG2D cells can mediate effective tumor rejection in the early stages of tumorigenesis. Sustained surface expression and shedding of MICA by late-stage tumors, though, can negatively imprint on the immune response and facilitate tumor immune evasion (33, 47). Shed MICA induces internalization and degradation of NKG2D and stimulates population expansions of normally rare NKG2D+CD4+ T cells that have negative regulatory functions. Shedding of MICA is triggered by endoplasmic reticulum protein 5 (ERp5) cleavage of a MICA disulfide bond at the cell surface (47). Mixed disulfide complexes between ERp5 and cell-surface MICA have been detected, which resolve following proteolytic cleavage of MICA in the membrane-proximal region (47). The MICA disulfide bond cleaved by ERp5 was identified using an ERp5-trapping mutant. The trapping mutant formed a mixed disulfide with recombinant α3 domain but not with recombinant α1α2 domain, identifying the α3 domain bond, Cys202-Cys259, as the substrate for ERp5. This bond links the β-sheets of the immunoglobulin-like fold (Fig. 6).
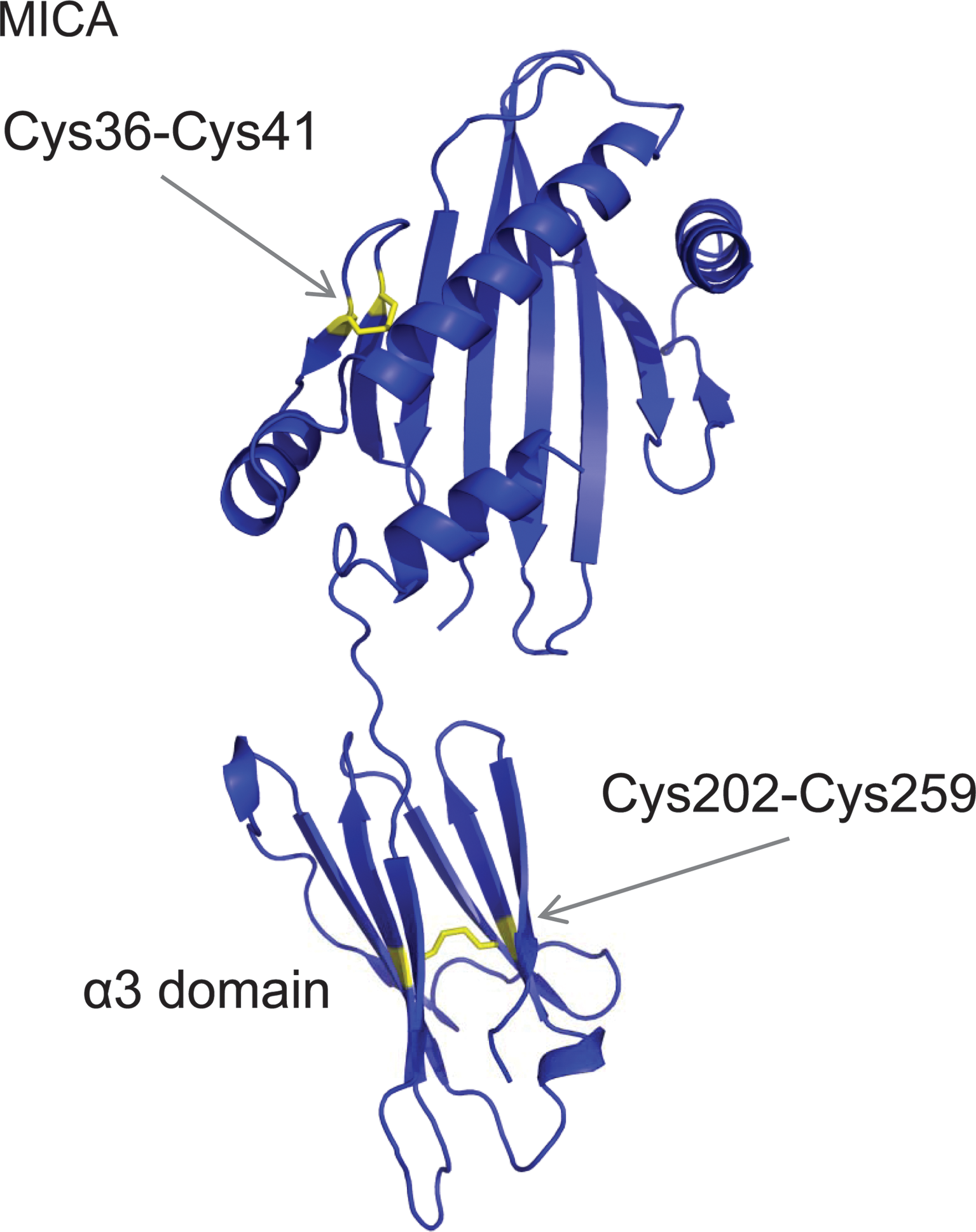
The Cys202-Cys259 bond is buried in the crystal structure so a major conformational change in the α3 domain is required for ERp5 to react with the disulfide (47). The extra disulfide bond in the MICA α1 domain, Cys36-Cys41, is intriguing. This bond is a −RHStaple disulfide that constrains a β-loop structure (Fig. 6), a feature typical of the allosteric bonds in gp120, CD4, and botulinum neurotoxins.
β2-Glycoprotein I
β2-Glycoprotein I (β2GPI) is a 50-kDa plasma protein that circulates in blood at a relatively high concentration of 4 μM. Its main physiological function remains uncertain, although it is thought to play a role in apoptotic cell clearance and coagulation through interactions with serine proteases, anionic phospholipids, and cell surface receptors (64). It has a clear role in the antiphospholipid syndrome, which is an autoimmune condition characterized by pathogenic circulating anti-β2GPI antibodies (32). This syndrome is associated with vascular thrombosis, recurrent miscarriages, accelerated atherosclerosis, and enhanced oxidative stress.
β2GPI consists of four complement control protein modules and an unusual fifth domain (7, 88) (Fig. 7A). The fifth domain contains a central β-spiral of four antiparallel β-sheets, two small helices, and an extended C-terminal loop region (Fig. 7B). Each of the four complement modules contains two conserved disulfide bonds, whereas the fifth domain contains three disulfide bonds. There are no unpaired cysteines in the protein. Of the 11 disulfide bonds in β2GPI, the domain 5 Cys288-Cys326 bond stands out as being exposed to solvent. Cys326 is the C-terminal residue that links to Cys288 in a β-loop structure (Fig. 7B). The Cys288-Cys326 bond has a −/+RHHook configuration (72a).
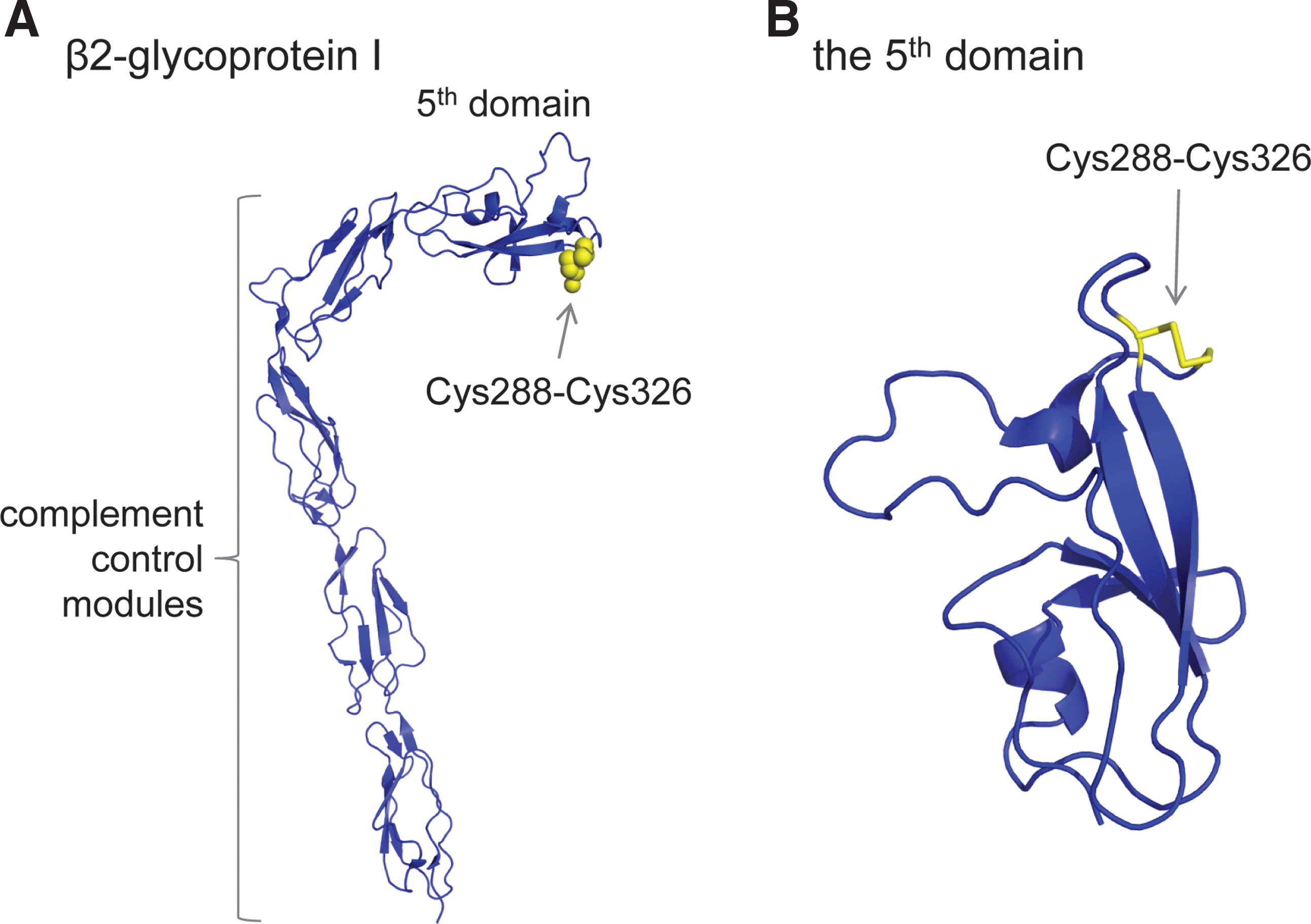
The Cys288-Cys326 disulfide bond is reduced by both thioredoxin and PDI and on the surface of endothelial cells and platelets in culture (42, 72a). Cleavage of the β2GPI Cys288-Cys326 disulfide occurs in vivo as circulating human blood β2GPI contains unpaired cysteine thiol(s) (42). The reduced β2GPI is very effective at protecting endothelial cells from injury by oxidative stress and S-nitrosylation of the unpaired β2GPI thiols may contribute to this protection. It may be that β2GPI's primary physiological function is protection of vascular cells from oxidative damage via the Cys288-Cys326 disulfide/dithiol (42). The high concentration of β2GPI in plasma is consistent with such a role.
Reduced β2GPI also binds to von Willebrand factor (VWF) in a thiol-dependent manner, which increases the binding of glycoprotein Ibα to VWF and platelets to activated VWF (72). These interactions may contribute to the redox regulation of platelet adhesion and thrombosis (20, 25).
Possible Allosteric Disulfide Bonds
PapD-like chaperones
P pili are adhesive fibers made by Gram-negative bacteria, which mediate specific recognition and attachment to host tissues. They are assembled from individual subunits in the periplasm by the immunoglobulin-fold PapD-like chaperones. The chaperones bind, stabilize, and cap subunits until they are assembled into the pilus. The pili subunits lack the seventh β-strand of their immunoglobulin-fold and the chaperone supplies this strand, capping the subunit until it reaches the assembly site at the growing pilus. Each subunit of the mature pilus is thought to supply the missing strand to complete the fold of its neighbor (85).
PapD and family members contain a −RHStaple disulfide bond linking the last two β-strands of its CD4-like C-terminal domain (Cys207-Cys212) (43, 75, 100). The −RHStaple bond is required for in vivo activity. Its formation is mediated by the DsbA/DsbB redox couple in the periplasm.
The question of whether the bond is redox active in the mature form of the protein has been addressed in the F1 antigen/Caf1M system of Yersinia pestis (100). This is analogous to the P pilus/PapD system of Escherichia coli (85). PapD and Caf1M have the same −RHStaple bond. Zav'yalov et al. (100) have suggested that the −RHStaple bond may undergo a reduction/reoxidation cycle relating to release of the Caf1 subunit on interaction with the outer-membrane usher protein, Caf1A. The Caf1–oxidized Caf1M complex interacts with the outer-membrane Caf1A, where Caf1M is released as the Caf1 subunits assemble at the cell surface to form the F1 capsule. The trigger for dissociation of Caf1M is possibly reduction of the −RHStaple disulfide bond by DsbA, for example. The reduced Caf1M could then re-enter the cycle following reoxidation. It is not yet clear, though, whether the bond is redox active in the mature form of the protein and hence its assignment to the possible allosteric disulfides.
Aryl sulfotransferase
Bacterial aryl sulfotransferases are periplasmic enzymes that transfer sulfuryl groups among phenolic compounds that mediate cell–cell and host–pathogen interactions. The uropathogenic E. coli aryl sulfotransferase contains a −RHStaple disulfide bond (Cys418-Cys424) that is required for activity (36, 59). It is located close to the surface in a β-loop of the sixth propeller blade, just 12 residues before the catalytic residue His436, which undergoes transient sulfurylation in the enzyme reaction. The disulfide bond may influence conformational equilibria in the protein.
The DsbL/Dsbl redox couple appears to have evolved specifically to control the redox state of this Cys418-Cys424 disulfide in the bacterial periplasm. DsbL, Dsbl, and the sulfotransferase are encoded in a tricistronic operon (36). As with the PapD–RHStaple bond, it is not clear whether the aryl sulfotransferase bond can break and/or form in the mature protein; therefore, it has been included as a possible allosteric disulfide.
AtFKBP13
Chloroplasts are the site of photosynthesis in plants and eukaryotic algae and the lumen of these organelles, the thylakoids, is the site of oxygen evolution. In addition to the proteins directly involved in oxygen evolution, there are more than a dozen resident immunophilins in the thylakoid lumen. These proteins were originally defined as targets for immunosuppressive drugs that were later found to have peptidyl-prolyl cis-trans isomerase activity. One of these is AtFKBP13, a representative of the FKBP immunophilin group.
AtFKBP13 consists of six β-strands that form an antiparallel β-sheet, and two α-helices (34). AtFKBP13 contains a −RHStaple disulfide bond (Cys106-Cys111) that is not present in FKBPs from animals or yeast. The disulfide links a β-loop motif that shifts position considerably when the bond is reduced. This bond can be reduced by chloroplast m-type thioredoxin (34), which results in loss of peptidyl-prolyl isomerase activity. It has not been proven that m-type thioredoxin regulates AtFKBP13 function in the chloroplast thylakoids; however, it has been listed as a possible allosteric disulfide.
Transglutaminase 2
Transglutaminase 2 (TG2), also known as tissue transglutaminase, functions both inside and outside the cell (35, 90). Inside the cell, TG2 operates as a G-protein in the phospholipase C signal transduction cascade. Outside the cell, TG2 catalyses Ca2+-dependent transamidation of glutamine residues to lysine residues, resulting in Nɛ(γ-glutamyl)lysyl isopeptide bonds. TG2 also binds tightly to fibronectin of the extracellular matrix and cell surface integrins to influence cell adhesion, motility, signaling, and differentiation.
Human TG2 contains an N-terminal β-sandwich, a catalytic domain, and two C-terminal fibronectin type III β-barrels (76). A −RHStaple disulfide bond forms between Cys370 and Cys371 in the catalytic domain. The bond is within an extended β-loop structure. There is evidence that formation of this bond inactivates the enzyme by interfering with the ability of TG2 to change shape upon binding to GTP (5, 14, 16). It has not been proven that the Cys370-Cys371 TG2 bond controls the function of the mature protein, so it has been included as a possible allosteric disulfide. Interestingly, the cysteine pair is also found in TG1, TG4, TG5, and TG7.
Carboxyl-terminal Src kinase
Src family kinases are involved in a number of cellular events, including division and differentiation, cell attachment and movement, and survival and death. The carboxyl-terminal Src kinase (Csk) negatively regulates Src family tyrosine kinases in the cytoplasm. Csk consists of an SH3, an SH2, and a kinase domain (68). The SH2 domain contains an unusual disulfide bond between Cys122 and Cys164 that can exist in two different configurations, +LHHook or +/−RHSpiral (PDB ID 3EAC), in a 1 Å resolution crystal structure.
SH2 domains are ∼100 residues in length and contain two central antiparallel β-sheets flanked by two α-helices. The Cys122-Cys164 disulfide bond links a β-loop (Cys164) to coil (Cys122) and is exposed to solvent. NMR analysis of the intact molecule indicates that reduction of the bond leads to change in residues that extend from the disulfide bond across the molecule to a surface that is in direct contact with the small lobe of the kinase domain, whereas normal mode and molecular dynamics analyses suggest that disulfide bond formation has effects on residues within the active-site cleft of the kinase domain. Comparison of the crystal structures of the oxidized (PDB ID 1K9A:C) and reduced (PDB ID 1K9A:B) SH2 domain shows that the positioning of the central antiparallel β-sheets are slightly different in the two structures.
The reduced protein has 10-fold higher kinase activity than the oxidized enzyme (63). This finding suggests that redox control of the disulfide bond in the cytoplasm may control enzyme activity. This has not been shown; however, the Csk disulfide has been listed as a possible allosteric bond.
Tissue factor
Injury to a blood vessel rapidly initiates events in the vessel wall and in blood that seal the breach (25). Circulating blood platelets are recruited to the site of injury where they incorporate into the developing thrombus. Meanwhile, blood coagulation culminates in the generation of thrombin and fibrin, which stabilizes the developing thrombus. Unwanted thrombosis is the precipitating event in myocardial infarction and stroke, whereas venous thrombosis is the second leading cause of death in patients with cancer (25).
Blood coagulation is triggered by the transmembrane glycoprotein, tissue factor. Tissue factor mostly resides on the cell surface in a cryptic or inactive configuration that must be de-encrypted or activated before it can trigger coagulation. Cryptic tissue factor binds coagulation factor VIIa and cleaves a peptidyl substrate but not coagulation factor X, whereas active tissue factor binds factor VIIa and cleaves both a peptidyl substrate and factor X (50, 77, 83). A characteristic feature of cryptic tissue factor is the slow rate of binding of factor VII/VIIa. Equilibrium binding of VII/VIIa to active tissue factor is established within 1 min, whereas binding to cryptic tissue factor takes 1–2 h to reach equilibrium. Tissue factor activation of factor X leads to a cascade of protease activations culminating in thrombin formation and production of fibrin.
Tissue factor contains an unusual disulfide bond in the membrane-proximal domain (Cys186-Cys209), which has been implicated in de-encryption of the cofactor (1, 10, 11, 54). The cofactor is a member of the class II cytokine superfamily and contains two fibronectin type III domains in the extracellular part, each with a disulfide bond. The N-terminal domain Cys49-Cys57 disulfide is a typical structural bond, whereas the C-terminal domain Cys186-Cys209 disulfide is a −RHStaple bond that straddles the F and G strands of the antiparallel β-sheet and is exposed to solvent (10).
We have hypothesized that the bond is reduced in cryptic tissue factor and de-encryption involves oxidation of the disulfide. Unpaired cysteine thiols have been detected in cryptic tissue factor that are reduced upon de-encryption and cysteine thiols are involved in the process as thiol alkylating compounds block tissue factor activation, whereas dithiol crosslinkers activate tissue factor (10). Moreover, an intact Cys186-Cys209 bond is required for tissue factor coagulant activity (1). However, this mechanism of tissue factor de-encryption is controversial, and others have been proposed (3, 73).
A major unanswered question is the identity of the tissue factor de-encrypter, that is, what triggers activation of tissue factor at the site of vessel injury. PDI (1, 12, 79), glutathione (79), and nitric oxide (1) have been implicated in redox control of the tissue factor Cys186-Cys209 disulfide, although in vitro studies using the purified compounds have yet to establish a role in tissue factor de-encryption (74, 94). Notably, though, PDI is on the surface of platelets (21) and platelet microparticles (78) and PDI ligands markedly impair tissue factor-mediated thrombus formation in vivo (12, 79).
Binary tissue factor-VIIa and ternary tissue factor-VIIa-Xa complexes also signal in inflammation, tumor progression, and angiogenesis by cleaving protease-activated receptor 2 (11). Interestingly, the Cys186-Cys209 disulfide is not required for binary tissue factor-VIIa complex signaling (1). The Cys209Ala tissue factor mutant retains signaling activity, whereas the Cys186Ala mutant is inactive in signaling (1).
The evidence that formation of the Cys186-Cys209 disulfide bond is involved in de-encryption of tissue factor is circumstantial at this time, so it has been listed as a possible allosteric disulfide.
von Willebrand factor
VWF is a plasma glycoprotein that mediates adhesion of platelets to damaged blood vessels in the high shear of flowing blood. VWF is made by endothelial cells and megakaryocytes and circulates as a series of multimers made up of disulfide-linked 500-kDa homodimers (17). When a blood vessel is damaged, ultra-large VWF molecules as big as 20,000 kDa are released from Weibel-Palade bodies in vascular endothelial cells. Plasma VWF multimer size is regulated by the shear forces in flowing blood (104). Unfolding of the VWF A2 domain exposes the Tyr1605-Met1606 peptide bond in the central β-sheet to cleavage by ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). Deficiency of ADAMTS13 causes thrombotic thrombocytopenic purpura (93), where microvascular thrombi form in arterioles and capillaries, whereas deficiency of the larger VWF multimers is associated with the bleeding disorder, type IIA von Willebrand disease (26).
The recent crystal structure of the A2 domain revealed an unusual disulfide bond between adjacent cysteines (Cys1669 and Cys1670) at the C-terminus of the α6-helix (103). Interestingly, ablation of the bond by mutagenesis results in increased susceptibility of the A2 domain and full-length VWF to cleavage by ADAMTS13 (56). It is possible that redox control of the Cys1669-Cys1670 disulfide bond in plasma is a means of regulating VWF multimer size. Disulfide bond reduction in VWF has been implicated in control of plasma multimer size (29, 98). Notably, the bond is a +/−LHStaple with a short α-carbon distance and a high dihedral strain energy. Both cysteines of the disulfide bond are exposed to solvent in the A2 domain crystal structure (103).
Strings or filaments of VWF with lengths of up to 1 mm have also been found in endothelial cells and in blood (18, 62). These structures are formed by lateral association and appear to be mediated by a thiol/disulfide exchange process (53). Unpaired cysteine thiols have been mapped to the D3 and the VWF type C (VWC) domain number 2 of plasma VWF (13). VWC domains are 60–80 amino acids in length and contain 10 conserved cysteine residues (101). More than 1000 proteins contain VWC domains and some proteins have been shown to self-associate or interact with other partners via their VWC domains (30, 101). The Cys3-Cys5 VWC disulfide is a −RHStaple bond that links adjacent β-strands (69, 102). It is possible that this bond is the one reduced in the VWC2 domain (Cys2451-Cys2468) (13) and is involved in formation of the VWF strings. This has not been shown; however, it has been included as a possible example.
β3 Integrin
Integrins are cell adhesion receptors that link the extracellular environment to the actin cytoskeleton (41). They consist of α and β type 1 membrane receptor subunits. Unpaired cysteine thiols appear in the β3 subunit of αVβ3 (92) and αIIbβ3 (22) when the integrins are activated. There is also evidence that thiol/disulfide exchange plays a role in the activation of αVβ3 (92) and αIIbβ3 (22) and that cell surface PDI is involved in the redox events.
The details of the redox change(s) in the β3 subunit are not known, although the current information points to a thiol/disulfide rearrangement in the epidermal growth factor (EGF)-like domains [reviewed by Essex (20)]. Disruption of disulfide bonds in the third EGF domain resulted in a constitutively active αIIbβ3 (67). The X-ray structure of the extracellular part of the β3 integrin subunit has been solved in complex with αV (99) and αIIb (105). Of the 27 disulfide bonds in the structure of β3 in αVβ3 (92), both the Cys523-Cys544 bond in the EGF3 module and the Cys663-Cys687 disulfide in the β-tail domain are −RHStaple disulfide bonds. Of the 29 disulfide bonds in the structure of β3 in αIIbβ3 (92), the Cys663-Cys687 disulfide is a −RHStaple disulfide bond, whereas the Cys523-Cys544 bond is a −LHHook. This Hook, though, has a very short α-carbon–α-carbon distance of 4.1 Å, which is more in keeping with −RHStaple bonds. Labeling of the activated platelet surface with a biotin-linked maleimide and mass spectrometry analysis of the alkylated peptides have identified the EFG3 Cys523-Cys544 bond as being cleaved (unpublished observations), which implies that this bond is redox active.
A −RHStaple Bond with a Catalytic Function
Disulfide bond formation in the E. coli periplasm is mediated by the Dsb proteins, where the DsbC/DsbD pathway catalyzes disulfide bond isomerization. DsbD is an inner-membrane protein that transfers electrons from thioredoxin in the cytoplasm to other oxidoreductases in the periplasm. DsbD contains a periplasmic N-terminal domain, a central transmembrane domain, and a periplasmic C-terminal domain (81). Each of these domains contain two cysteine residues that transport two electrons. Cytoplasmic thioredoxin donates two electrons to the transmembrane domain cysteines, which are shuttled to the catalytic +/−RHHook disulfide of the periplasmic C-terminal domain and then to the −RHStaple disulfide of the periplasmic N-terminal domain. The reduced −RHStaple donates its electrons to periplasmic DsbC, DsbG, and CcmG.
Redox Potentials of −RHStaple Disulfide Bonds
The redox potentials of four −RHStaple bonds are known (Fig. 8): the Cys186-Cys209 tissue factor bond (H.P.H. Liang, T. Brophy, and P.J. Hogg, unpublished observations), the Cys149-Cys202 bond engineered into green fluorescent protein (70), the Cys130-Cys159 CD4 bond (L.J. Matthias, H.P.H. Liang, and P.J. Hogg, unpublished observations), and the Cys103-Cys109 DsbD bond (15). The redox potentials of the catalytic disulfides of PDI (57) and thioredoxin (48) are shown for comparison in Figure 8. The redox potentials of these four −RHStaple bonds are closer to that of the strong protein reductant thioredoxin than the more oxidizing PDI.

Stereochemistry of Disulfide Bond Reduction
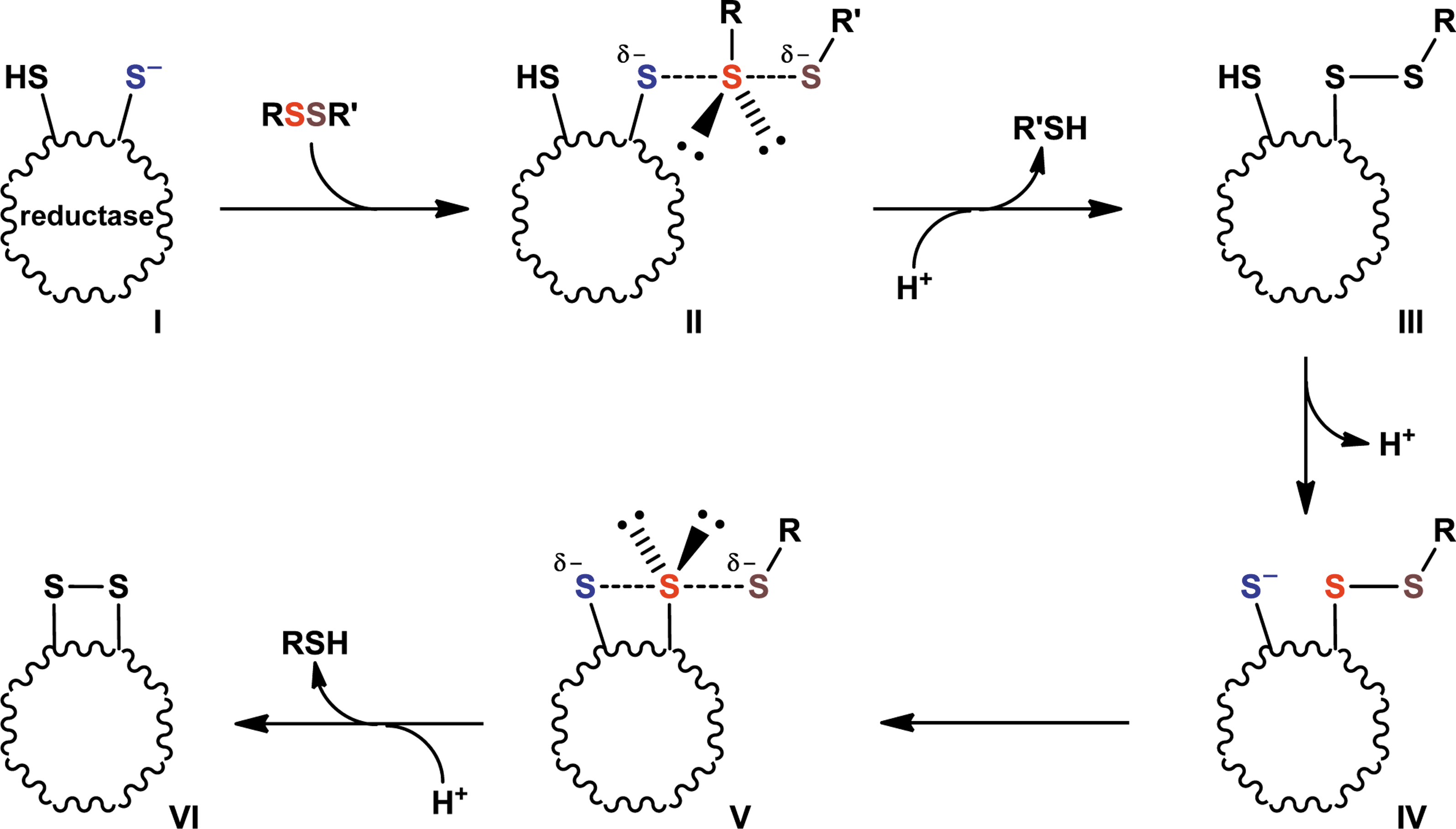
Disulfide bond reduction occurs via a bimolecular nucleophilic substitution (SN2) reaction mechanism (23, 95). This reaction is highly directional and proceeds via a transition state in which the three sulfur atoms involved, the sulfur ion nucleophile of the oxidoreductase and the two sulfur atoms of the disulfide bond, must form an ∼180° angle (Fig. 9). Steric factors that prevent proper positioning of the three sulfur atoms will preclude any reaction. Therefore, it follows that allosteric disulfide bonds must have a favorable stereochemistry to be reduced by oxidoreductases. It may be that the −RHStaple (or −LHStaple) configuration provides for the correct positioning of the three sulfur atoms involved.
Disulfide Bond Analysis Tool
To help with identification of possible functional disulfide bonds in proteins, we have generated a simple interface to obtain structural information about disulfide bonds in X-ray or NMR structures. The tool is found at:
Conclusions
Allosteric disulfides control the function of the protein in which they reside when they undergo redox change. There are currently five examples of allosteric disulfide bonds that clearly control the function of the mature protein in which they reside, and there are several other potential examples at various stages of characterization. Most of the bonds discussed herein have a −RHStaple configuration and a common feature of all bonds is that they link β-strands or β-loop structures. This means of posttranslational control of protein function may prove to be a generally important mechanism in prokaryotes and eukaryotes.
Footnotes
Acknowledgments
The authors thank Tobias Dick and Neil Donoghue for discussions on the stereochemistry of disulfide bond cleavage.