
Abstract
Several S100 Ca2+-binding proteins are considered damage-associated molecular pattern molecules (DAMPs). They are actively secreted or released from necrotic cells in response to tissue injury or stress and have various functions important in innate immunity. Here, we review several DAMPs, with particular focus on S100A8 and S100A9, which are susceptible to oxidative modifications by various forms of reactive oxygen species. We discuss the unique posttranslational modifications generated in S100A8 by hypochlorite and the likely structural consequences that alter function. We propose that some reversible modifications act as regulatory switches, representing a mechanism to arrest their novel antiinflammatory activities. These may be important in dampening mast cell activation and altering properties of the activated microcirculation to limit leukocyte adhesion, transmigration, and accumulation. S-nitrosylation of S100A8 in the vasculature could regulate nitric oxide transport and contribute to vessel reflow during resolution of inflammation. Antioxid. Redox Signal. 15, 2235–2248.
Introduction
A group of endogenous stimuli known as damage-associated molecular pattern molecules (DAMPs) also trigger inflammatory responses by binding PRRs. DAMPs are released from dying or damaged cells in response to tissue injury or stress, serving as early-warning signals to activate the innate and adaptive immune systems and to promote tissue repair. They include diverse multifunctional host proteins, including high-mobility group box 1 protein (HMGB-1), defensins, galectins, and several S100s (5). Various types of ROS are generated as a consequence of immune activation, and although low levels mediate numerous cell-signaling pathways, excessive production can directly damage tissue and generate several oxidation products, particularly in lipids and proteins, and these can contribute to chronic inflammation (46). Specifically, ROS activate monocytes/macrophages via TLR-2 in synergy with TLR-1 or -6, and in concert with exogenous stimulants, such as cigarette smoke (60).
Several DAMPs are susceptible to oxidative modifications that can alter some functions, and oxidation-mediated inactivation is suggested as a mechanism to terminate the proinflammatory effects of DAMPs, thereby limiting inflammation (10). We propose that some oxidative modifications cause structural changes in proteins, or generate adducts that can shuttle certain oxidants, allowing them to become regulatory switches that activate antiinflammatory properties. These may constitute a novel group of antiinflammatory DAMPs involved in resolution and restoration of homeostasis, likely by suppressing particular activation processes, protecting tissues from excessive damage by scavenging ROS, and mediating tissue repair.
Excess ROS and reactive nitrogen species (RNS) production is counteracted by antioxidant defense mechanisms, including biologic and enzymatic scavengers [reviewed in (12)], and antioxidant proteins [e.g., heme oxygenase; reviewed in (43)]. Although these antioxidant processes contribute to the maintenance of intracellular redox, they are easily compromised and are not completely efficient in preventing extensive ROS-induced damage (12). For example, oxidative modifications of important enzymes involved in antioxidant defense, such as manganese superoxide dismutase (MnSOD) and catalase, inactivate their function. In asthma, this may contribute to airway remodeling and epithelial apoptosis, leading to denudation of the airway surface and predisposing to greater airway hyperreactivity. Reduced levels of active SOD in the circulation may also compromise antioxidant defense in the vasculature in inflammatory conditions [reviewed in (12)].
We propose that some DAMPs, particularly S100A8 and S100A9, generated by an active, ongoing inflammatory response, act as oxidant scavengers, representing a compensatory mechanism that may function in concert with products of antioxidative genes produced through nuclear factor erythroid 2–related factor-2 (Nrf2) activation. Nrf2 is a redox-sensitive transcription factor that plays a key role in induction of genes important in cellular defense against oxidative insult, detoxification, and stress responses [reviewed in (43)]. Oxidant-scavenging activity of S100A8 and S100A9 may be particularly important in restricting excessive tissue damage, limiting leukocyte recruitment, and promoting tissue repair within inflammatory foci.
At first glance, the oxidant-scavenging properties of S100A8 and S100A9 may be seen as possibly compromising antimicrobial functions of activated phagocytes and the antimicrobial activity of the S100A8/S100A9 complex (calprotectin) (35). However, some effects of DAMPs may be concentration dependent. Large quantities of calprotectin are released by necrotic neutrophils, and the high levels in the circulation of individuals with many inflammatory diseases may offer additional protection, whereas at very low concentrations, they may exhibit proinflammatory functions that will also protect the host. Pleiotropic properties of some mediators are not uncommon; transforming growth factor-β (TGF-β) is an example of a protective protein, even though it has potent chemotactic activity at low concentrations [reviewed in (14)]. This review discusses the effects of oxidative modifications that may regulate particular functions of S100A8 and S100A9, and the implications of these in limiting inflammation to restore homeostasis. We propose that some oxidative modifications of DAMPs may cause irreversible structural changes that could result in their accumulation in lesions, thereby contributing to the pathogenesis of chronic inflammatory conditions.
Oxidative Modifications of Proteins by ROS/RNS
Transient levels of ROS are generated by exogenous sources or endogenously as by-products of electron-transfer reactions in mitochondria. However, large quantities of superoxide (O2 .-) are produced by the NADPH oxidase system after phagocyte activation by various soluble mediators (e.g., chemoattractants and chemokines) and particulate stimuli (e.g., PAMPs and DAMPs). O2 .- is rapidly converted to hydrogen peroxide (H2O2) and other oxidants, including hydroxyl radical (OH·) and hypochlorous (HOCl) or hypobromous acids (HOBr); the halide oxidants are catalyzed by heme peroxidases, myeloperoxidases, or eosinophil peroxidases released from activated leukocytes and granulocytes (22). Generation of HOBr is largely mediated by eosinophils, although low levels are produced by activated neutrophils (11).
Nitric oxide (NO) is the predominant RNS, and the main source of RNS in phagocytes. NO is generated by inducible nitric oxide synthase (iNOS), endothelial NOS (eNOS), or neuronal NOS (nNOS); nNOS and eNOS are constitutively expressed in several cell types, whereas iNOS is induced in macrophages and other cells by specific stimuli, including LPS, TNF-α, IFN-γ, and IL-1β (57). This is particularly relevant in inflammation, in which iNOS induction in activated macrophages is regulated by NF-κB and is essential for microbial killing and regulation of immune responsiveness. Induction of iNOS can produce large amounts of NO for prolonged periods, and excess production may lead to nitrosative stress.
Proteins are extremely sensitive to oxidation by ROS and RNS, which promote various reversible and irreversible modifications, depending on the type of oxidant, the duration of exposure, and the mechanism whereby proteins are oxidized (e.g., one- versus two-electron oxidation). Cys, Met, Lys, His, Arg, and Pro residues are particularly susceptible, and their modification can have different structural consequences, varying from slight changes that may or may not affect function, to large global alteration, and in some cases, protein fragmentation or denaturation (7).
Reversible modifications under moderate oxidative stress, including Met oxidation to Met sulfoxide, and Cys oxidation to Cys-sulfenic acid and intra/intermolecular disulfides, may protect proteins from irreversible oxidation and permanent damage, and these modifications also regulate the functions of some susceptible proteins. Disulfide-bond formation between low-molecular-weight thiols such as glutathione (GSH), in a process known as S-thiolation (6), can also be protective. GSH is an abundant intracellular thiol, and S-glutathionylation, the coupling of GSH to Cys thiols, is the most prevalent S-thiolation reaction in cells. This adduct can be reversed by glutaredoxins and thioredoxins, or by changes in intracellular redox (25). S-nitrosylation is another S-thiol modification that readily disassociates or is reversed by specific enzymes (31). Reversibility of functionally critical Cys residues by specific oxidoreductases is the key to the physiologic relevance of susceptible proteins in regulating function and signaling. For example, glutathionylation of some signal-transduction proteins under mild oxidative stress can initiate signaling events that promote adaptive responses to injury. However, when injury diminishes and redox is restored, these proteins are deglutathionylated, and signaling is abrogated (27).
Excessive ROS/RNS can cause irreversible modifications that are generally associated with loss of function (6). For example, Phe and Tyr residues are modified in MnSOD from human asthmatic airways. In particular, Tyr nitration and chlorination caused by myeloperoxidase-catalyzed halogenation and oxidative cross-linking of Tyr lead to functional impairment (12). Another example is Met sulfoxide, which can be further oxidized to Met sulfone, an irreversible adduct that is quite rare in biologic systems (34). Susceptible Cys residues in some proteins can be oxidized to Cys-sulfenic acids that react readily in the presence of high concentrations of oxidants to form Cys-sulfinic and sulfonic acids, and covalent sulfinamide bonds between Cys and Lys residues, the latter arising from sulfenic acid intermediates reacting with ɛ-amine groups in Lys residues (61, 65). Protein carbonylation is another well-characterized modification that can occur as a consequence of direct oxidation of amino acid side chains. Formation of irreversible modifications can trigger oxidation-induced peptide cleavage, or increase susceptibility to proteolysis, and lead to degradation by the proteasome (7). Irreversible modifications may also promote complex formation, leading to protein aggregation. For example, low-density lipoprotein (LDL) treated with increasing molar excesses of HOCl forms high-molecular-weight complexes resistant to reduction by DTT. These are not generated when Lys residues are blocked (30), suggesting intermolecular sulfinamide formation. Sulfinamides are generated in HOCl-oxidized S100A8 and peptides (65). Formation of these types of cross-linked complexes, and their consequential aggregation, may contribute to the pathogenesis of chronic disorders, particularly cardiovascular and neurodegenerative diseases occurring later in life, such as in Alzheimer and Parkinson diseases, and amyotrophic lateral sclerosis (3). Oxidative and nitrosative stress increases with age, and oxidative injury may be exacerbated by long-term accumulation of irreversible protein complexes, ROS/RNS-induced cellular damage, and diminished antioxidant defense and repair mechanisms (3).
Oxidative Modifications of DAMPs
Several DAMPs undergo oxidative modifications, likely when secreted through nonclassic pathways, or released passively from necrotic cells into the oxidizing extracellular environment. Modifications of these molecules are proposed to mediate their activity (10) and may contribute to reduced cell activation that would ultimately limit inflammation and ROS production. One example is HMGB-1, expressed in most cells as a nuclear protein that binds nucleosomes and promotes binding for several transcription factors. HMGB-1 is released from necrotic cells or is secreted by activated myeloid, smooth muscle cells (SMCs) and endothelial cells (ECs). Extracellularly, HMGB-1 is chemotactic for myeloid cells, fibroblasts, dendritic cells (DC), SMCs, ECs, and stem cells, and promotes proinflammatory cytokine production in neutrophils, monocytes, and macrophages by binding bacterial CpG or endotoxin (53). Posttranslational modifications of HMGB-1, including oxidation, acetylation, methylation, glycosylation, and phosphorylation (20), may regulate its ability to induce cytokines. HMGB-1 has three Cys residues (Cys23,45,106) that are readily oxidized, and, depending on the structural change, these modifications can promote distinct functional consequences.
Under mild oxidative conditions, HMGB-1 forms intramolecular disulfide-linked complexes between Cys23 and Cys45, whereas Cys106, which is critical for nuclear localization of HMGB-1 and its interaction with TLR4 (89), is not oxidized (33). Oxidized HMGB-1 complexes were isolated from UV-irradiated HeLa cells, likely generated after caspase-dependent ROS production during apoptosis. Unlike reduced HMGB-1, these complexes do not activate DCs, and it is likely that oxidation impairs its nuclear distribution or TLR4 binding, and abrogates proinflammatory effects. ROS scavengers or DTT treatment prevented HMGB-1 oxidation in vitro and restored its immunostimulatory effects (41). The redox state of HMGB-1 is also a crucial determinant of its effect on tumor cells; reduced HMGB-1 promoted Beclin1-dependent autophagy, likely via RAGE binding, whereas the oxidized form induced apoptosis in a caspase-3 and -9–dependent manner (MT Lotze, personal communications).
Galectins comprise a diverse family of lectins with broad immunomodulatory functions, including regulation of cell adhesion, growth, and apoptosis; these proteins are also considered DAMPs. Galectin-1 is expressed predominantly in immune cells (e.g., macrophages, activated T cells, DCs) and contains six Cys residues that form three intramolecular disulfides when oxidized. Reduced galectin-1 is suggested to modulate cell adhesion, proliferation, and differentiation by cross-linking β-galactoside–containing glycoconjugates on cell surface (73), whereas oxidation reduces activation, possibly because the structural changes generated affect its ability to bind these receptors. Conversely, oxidized galectin-1 has protective functions not seen with the reduced form, by promoting axonal regeneration for initial repair of peripheral nerves after axonomy (39).
Defensins are another group of DAMPs and are abundantly expressed in neutrophils. These antimicrobial peptides contribute to elimination of a range of pathogens and promote innate immunity by recruiting monocytes, DCs, and T lymphocytes. Defensin peptides contain six conserved Cys residues and typically form three intramolecular disulfides; the chemotactic activity of human β-defensin 3 may depend on these, although its antimicrobial activity is unaffected by oxidation (86), indicating that specific structural conformations caused by oxidation may mediate distinct functions.
S100 Proteins as DAMPs
S100 proteins belong to the EF-hand superfamily of Ca2+-binding proteins and have diverse intracellular and extracellular functions. S100A8, S100A9, and S100A12 are designated inflammation-associated S100s because their expression is associated with a range of acute and chronic inflammatory disorders, and they are considered prototypic DAMPs (52). They are associated with innate immune functions because of their prominent expression in myeloid cells, and are secreted independent of the classic ER-Golgi route, a property described for most DAMPs. S100A8 and S100A9 are released passively from necrotic cells or are secreted via a novel tubulin-dependent pathway involving PKC activation (67). In the extracellular space, S100A8, S100A9, and S100A12 may exert proinflammatory activities, some of which are proposed to be mediated by interactions with PRRs such as TLRs and RAGE.
S100B can also be considered a DAMP. It is expressed by astroglial cells, and elevated levels, possibly released from damaged astrocytes, are found in blood and cerebrospinal fluid from patients after brain injury or ischemic stroke, and in neurodegenerative (e.g., Alzheimer disease), inflammatory (e.g., multiple sclerosis), and neoplastic disorders (e.g., malignant melanoma) [reviewed in (70) and (72)]. S100B has neurotrophic and neurotoxic effects that are dependent on its concentration and oxidative state, and many of its functions, such as regulation of neurite outgrowth and cell survival, and stimulation of neuronal apoptosis via excessive activation of extracellular signal-regulated kinase (ERK) 1/2 and NF-κB, and ROS generation, may depend on its interaction with RAGE [reviewed in (18)].
Functions of myeloid-associated S100s
S100A8 and S100A9 compose ∼40% of cytoplasmic proteins in neutrophils and are induced in other cells, including monocytes, macrophages, epithelial cells, keratinocytes, and fibroblasts by inflammatory mediators (23, 36, 87) or oxidative stress [reviewed in (35, 52)]. S100A8 and S100A9 form noncovalent homo- or hetero-complexes and have distinct functions, depending on their oligomeric forms; some functions are independent of heterocomplex formation. This is underscored by differences seen in S100A8 and S100A9 knockout mice. Targeted deletion of the mS100A8 gene causes embryo resorption at 9.5 dpc (59), providing strong evidence that it plays a nonredundant role in embryogenesis, whereas mS100A9-null mice develop normally and lack any obvious phenotype (32). Moreover, murine macrophages activated with TLR3 or TLR4 ligands (23) and keratinocytes in skin of mice treated with UVA, express only S100A8 (28). Thus S100A9 is not obligatory for structural stability of S100A8, and our results suggest that S100A9 may be more important in neutrophil responses in innate immunity in the mouse.
Intracellularly, S100A8 and S100A9 may contribute to maintenance of normal cell function by regulating some enzymes (e.g., inhibition of casein kinase I and II) and may mediate cytoskeletal rearrangement or dynamics (or both) important in processes such as phagocytosis, exocytosis, and migration by interacting with cytoskeletal components such as F-actin and microtubules [reviewed in (21)].
Several extracellular activities of S100A8 and S100A9, including their chemotactic function, can be described as proinflammatory. However, leukocyte recruitment in infection is the first line of defense and is critical for host protection. Murine (m)S100A8 promotes chemotaxis of monocytes and neutrophils at picomolar levels via a “nonclassic,” pertussis toxin–sensitive, G protein–coupled pathway that causes actin polymerization and profound shape changes without provoking a Ca2+ flux, degranulation, or an oxidative burst (14). These properties are similar to those of TGF-β, which is generally regarded as antiinflammatory or immunomodulatory. The mS100A8 causes mild transient leukocyte infiltration when injected into mice, with early recruitment of neutrophils followed by monocytes; the kinetics of this reaction is similar to delayed-type hypersensitivity responses when antigen is injected into a sensitized host. Neutrophils recruited by mS100A8 expressed high levels of β2 integrin Mac-1 (CD11b/CD18), suggesting that it may also modulate leukocyte adhesion (17). Chemotactic activity of mS100A8 is attributed to its hinge region (mS100A842-55), as the synthetic peptide is chemotactic for leukocytes and stimulated leukocyte recruitment after injection into mice, although responses were less potent (47). Conversely, the chemotactic activity of human (h)S100A8 is contentious; Lackmann et al. (47) found that the hinge region of hS100A8 was not chemotactic for monocytes, a finding supported by another study showing that hS100A8 and hS100A9 did not stimulate neutrophil chemotaxis (58). However, Ryckman et al. (71) demonstrated that hS100A8, hS100A9, and hS100A8/A9 were chemotactic for neutrophils in vivo in mice and in vitro, stimulating L-selectin shedding, upregulating CD11b, inducing neutrophil adhesion to fibrinogen via CD11b, and increasing Mac-1 affinity. The hinge domain of S100A12, implicated in its chemotactic activity, shares significant amino acid sequence homology with the hinge region of mS100A8, particularly residues Asn46 and Ile47, which are conserved at similar positions and are essential for activity. However, these are not conserved in hS100A8, suggesting that S100A12 may be the human chemotactic homologue (88). This is an interesting possibility, particularly because S100A12 has no Cys or Met residues and is relatively stable to oxidation, whereas S100A8 is highly sensitive. Thus, leukocyte recruitment mediated by S100A12 would be sustained in an oxidizing milieu.
hS100A9 stimulates neutrophil adhesion to extracellular matrix (ECM) components, and monocyte adhesion to the endothelium in vitro, possibly by upregulating β2 integrins and inducing a high-affinity Mac-1 epitope through a pertussis toxin–sensitive, G protein–coupled receptor (58), and may be important in neutrophil transmigration and extravasation. Mice lacking S100A9 have compromised leukocyte migration to some chemoattractants and are protected against endotoxin shock [reviewed in (21)].
S100A8 and S100A9 are ideally located to support epithelial barrier function, and the calprotectin complex is expressed in squamous mucosal keratinocytes and innate immune cells at mucosal surfaces. We recently reviewed the important antimicrobial and antiinvasive properties of calprotectin (35) and proposed that the functions displayed by these proteins may reflect their concentration, posttranscriptional modifications, oligomeric forms, and their proximal intracellular or extracellular environments.
ROS/RNS-dependent functions mediated by S100A8 and S100A9
S100A8/A9 has other concentration-dependent effects on some cells. At high extracellular concentrations (micromolar amounts), S100A8/A9 induces apoptosis of ECs, fibroblasts, and several tumor cell lines; activity is attributed in part to Zn2+ exclusion by S100A8/A9 from target cells, which may promote caspase-3 and -9 activation. S100A8/A9 also decreases mitochondrial membrane potential and expression of antiapoptotic Bcl2 and Bcl-XL proteins, and induces release of proapoptotic proteins from mitochondria of MCF-7 (human breast cancer) and SHEP (human neuroblastomas) cells. The antioxidant N-acetyl-
S100A8/A9 binds arachidonic acid (AA) intracellularly, and its delivery to membrane-bound gp91 phox is proposed to facilitate activation of NADPH oxidase in phagocytes; an S100A8/A9 mutant with a truncated C-terminal domain (S100A91-100) does not bind AA, and cells expressing this complex had reduced NADPH oxidase activation (42). S100A8/A9 may enhance this activity by acting as a scaffold for cytosolic phox proteins to facilitate their interactions. S100A8 binds p67 phox and Rac-2 directly (42), and binding to p67 phox increases its affinity for flavocytochrome b558, thereby increasing its turnover rate (19).
Increased NADPH oxidase (Nox1 and Nox5) activity and ROS production were observed in HaCaT keratinocytes overexpressing S100A8/A9 after ionomycin stimulation (4). Furthermore, NADPH oxidase activity is impaired in neutrophil-like NB4 cells after S100A9 gene silencing and in neutrophils from S100A9 knockout mice (42). Interestingly, NADPH oxidase activity is suppressed in cells transfected with S100A9 mutants lacking Thr113, suggesting that phosphorylation of this residue, which promotes S100A8/A9 translocation to the plasma membrane after phagocyte activation, may affect activity in response to some stimulants (4). S100A8/A9 may also contribute to RNS generation by stimulating iNOS expression in murine macrophages (62), although we found no proinflammatory cytokine or iNOS production in macrophages treated with S100A8, S100A9, or the complex.
S100A8 is a two-electron oxidant scavenger, and protection against oxidative stress may be an important extracellular function, particularly if the protein is present at high concentrations. S100A9 is somewhat less efficient (54). These proteins are readily oxidized by H2O2 and are far more susceptible to HOCl oxidation than serum albumin or LDL (54), indicating that they may be particularly important in acute inflammation, characterized by prominent neutrophil infiltration and necrosis that facilitates release of large amounts of constitutive S100s (13). S100A9 is released from epithelioid macrophages and inhibited the respiratory burst and subsequent ROS production by bacillus Calmette-Guerin–activated macrophages (1). S100A9 suppressed macrophage activation after uptake of apoptotic neutrophils and inhibited production of NO, H2O2, and TNF-α (16). Relatively high amounts of S100A8 and S100A9 also inhibit ROS production in resting and PMA- and LPS-activated neutrophils. These proteins are released by activated neutrophils, and blocking antibodies to S100A8 or S100A9 increased ROS generation in this system, although the source of ROS was not identified (80).
Putative S100 receptors
Several putative receptors, including some glycosaminoglycans, G protein–coupled receptors, CD36, RAGE, and TLR-4 have been implicated in S100A8 and/or S100A9 binding. mS100A8 is a proposed TLR-4 ligand and bound recombinant TLR4; no TNF-α was produced in mS100A8-stimulated bone marrow cells from mice with a nonfunctional TLR-4. S100A8 binding to TLR-4 promoted MyD88-dependent NF-κB activation, and activation of IRAK1, ERK, p38 MAPK, and PKC pathways. The S100A8/A9 heterocomplex was inactive, and S100A9 suppressed activation by mS100A8 (85). However, these findings could not be replicated in our laboratory by using pure human monocytes or macrophages, or murine macrophages activated with S100A8 (unpublished observations). We showed that the chemotactic activity of mS100A8 was likely mediated by a G protein–coupled receptor (14), although the identity of this remains obscure. Hence, the role of TLR-4 in mediating activation of a proinflammatory response by S100A8 needs further clarification.
Binding of some S100s to RAGE is also controversial. Some studies indicate that S100A8/A9 colocalizes with RAGE and triggers RAGE-dependent functions. RAGE was co-immunoprecipitated from lysates of LPS-stimulated cardiac myocytes by using anti-S100A8 and anti-S100A9 antibodies, and RAGE-specific antibodies abrogated S100A8/A9-mediated cardiomyocyte dysfunction (8). However, surface plasmon resonance studies indicate that S100A8/A9 does not interact directly with recombinant RAGE (85) or bind to CHO cells transfected with RAGE (69). Interaction of S100A8/A9 with RAGE may be cell specific or influenced by contaminants such as LPS in S100 preparations used in functional studies. Furthermore, S100A8/A9 binds carboxylated N-glycans on human microvascular endothelial cells (HMEC-1) and, because RAGE expresses these moieties, they may mediate S100A8/A9 binding to RAGE, and presumably to other receptors that are similarly N-glycated. Binding to N-glycans on RAGE is a proposed mechanism for the recruitment of myeloid-derived suppressor cells that are important in regulating tumor growth (82).
Gene regulation of S100A8 and S100A9
In human neutrophils, S100A9 mRNA levels are some fourfold greater than those of S100A8, and both are among the highest expressed genes in these cells; the same ratio is reflected in blood monocytes, although gene expression is some 50-fold less than that in neutrophils. Comparatively, the genes are expressed in about the same ratio in murine bone marrow cells, although levels are some ∼1,500-fold less than in human neutrophils (68). Interestingly, we have never found S100A9 induction in murine macrophages (36, 87), although S100A8 and S100A9 are induced to varying degrees in stimulated human monocytes/macrophages (61). Other cells may have similar distinctions. For example, keratinocytes in UVA-irradiated murine skin do not express S100A9, whereas S100A8 is strongly upregulated (28). Clear distinctions are found in their expression patterns in human diseases. For example, atherosclerotic lesions of ApoE-/- mice do not express S100A8, and little S100A9 is present, whereas foam cells and neovessels in human atheroma strongly express both proteins (54). These observations indicate important differences in the requirements for S100A8 and/or S100A9 in mouse and human. Intriguingly, macrophages in murine atheroma express little myeloperoxidase (55) and so would generate negligible HOCl. It is tempting to speculate that the large differences in susceptibility of humans and mice to endotoxin, and/or to the oxidative capacities of phagocytes from each species, may be reflected in expression levels of S100A8 or of S100A9 or both.
The mode of S100A8 gene regulation in monocytes/macrophages supports a protective role in inflammation and seems contrary to its proposed proinflammatory functions. The proteins are not expressed by tissue macrophages but are upregulated in macrophages in inflammatory lesions (54). Induction of S100A8 in murine macrophages, and human S100A8 and S100A9 in monocytes and macrophages is IL-10 dependent (87). S100A8 is induced by PAMPs, including LPS and Poly I:C. Induction is delayed, abrogated by neutralizing anti-IL-10 antibodies, and absent in IL-10-/- murine macrophages (23, 37). It is important that antiinflammatory corticosteroids directly induce S100A8 and S100A9 in human monocytes and enhance LPS-induced S100A8 expression in murine macrophages; enhancement is dependent on IL-10 generation (36). Corticosteroids also increased S100A8 production in activated murine fibroblasts, microvascular endothelial cells (36), and keratinocytes (2), suggesting global upregulation. Increased mS100A8 levels were detected in bronchoalveolar lavage fluid of mice treated with corticosteroid after LPS challenge, and S100A8- and S100A9-positive macrophages were more plentiful in the synovium of rheumatoid arthritis patients undergoing high-pulse steroid treatment (36), supporting our in vitro findings. IL-10 and corticosteroids are considered antiinflammatory mediators with broad immunosuppressive functions, such as inhibition of macrophage activation by downregulating proinflammatory cytokine production, inducing antiinflammatory cytokines (e.g., IL-4, IL-10, and TGF-β), and inhibiting adaptive T cell–mediated responses. Dependence of S100A8 expression on IL-10 is in keeping with its protective activities, rather than the proinflammatory roles generally proposed.
S100A8 and S100A9 are not expressed in normal skin, but UVA, an important initiator of sunburn, induces S100A8 in murine keratinocytes; the protein is also induced by UVA or H2O2 exposure of keratinocytes in vitro (28). Responses to UVA irradiation are predominantly oxygen-dependent processes driven by formation of ROS, including O2 .-, OH · , H2O2, and singlet oxygen. Induction in keratinocytes is suppressed by SOD and catalase during irradiation in vitro, or by Tempol (an SOD mimetic) application to skin before UVA irradiation (28). X-ray irradiation, which elevates ROS generation in the cell membrane, also induced S100A8 in keratinocytes, and the overexpression of S100A8 significantly protected against x-ray–induced apoptosis (49), indicating that like some antioxidants, S100A8 is upregulated in response to oxidative stress. S100A8 and S100A9 are strongly induced in human keratinocytes in response to UVB irradiation (15) and are overexpressed in keratinocytes in inflamed skin of several chronic inflammatory dermatoses, including psoriasis, systemic lupus erythematosus, and lichen planus (21). ROS production increases during the course of these conditions, and the elevated expression of these proteins supports a role in oxidant protection.
S100A8 is upregulated in IL-1β and FGF-2–activated fibroblasts, factors associated with tissue healing, and is expressed in macrophages, extravasating neutrophils, and spindle-shaped fibroblasts in tissues from rat dermal wounds 2 to 4 days after injury, but is absent in healed wounds. S100A8 induction in fibroblasts is downregulated by TGF-β, indicating that it is unlikely to play a role in skin fibrosis (66). Its pattern of gene induction and temporal expression at sites of repair/remodeling suggest an additional protective role in regulating fibroblast proliferation and differentiation, and repair/remodeling during the resolution phase of inflammation. S100A8 (but not S100A9) is induced in lung epithelial cells by dsRNA in vitro, but is not seen in lungs of mice infected with influenza virus during the acute phase, although mRNA levels increase during epithelial destruction and then decline on recovery (23). In chronic human rhinosinusitis, S100A8/A9 expression in nasal mucosal epithelium correlates with resistance, and reduced expression is linked to increased susceptibility [reviewed in (35)].
Oxidation generates reversible and irreversible modifications in S100A8 and S100A9
H2O2 or HOCl generates distinct structural modifications in S100A8 or S100A9 or both, principally involving Cys and Met residues that have reactive sulfur-containing functional groups. Amino acid sequence comparisons of human and murine S100A8 (Fig. 1) show the single conserved Cys residue (Cys42 and Cys41, respectively) and a Met residue (Met78 and Met74, respectively) that are most susceptible; mS100A8 has an additional Met at position 36. Various modifications of Cys41 and Met74 in mS100A8 that we have characterized by peptide mapping and mass spectrometry are shown in Fig. 2.

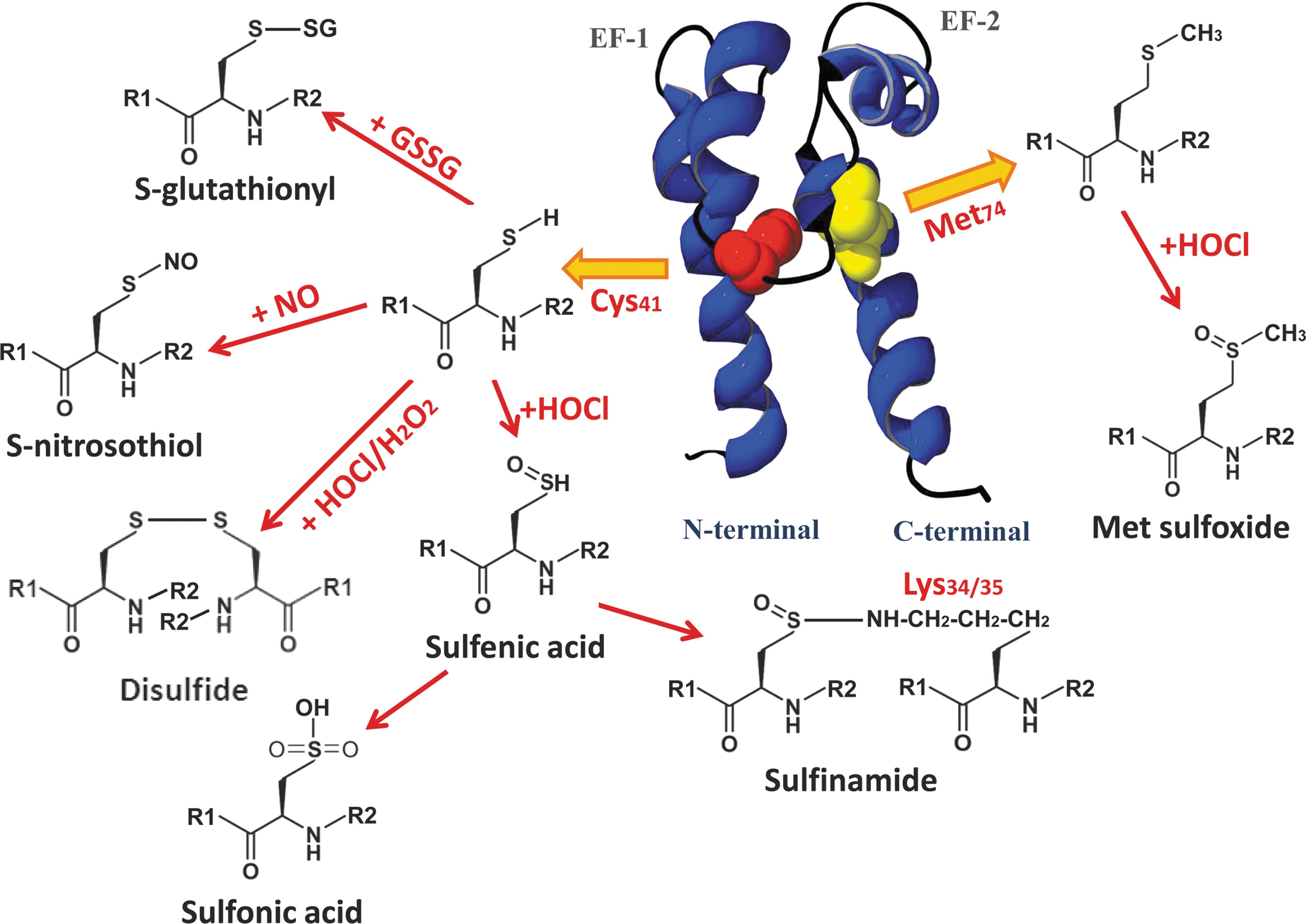
Mild oxidation by low amounts of H2O2 or HOCl generates disulfide-linked S100A8 dimers (29). Disulfide-linked mS100A8 dimers were identified in PMA-stimulated HL-60 granulocytic cells (65) and in lung lavage fluid from LPS-treated mice (29), confirming their formation in vivo. mS100A8 is preferentially oxidized to the homodimeric form with low molar ratios of HOCl, and this may prevent heterocomplex formation with mS100A9 and thereby inhibit or regulate S100A8/A9-dependent functions (54). Molar ratios of >10-fold HOCl generate Met sulfoxides, sulfonic acid, and intra- and intermolecular Cys-Lys sulfinamide bonds in mS100A8 (65). Several Lys residues (Lys6/34/35/76/83/87) in mS100A8 readily react with the sulfenic acid intermediate to form sulfinamide bonds, but Lys34/35 are structurally the most favorable for intermolecular sulfinamide cross-linking (64, 65). Analysis of sulfinamide cross-linked S100A8 peptides by electrospray low-energy collisionally induced (CID) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) postsource decay spectra indicate that these bonds have unusual characteristic fragmentations corresponding to cleavage of nitrogen–sulfur bonds (64).
Similar to its murine counterpart, hS100A8 forms disulfide-linked dimers in the presence of H2O2 and HOCl (29), and Met78 is rapidly oxidized to Met sulfoxide by HOCl. By using a monoclonal antibody that preferentially recognized the disulfide S100A8 dimer, we clearly demonstrated increased levels of this form in the cytosol after activation of human neutrophils with activated zymosan, supporting an intracellular role in redox regulation (45). We also detected S100A8 with Met sulfoxide and sulfone oxidation products in the cytosol of neutrophils activated with PMA and ionomycin (51), suggesting that these may be generated in vivo during infection or tissue injury or both. Interestingly, Met oxidation of hS100A8 was also detected in normal granulocytes (84), indicating that it is readily oxidized once exposed to air, and we have validated this. Recently, we identified Cys-sulfenic and sulfonic acid adducts in hS100A8 treated with equimolar levels of HOCl; the human protein is some 50-fold more sensitive to modification than is the murine counterpart (unpublished observation).
We produced antibodies that preferentially recognize HOCl-oxidized hS100A8 (anti-S100A8ox) (54). DTT-resistant complexes of S100A8 and S100A9, strongly reactive to anti-S100A8ox, were found in extracts of human atherosclerotic carotid arteries (54), indicating formation of sulfinamide-containing complexes. Immunohistochemistry confirmed high levels of oxidized S100A8 in human atheroma, particularly in foam cells and neovessels (54). Interestingly, the S100s are not expressed by larger vessels, but are induced in microvascular ECs by IL-1β and LPS (90), and their ability to regulate redox may be important in the inflamed microcirculation.
S100A8 is also susceptible to S-glutathionylation and S-nitrosylation. S-nitrosylated (S100A8-SNO) and S-glutathionylated S100A8 (S100A8-SSG) adducts were generated by reaction with S-nitrosoglutathione (GSNO), a physiological NO donor (50). Few proteins are targets of S-nitrosylation as the reactivity of particular Cys residues is precisely regulated by the amino acids surrounding the reactive Cys residue, and by the overall protein structure (31). The proximal basic Lys and acidic Glu residues surrounding Cys41 (Fig. 1) are likely to facilitate S-nitrosylation of mS100A8, causing acid-base–catalyzed SNO/SH exchange for SNO generation. S100A8-SSG is also generated, likely after reaction of the SNO intermediate with endogenous GSH (50). In hS100A8, selectivity of S-nitrosylation may depend on the basic Arg or Lys, and the conserved acidic Glu residues surrounding Cys42 (Fig. 1). Basal levels of S-nitrosylated S100A8 were detected in resting human neutrophils, but increased by >30% when cells were treated with the NO donors SNAP or GSNO (50).
In contrast to S100A8, amino acid sequence comparisons of human and murine S100A9 indicate some conservation of Met but not of Cys residues (Fig. 3). mS100A9 has three Cys residues (Cys79/90/110) and seven Met residues (Met9/41/50/64/81/82/84), whereas hS100A9 has five Met residues (Met5/63/81/83/95) but only a single Cys residue at position 3. Native mS100A9 has an intramolecular disulfide bond between Cys79 and Cys90; HOCl did not induce disulfide bond formation, or sulfinamide complexes, at the free Cys110 (63). Oxidative modifications of the Met residues in S100A9 have not been characterized (29), but higher-mass monomers of S100A9 are generated with increasing amounts of HOCl, ranging from 14 to 16 kDa (29), indicating sequential, concentration-dependent Met oxidation.

Oxidative modifications of hS100A9 are studied in more detail and are depicted in Fig. 4. Cys3 is not always expressed because of an alternate Met start site at codon 5 in the human S100A9 gene. Hence, full-length and truncated S100A9 are both expressed in monocytes and neutrophils; the latter lacks Cys3, but little is known concerning mechanisms that regulate translation of these isoforms. Relative expression levels of the two S100A9 forms would affect the protein's susceptibility to oxidation, and given its high amounts in neutrophils, possibly the redox balance within the cell. It is reasonable to suggest that each form has distinct and common functions. We found that full-length S100A9 comprises ∼70% of total S100A9 in neutrophil cytosol from five healthy donors (51). S-nitrosylated (S100A9-SNO) and S-glutathionylated S100A9 (S100A9-SSG) were identified by mass spectrometry after treatment of the recombinant protein with GSNO, although S100A9-SSG was more abundant. More important, S100A9-SSG was detected in cytosol of neutrophils activated with PMA, suggesting that it is generated in vivo. Additional modifications include mass additions likely corresponding to disulfide-linked S100A9 and Cys-sulfonic acid (51); the latter may be generated via sulfenic acid intermediates in a reaction similar to that seen in S100A8 (65).
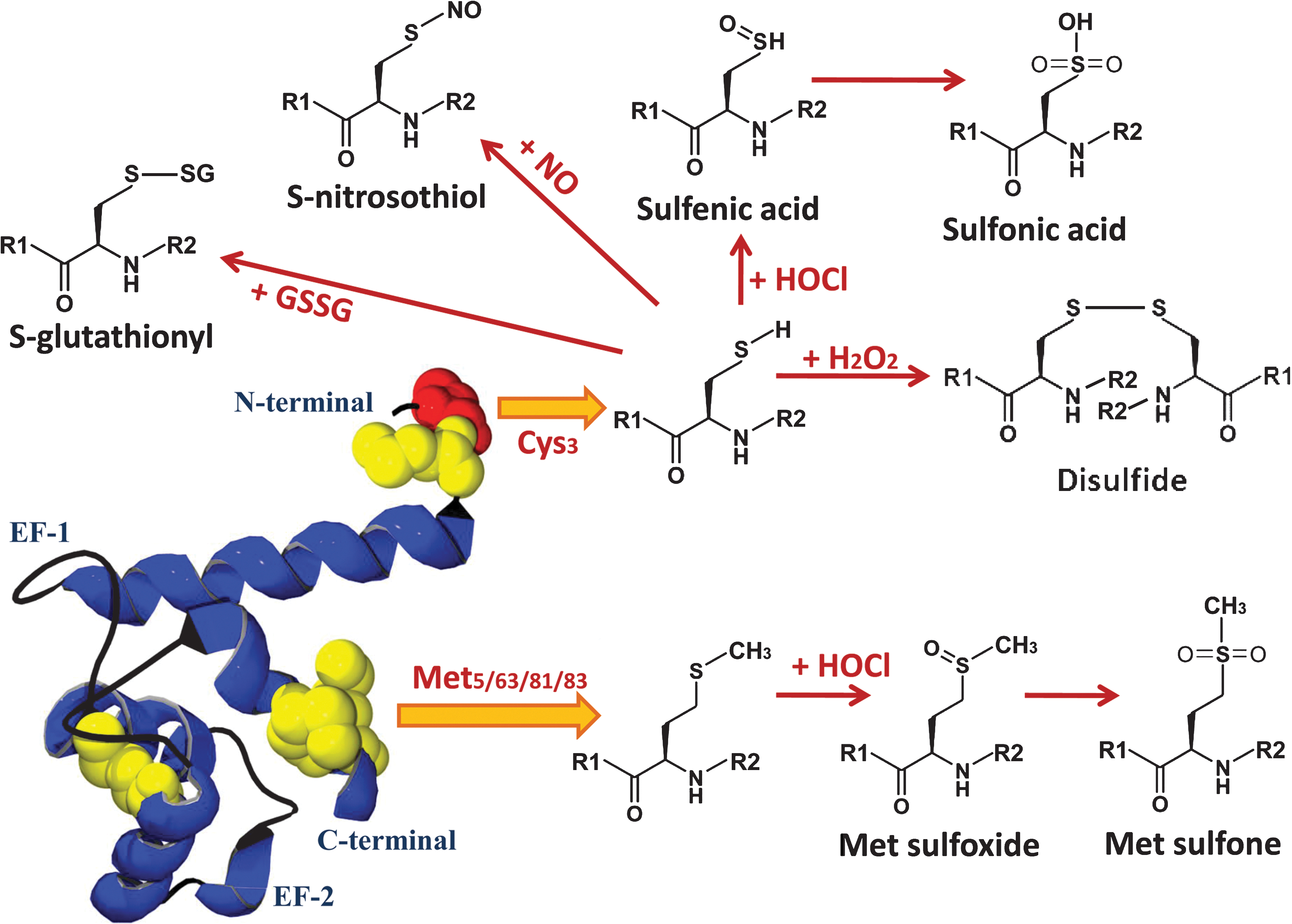
We also observed Met sulfoxides and an unusual Met sulfone derivative in S100A9 from the cytosol of activated neutrophils (51). Met residues are exceptionally susceptible to oxidation, but further oxidation to Met sulfone is rare (34), although its generation is linked to the pathogenesis of Parkinson and Alzheimer disease. The functional implication of this modification is unclear, and more-detailed investigation is currently under way in our laboratory.
Functional regulation by oxidation
Oxidation of some S100s has important functional consequences. For example, S100B promotes neuronal and glial cell proliferation and survival, likely by modulating ERK activity. Its neurotrophic effects are enhanced by oxidation of Cys68 and Cys84 to intramolecular and intermolecular disulfide-linked monomers and dimers (74), and activity is abrogated when the Cys residues are mutated to Ser (44).
Some oxidative modifications of S100A8 and/or S100A9 not only inhibit their activities, but may promote antiinflammatory functions. The chemotactic activity of mS100A8 is modulated by particular oxidants and the extent of oxidation. Disulfide-linked mS100A8 dimers and intermolecular sulfinamide-linked (between Lys34/35 and Cys41) oligomers are not chemotactic (29, 65). However, oxidized Met and Cys residues, to Met sulfoxide and Cys-sulfenic acid, respectively, or generation of intramolecular sulfinamide-containing mS100A8 monomers (between Lys6, Lys34/35, Lys87, and Cys41), retain chemotactic activity characteristic of the native monomer (65). Mutation of Cys41 in mS100A8 did not affect its chemotactic activity (29). Hence, Cys41 is not essential for chemotaxis, but covalent oligomerization promoted by oxidation apparently reduced accessibility of the hinge domain (adjacent to Cys41), and hindered receptor interactions instead of directly modifying amino acids within the chemotactic domain. The chemotactic activity of hS100A8 for neutrophils was similarly inactivated by HOCl oxidation, likely due to disulfide dimer formation (71). S100A8 and S100A9 have fugetactic activity (cell repulsion) for neutrophils, and this activity also depends on the oxidation state, as Ala substitution of Met63 and Met83 in hS100A9 (78), and of Cys42 in hS100A8 (77) abolished activity. Likewise, the antifungal activity of S100A8/A9 is abrogated by oxidation, confirmed by Ala substitution of Met63 and Met83 in hS100A9, and of Cys42 in hS100A8 (79). A recent study showed that the Ala-Cys42 hS100A8 mutant can more effectively ameliorate impaired wound healing than the native form (81), indicating another antiinflammatory activity that may be mediated by oxidative modification.
Some modifications, such as generation of sulfinamide-linked S100A8 complexes with/without S100A9, would be irreversible, and their formation in locations of high oxidative stress generated by HOCl, such as seen in human atheroma (54), may permanently suppress putative proinflammatory functions and negatively affect other functions such as antimicrobial activity.
Levels of S100A8 and S100A9, either as the S100A8/A9 complex, or the individual proteins, are elevated in the circulation of patients with many inflammatory diseases, and in some cases, correlate with levels of the acute-phase reactant, C-reactive protein (48). Increases seen in inflammatory conditions, or in association with some tumors, led to the proposal that these represent alternate inflammatory markers. However, their elevated levels in many disease conditions probably gives them no more predictive power of disease association or progression than does C-reactive protein, which is commonly used. The question concerning the role of these proteins in circulation is interesting. Oxidative stress and impaired antioxidant systems induce vascular dysfunction that contributes to aging, diabetes, and cardiovascular disease and is an important factor in lung disorders. In the latter, the diversity of environmental oxidants and the possibility of disease-specific pathways are likely to contribute. Reduced antioxidant systems in such conditions are reflected in the circulation (12).
Plasma proteins are important in antioxidant defense, and levels of some antioxidant enzymes such as SOD decrease under oxidative stress. Albumin has a key role in maintaining the redox balance in the circulation because of its high concentrations (9). Albumin can be extensively modified by ROS; in particular, the free thiol (Cys34) forms reversible derivatives, sulfenic and sulfinic acid intermediates, and the stable adduct, albumin-sulfonic acid. These are generated in vitro by chloramines or the myeloperoxidase system or both. The sulfenic and sulfinic adducts convert back to the nonoxidized form by changes in redox potential, or by reacting with GSH or homocysteine to form nonmercaptoalbumin, which composes 5% to 15% of serum albumin. Recent proteomic studies with serum from patients with focal segmental glomerulosclerosis confirmed generation of the stable Cys-sulfone adduct (9). Albumin-sulfonic acid does not dimerize but has altered structural properties, is more susceptible to protease digestion than is nonoxidized albumin, and may be most readily removed from plasma by this mechanism. Similar adducts are generated in S100A8 by HOCl, and we showed that S100A8 is several hundred-fold more susceptible to HOCl-mediated oxidation than is albumin (54). Together with the fact that S100A8 is markedly more sensitive to oxidation than is LDL, it is possible that the elevated levels of the S100s in the circulation of individuals with inflammatory disorders may form a component of the antioxidant defense system.
Implications of S-nitrosylation of S100A8
Low levels of NO are generally regarded as protective with antiproliferative, antiatherogenic, and antiinflammatory functions, although excess production can cause tissue injury and exacerbate inflammation. NO modulates numerous important processes including neuronal development, differentiation and signaling, and cellular proliferation and apoptosis, and is particularly important in vascular homeostasis, regulating vasodilation, vascular resistance, and blood flow. NO also reduces leukocyte adhesion to the endothelium, suppresses O2 .- production via NADPH oxidase, and inhibits mast cell (MC) degranulation and MC-mediated leukocyte recruitment (24). S-nitrosylated proteins, including kinases, transcription factors, structural proteins, and metabolic enzymes may regulate some NO-derived effects. Because NO has a relatively short half-life, S-nitrosylation of proteins may stabilize NO to preserve its functions (40); denitrosylation in the presence of ascorbate or GSH, or transnitrosylation via transfer of NO to more susceptible targets may protect critical protein thiols from irreversible oxidation under oxidative stress (40).
S100A8 is more susceptible to S-nitrosylation than S100A9, and higher levels of S100A8-SNO were detected in neutrophils treated with GSNO. Intriguingly, even though LPS or IFN-γ stimulation of murine macrophages induces S100A8, when both agents were combined, no S100A8 was produced, whereas iNOS was markedly elevated. Thus, in circumstances requiring antimicrobial defense mediated by activated macrophages, S100A8 is unlikely to be generated, and thus would not compromise bacterial killing by scavenging the NO generated. Conversely, S100A8 induction by LPS is dependent on IL-10, and S100A8 may scavenge excessive NO under appropriate circumstances. Serum nitrite levels are significantly reduced in LPS-treated rats injected with S100A8/A9 (38).
We propose that S100A8-SNO may regulate particular processes in the microcirculation that are important in restoring blood flow and suppressing inflammation. S100A8-SNO is relatively stable in solution and transnitrosylates hemoglobin as effectively as does GSNO (50), indicating a potential role in regulation of blood-vessel homeostasis. Thus S100A8-SNO may stabilize NO and act as a carrier to preserve its antiinflammatory effects. S100A8-SNO inhibited MC degranulation in vitro, and this would affect vessel permeability by reducing histamine release. Moreover, MC-mediated leukocyte adhesion and extravasation in the rat mesenteric microcirculation was almost totally suppressed, in a manner similar to that displayed by NO donors such as GSNO (50).
Because S100A8 is induced in microvascular EC, but not larger-vessel endothelium, by proinflammatory stimulants (90) or is deposited on the endothelial cell surface by transmigrating leukocytes, or both, it could be S-nitrosylated in these cells by NO generated by eNOS. MCs are critically located around blood vessels, and generation of S100A8-SNO would likely halt their activation and suppress further leukocyte recruitment. NO downregulates intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and P-selectin expression on endothelial cells (24), and it is tempting to speculate that S100A8-SNO would have similar antiinflammatory effects. Our preliminary experiments indicate that S100A8-SNO reduced P-selectin expression provoked by activated MC (unpublished observation).
S-glutathionylation of S100A8 and S100A9
S-glutathionylation can persist during oxidative/nitrosative stress but is also apparent, albeit to a lesser extent, under basal conditions. Glutathionylation of several proteins including transcription and translation factors (Jun, NF-κB), enzymes [creatine kinase, HIV-1 protease, glyceraldehydes-3-phosphate dehydrogenase (GAPDH), cytoskeletal proteins (actin, tubulin), and heat-shock proteins (HSP27)] affects numerous important physiologic processes. Altered levels of some glutathionylated proteins are associated with pathologies such as hyperlipidemia, diabetes, Friedreich ataxia, atherosclerosis, and cancer, and some adducts are suggested markers of oxidative stress [reviewed in (25)].
Although S100A8 and S100A9 are both susceptible to glutathionylation, only S100A9-SSG was isolated from neutrophils activated with PMA and ionomycin. Glutathionylation induced structural changes that increased surface hydrophobicity on Zn2+ binding and reduced the affinity of S100A9 for non-covalent complex formation with S100A8, although it did not affect the ability of the S100A8/S100A9 heterocomplex to bind AA. Glutathionylation also decreased the capacity of S100A9 to bind HMEC-1 cells, although we found no effects of S100A9 or S100A9-SSG on ICAM-1, VCAM-1, and IL-8 induction in these cells (51). However, S100A8/A9 downregulates endothelial cell junction proteins α-catenin and VE-cadherin (83), and effects on monolayer integrity are worthy of investigation. S100A9 has high affinity for heparin/heparan sulfate, which is proposed to mediate the interaction between S100A9 and EC surfaces. S100A9 also binds fibronectin, another ECM component, and its binding to ECM components in atherosclerotic lesions is evident by immunohistochemical staining (54). In the presence of S100A8, S100A9-SSG almost totally suppressed neutrophil adhesion to fibronectin, indicating that it may modulate transmigration and accumulation of neutrophils in the extravasculature. Interestingly, S-glutathionylation did not alter the ability of S100A9 to increase neutrophil adhesion to fibronectin, suggesting an important function of the modified complex (51). Taken together, modifications of S100A8 and S100A9 to their S-nitrosylated or S-glutathionylated forms, which are generated as a result of activation of leukocytes or microvascular ECs or both, may mediate leukocyte–endothelial cell interactions in the vasculature to decrease inflammation and facilitate resolution.
Concluding Remarks
Oxidative injury during inflammation is attributed in part to the direct attack of excess ROS/RNS, produced by activated phagocytes, on proteins, DNA, and lipid membranes, and this can propagate inflammation, ultimately resulting in tissue damage. Most DAMPs, including S100A8 and S100A9, have proinflammatory functions such as recruitment of leukocytes, promotion of cytokine and chemokine production, and regulation of leukocyte adhesion and migration, as shown in Fig. 5. These effects may depend on their relative concentrations in the extracellular environment. A critical function of DAMPs is to restore cellular homeostasis, in part by promoting tissue reconstruction (10). Hence, activities of most DAMPs must be regulated to prevent excessive inflammation and tissue damage, and subsequently, to initiate tissue repair. Here, we propose that oxidation of some DAMPs may represent a regulatory switch to suppress inflammation and restore homeostasis.
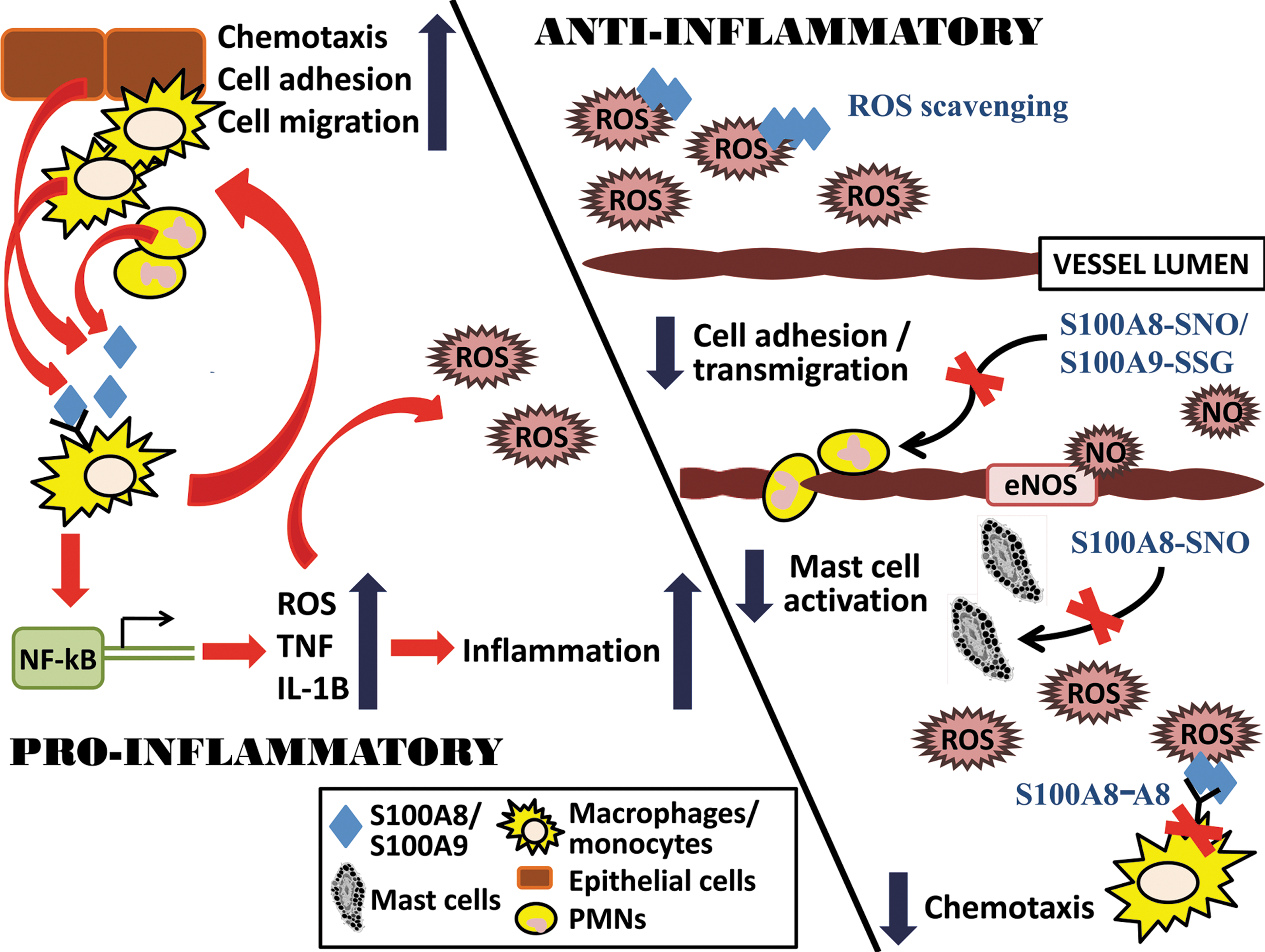
In conditions in which high levels of ROS and RNS are produced, such as during infection or inflammation, specific oxidative modifications of S100A8 or S100A9 or both arrest their chemotactic properties (29, 65), which could limit leukocyte recruitment. S100A8 and S100A9 effectively scavenge oxidants, and high extracellular levels may represent a compensatory mechanism to increase antioxidant defense and mop up excess ROS/RNS at inflammatory sites to promote tissue repair. Importantly, reversible modifications such as S-nitrosylation and S-glutathionylation of S100A8 or S100A9 or both in vitro trigger novel antiinflammatory activities that may affect events in the microcirculation. However, some modifications, including oxidation of Met residues to Met sulfones and Cys-Lys sulfinamide formation are resistant to reduction by oxidoreductases, and excessive accumulation of these, such as the S100A8 or S100A9 complexes or both found in human atheroma (54) and in brain extracts from patients with Alzheimer disease (75), may contribute to the pathogenesis of chronic inflammatory conditions.
Footnotes
Acknowledgments
Research contributing to this article was funded by the National Health and Medical Research Council of Australia (Project grants 455306, 455307). The authors acknowledge Dr. Jesse Goyette for his assistance with structural analysis and Lincoln Gomes for sharing some unpublished data.