
Abstract
Mammalian aging is associated with elevated levels of oxidative damage of DNA, proteins, and lipids as a result of unbalanced prooxidant and antioxidant activities. Accumulating evidence indicates that oxidative stress is a major physiological inducer of aging. p53, the guardian of the genome that is important for cellular responses to oxidative stresses, might be a key coordinator of oxidative stress and aging. In response to low levels of oxidative stresses, p53 exhibits antioxidant activities to eliminate oxidative stress and ensure cell survival; in response to high levels of oxidative stresses, p53 exhibits prooxidative activities that further increase the levels of stresses, leading to cell death. p53 accomplishes these context-dependent roles by regulating the expression of a panel of genes involved in cellular responses to oxidative stresses and by modulating other pathways important for oxidative stress responses. The mechanism that switches p53 function from antioxidant to prooxidant remains unclear, but could account for the findings that increased p53 activities have been linked to both accelerated aging and increased life span in mice. Therefore, a balance of p53 antioxidant and prooxidant activities in response to oxidative stresses could be important for longevity by suppressing the accumulation of oxidative stresses and DNA damage. Antioxid. Redox Signal. 15, 1669–1678.
p53 Is a Critical Tumor Suppressor
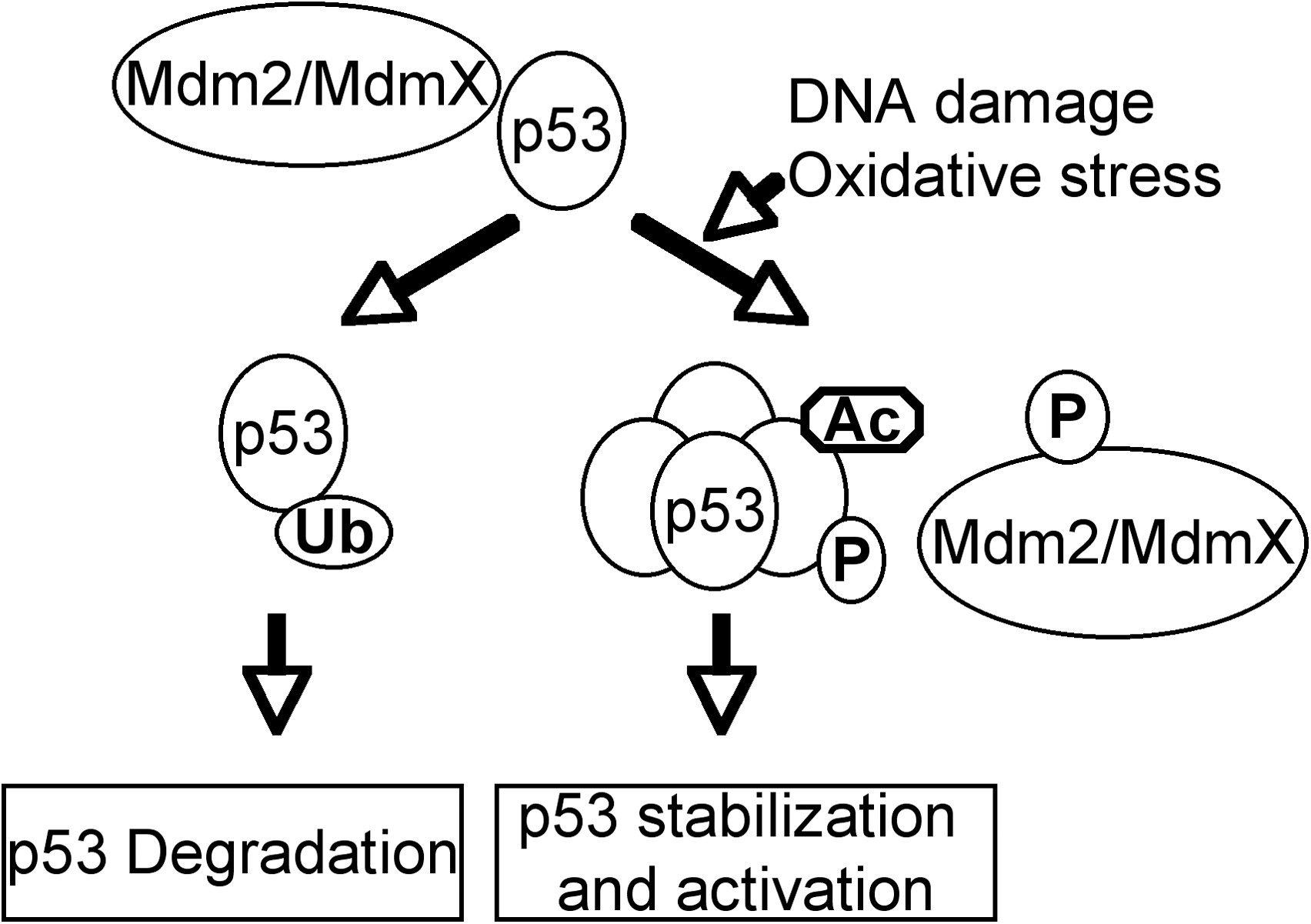
As a transcription factor, p53 can directly regulate the expression of hundreds of genes, products of which mediate various p53-dependent functions (Fig. 2) (43, 53, 69). For example, p21 and 14-3-3σ are responsible for p53-dependent cell-cycle arrest (30, 31, 50); p53 can also induce embryonic stem (ES) cell differentiation by suppressing the expression of Nanog, which is required for the self-renewal of ES cells (64). In response to high levels of DNA damage, p53 induces apoptosis and senescence by upregulating apoptotic genes such as Noxa and Puma (66, 71). These functions of p53 prevent the passage of DNA damage to the daughter cells and thus maintain genomic stability. In response o oxidative stresses, p53 activates the transcription of a number of genes involved in regulating oxidative stress, such as Sestrin, glutathione peroxidase (GPX), aldehyde dehydrogenase (ALDH), and tumor protein 53–induced nuclear protein 1(TP53INP1) (14, 16, 115, 130). p53 can also regulate the cellular oxidative stress levels by modulating glycolysis through inducing the expression of TIGAR (TP53-induced glycolysis and apoptosis regulator) and suppressing the expression of phosphoglycerate mutase (PGM) (9, 58).

p53 and Aging
Recent studies have functionally linked p53 to aging in various organisms (Fig. 3). The p53 orthologue in Caenorhabditis elegans, Cep-1, is involved in negatively regulating the life span of the worm, because the reduced expression of Cep-1 results in increased longevity (4). Expression of dominant-negative versions of Drosophila melanogaster p53 (Dmp53) in adult neurons extends the life span and increases the genotoxic stress resistance in the fly (8). Because the expression of the dominant-negative Dmp53 does not further increase the life span of flies that are calorie restricted, these findings suggest that p53 is involved in mediating the calorie-restricted life span in flies. However, mutagenesis studies in C. elegans show that certain mutations extending the life span increase activities of p53 and cancer resistance (94). Therefore, increased p53 activities are associated with both accelerated aging and increased life span in C. elegans.
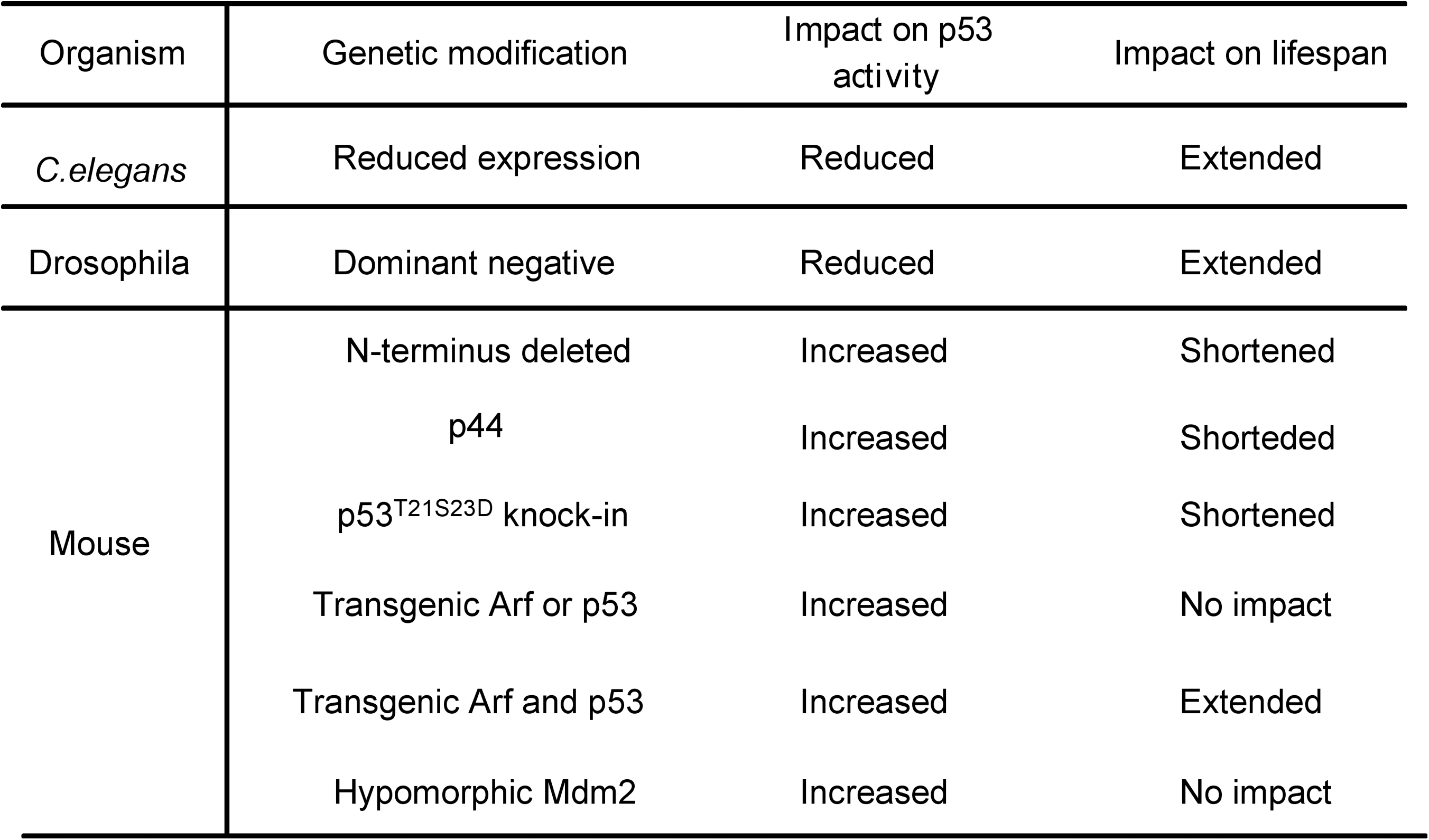
A similarly complicated scenario is also observed when studying the roles of p53 in mammalian aging. One mouse model, in which the N-terminus of p53 is truncated, exhibits increased p53 activities and accelerated aging (119). However, because of the large deletion of the genomic DNA upstream of p53 that contains 24 genes (40), it remains unclear whether any of these deleted genes is responsible for these aging phenotypes. The potential involvement of N-terminus–truncated p53 in aging is further supported by the overexpression of the N-terminus–deleted p53 isoform p44 in mice, leading to accelerated aging (72). This study suggests that p44 modulates the life span by inhibiting the PTEN and IGF signal pathways (39, 75, 110). To link p53 to aging in humans, a recent study shows that polymorphism of p53 at codon 72 (arginine-to-proline substitution) reduces p53 activities, correlating with increased life span but also with higher cancer risk in older individuals (120). Therefore, it has been suggested that p53 might suppress cancer at the cost of longevity.
The notion that increased p53 activity induces aging in mice is challenged by recent studies of mouse models with increased p53 activities. For example, mice with a hypomorphic mutation in Mdm2 exhibit increased p53 activity but normal life span (78). In addition, mice with an additional copy of p53 and ARF exhibit an enhanced expression of antioxidant activity and decreased levels of endogenous oxidative stresses, correlating with increased life span (74). Therefore, the increased antioxidant activity of p53 in these transgenic mice prevents the accumulation of oxidative stresses to the high levels required to induce p53-dependent apoptosis and senescence, thus delaying aging in these mice. In summary, the functions of p53 in aging are complex and could be context dependent. In this context, mild and transient activation of p53 in response to a low dosage of oxidative stress could protect cells from oxidative damage. In contrast, persistent activation of p53 in response to high levels of oxidative stresses can result in cell death and organismal aging. In further support of this notion, persistent activation of p53 depletes adult stem cells primarily through p53-dependent apoptosis (64).
Oxidative Stress and Aging
The free radical hypothesis remains the most well-established theory on the mechanism of aging (46). The increased ROS production and a decreased antioxidant capacity are thought to contribute to the aging process by oxidative modification of different macromolecules, such as lipids, proteins, and genomic DNA (12, 20, 25, 62, 63, 65, 96, 109, 117). In the context of DNA, oxidative damage to mitochondrial and nuclear DNA is significantly increased in different tissues in old rats and mice (20, 45, 61, 67, 76, 82, 116). Levels of lipid-peroxidation products are also increased with aging (44, 83, 87, 97, 108, 113, 119, 123). In addition, aging-related oxidative modification of different proteins causes changes in protein structure, enzyme activities, transcriptional activities, and signal-transduction pathways (32, 70, 103, 111, 112, 124), leading to age-related diseases. In summary, the levels of oxidative damage are increased during aging in various organisms, including C. elegans (11, 52, 121), flies (3, 64), and mice (22, 74, 79).
Free radicals are physiologic byproducts of metabolism and are rapidly eliminated by various antioxidant enzymes in cells (23). For example, the antioxidant enzymes, including superoxide dismutase (SOD), catalase, and peroxiredoxins, convert superoxide to hydrogen peroxide and eventually to water (5, 19, 99). SODs catalyze the breakdown of the superoxide anion into oxygen and hydrogen peroxide. Mice lacking SOD2 develop neurologic defects and die soon after birth because of excessive mitochondrial production of ROS (77); mice lacking SOD1 are viable but have numerous pathologies and a reduced life span (98). Catalase converts hydrogen peroxide into water and oxygen (19, 132). Humans and mice deficient in catalase can still efficiently remove H2O2, implying that other enzymes are also involved in this reaction (72, 88). Peroxiredoxins catalyze the reduction of hydrogen peroxide, organic peroxide, and peroxynitrite (99). These enzymes can be divided into there classes: typical 2-cysteine peroxiredoxins, atypical 2-cysteine peroxiredoxins, and 1-cysteine peroxiredoxins (128). Mice lacking peroxiredoxins 1 and 2 have a shortened life span (55, 86). Together, these findings underscore the importance of antioxidant enzymes in preventing aging processes. In further support of this notion, a diet rich in the building-block nutrients of antioxidant enzymes, including cofactors for SOD (manganese, zinc, and copper), show beneficial effects on delaying aging (1, 24, 49, 59, 81, 106).
In further support of the notion that oxidative stress is an inducer of aging, treatment with antioxidants can increase the life spans of various organisms and has a beneficial impact on aging-related diseases (6, 29, 38, 57, 114, 119). A low dose of dietary supplement with antioxidants partially mimics the effects of caloric restriction and delays aging in mice (6), and long-term treatment with free radical scavenging Schisandrin B, a dibenzocyclooctadiene derivative isolated from the fruit of Schisandra chinensis, delays aging-related functional impairment in various organs and improves the survival rate of aging mice (114). A dietary supplement of cysteine, which is required for the synthesis of the primary antioxidant glutathione, has clear benefits in delaying some aspects of aging (29). However, clinical trials have also found no significant beneficial effects of supplementation with antioxidant vitamin E, indicating that not all antioxidants have antiaging activities (55, 107, 125).
p53 and Oxidative Stress
ROS levels have a significant impact on cell growth, survival and development, and tumorigenesis (17). p53 plays key and complex roles in cellular responses to oxidative stresses (84, 100). In response to low levels of oxidative stresses, p53 plays primarily antioxidant roles. In this context, a number of p53 target genes, including Sestrin, glutathione peroxidase (GPX), and aldehyde dehydrogenase (ALDH), are involved in reducing oxidative stresses (Fig. 4). For example, Sestrin protects the cells from hydrogen peroxide–induced damage by generation of peroxiredoxins (14). GPX is a primary antioxidant enzyme that scavenges hydrogen peroxide or organic hydroperoxides (115). Aldehyde dehydrogenase (ALDH) also contributes to the antioxidant function of p53 (130).
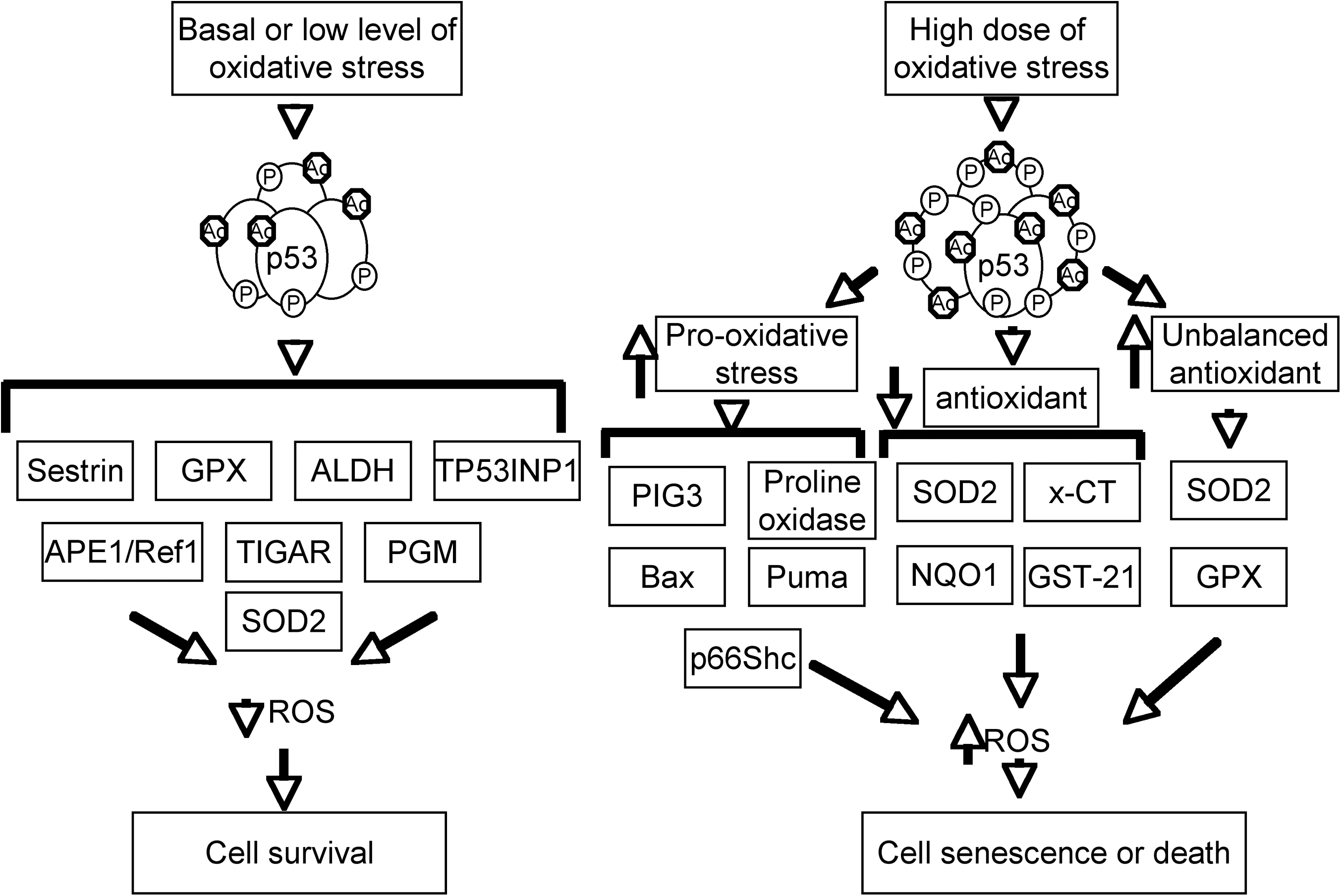
p53 can also reduce the intracellular levels of ROS by regulating cellular metabolism. In this context, p53 induces the expression of TIGAR (TP53-induced glycolysis and apoptosis regulator), which slows glycolysis and promotes the production of NAPDH to decrease ROS levels (9). In addition, p53 suppresses the expression of phosphoglycerate mutase (PGM), leading to a decrease of pyruvate required for oxidative respiration in mitochondria and thus reduced ROS production (10, 74).
In response to high levels of oxidative stress, p53 exhibits prooxidative activities by turning on prooxidative genes such as PIG3 and proline oxidase (27, 95). Overexpression of these genes leads to higher levels of oxidative stress. In addition, p53 induces the expression of BAX and PUMA, which induce apoptosis through the release of cytochrome c from mitochondria (66, 71). The prooxidative activities of p53 also include the inhibition of the expression of antioxidant genes, leading to increased cellular oxidative stresses to induce apoptosis. For example, p53 could repress the expression of SOD2 and Nrf2, resulting in sensitivity to oxidative stress or inducing apoptosis (28, 34, 91). Interestingly, p53-induced upregulation of MnSOD and GPX, but not catalase, increases oxidative stress and apoptosis (54), suggesting that the balance of antioxidant enzyme and oxidative stress is important for cell survival. In summary, p53 plays important but context-dependent roles in regulating cellular oxidative stresses, and the levels of oxidative-stress damage dictate whether the p53 behavior is that of a protector or a killer (100).
p53 Interacts with Other Pathways Involved in Oxidative Stress and Aging
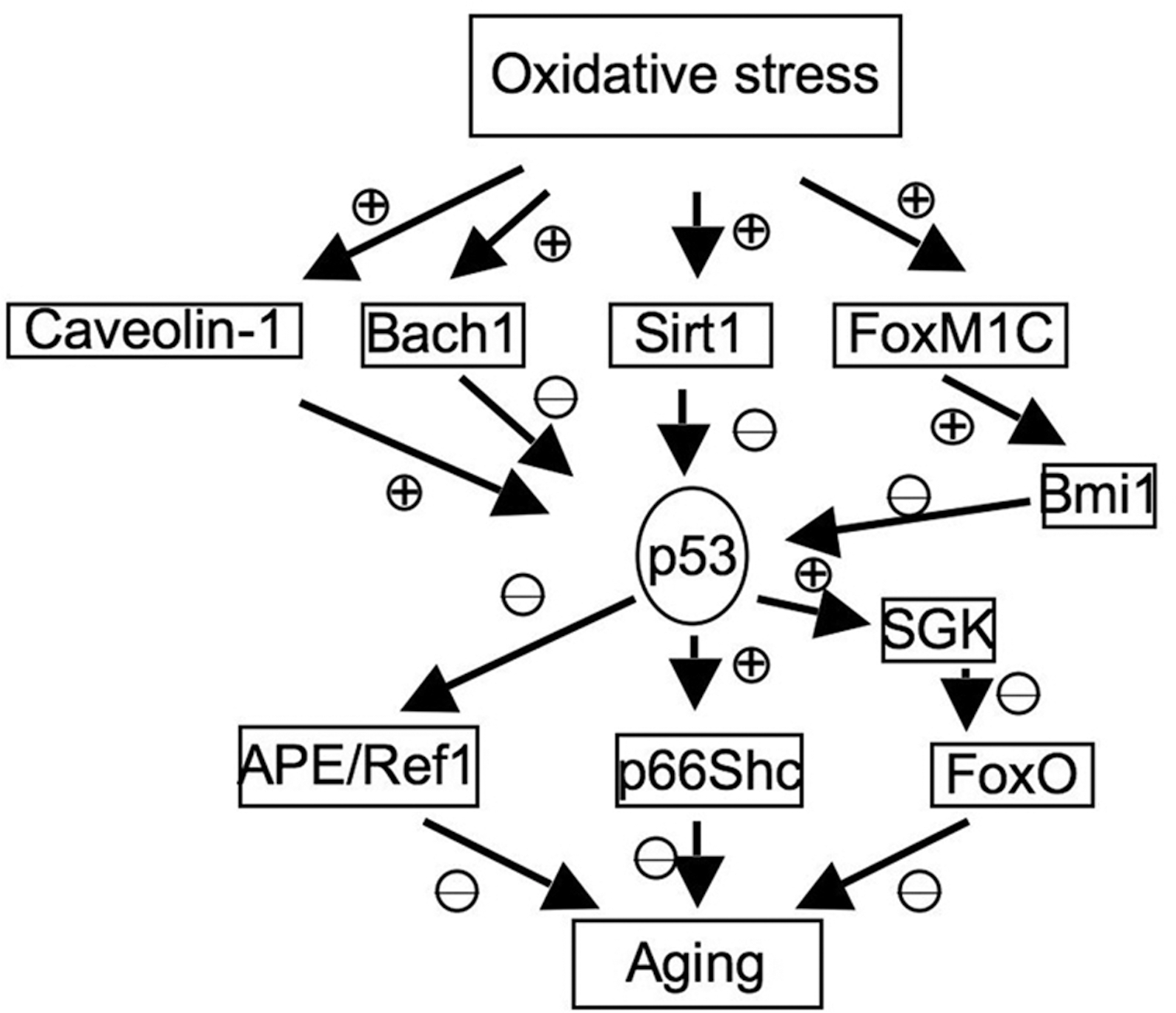
In addition to its direct regulation of genes involved in oxidative stresses, p53 also interact with other pathways that are involved in aging and oxidative stresses, which are summarized here (Fig. 5).
Sirt1
The Sirt1 gene encodes the NAD-dependent histone deacetylase, which is important for the longevity in yeast and mammalian species by calorie restriction (42, 60, 64, 122). Sirt1 can deacetylate and inactivate p53, leading to impaired cell growth arrest and apoptosis in response to oxidative stresses (101). In addition, the expression of a dominant-negative version of Sirt1 increases the cellular sensitivity to oxidative stress, further indicating its antioxidant roles in cellular responses to oxidative stresses. However, the roles of Sirt1 in suppressing p53 in response to oxidative stresses remain to be fully established. In contrast to the prediction that Sirt1 deficiency would increase p53 activity, recent studies indicate that deficiency of Sirt1 extends the replicative capacity of mouse embryonic fibroblasts (MEFs) under the conditions of chronic oxidative stress due to the inefficient activation of p53 (21). However, the physiological relevance of replicative senescence in aging is not clear, because it primarily reflects a cell-culture phenomenon in the presence of nonphysiologically high levels of oxygen. Because Sirt1 is an NAD-dependent deacetylase, and NAD levels are regulated by cellular metabolism and levels of ROS, these findings implicate a complex functional interaction of p53, Sirt1, oxidative stresses, and aging.
p66Shc
p66Shc, a downstream target of p53, is indispensable for p53-dependent elevation of intracellular oxidative stresses and apoptosis (118). p66Shc is a splice variant of p52Shc/p46Shc, a cytoplasmic signal transducer involved in the transmission of mitogenic signal from activated receptors to Ras (93). However, p66Shc is not involved in regulating Ras signal but instead is involved in inducing apoptosis in response to oxidative stresses (80). The important role of p66Shc in oxidative stresses and aging is indicated by the findings that ablation of p66Shc enhances cellular resistance to apoptosis induced by oxidative stresses and extends the life span of p66Shc-deficient mice (79). In this context, cytochrome c release after oxidative signals is impaired in p66Shc-deficient cells (90). Therefore, p66Shc functionally links p53 to oxidative stress response and aging.
FoxO
Forkhead box O (FoxO) transcription factors are important mediators of the PI3K/Akt signaling pathway and regulate the cellular responses to oxidative stresses and the life span (56, 105). p53 negatively regulates the activities of FoxO by inducing the expression of serum- and glucocorticoid-inducible kinase (SGK), a negative regulator of FoxO and PTEN (37). In addition, Sirt1 can deacetylate FoxO3 and FoxO4, thus attenuating FoxO-induced apoptosis and cell-cycle arrest (41). Therefore, the balance of the functional interaction among Sirt1, FoxO, and p53 might play important roles in regulating oxidative stresses and aging.
APE/Ref1
The expression of APE/Ref1 is decreased in senescent human bone marrow–derived mesenchymal stem cells (hBMSCs) with increased endogenous ROS levels. Overexpression of APE1/Ref-1 suppresses superoxide production and decreases senescence in hBMSCs (48). In addition, aging mice have an impaired induction of APE in response to oxidative damage (15). The activities of APE/Ref1 are negatively regulated by p53 (131), implicating another pathway for p53 to modulate oxidative stresses and aging.
Caveolin-1
Expression of Caveolin-1 is induced in fibroblasts undergoing oxidative stress–induced senescence, and the antioxidant prevents the senescence and upregulation of Caveolin-1 (36, 126). Overexpression of Caveolin-1 in MEF induces the premature senescence through a p53-p21–dependent pathway, suggesting that Caveolin-1 could activate p53-dependent premature senescence after oxidative stresses (36). In this context, Caveolin-1 binds to Mdm2 and disrupts the binding of Mdm2 to p53, leading to the activation of p53 in response to oxidative stresses. The activation of p53 and induction of premature senescence are compromised in the Caveolin-1–null MEFs, confirming that Caveolin-1 is an upstream activator of p53 in response to oxidative stresses (7).
FoxM1C-Bmi1 pathway
Bmi1 is a negative regulator of the Ink4a/Arf and p53; FoxM1C induces the expression of Bmi1 to prevent the oxidative stress–induced cellular senescence by inhibiting the expression of p53 (13, 18, 33, 89). Bmi1 is important to repress the prooxidant activities of p53 in neurons and to suppress oxidative stress–induced apoptosis and premature aging-like phenotypes (18). In addition, targeted depletion of Bmi1 sensitizes tumor cells to p53-mediated apoptosis in response to radiation therapy (2).
Bach1
For transcription factors, the recruitment of co-activators or co-repressors to p53 target promoters is critical for p53-dependent transcription. Bach1 is induced by oxidative stresses and forms a complex with p53, histone deacetylase 1, and nuclear co-repressor N-coR, promoting histone deacetylation and suppression of certain p53 target genes (26). In this context, Bach1 inhibits oxidative stress–induced cellular senescence by disrupting p53-dependent gene expression (26).
Conclusion
The accumulation of oxidative stress and oxidative damage is a major inducer of aging. Many pathways involved in cellular responses to oxidative stresses regulate the aging process and the life spans of various organisms. p53 plays important but context-dependent roles in cellular responses to low or high levels of oxidative stresses. In response to low levels of oxidative stresses, p53 exhibits antioxidant activities and promotes cellular survival; in response to high levels of oxidative stresses, p53 exhibits prooxidative activities to induce cellular apoptosis. Both functions of p53 can prevent the accumulation of oxidative damage in cells and thus maintain genomic stability. p53 accomplishes these functions by direct transcriptional regulation of genes involved in oxidative-stress responses or modulating other pathways important in oxidative-stress responses.
Consistent with its context-specific roles in oxidative-stress responses, the roles of p53 in aging appear to be complex as well. In this context, increased p53 activities can accelerate aging in some transgenic mouse models but not in others (72, 74, 78, 119). In addition, the increase of the gene dosage of ARF and p53 does not promote aging but increases the life span of transgenic mice (74). Therefore, the roles of p53 in aging could also be context dependent. The accumulation of oxidative stresses in old mice could turn on the apoptotic or senescent roles of p53, thus promoting the aging process. However, increased dosages of p53 and ARF could ensure more efficient elimination of oxidative stress and thus prevent the accumulation of oxidative stresses to high levels in old mice. In support of this notion, a significant reduction of DNA damage occurs in old transgenic mice with additional copies of p53 and ARF (74). p53 primarily plays a protective role to increase the life span in these transgenic mice. Therefore, further elucidation of the mechanism that governs the context-dependent roles of p53 in oxidative-stress responses and the functional outcomes of the interaction between p53 and other pathways involved in cellular responses to oxidative stresses will shed light on its role in aging.
Footnotes
Acknowledgment
This work is supported by an NIH grant to YX (R01 CA94254).