
Abstract
The folding pathways of disulfide proteins vary substantially (Arolas et al., Trends Biochem Sci 31: 292–301, 2006). The diversity is mainly manifested by (a) the extent of heterogeneity of folding intermediates, (b) the extent of presence of native-like intermediates, and (c) the variation of folding kinetics. Even among structurally similar proteins, the difference can be enormous. This is demonstrated in this concise review with two structurally homologous kunitz-type protease inhibitors, bovine pancreatic trypsin inhibitor and tick anticoagulant peptide, as well as a group of cystine knot proteins. The diversity of their folding mechanisms is illustrated with two different folding techniques: (a) the conventional method of disulfide oxidation (oxidative folding), and (b) the novel method of disulfide scrambling (Chang, J Biol Chem 277: 120–126, 2002). This review also highlights the convergence of folding models concluded form the conventional conformational folding and those obtained by oxidative folding. Antioxid. Redox Signal. 14, 127–135.
Introduction
Diversity of protein conformational folding
There are two favored models of conformational folding. The framework model (40,41,48) proposes that secondary structures (α-helix, β-strand, etc.) form first during the early stage of folding. This is followed by docking and packing of preformed secondary structural units to form the native tertiary structure. The hydrophobic collapse model (6,31,39,57) stipulates that a rapid hydrophobic collapse (interaction) accounts for the major driving force of folding, which is followed by searching and fine-tuning of conformation in a confined volume to reach the native structure. A basic distinction between these two models is the relative kinetics of formation of the secondary structure and the tertiary structure (compaction) of the polypeptide chain during the process of folding. A strong preference of localized conformation would favor the framework model and lead to the rapid formation of secondary structure before organization of tertiary structure is initiated.
The framework model and the hydrophobic collapse model apparently correspond to two extreme folding models. Results obtained from conformational folding of many proteins have shown that extreme mechanisms fitting either model are rare. The formation of secondary structure and compaction of the protein occur usually in parallel for most proteins during the course of folding. In other words, folding of most proteins follows the middle-of-the-road between the framework and hydrophobic collapse models. Uversky and Fink (58) have analyzed data on the conformational properties of 41 native and partially folded states and concluded a valid correlation between the increase of secondary structure and the decrease of hydrodynamic volume during folding. Essentially, they found little evidence among analyzed proteins for either compact intermediates lacking secondary structure or unfolded intermediates comprising highly ordered secondary structure. These studies have led to the proposal of a third folding model, the nucleation–condensation model (29,36), which invokes the importance of interplay and interdependence of the secondary and tertiary structures during protein folding. This model essentially stipulates that secondary structure is inherently unstable and its stability can be enhanced by protein compaction and tertiary interactions.
Diversity of protein oxidative folding
The pathway and mechanism of protein oxidative folding exhibit comparable extent of diversity as those observed in the conformational folding. This has been reviewed recently (3,45). There are two well-characterized extreme models of disulfide folding pathways, represented by two 3-disulfide proteins, bovine pancreatic trypsin inhibitor (BPTI) and hirudin, respectively. The folding pathway of the BPTI model (27,28,38,60) is characterized by a limited number of folding intermediates adopting exclusively native-like structures; a folding mechanism that resembles that of framework model. In contrast, the folding pathway of the hirudin model (11,15,23,24) is illustrated by a rapid accumulation of fully oxidized, compact isomers as intermediates; a folding mechanism that bears a resemblance to that of hydrophobic collapse model. In addition, there are protein models that exhibit folding properties of both BPTI and hirudin. The folding mechanisms of these proteins, including epidermal growth factor (22,62) and RNase A (46,49,53,61), are consistent with the nucleation–condensation model (29,36). Thus, the compatibility between the models of conformational folding and oxidative folding supports a unified, although diverse mechanism of protein folding, independent of the distinct physicochemical signals utilized to monitor the folding.
Scope of this review
In this concise review, we intent to further highlight the diversity of protein folding mechanism by presenting the divergent folding pathways of structurally similar proteins. It will narrow the subjects to small homologous disulfide proteins elucidated by the technique of disulfide oxidation (26) and disulfide scrambling (18,14).
Methods for Analysis of Folding Pathway of Disulfide Proteins
The folding pathway of disulfide proteins can be elucidated by three distinct methods.
Conformational folding
This is the most commonly applied folding technique (7,32,37), for both disulfide and nondisulfide containing proteins. In this approach, native disulfide bonds are kept intact throughout the folding experiments. The step of unfolding is achieved by using denaturant, extreme pH, or temperature. The folding is then initiated by removal of denaturant (e.g., by gel filtration, dilution, or dialysis), pH adjustment, or temperature jump. Most unfolded proteins usually refold spontaneously to form the native structure. The pathway of protein refolding is then observed and characterized by the mechanism of restoration of selective physicochemical signals that differentiate between the native and unfolded states. These signals include spectra of fluorescence, circular dichroism, infrared, and NMR (34,35,40,55).
Oxidative folding
Application of this method is limited to disulfide containing proteins. In this approach, disulfide bonds of native proteins (N-protiens) are first reduced and denatured by a reducing agent (e.g., dithiothreitol [DTT]) and a denaturant (e.g., guanidine hydrochloride [GdmCl]). After removal of the reductant and denaturant (e.g., by gel filtration or sample dilution), the reduced and denatured protein is allowed to refold in the presence of redox buffer via disulfide formation (oxidation) that leads to the formation of the species containing the intact native disulfide bonds. The disulfide folding pathway is then characterized by heterogeneity, structures, and kinetic properties of disulfide isomers accumulated along the folding pathway, which are fractionated, quantified, and analyzed by high-performance liquid chromatography (HPLC). This technique, pioneered by Creighton (26 –28), has been applied to the extensive elucidation of folding pathways of numerous disulfide containing proteins (3,45), including among many others, BPTI (27,28,38,60), ribonuclease A (46,49), and hirudin (11,24).
Scrambling folding
Application of this method is also limited only to the disulfide-containing proteins. It can be considered as a hybrid of conformational folding and oxidative folding. In this approach, a native protein is unfolded in the presence of denaturant and thiol catalyst (e.g., 0.1 mM β-mercaptoethanol). Under these conditions, the native protein unfolds by shuffling its native disulfide bonds and converts to a mixture of fully oxidized scrambled isomers that are amenable to fractionation, isolation, and further structural analysis (20,12). These unfolded isomers are collectively termed as X-isomers. The heterogeneity of X-isomers is determined by the number of disulfide bonds. For proteins that contain 3- and 4-disulfide bonds, there are 14 and 104 possible unfolded states of X-isomers, respectively. The folding experiment is initiated by incubation of isolated X-isomers in the alkaline buffer containing a thiol catalyst. Under these conditions, all X-isomers refold (also via disulfide shuffling) to form the native structure. The folding pathway is then characterized by the heterogeneity, structures, and kinetic properties of X-isomers accumulated along the folding pathway (18,14).
The technique of scrambling folding differs from that of oxidative folding in one major aspect—the nature of the starting material of the folding experiment. The starting material of oxidative folding is fully reduced protein (R-isomer or R-protein), which can be generated by using reducing agent alone, in the absence of denaturant or heat (reductive unfolding). In contrast, preparation of the starting material of scrambling folding (X-isomers or X-protein) requires strong denaturing conditions (e.g., 6 M GdmCl), similar to that used in preparation of the starting material of conformational folding. In addition, GdmCl-unfolded proteins often still comprise heterogeneous conformational isomers possessing diverse states of free energy (33,56). This has been verified in the production of X-isomers using the method of disulfide scrambling (20,12). These diverse X-isomers can be fractionated and isolated. Thus, unlike the method of oxidative folding, scrambling folding can be initiated with a structurally defined X-isomer that possesses the highest free energy among all denatured isomers (16,18,14).
BPTI and Tick Anticoagulant Peptide Are Two Structurally Homologous Disulfide Proteins
BPTI and tick anticoagulant peptide (TAP) both belong to the family of kunitz-type protease inhibitors (42). TAP is a factor-Xa-specific inhibitor isolated from the blood-sucking tick Ornithodoros moubata (59). The two proteins are nearly identical in size (58 a.a. for BPTI vs. 60 a.a. for TAP). They also share modest sequence homology (44), the same disulfide pattern (52), similar content of secondary structures (2), and almost superimposable three-dimensional structures (Fig. 1) (44). Despite the overall similarity of their structures, BPTI and TAP fold via very different pathways. This will be illustrated here using the two folding techniques specific for disulfide proteins, folding via disulfide oxidation and disulfide scrambling.
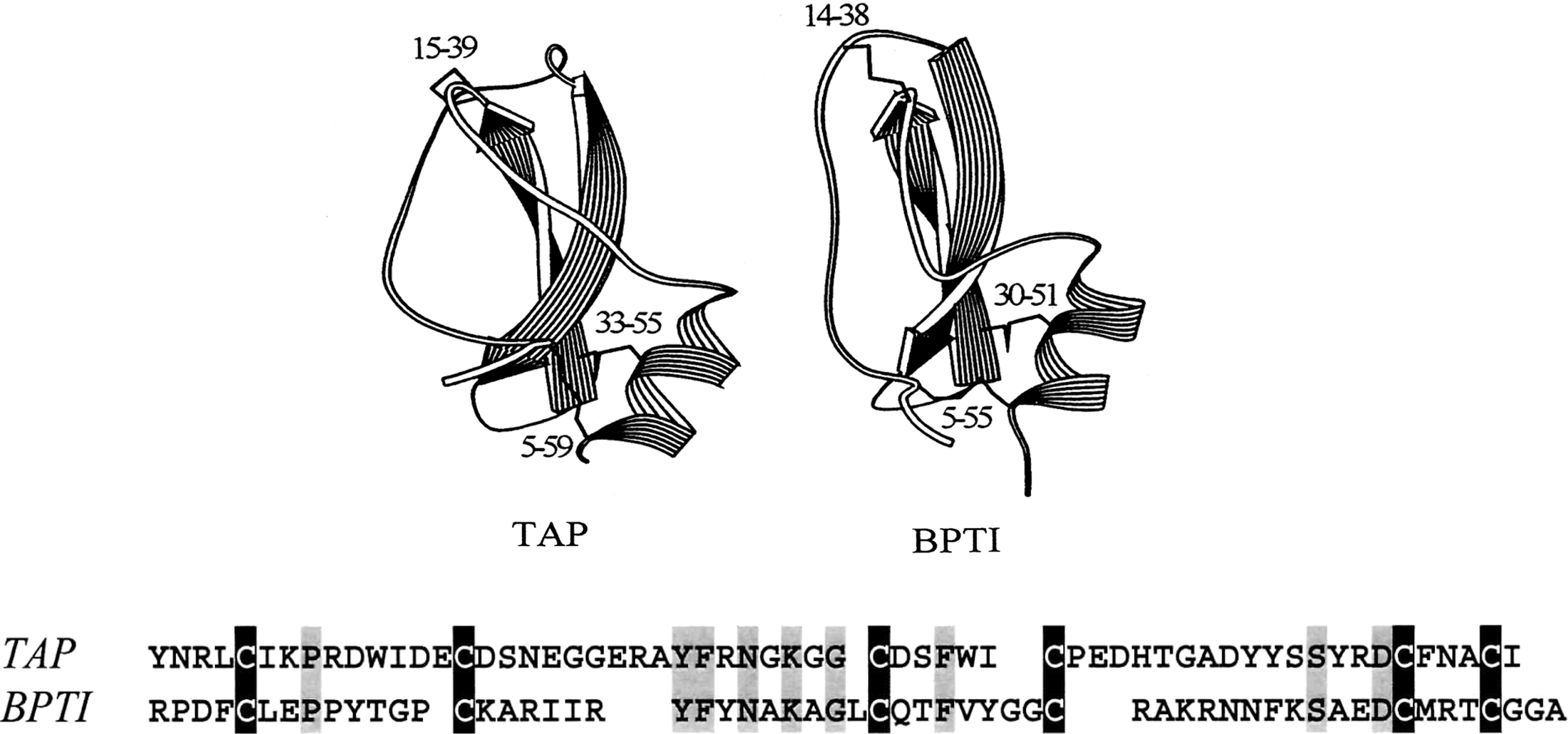
Diversity of Folding Pathways of BPTI and TAP Elucidated by the Method of Disulfide Oxidation (Oxidative Folding)
BPTI is the first and most extensively investigated model for elucidation of disulfide folding pathway. The details of BPTI folding, pioneered by Creighton, have been documented and reviewed in a number of articles (27,28,38,60). In short, oxidative folding of BPTI proceeds via a limited number of intermediates comprising mainly native disulfide bonds. Out of the 74 possible disulfide isomers (15-1SS, 45-2SS, and 14-3SS) that may serve as folding intermediates, only five (two 1SS and three 2SS) were shown to accumulate along the folding pathway of BPTI and all adopt native disulfide bonds. Folding of BPTI proceeds via two predominant native 1SS intermediates (Cys30-Cys51 and Cys5-Cys55), followed by formation of two 2SS kinetic traps (Cys30-Cys51, Cys14-Cys38 and Cys5-Cys55, Cys14-Cys38) and one 2SS intermediate (Cys5-Cys55, Cys30-Cys51), which serves as a direct precursor of the native BPTI (Cys5-Cys55, Cys30-Cys51, Cys14-Cys38). The flow chart of folding pathway of BPTI and the original HPLC data of intermediates analysis are shown in Figure 2A and 2B, respectively. The unique folding pathway of BPTI clearly indicates that stable subdomain structures dictate the formation of native-like intermediates and limit the heterogeneity of folding intermediates.

Thus, the folding mechanism of BPTI is congruent with the framework model (29), in which secondary structures (α-helix, β-strand, etc.) form early during the folding, which is followed by docking and packing of preformed secondary structural units to form the native tertiary structure.
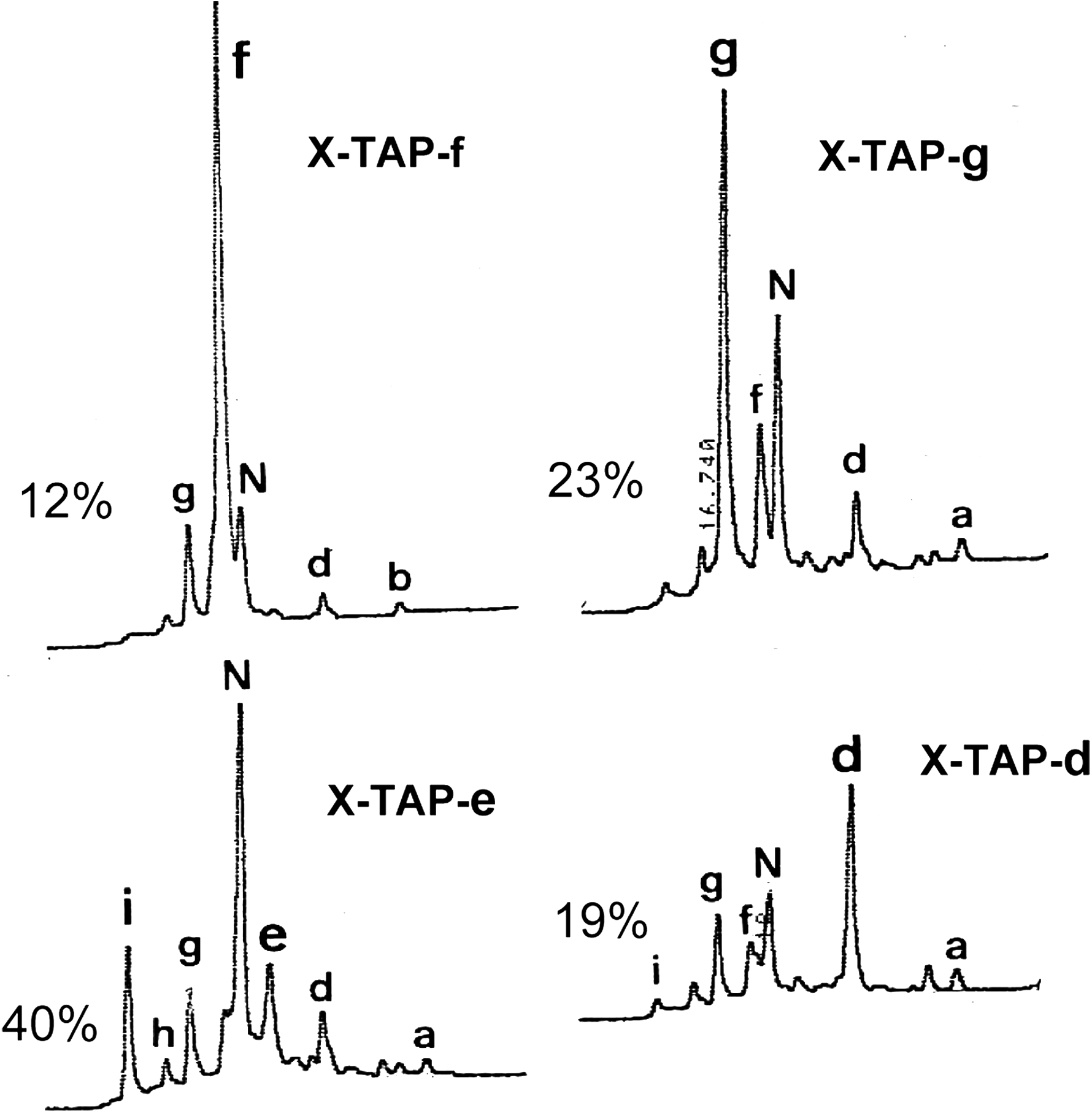
The folding pathway of TAP differs significantly from that of BPTI. The folding intermediates and folding pathway of TAP are far more complex than that of BPTI (Fig. 2A, C). The folding intermediates consist of a highly heterogeneous population of 1SS and 2SS isomers (16,13). Although one 1SS (Cys33-Cys55) and one 2SS (Cys33-Cys55, Cys15-Cys39) intermediate containing exclusively native disulfide bonds are also predominant along the pathway (16), they are among at least 25–30 species of well populated intermediates identified. Most importantly, scrambled 3SS X-TAP isomers, not observed with BPTI folding, were shown to serve as essential folding intermediates of TAP (Fig. 2C) (13). Among the 14 possible X-TAP isomers, four have been found to predominate at the late stage of oxidative folding (see fractions g, f, k, and d in Fig. 2C). Their disulfide structures were determined and shown in Figure 4. Indeed, the folding pathway of TAP resembles that of hirudin (11,24). The mechanism consists of an initial stage of nonspecific packing that leads to the formation of scrambled 3SS isomers as folding intermediates. This is followed by consolidation of scrambled 3SS isomers to reach the native structure via disulfide shuffling.
Thus, the folding mechanism of TAP is more compatible with the hydrophobic collapse model (29,31), in which a rapid hydrophobic collapse accounts for the major driving force of folding, which is followed by searching and fine-tuning of conformation in a confined volume to reach the native structure.
Diversity of Folding Pathways of BPTI and TAP Elucidated by the Method of Disulfide Scrambling
The technique of disulfide scrambling permits folding experiments to be initiated with an isolated X-isomer that represents the most extensively unfolded state and possesses the highest free energy among all unfolded X-isomers (18,14). Such X-isomers can be identified through systematic unfolding experiments with increasing strength of denaturant using the technique of disulfide scrambling. In the cases of BPTI and TAP, these two specific X-isomers were identified as X-BPTI-a (17) and X-TAP-a (12), which share the same beads-form disulfide pattern (Fig. 4). X-BPTI-a and X-TAP-a were allowed to refold in parallel under identical conditions via disulfide shuffling to reach the native BPTI and TAP. Folding intermediates were trapped by acid quenching and analyzed by HPLC. The results are illustrated in Figure 3. Of the 13 possible X-isomers that may serve as folding intermediates, 7 were found in the folding pathway of X-BPTI-a → N-BPTI and 9 were found in the pathway of X-TAP-a → N-TAP. All folding intermediates have been isolated and characterized (18). Their disulfide structures are also shown in Figure 4.

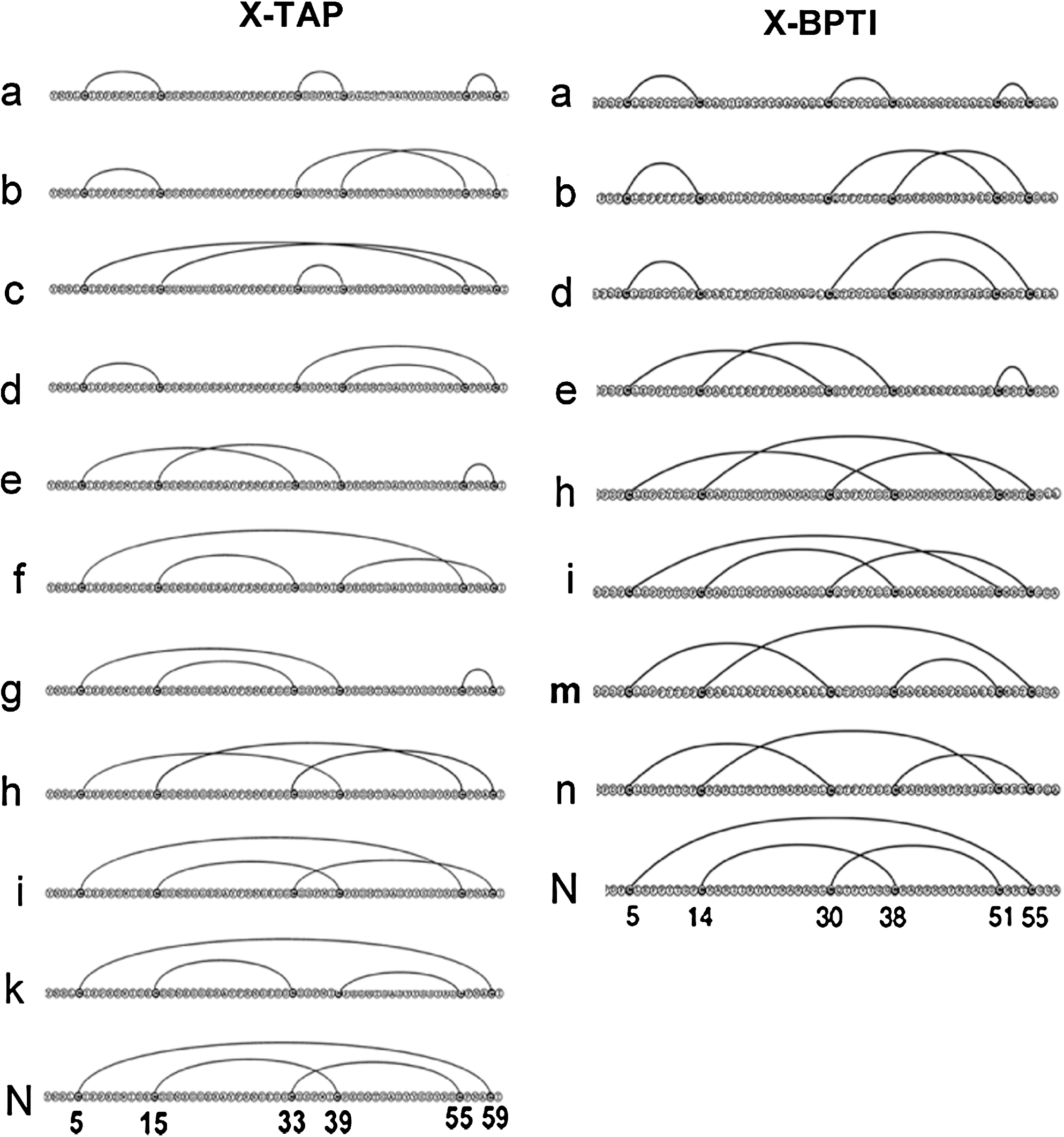
Folding activity of both proteins appears to initiate at the C-terminal domain, resulting in rapid formation of “X-TAP-d” and “X-BPTI-d” during the early stage of folding (Fig. 3). However, this is essentially the only observed similarity. There are several striking dissimilarities that distinguish the folding pathway of X-BPTI-a from that of X-TAP-a. (a) One of the predominant BPTI intermediates X-BPTI-b (Cys5-Cys14, Cys30-Cys51, Cys38-Cys55) contains a native disulfide bond Cys30-Cys51, and constitutes about 34% of the total BPTI folding intermediates. The counterpart, X-TAP-b (Cys5-Cys15,Cys33-Cys55,Cys39-Cys59), is barely detectable in TAP folding. This difference may be due to the unique BPTI subdomain structure that stabilizes Cys30-Cys51, a similar mechanism that favors the formation of predominant 1SS intermediate (Cys30-Cys51) during the oxidative folding of BPTI (Fig. 2B). The same factor apparently does not exist in TAP structure. (b) Three X-TAP isomers (X-TAP-f, X-TAP-g, and X-TAP-k) sharing a stable non-native disulfide bond Cys15-Cys33 are shown to act as kinetic traps of TAP folding. Their counterparts are completely absent in the BPTI folding pathway. (c) Folding intermediates of BPTI are energetically compartmentalized (Fig. 5), whereas most folding intermediates of TAP are interconvertible and exist in equilibrium (Fig. 6). This is demonstrated by the energy booth accommodating isomers X-BPTI-e, n, and m (18). Stop/go folding experiments show that X-BPTI-e, X-BPTI-n, and X-BPTI-m equilibrate rapidly among each other (Fig. 5), but these three isomers are unable to cross energy barriers and convert to X-BPTI-b, X-BPTI-d, or X-BPTI-i. Structurally, X-BPTI-e, n, and m share Cys5-Cys30, implying that stability of this non-native disulfide bond accounts for the energy compartment of these three isomers.
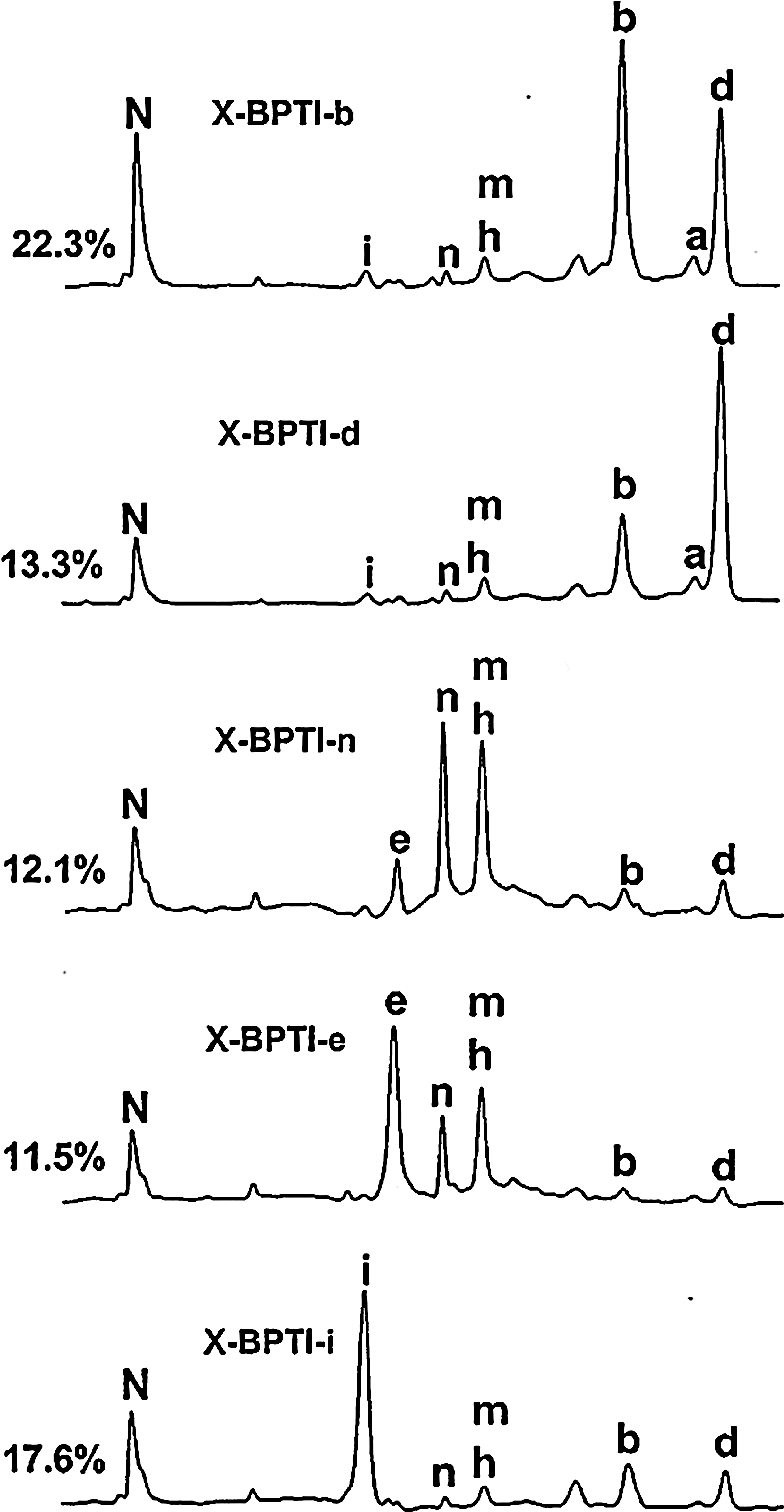
The remarkable diversity of folding mechanism between X-BPTI-a and X-TAP-a may provide an invaluable model in further understanding why structurally homologous proteins fold differently. Specifically, detailed NMR analysis can be carried out with X-isomers that constitute the predominant intermediate (X-BPTI-b), the kinetic traps (X-TAP-f, X-TAP-g, and X-TAP-k), and the energy compartments (X-BPTI-e, X-BPTI-n, and X-BPTI-m). The results should provide structural basis as to how these intermediates are stabilized.
A Group of Homologous Cystine Knot Protease Inhibitors also Exhibit Diversity of Disulfide Folding Pathways
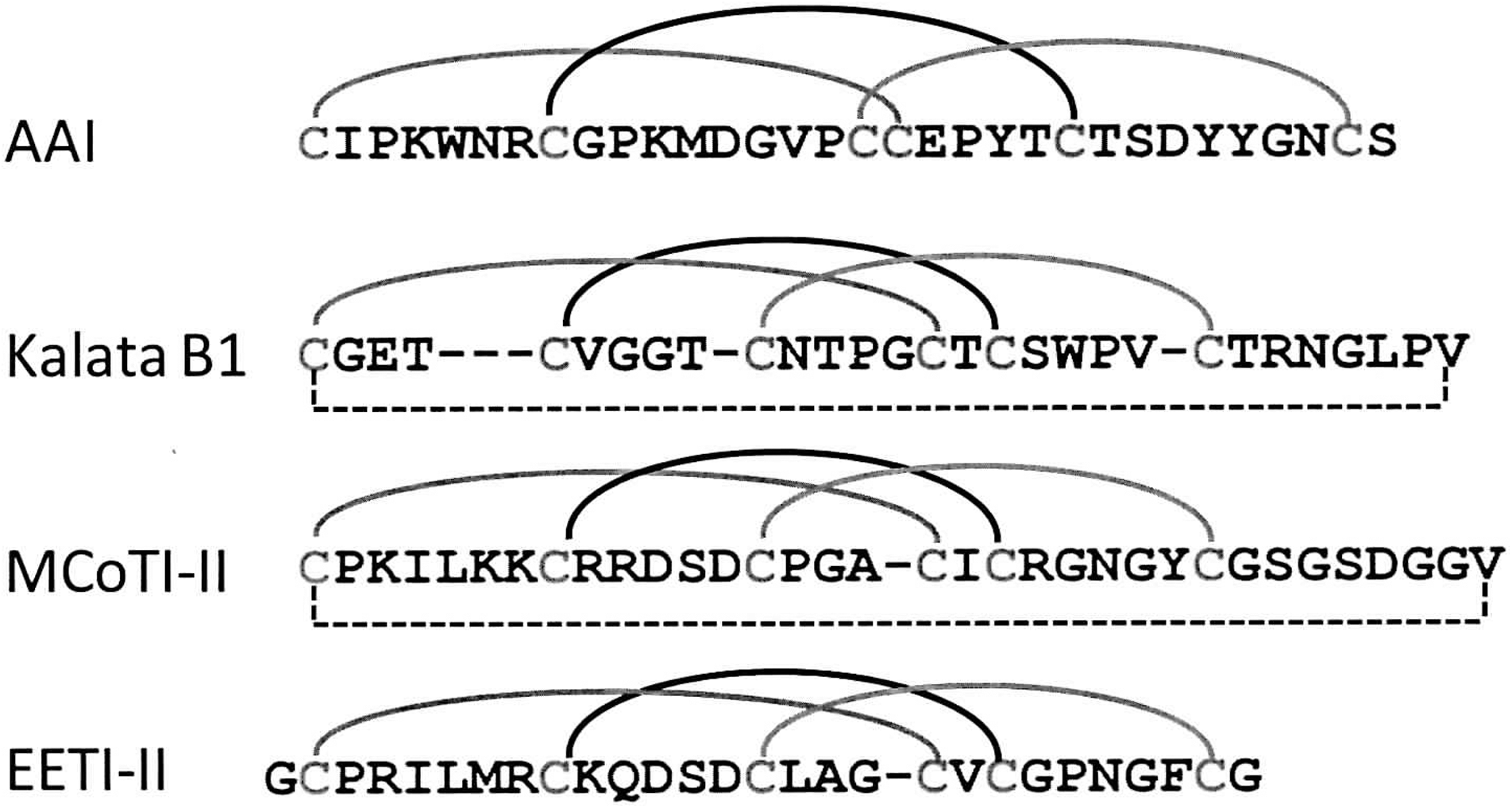
It is apparent that BPTI and TAP will not be the only examples. MCoTI-II, EETI-II, kalata B1, and AAI all belong to a group of homologous cystine knot protease inhibitors, comprising roughly 30 amino acids and 3 disulfide bonds. They share some sequence homology and similar disulfide pattern [1–4,2–5,3–6] (Fig. 7). The disulfide folding pathways of these four small proteins have been investigated in detail in the laboratory of Craik and Cemazar (8,10,25,30,43). MCoTI-II, EETI-II, and kalata B1-fold via pathways resembling that of BPTI, with an instant accumulation of single predominant 2SS intermediate comprising two native disulfide bonds. However, their folding kinetics are different. For MCoTI-II and EETI-II, both native 2SS intermediates fold to form corresponding native proteins directly (8), similar to the role of BPTI[Cys5-Cys55, Cys30-Cys51] in BPTI folding (Fig. 2). In the case of kalata B1, the native 2SS intermediate needs to unfold and revert back to the state of 1SS isomer in order to reach native kalata B1 (8). In a sharp contrast, AAI folds via the pathway similar to that of TAP. Oxidative folding of AAI is characterized by a high heterogeneity of 1SS, 2SS, and 3SS intermediates. Among the 14 possible 3SS X-isomers of AAI, seven have been isolated and structurally characterized by Cemazar and colleagues (10). These results demonstrate that the diversity of disulfide folding pathways of structurally homologous proteins may be a common phenomenon.
The Underlying Cause for the Diversity of Disulfide Folding Pathway: A Protein May Fold via BPTI-Like Pathway or TAP-Like Pathway Depending on the Stability of Its Subdomain
The predominance of 1SS intermediate (Cys30-Cys51) (Fig. 2) and X-BPTI-b (Cys5-Cys14, Cys30-Cys51, Cys38-Cys55) (Fig. 3) along the folding pathways of BPTI suggest that structural elements stabilizing the subdomain comprising Cys30-Cys51 bond play a critical role in dictating the folding pathway of BPTI. Similar structural features are apparently much less evident in the folding of TAP. Indeed, the stable subdomain of BPTI may also account for the distinct pathways of reductive unfolding of BPTI and TAP. The experiment of reductive unfolding is designed to evaluate the relative stability and interdependency of disulfide bonds in the native protein. The three native disulfide bonds of TAP were reduced by DTT in an all-or-none manner without accumulation of partially reduced intermediates, whereas those of BPTI were reduced in a sequential and stepwise manner, with preferential cleavage of Cys14-Cys38 at a very low concentration of DTT (21,63). We have previously shown that there exists a striking correlation between the mechanism(s) of reductive unfolding and that of oxidative folding (21). Those with their native disulfide bonds reduced in a collective all-or-none manner tend to exhibit both a high degree of heterogeneity of folding intermediates and the accumulation of fully oxidized scrambled isomers along the folding pathway, such as TAP and hirudin. A sequential reduction of the native disulfide bonds is associated with the presence of predominant intermediates with native-like structures, such as BPTI.
To corroborate the hypothesis that the extent of stability of subdomain structure accounts for the divergent folding pathways of BPTI and TAP, we have analyzed oxidative folding of a 3-disulfide variant of α-lactalbumin (αLA-IIIA) (Fig. 8) (19,51). αLA-IIIA lacks one native disulfide bond Cys6-Cys120 of αLA, which is reduced and carboxymethylated (see Fig. 8C). The remaining three native disulfide bonds of αLA-IIIA (Cys28-Cys111, Cys61-Cys77, Cys73-Cys91) adopt the disulfide pattern [1–6,2–4,3–5] similar to that of BPTI and TAP. Most importantly, αLA-IIIA also retains a β-sheet domain surrounding Cys61-Cys77, Cys73-Cys91 that can be stabilized upon calcium binding. We demonstrated that αLA-IIIA can fold via either BPTI-like pathway or TAP-like pathway, depending on whether its subdomain is stabilized (19,50,51).
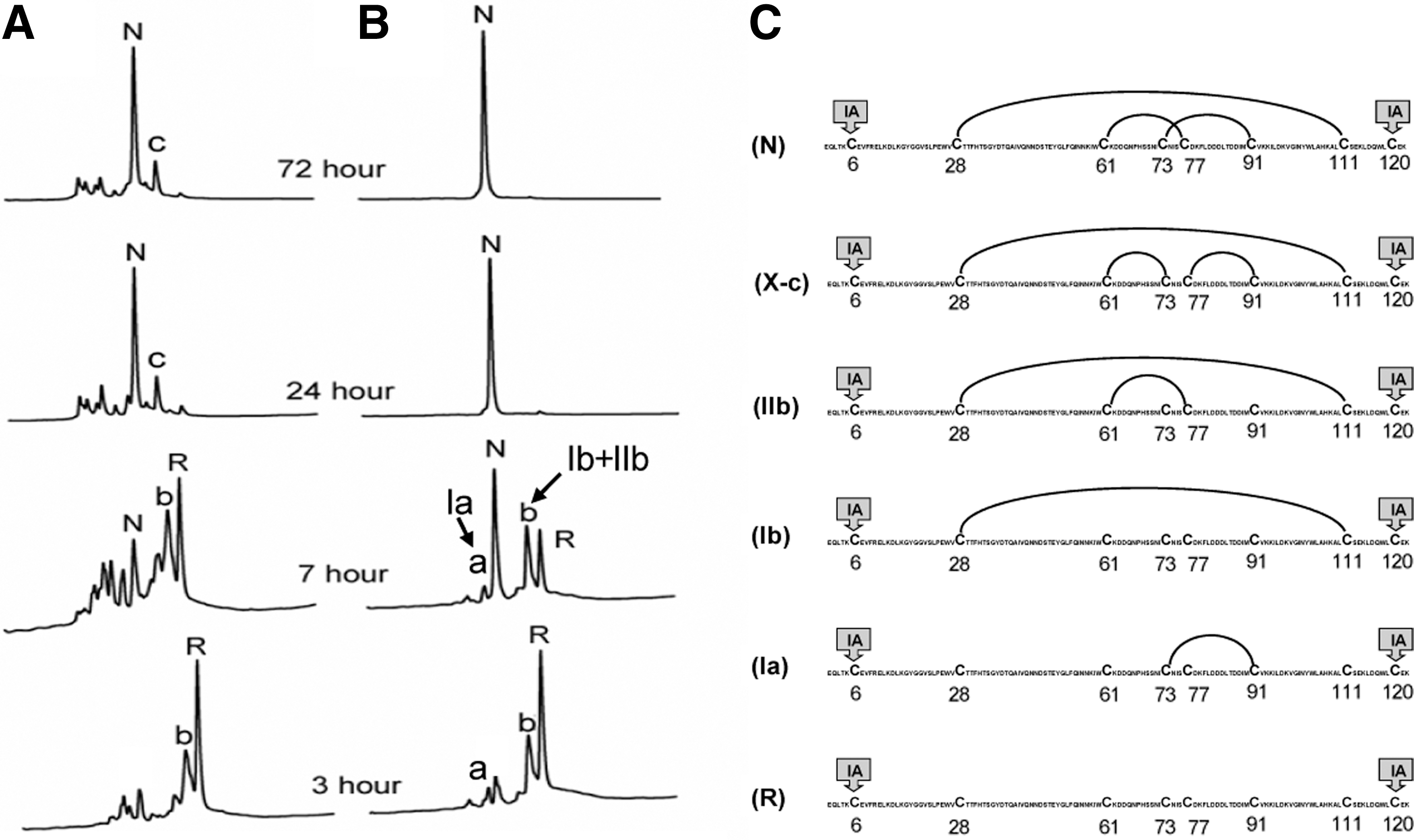
Fully reduced αLA-IIIA (R) was allowed to refold with and without stabilization of its β-sheet domain by calcium binding. The results showed that in the absence of CaCl2 (Fig. 8A), the folding pathway of αLA-IIIA resembles that of TAP (16,13) and hirudin (11,24), which is characterized by a high heterogeneity of folding intermediates and the accumulation of scrambled 3SS isomers along the pathway. The fractions appeared in the 24 and 72 h samples are all 3SS isomers of αLA-IIIA. Among them, the disulfide structure of X-αLA-IIIA-c is known (Fig. 8C) (50). In the presence of CaCl2 (Fig. 8B), αLA-IIIA folded via BPTI-like pathway (27,38,60), with limited intermediates comprising exclusively native disulfide bonds. Two 1SS and one 2SS intermediates connected by native disulfide bonds were identified (Fig. 8C).
Concluding Remarks
This diversity of disulfide folding pathway has been broadened during the past decade through analysis of oxidative folding of numerous small disulfide-rich proteins (3,45). It has been further highlighted by the significant differences of folding mechanisms observed among structurally similar proteins demonstrated in this review. It has been proposed (21) and experimentally demonstrated (19,51) that one of the underlying causes of the diversity is the relative stability of protein subdomains. This information, together with elucidation of new NMR structural data of several native-like folding intermediates (2a,4,5,9,47), has provided a more detailed picture of folding mechanism of disulfide proteins. Finally, it is important to stress that the diversity of protein oxidative folding is fundamentally consistent with that of conformational folding. The folding models of BPTI and TAP are compatible with the framework model and hydrophobic collapse model (29), respectively.
Footnotes
Acknowledgments
The author wishes to acknowledge the support of Robert Welch Foundation and IsoVax Therapeutics Inc.