
Abstract
Cellular redox status, regulated by production of reactive oxygen species (ROS), greatly contributes to the regulation of vascular smooth muscle cell contraction, migration, proliferation, and apoptosis by modulating the function of transient receptor potential (TRP) channels in the plasma membrane. ROS functionally interact with the channel protein via oxidizing the redox-sensitive residues, whereas nitric oxide (NO) regulates TRP channel function by cyclic GMP/protein kinase G-dependent and -independent pathways. Based on the structural differences among different TRP isoforms, the effects of ROS and NO are also different. In addition to regulating TRP channels in the plasma membrane, ROS and NO also modulate Ca2+ release channels (e.g., IP3 and ryanodine receptors) on the sarcoplasmic/endoplasmic reticulum membrane. This review aims at briefly describing (a) the role of TRP channels in receptor-operated and store-operated Ca2+ entry, and (b) the role of ROS and redox status in regulating the function and structure of TRP channels. Antioxid. Redox Signal. 15, 1549–1565.
Introduction
Under pathological or pathophysiological conditions, ROS levels are increased and lead to endothelial dysfunction, VSMC proliferation, and increased contractility (86). These changes may be involved in the pathogenic mechanisms of various cardiovascular diseases (e.g., systemic and pulmonary arterial hypertension). ROS-induced vascular changes occur though redox-sensitive signaling pathways involving mitogen-activated protein kinases, tyrosine kinases, various transcription factors (e.g., NF-κB, AP-1, HIF-1α), and many different steps of the Ca2+ signaling cascade (9, 68). Although these are all important signaling mechanisms, for the purposes of this review, we will focus on the role of ROS in regulating Ca2+ signaling particularly the effect of ROS on the function of transient receptor potential (TRP) channels. ROS and Ca2+ signaling are both relevant to the physiology and pathophysiology of the vasculature, particularly in the function of vascular smooth muscle and endothelial cells. However, we will focus on discussing the effect ROS and redox status on regulating TRPC channels and Ca2+ signaling in general rather than on the signaling mechanisms in a specific cell type.
Recent work by many investigators has elucidated much on the effect of redox state on Ca2+ signaling. There likely exists an overall trend where increased ROS inhibits Ca2+ pumps and opens Ca2+-permeable channels in the SR/ER membrane and plasma membrane, respectively. Inhibition of Ca2+ pumps in the plasma membrane attenuates the extrusion of Ca2+ out of the cytoplasm, whereas opening of Ca2+-permeable channels on the SR/ER and plasma membranes allows Ca2+ to flow down its concentration gradient and into the cytoplasm and increase cytosolic Ca2+ concentration ([Ca2+]cyt). Therefore, the net effect of ROS has been demonstrated to increase [Ca2+]cyt.
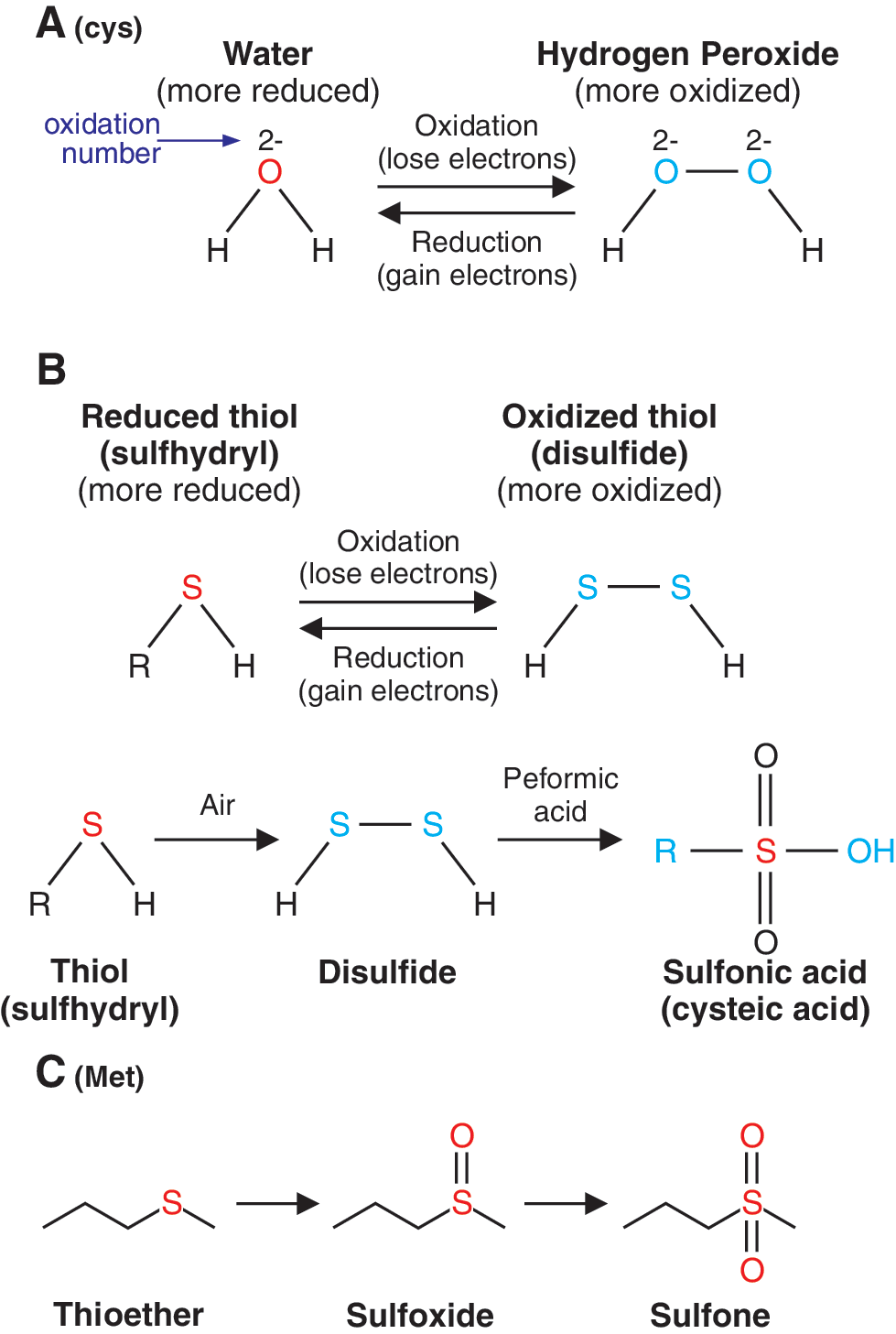
ROS Chemistry
Oxygen (O2) is essential for generating the energy needed to live by reducing molecular oxygen to water. During this process, oxygen undergoes a series of one-electron reductions in the mitochondrial electron transport chain to produce superoxide radical (
The superoxide radical (
Hydrogen peroxide (H2O2) is not a radical because it has no unpaired electrons. Hydrogen peroxide, which is relatively stable and can easily pass through cell membranes, is produced from the superoxide radical by superoxide dismutase from microsomes, mitochondria, and phagocytic cells:
Most of the effects or damage produced by superoxide and hydrogen peroxide in vitro is due to the production of hydroxyl radicals via the metal catalyzed Haber-Weiss reaction or the superoxide radical-driven Fenton reaction:
In addition, the superoxide can react with nitric oxide (NO) to produce peroxynitrite (ONOO−):
The products (
All these highly reactive molecules (
Production of ROS in the Vasculature
In the vascular cells (including adventitial, smooth muscle, and endothelial cells), ROS are produced primarily via an NADPH oxidase (NOX) and the mitochondrial electron transport chain (Fig. 2A) (11). Other mechanisms, including xanthine oxidase, cytochrome P-450, cyclooxygenase, and NO synthase (NOS), also contribute to ROS level (8, 21). NADPH catalyzes superoxide (
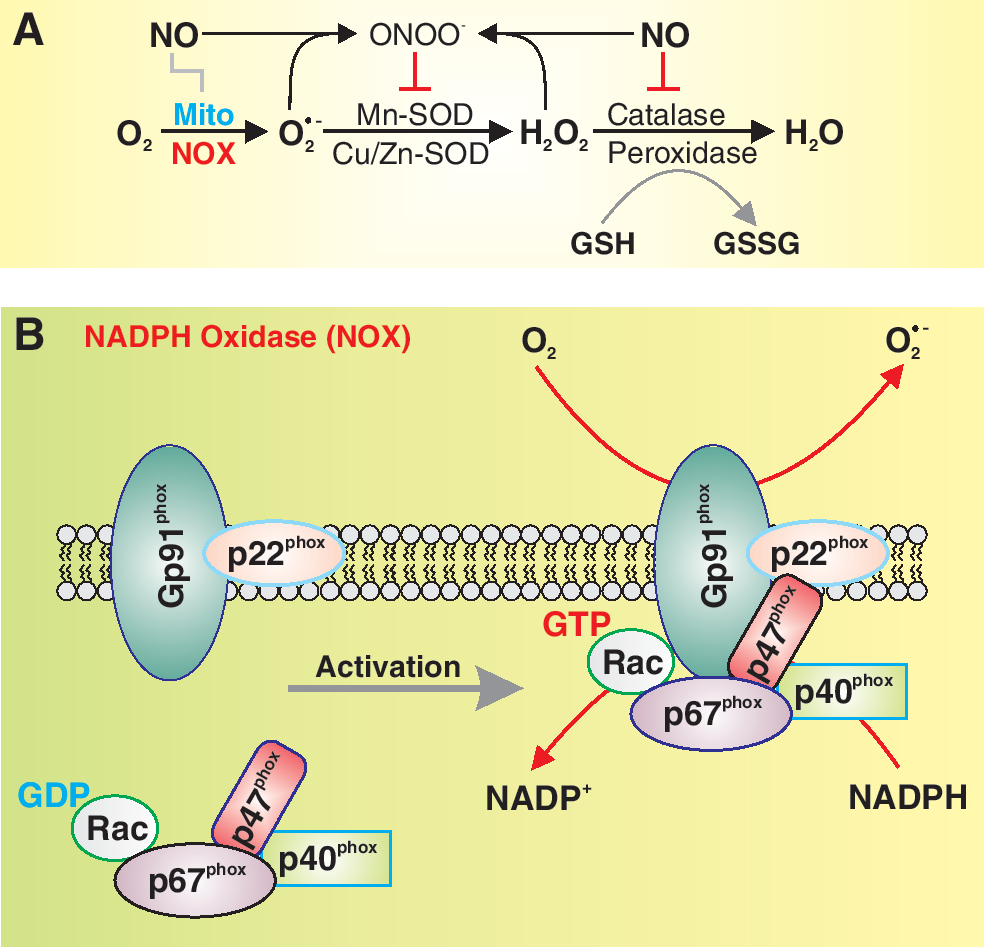
The NOX family is composed of NOX1-5 and Duox1-2. All five NOX isoforms are predominantly localized on the plasma membrane. NOX2 produces large amounts of ROS in phagocytes in a process termed “respiratory burst” for the purpose of mediating host defense against invading microorganisms. NOX 1, 2, 4, and 5 are important in the physiology and pathophysiology of the cardiovascular, pulmonary, and renal system. NOX3 is involved in the function of the vestibular system. NOX5 is expressed in spleen, testis, and vascular tissues. NOX5 has been found in both the smooth muscle and the endothelial cells of the vasculature (12, 26, 89). Interestingly, coronary artery disease has been correlated with the increased expression of NOX5 in vascular cells. The vascular NOX, which is constitutively active and a major source of vascular superoxide production, is comprised of two cytochrome b558 subunits, p22 phox and gp91 phox (Fig. 2B), which have been demonstrated to be important for electron transport and the reduction of molecular oxygen to superoxide. Interestingly, the voltage-gated K+ (Kv) channel β subunit, a cytoplasmic regulatory subunit that interacts with the pore-forming α subunit to form functional Kv channels, has >60% homology to the NOX, implying that (a) the Kv channel β subunits may have NOX activity and/or (b) the vascular NOX can function as a regulatory β subunit for certain Kv channels (34, 35).
Regulation of NOX
In the vasculature, NOX is regulated by chemical means such as cytokines, growth factors, and vasoactive agents as well as physical means such as stretch, strain, pressure, and shear stress. These different factors tightly control the level of ROS at a low level such that it can function as a signaling molecule to regulate endothelial function and vasorelaxation and vasocontraction. Various vasoconstrictors and mitogens such as angiotensin II, serotonin, endothelin-1, platelet-derived growth factor, and tumor necrosis factor-α stimulate the production of superoxide (27, 48, 80).
Not only do ROS regulate Ca2+ signaling mechanisms, but Ca2+ can also regulate the activity of NOX. NOX5, for instance, is distinguished from NOX1-4 by four Ca2+ binding EF hand domains on an extended intracellular N-terminal domain. It has been shown that elevation of [Ca2+]cyt promotes the binding of Ca2+ to the NOX5 EF hand domains, changing the conformation of NOX5 and enabling the protein to generate superoxide (27). That is, a rise in [Ca2+]cyt is an important trigger for production of ROS. This positive “chain reaction” circle of ROS and [Ca2+]cyt increase in VSMC may lead to sustained vasoconstriction and excessive cell proliferation and migration via Ca2+-sensitive signaling cascades (e.g., MLCK, CaMK, and MAPK) and transcription factors (e.g., AP-1, NF-κB, CREB, and NFAT).
Regulation of Redox Status by Thioredoxin and Glutathione Systems
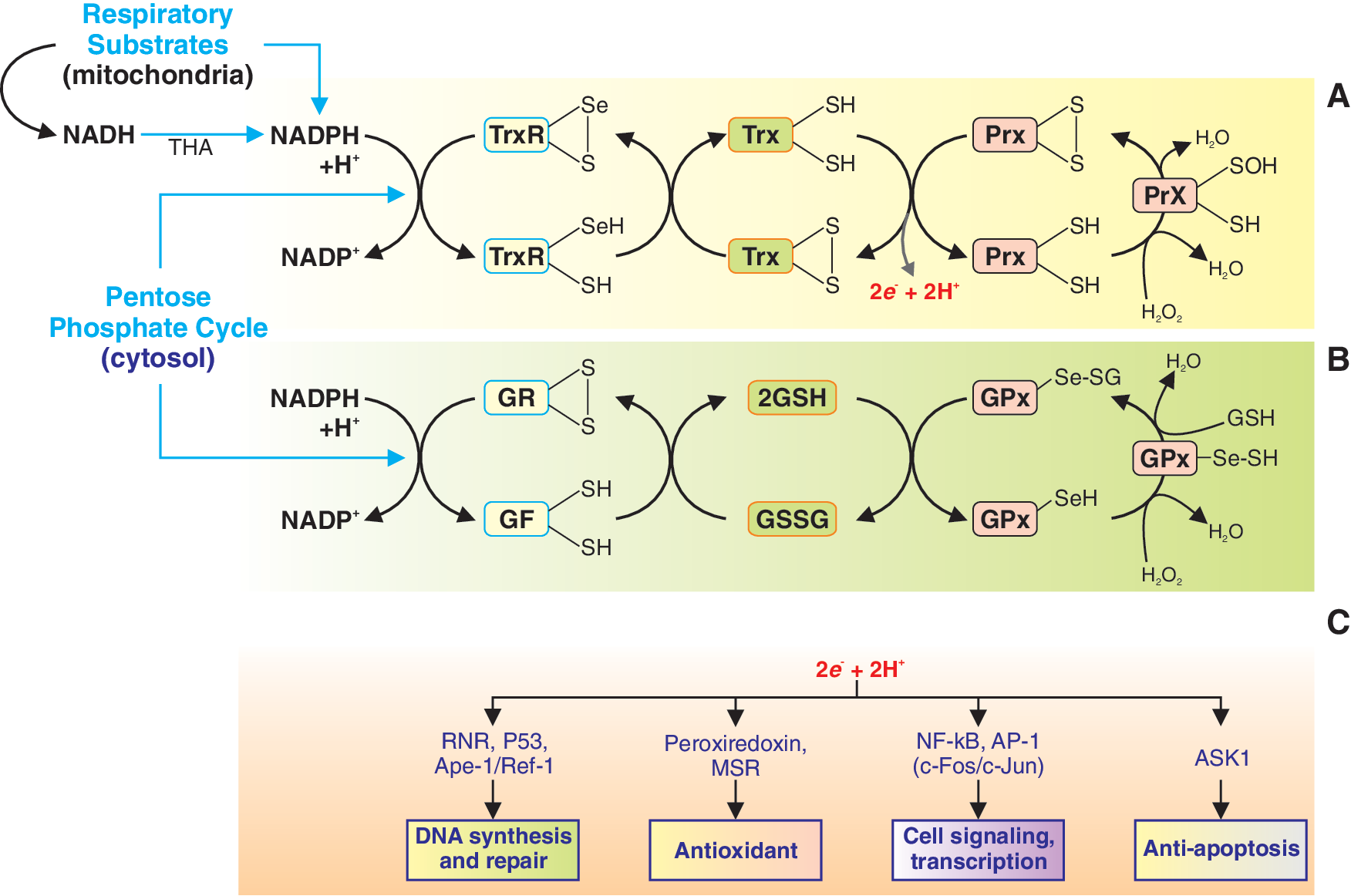
Redox status in cells is not solely determined by the concentration of ROS. The major determinants of redox status are the thioredoxin (Trx) and glutathione (GSH) systems (41). These two regulatory systems of redox status are the primary control mechanism of the status of thiols in the cell and are therefore very important in the regulation of cellular functions. These two systems are present in the cytosol and mitochondria, which are the main sources of ROS in the cell. In the cytosol, NADP+ is reduced in the pentose phosphate pathway to NADPH, and in mitochondria NADPH is generated through oxidative phosphorylation. NADPH functions as a cofactor in the Trx and GSH systems. The Trx and GSH systems are regulated by hydrogen peroxide through peroxiredoxins (Prx) and GSH peroxidases, respectively (Fig. 3). Under hypoxic or hyperoxic conditions, redox status may change in different directions in systemic and pulmonary vascular smooth muscle and endothelial cells.
Trx System
Trx is ubiquitously found in organisms from prokaryotes to mammals. They act as an antioxidant by reducing disulfide bonds to thiols (4, 51, 69). Trx is a 12-kDa enzyme that itself contains a active site composed of two cysteines that are capable of redox chemistry and are integral in the enzymes ability to reduce other proteins such as ribonuclease, choriogonadotropins, coagulation factors, glucocorticoid receptor, and insulin (50, 55). Thioredoxin reductase (TrxR) regulates the redox status of Trx in a NADPH/NADP+-dependent manner. The active site of mammalian TrxR includes a selenocysteine (-Cys-SeCys-), which participates in redox chemistry. TrxR, which include TrxR1 (cytosol and nucleus) and TrxR2 (mitochondria), catalyze the oxidation–reduction of Trx. TrxR's own redox state is mediated by the interconversion of NADPH and NADP+. TrxR act as electron donors to a number of enzymes, including Prx. Prx can act as an antioxidant to catalyze the conversion of hydrogen peroxide to water. Trx, TrxR, and Prx form a system of enzymes that regulate the redox state in cells (Fig. 3A) and regulate cell proliferation and apoptosis, as well as intracellular signaling cascades by modulating various signal transduction proteins, transcription factors, and intracellular enzymes (Fig. 3C).
Glutatione System
Glutathione is the main nonprotein thiol in cells and exists at a high concentration of up to 5 mM (19). Glutathione is a tripeptide with a reactive thiol group that allows it to exist as reduced glutathione (GSH) and oxidized glutathione (GSSG) states. In the reduced state, GSH is able to convert a disulfide bond to two thiols. In addition, after donating an electron, GSH readily reacts with other reactive GSHs to from a disulfide, which is the reduced form of glutathione. The enzyme GSH reductase regulates the redox status of GSH analogous to how TrxR regulates Trx. GSH can transfer its reducing equivalents to GSH peroxidases, which catalyze the conversion of hydrogen peroxide to water. An increased GSSG/GSH ratio is considered indicative of oxidative stress (Fig. 3B) (9). The changes in redox status mediated by GSH system also contribute to regulating DNA synthesis and repair, cell signaling, cell proliferation, and apoptosis (Fig. 3C).
Importance of Intracellular Ca2+
The presence and regulation of the divalent cation calcium (Ca2+) is fundamentally important to the functions of that constitute life at the cellular level. Much energy is expended in extruding Ca2+ from the cytoplasm and sequestering Ca2+ into the sarcoplasmic (or endoplasmic) reticulum, mitochondria, and extracellular space (to maintain a low concentration of Ca2+ in the cytosol). Ca2+ gradients across the plasma membranes allow for the function of metabolism, nerve propagation, muscle contraction, and other important processes. Ca2+ is also fundamentally important in the regulation of transcription, translation, cellular division, and apoptosis (23, 28). It is then unsurprising that the regulation of intracellular Ca2+ levels in the cytosol and organelles is fundamentally involved in many forms of pathophysiology, including diseases involving the vasculature (23, 72, 76).
The concentration of Ca2+ in different intracellular compartments and the flux of Ca2+ between compartments are of great importance in the function of smooth muscle cells and endothelial cells. Since smooth muscle and endothelial cells are the principle cells in determining vascular tone and blood vessel diameter, the regulation of cytosolic Ca2+ is of great importance to the physiological function of the vasculature. Abnormalities in intracellular Ca2+ regulation and chronic elevation of resting [Ca2+]cyt have been linked to pulmonary and systemic arterial hypertension. Therefore, understanding the function and regulation of the ion channels and pumps, which mediate Ca2+ transportation across the plasma membrane and intracellular organelle membranes, is of critical importance.
Intracellular Ca2+ Homeostasis
The lipid membrane is impermeable to Ca2+. Transportation of Ca2+ across the plasma membrane (and the SR/ER membrane) takes place through various Ca2+ channels, pumps, and exchangers. The Ca2+-permeable channels allow Ca2+ to flux through the membrane based on its electrochemical gradient, the Ca2+ pumps (e.g., Ca2+/Mg2+ ATPase or Ca2+ pump) transport Ca2+ against its concentration gradient using energy produced by ATP, and the Ca2+ exchangers (e.g., Na+/Ca2+ exchanger, NCX) can transport Ca2+ across the membrane using the transmembrane electrochemical gradients of other ions (e.g., the transmembrane Na+ gradient for NCX) (Fig. 4).

The extracellular Ca2+ concentration is 1.6–1.8 mM, whereas the cytosolic Ca2+ concentration ([Ca2+]cyt) is ∼100 nM. Various Ca2+ transporters are functionally involved in maintaining the concentration gradient of Ca2+ across membranes and the low resting [Ca2+]cyt (29). The Ca2+ concentration in the intracellular stores, such as SR/ER, can be as high as 1 mM because of the active Ca2+ pumps on the SR/ER membrane (i.e., sarcoplamic endoplasmic reticulum Ca2+ ATPase [SERCA]), which uses the energy produced by ATP to constantly transport (or uptake) Ca2+ from the cytosol to the SR/ER (Fig. 4). In addition, intracellular Ca2+ level is also regulated by many other Ca2+ transporters, such as Na+/Ca2+ exchangers (NCX) and H+/Ca2+ exchangers in the plasma membrane and SR/ER membrane, and Ca2+ uniporters on the mitochondrial membrane (29).
[Ca2+]cyt can be increased by Ca2+ release from the SR/ER through inositol-1,4,5 phosphate receptors (IP3R) and ryanodine receptors (RyR) on the SE/ER membrane, and Ca2+ influx through different Ca2+ channels on the plasma membrane. There are three major Ca2+-permeable channels in the plasma membrane that are functionally expressed in VSMC: (a) voltage-dependent Ca2+ channels (VDCC) that are opened by membrane depolarization, (b) receptor-operated Ca2+ channels (ROC) that are opened by diacylglycerol (DAG) synthesized via PIP2 when membrane receptors are activated by ligands, and (c) store-operated Ca2+ channels (SOC) that are opened when Ca2+ in the intracellular Ca2+ stores (e.g., SR/ER) is depleted by, for example, activation of IP3R/RyR and inhibition of SERCA on the SR/ER membrane (Fig. 4).
While the concentration of Ca2+ and proportion of Ca2+ bound to calmodulin (CaM) in the nucleus are important in regulating various transcription factors involved in VSMC physiology and pathophysiology, the concentration of Ca2+ in the nuclear compartment is unregulated and roughly equal to the cytosolic compartment due to free flow though large nuclear pores. By sequestering Ca2+ into the SR/ER and, to a lesser extent, into mitochondria, and by extruding Ca2+ from the cytosol to the extracellular space, the concentration of Ca2+ in the cytoplasm is maintained at a low level such that influx of Ca2+ from outside the cell or release of Ca2+ from intracellular stores requires no energy and functions as a signaling event for cell contraction, migration, and proliferation. In smooth muscle cells, a rise in [Ca2+]cyt leads to cell contraction, increases cell migration, and stimulates cell proliferation. Therefore, an acute rise in [Ca2+]cyt can cause vasoconstriction, whereas chronically elevated [Ca2+]cyt leads to thickening of the vessel wall (due to its stimulating effect on cell proliferation and migration), thus decreasing the lumen of the vessel and the ability of the vessel to expand to meet increased flow.
Plasma Membrane Ca2+ ATPase and ROS
Both the Ca2+/Mg2+ ATPase (or Ca2+ pump) in the plasma membrane and CaM in the cytoplasm are regulated by redox state. ROS inactivates the Ca2+ pump in the plasma membrane (PMCA), which leads to less pumping of Ca2+ out of the cytoplasm into the extracellular space and an increase in Ca2+ concentration in the cytoplasm. While the specific amino acids that mediate inactivation of PMCA by ROS have not been determined experimentally, an in silico analysis suggests that residues Tyr589, Met622, and Met831 are responsible (54). CaM, which is also regulated by oxidative modification, binds to PMCA and induces the dissociation of the autoinhibitory domain from the active site of the enzyme and causes a several fold increase in PMCA's Ca2+ transportation rate. Oxidation of CaM methionine residues 144 and 145 by ROS may lead to the inhibition of PMCA and increased [Ca2+]cyt (3, 13).
SERCA and ROS
The Ca2+ pump on the SR/ER membrane (SERCA) sequesters Ca2+ from the cytoplasm to the SR/ER in many cell types, including vascular smooth muscle and endothelial cells. It has been reported that SERCA is inhibited by ROS or reactive nitrogen species ROC receptor-operated channel (RNS). Early reports suggested that NO activated SERCA in VSMC, thus leading to decreased [Ca2+]cyt and vasorelaxation (2). While this finding conflicts with the general trend that ROS inhibits Ca2+ pumps, other studies have shown that ROS inhibit SERCA activity by modifying cysteine residues (6, 33, 97). The different SERCA isoforms contain between 22 and 28 cysteine residues, and it is currently believed that the redox state of these residues is crucial for SERCA function. In fact, in atherosclerotic arteries, the intracellular ROS is increased and up to 9 SERCA cysteine residues are irreversibly sulfonylated (2).
IP3R and RyR and ROS
Activation of IP3R and RyR on the SR/ER membrane allows Ca2+ release from the SR/ER to the cytosol through the Ca2+ release channels and, upon stimulation or activation of receptors in the plasma membrane, lead to an increase in [Ca2+]cyt and vasoconstriction. In accordance with the trend of ROS elevating [Ca2+]cyt, ROS stimulate the activity of IP3R and RyR. Early reports from several groups demonstrated that oxidation of thiol groups by endogenous redox agents, such as superoxide and GSSG, promotes IP3R-mediated Ca2+ release in different cell types as well as from sarcoplasmic reticulum isolated from VSMC (10, 58, 79, 82). The thiol reagent thimerosal caused elevation of [Ca2+]cyt by increasing the sensitivity of IP3R to the resting levels of IP3 (38). Further, endogenous hydrogen peroxide could also increase the sensitivity of IP3R to IP3, demonstrating the physiological relevance of the effect of ROS on IP3R activity in human aortic endothelial cells (43). ROS modify RyR activity by targeting hyperreactive cysteine thiol residues (36, 37, 65). Skeletal muscle primarily contains the RyR1 isoform, cardiac muscle the RyR2 isoform, and brain and smooth muscle the RyR3 isoform although small amounts of the other two isoforms are also expressed in VSMC. Modification of RyR cysteine residues by ROS results in the activation of RyR and Ca2+ mobilization from the SR/ER to the cytoplasm. It is believed that Cys3635 is involved in RyR1 redox sensing (65).
VDCC and ROS
The membrane potential of VSMCs is critical in controlling [Ca2+]cyt. L-type VDCC on the plasma membrane are opened by membrane depolarization and allows the influx of Ca2+ to the cytoplasm. This increase of [Ca2+]cyt activates Ca2+/CaM-dependent myosin light chain kinase (MLCK) and ultimately causes smooth muscle contraction. In isolated arteries, high K+ solution causes a sustained contraction, which is mainly due to membrane depolarization and opening of VDCC in VSMC. In addition to activating MLCK and causing contraction, a localized rise in cytosolic [Ca2+] due to Ca2+ release through RyR on the SR/ER membrane leads to Ca2+ sparks, which subsequently opens Ca2+-activated K+ (KCa) channels in the plasma membrane, induces membrane hyperpolarization, and causes relaxation of VSMCs. Further, extrusion of Ca2+ by the PMCA or reuptake of Ca2+ by SERCA can also decrease [Ca2+]cyt and lead to vasorelaxation.
VDCC on the plasma membrane are affected by exogenous and endogenous redox compounds (42). However, there is controversy regarding the effect of oxidation on L-type VDCC activity. A study in isolated guinea pig ventricular myocytes demonstrated that oxidation of thiol groups in L-type VDCC inhibited Ca2+ entry (47). Further, exogenous reagents inhibited Ca2+ currents though these channels when expressed in HEK293 cells and DTT reversed this inhibition (14). In contrast to these findings, however, thiol reducing agents decreased basal L-type Ca2+ currents in ventricular myocytes and oxidizing agents increased these currents independent of cAMP and G-protein, suggesting the involvement of thiol groups (99). Also, transient exposure of cardiac myocytes to hydrogen peroxide increased basal L-type channel activity (93).
ROC and SOC
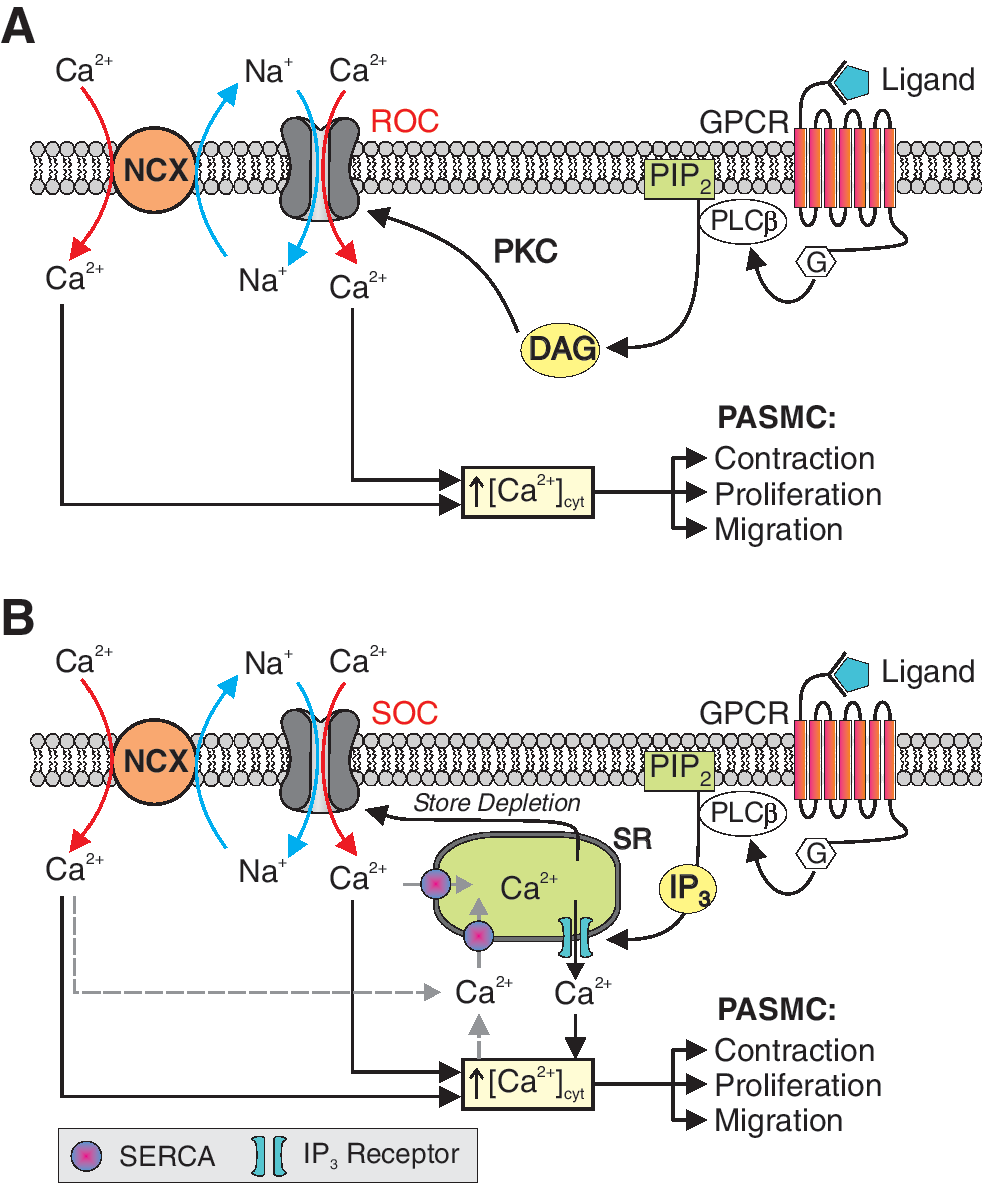
[Ca2+]cyt is also affected by influx of Ca2+ through ROC and SOC on the plasma membrane (Fig. 5). Activation of membrane receptors, for example, G protein-coupled receptors (GPCR) and tyrosine kinase receptors, leads to increased synthesis of DAG and IP3. DAG opens ROC, promotes receptor-operated Ca2+ entry, and increases [Ca2+]cyt (Fig. 5A). IP3 activates IP3R on the SR/ER membrane, mediates Ca2+ release or mobilization from the intracellular stores to the cytosol, and increases [Ca2+]cyt. Depletion of Ca2+ in the intracellular stores (e.g., SR/ER) by IP3-mediated Ca2+ mobilization opens SOC on the plasma membrane and induces store-operated Ca2+ entry (SOCE), which is also referred to as capacitative Ca2+ entry (Fig. 5B). In addition to active depletion of Ca2+ from the intracellular stores via IP3-mediated Ca2+ release, passive depletion of Ca2+ from the SR/ER by inhibition of SERCA with cyclopiazonic acid or thapsygargin can also activate ROC and induce receptor-operated Ca2+ entry in vascular smooth muscle and endothelial cells. It has been demonstrated that functional ROC and SOC are formed by TRP channel subunits.
TRP Channels
TRP channels are cation channels that mainly mediate the influx of Na+ and Ca2+ across the plasma membrane and into the cytoplasm (7). TRP channels were first discovered due to a mutation in Drosophila photoreceptor, which resulted in inhibited Ca2+ permeability and sensitivity to light (70). The influx of cations into the cytoplasm depolarizes cells and is necessary for action potentials in excitable cells such as neurons (85). In nonexcitable cells, membrane depolarization by TRP channels stimulates voltage-dependent channels (Ca2+, K+, Cl−) and influences many cellular events such as transcription and translation (15), whereas increased Ca2+ entry through TRP channels is involved in ligand-mediated cell contraction, migration, and proliferation. TRP channels and their regulation are thus fundamentally important in cellular function and disease (7).
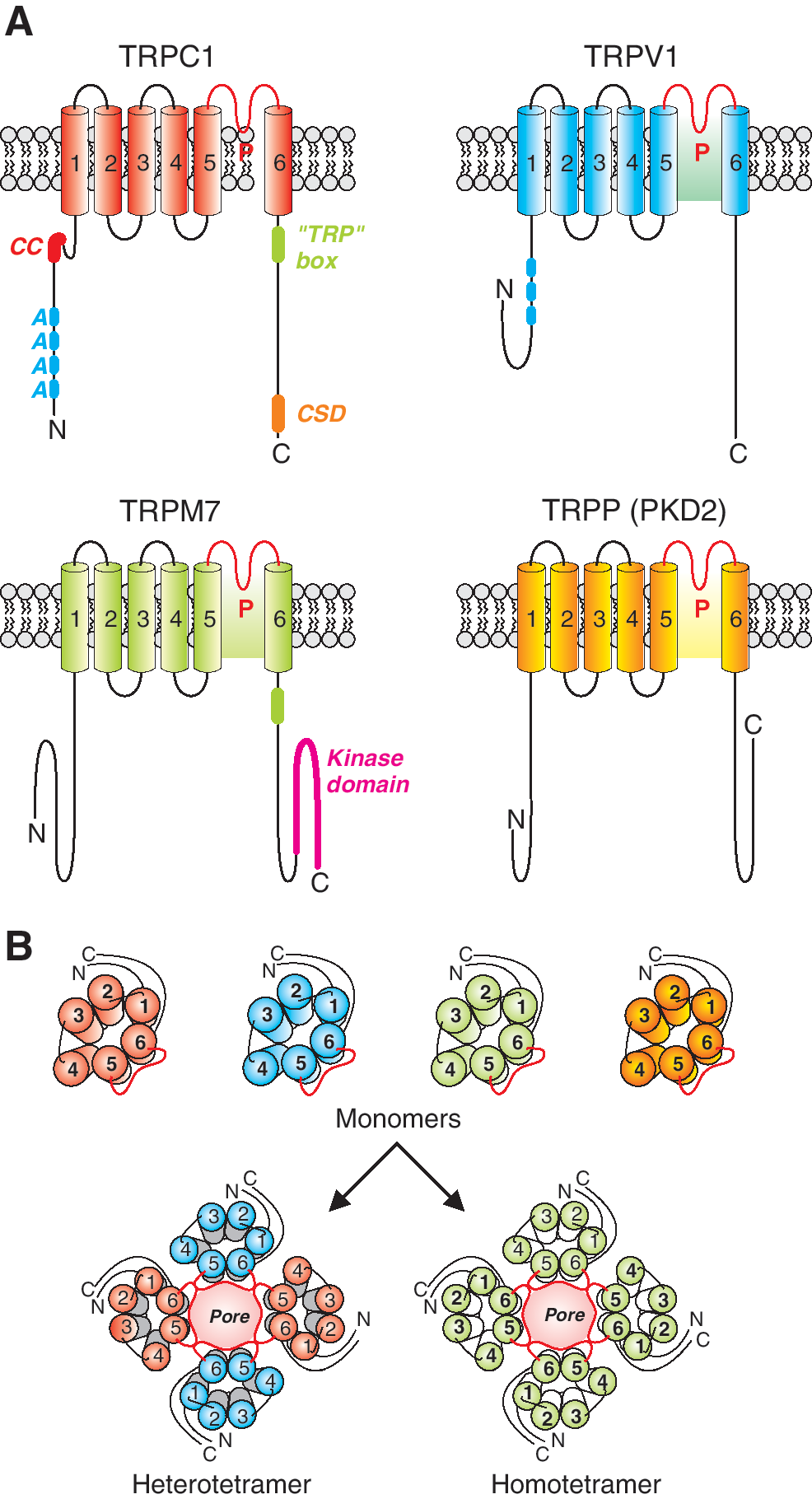
TRP genes are expressed in organisms from archaea to plants to animals (18). In animals, TRP is expressed in brain, heart, lung, and other tissues (18). The expression profile of TRP proteins in vascular tissues is shown in Table 1. Mammalian TRP channels possess a high degree of sequence homology particularly in the putative functional regions (59). The TRP superfamily of genes is categorized into two groups based on sequence and topological similarities (59). Group 1 includes TRPC, TRPV, TRPM, TRPA, and TRPN. Group 2 includes TRPP and TRPML (Fig. 6).
Cereb., cerebral; CA, coronary artery; MA, mesenteric artery; PA, pulmonary artery; RA, renal artery; VEC, vascular endothelial cells (isolated from the middle cerebral arteriole, coronary and mesenteric artery, and aorta); PAEC, pulmonary artery endothelial cell; R, rat; M, mouse; H, human; C, canine; B, bovine; F, frog.; m, mRNA identified only; p, protein identified only; m/p, both mRNA and protein identified; –, no data available.
Structure, Function, and Regulation of TRP Channels
TRP proteins form cation channels with varying selectivity to different cations (63). TRP proteins are transmembrane proteins with six transmembrane domains with a pore domain wedged between the fifth and sixth transmembrane domains (Fig. 6). The N- and C-terminal domains are intracellular and believed to be involved in regulation of TRP channel function and in channel assembly. It is believed that TRP channels are homo- or hetero-tetramers of TRP proteins with each subunit contributing to selectivity of the ion-conducting pore (17). Allosteric interactions between subunits are thought to contribute to gating of TRP channels; however, the location and structure of these gates are unknown. Amino acid sequences flanking the pore-forming regions of TRP proteins are strongly conserved across the various TRP channel families highlighting their importance in pore formation and/or pore gating (61).
TRP channel function is regulated by four basic mechanisms: (a) plasma membrane receptor activation, (b) ligand activation, (c) direct activation, and (d) indirect activation (71). GPCR and receptor tyrosine kinase act through DAG (as well as IP3-mediated store depletion) to activate TRP channels on the plasma membrane (39, 64). Further, activation of GPCRs and receptor tyrosine kinases also increase synthesis of IP3, depletes Ca2+ from the intracellular Ca2+ stores (i.e., SR/ER) and leads to SOCE through TRP channels on the plasma membrane (95). The TRP channels that are activated by DAG are termed ROC, whereas the TRP channels that are activated by IP3-mediated store depletion are termed SOC. Most of TRP channel subunits can form both ROC and SOC in many cell types, including vascular smooth muscle and endothelial cells.
Various ligands can activate TRP channels. Ligand activation includes activation by (a) exogenous small molecules such as capsaicin, icilin, 2-APB; (b) endogenous lipids such as DAG, phosphoinosidides, eicosanoids, purine nucleotides; (c) ions such as Ca2+ and Mg2+; and (d) the Ca2+/CaM complex (92, 105, 107). TRP channel activity can be stimulated through direct activation as well. Examples of direct activation of TRP channels include temperature change (16), mechanical stimulation (56), and conformational coupling with other proteins such as stromal interacting molecule 1 (STIM1) or IP3R. Indirect activation refers to transcriptional control or insertion of vesicles containing TRP proteins into the plasma membrane.
Over 10 TRP isoforms are expressed in the vasculature. Expression of mRNA, protein, or both of TRPC1-7, TRPV1-4, TRPP2, TRPM2-4, -7, -8, has been implicated in different vascular smooth muscle and endothelial cells. Of the different TRP isoforms expressed in the vasculature, much work has been done on the canonical class of TRP channels, or TRPC channels, and this review will focus on this class of proteins.
TRPC
TRPC1-7 are categorized into three categories based on sequence and functional characteristics (18). TRPC1, 4, and 5 form one group. TRPC1 was the first mammalian TRP protein discovered. It is widely expressed in many tissues and thought to form heteromeric channels with TRPC4 and TRPC5 (96). TRPC4 and TRPC5 are believed to form homomeric channels. When expressed together, TRPC1, 4, and 5 form nonselective cation channels that are activated by Gq signaling through a phospholipase C β1 pathway (66). Growth factor stimulates rapid translocation of TRPC5 into the plasma membrane from vesicles located near the plasma membrane (44).
TRPC3, 6, and 7 have roughly 75% sequence homology and when co-expressed reconstitute nonselective, inward and outward rectifying, cation channels (71). These channels are activated by receptor-mediated pathway involving DAG and are believed to be important in vascular and airway smooth muscle (40). Channels formed by TRPC3 or TRPC6 are also regulated by N-linked glycosylation and Ca/CaM (20). TRPC3 is activated by phosphorylation by protein kinase G (PKG) (45), whereas TRPC6 can be phosphorylated by the Src family of tyrosine kinases (91). TRPC2 shares roughly 30% sequence homology with TRPC3/6/7 (90). TRPC2 full-length mRNA is expressed in mouse and rat tissues (53). However, TRPC2 is a pseudogene in humans (71).
TRPC1 is expressed in the vasculature and is linked to Ca2+ entry as an ROC and an SOC. Expression and knockdown data support the theory that TRPC1 forms heteromultimeric channels with TRPC3-5 and TRPP2 in the vasculature. Further, TRPC1 may be the key isoform responsible for SOCE that causes pulmonary vasoconstriction and stimulates PASMC proliferation (14, 17, 18). Our lab has previously demonstrated that siRNA-mediated knockdown of TRPC1 in PASMC mitigates the SOCE phenomenon, thus lending credence to the theory that TRPCs can function as SOCs as well as ROCs (13, 19).
TRPC3 and 6 are expressed in vascular tissues and are believed to function primarily as ROCs, although TRPC6 may also form heterotetrameric channels with other TRP isoforms that can be activated by store depletion in pulmonary vascular smooth muscle and endothelial cells. TRPC6 is expressed in VSMCs in the systemic and pulmonary vasculature. Knockout of TRPC6 abrogates the hypoxic pulmonary vasoconstriction and hypoxia-induced cation influx. TRPC4 is predominantly expressed in the endothelium and it is important in regulating lung microvascular permeablilty (44), agonist-dependent vasorelaxation (43), and gene transcription (45). While TRPC4 is expressed at a lower level in VSMC, it may play a role in regulating contraction and proliferation in both store and receptor-mediated manners. TRPC5 expression in VSMC is unclear. Some researchers describe TRPC5 protein and transcript expression in PASMCs and pulmonary artery endothelial cells, whereas other researchers show conflicting data. TRPC7 is thought to contribute to both ROC and SOC formation.
TRPC Channels and Contraction and Proliferation of SMCs
TRPC channels are important in the regulation of vascular tone since they mediate the Ca2+ influx that mediates agonist-induced vasoconstriction and mitogen-mediated smooth muscle cell proliferation. Further, the ability of TRPC channels to alter [Ca2+]cyt with a change in membrane potential lends them the ability to modulate vasoconstriction and vasorelaxation through a voltage-independent mechanism. Agonist- and hypoxia-induced pulmonary vasoconstriction is believed to be, at least in part, mediated though Ca2+ influx through TRPC1 and TRPC6 channels. Upregulated TRPC channel expression, enhanced SOCE, and increased [Ca2+]cyt are associated with enhanced proliferation of PASMCs isolated from patients with idiopathic pulmonary arterial hypertension (102).
TRPC and ROS
TRPC3 and TRPC4 expressed in HEK293 cells formed redox-sensitive cation channels that were activated by hydrogen peroxide (32). It is, however, unknown if the activation of TRPC3 and TRPC4 is mediated through the oxidation–reduction of cysteine thiol groups. It has been suggested that hydrogen peroxide may indirectly activates TRPC3 and TRPC4 channels though activation of phospholipase C. In endothelial cells, TRPC3 and TRPC4 are believed to be able to form heteromeric channels that are redox sensitive. In these cells, tert-butylhydroperoxide leads to membrane depolarization with currents that resembled TRP currents in terms of cation selectivity, La3+ sensitivity, and a lack of voltage dependence. Further, expression of the N-terminal fragment of human TRPC3, but not the C-terminal fragment, abolished the oxidant-induced cation current. These data suggest that TRPC3 forms channels that are activated by ROS in porcine aortic endothelial cells (31). Later, the same group of investigators showed that TRPC3 and TRPC4 associate to form heteromeric channels in porcine aortic endothelial cells and HEK293 cells (67). These TRPC3/4 heterometic channels were activated by cholesterol oxidase, suggesting that they are regulated by redox state. However, no direct evidence links TRPC4 itself to redox regulation. TRPC5 homotetrameric channels and TRPC5/TRPC1 heterotetrameric channels, expressed in HEK293 cells, are activated by extracellular Trx. Reduced Trx breaks a disulfide bond in the extracelluar loop adjacent to the ion selectivity filter of TRPC5 and increase the ion conductivity of the channel (Fig. 7) (98).

TRPC and NO
NO is a cell signaling molecule involved in a number of physiological and pathophysiological processes. In the nervous system, NO is a neural transmitter, whereas in the vascular system, NO is mainly an endothelium-derived relaxing and hyperpolarizing factor. NO is synthesized by NOS, which include at least three isoforms: endothelial NOS, inducible NOS, and neuronal NOS. Each of the enzymes is a homodimer requiring three co-substrates (L-arginine, NADPH, and O2) and five cofactors (FAD, FMN, Ca2+-CaM, heme, and tetrahydrobiopterin) for activity. The redox biochemistry of NO involves the action of three redox species: NO+ (nitrosonium), NO− (nitroxyl anion), and NO• (the free radical gas). NO can react with oxygen (O2), superoxide anion (
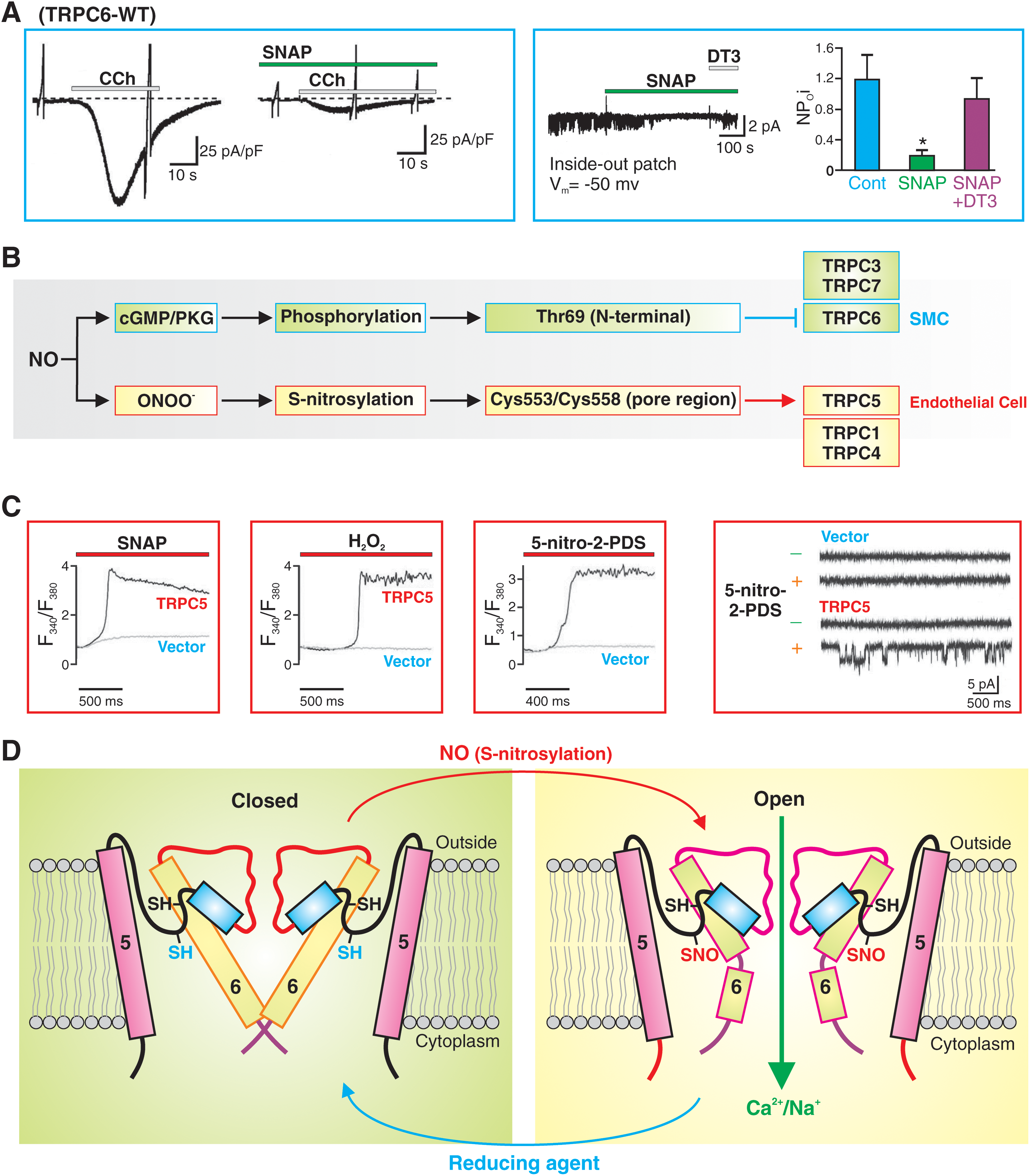
In VSMC, NO mainly acts through the stimulation of the soluble guanylate cyclase, which is a heterodimeric enzyme with subsequent formation of cyclic GMP (cGMP). cGMP activates PKG, which causes phosphorylation of myosin light chain phosphatase and therefore inactivation of MLCK and leads ultimately to the dephosphorylation of the myosin light chair, causing smooth muscle relaxation. In addition to phosphorylation of myosin light chain phosphatase, cGMP/PKG also causes phosphorylation of other proteins, such as TRPC6 channels (84). Indeed, NO/cGMP/PKG-mediated phosphorylation of threonine (Thr69) in the N-terminal of TRPC6 protein significantly attenuates receptor-mediated Ca2+ entry through TRPC6 channels (Fig. 8A, B) (84). These observations imply that PKG-mediated phosphorylation of specific amino acid residues (e.g., threonine and serine residues) in the TRPC6 protein causes inhibition of the channel activity. In contrast, NO-mediated cGMP-independent S-nitrosylation of certain cysteine residues in the TRPC1/4/5 and TRPV1/3/4 proteins significantly enhances Ca2+ influx through these channels (25, 101). Specifically, for example, Cys553 and Cys558 of TRPC5 are localized in the cytoplasm and mediate the channel's sensitivity to NO/S-nitrosylation in endothelial cells (Fig. 8B–D) (25, 101). S-nitrosylation of TRPC5 in endothelial cells upon G protein-coupled ATP receptor stimulation elicits Ca2+ entry. These observations imply that cGMP/PKG-dependent phosphorylation and cGMP/PKG-independent S-nitrosylation are two different pathways for NO-mediated effect on TRP channels. Whether NO-mediated effect is to activate or inhibit the channels depends on (a) which pathway (PKG-dependent phosphorylation vs. PKG-independent S-nitrosylation) occurs faster to reach the target protein (or which pathway is predominant in a given cell), (b) which TRP channel subtype (e.g., TRPC5 vs. TRPC6) is predominantly expressed in a given cell as the target, and (c) whether the target amino acids (e.g., threonine and serine for phosphorylation, and cysteine residues for S-nitrosylation) for phosphorylation or S-nitrosylation are present in the predominant TRP channels in a given cell.
SOCE
As mentioned earlier, SOCE is the phenomenon through which depletion of intracellular Ca2+ stores opens SOC on the plasma membrane. Upon depletion of Ca2+ in the SR/ER, this Ca2+ deficiency is sensed and transmitted to channels on the plasma membrane to allow Ca2+ to flow into the cytoplasm where it is then sequestered into the SR/ER by SERCA, thus replenishing Ca2+ stores. The SOCE is necessary for long-term regulation of Ca2+ signaling. Without SOCE, SR/ER Ca2+ stores would become depleted when cells are stimulated with ligands and the membrane receptors are activated.
A mutation in Orai1, an important molecular component of SOCE, has been demonstrated to be associated with severe combined immune deficiency in humans. Further, upregulation or downregulation of SOCE can chronically increase or decrease the rate and magnitude of the other intracellular Ca2+ signaling mechanisms and have a chronic effect on cellular biology (29). Much evidence links SOCE to altered proliferation and contraction in PASMCs, which are intimately involved in the function and pathophysiology of the pulmonary circulation. Our lab has demonstrated that SOCE can increase resting [Ca2+]cyt in PASMC independent of receptor- or voltage- dependent mechanisms. Inhibition of SOCE decreases [Ca2+]cyt and attenuates proliferation in PASMC. Further, PASMC isolated from patients with idiopathic pulmonary arterial hypertension demonstrate higher resting [Ca2+]cyt and greater SOCE than cells isolated from normal subjects and normotensive control patients (102, 103).
Mechanism of SOCE: STIM1 as the Ca2+ Sensor
Recently, much has been discovered on the mechanism of SOCE. Through genome-wide RNAi screening, investigators have identified the sensor that detects [Ca2+] in the SR/ER ([Ca2+]SR/ER) as STIM1 (1). STIM1 is a 685-amino acid-long single transmembrane protein that is expressed on the SR/ER membrane and plasma membrane. Near the N-terminus of STIM1 is an EF-hand domain that senses [Ca2+]SR/ER (which is in the range of 0.5–2 mM). When Ca2+ is not bound to the EF-hand domain when, for example, the SR/ER Ca2+ store is depleted, STIM1 undergoes a conformational change, which allows it to multimerize, translocates to the SR/ER-plasma membrane junction (or puncta), binds with Orai1 tetramers on the plasma membrane, activates SOC, and induces SOCE (Fig. 9) (78). Mutagenesis of the EF-hand domain (mimicking low Ca2+) leads to STIM1 multimerizing in distinct regions of the plasma membrane, called puncta, in the absence of store depletion and to activating SOC. Examination by total internal reflection (TIRE) microscopy shows that store depletion-induced puncta, which contains STIM1, localizes 10–20 nm from the plasma membrane, allowing for SR/ER STIM1 to interact with SOC in the plasma membrane (60).

SOC: Orai1
Strong evidence suggests that Orai1 interacts with STIM1 and functions as a SOC. Orai1 was discovered through genome-wide RNAi screening. A mutation in Orai1 in patients with severe combined immune deficiency eliminates the Ca2+ release-activated Ca2+ currents (I CRAC), which can then be reconstituted by expressing wild-type Orai1 (22, 57). Orai1 spans the plasma membrane four times with both the C- and N-terminus in the cytoplasm. Co-expression of STIM1 with Orai1 in HEK293 cells resulted in large increases (roughly 25–30 times control) in SOCE and I CRAC compared to vector control cells (81). Co-immunoprecipitation data show that STIM1 and Orai1 interact, and store depletion significantly increases the amount to interaction (100). Further, other isoforms of Orai (e.g., Orai2 and Orai3) are also thought to play a role in SOCE (57). It is unclear, however, what the role of these Orai isoforms are in the function of smooth muscle cells.
TRPC as SOC
As mentioned earlier, another category of channel subunits that have been proposed as store operated are the TRP channels (71). Of the TRPC's, TRPC1, 3, 4, 6, and 7 have shown promise as SOC. In fact, our lab has demonstrated that inhibition of endogenous TRPC1 in PASMC decreases SOCE (83). Researchers also suggest that TRPC1, STIM1, and Orai1 form a ternary complex, which constitutes an active SOC (104). However, while SOCE is a fundamental process in PASMC (30, 52, 75, 83, 103, 106), pulmonary artery endothelial cells (23, 46), and intestinal epithelial cells (62, 73), it is still unclear how TRP, Orai, and STIM interact precisely to form SOC and sense store depletion in vascular smooth muscle and endothelial cells.
SOCE and ROS
Not much is known on the effect of redox state on SOCE. In thyroid cells, hydrogen peroxide seems to inhibit SOCE. However, this is likely due to activation of protein kinase C and not modification of proteins directly involved in SOCE such as the STIM, Orai, or TRP families of proteins. Both STIM and Orai proteins contain cysteine residues and therefore may in fact be regulated by redox state. TRPM2 channel is believed to be activated by hydrogen peroxide to secrete insulin in pancreatic B cells; however, this may be due to the effect of hydrogen peroxide on mitochondria (24). TRPA1 channels in peripheral nociceptive neurons are activated by covalent modification of cysteine thiol residues (5). As discussed earlier, ROS and NO regulate TRPC channels, as well as other molecular components of SOCE (e.g., Orai and STIM), through different pathways, such as direct oxidation, nitration, and S-nitrosylation of the cysteine thiols and indirect phosphorylation of the threonine and serine residues mediated.
Conclusion
The regulation of intracellular [Ca2+] in different cellular compartments is of integral importance in the physiology and pathophysiology of the vasculature. Changes in [Ca2+]cyt, [Ca2+]SR/ER, and [Ca2+]Mit all play important roles in regulating cell contraction, proliferation, migration, and apoptosis. The concentration of Ca2+ in these cellular compartments is tightly regulated by Ca2+ channels and transporters functionally expressed in the plasma membrane and intracellular organelle membranes. TRP channels are a family of proteins that participate in forming functional ROC and SOC in vascular smooth muscle and endothelial cells, and thus play a critical role in the regulation of vascular tone, remodeling, and injury. ROS and redox state, in general, has emerged as an important system for regulating Ca2+ signaling, partially through their modulatory effect on TRP channels as well as ROC and SOC. More and more studies have been focused on examining whether and how ROS and redox status regulate these channels’ function, kinetics, expression, and trafficking in vascular smooth muscle and endothelial cells. Regulation of intracellular Ca2+ homeostasis by ROS/RNS offers explanations to the important role of ROS/RNS signaling in the vasculature physiology and pathophysiology. Further, the fact that many proteins and enzymes involved in regulating the redox status of cells are actually regulated, per se, by Ca2+ and Ca2+/CaM complex makes for a complicated and interesting intracellular regulatory system; derangement of this precisely controlled system would lead to the development of cardiovascular diseases.
Much work still needs to be done to elucidate the relationship between ROS and Ca2+ regulation. TRPC channels have been known to be important in regulating [Ca2+]cyt and [Ca2+]SR/ER in the vasculature. While researchers have made some headway in determining how ROS affect TRPC channels, more investigation needs to be conducted to, for example, (a) define the effect of ROS and RNS on STIM and Orai, two important families of proteins that mediate SOCE, (b) specify the domains and amino acid residues in TRP proteins that are differentially regulated by ROS-mediated oxidation, nitration, S-nitrosylation, and protein kinase-mediated phosphorylation, and (c) compare the ROS/redox status-mediated regulation of TRP channels, Orai and STIM, between normal and diseased tissues and cells.
Footnotes
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (HL054043 and HL066012 to J.X.-J.Y., and DK083506 to A.M.). M.Y.S. is supported by an NIH training grant (T32 DK007202).