
Abstract
Introduction
Bcl-2 portrait
Bcl-2 is the acronym for the B-cell lymphoma/leukemia 2 gene. As its name implies, this gene was first discovered in B-cell malignancies (95) where the Bcl-2 gene, normally localized to chromosome 18q21, was fused with the immunoglobulin heavy chain gene promoter and enhancer on chromosome 14q32 [t(14,18) chromosomal breakpoint], thereby excessively deregulating its transcription and inducing aberrant overexpression of Bcl-2 (24). This observation was associated with drug resistance due to the inherent pro-survival function of Bcl-2 through its ability to keep pro-apoptotic proteins at bay and thereby inhibiting mitochondria-dependent apoptotic signaling (86).
Structurally, Bcl-2 possesses four BH domains as well as a carboxyl terminal hydrophobic transmembrane domain (TM), which is responsible for membrane localization. With the use of immunofluorescence, electron microscopy, and subcellular fractionation studies, Bcl-2 has been identified in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes (54). The three-dimensional structure of Bcl-2 contains two central helices that are mostly hydrophobic, surrounded by four amphipathic α-helices (Fig. 1). A hydrophobic groove that consists of BH1, BH2, and BH3 domains is present on the surface of the protein and is essential in its heterodimerization with pro-apoptotic family members (Fig. 1) (84). In addition, a putative unstructured loop is also present in Bcl-2 between the first and second helix based on X-ray crystallographic and NMR spectroscopic studies on Bcl-xL (69), which offers ample opportunities for post-translational modifications (Fig. 1) (7, 8, 12, 16, 40, 78).

Bcl-2 and Intracellular Redox Status
Bcl-2, a pro-oxidant in disguise
The involvement of Bcl-2 in redox regulation was initially unearthed when Hockenbery et al. demonstrated the protection conferred by Bcl-2 overexpression against reactive oxygen species (ROS)-mediated apoptosis induced by oxidants such as H2O2 and menadione (42). This protection was echoed almost simultaneously by a separate observation, whereby neural cell death as a consequence of ROS elevation and glutathione depletion was reportedly reversed by Bcl-2 overexpression (48). Together, these studies provided the impetus for a series of studies revealing the protective capacity of Bcl-2 against a plethora of ROS triggers (γ-irradiation, H2O2, cyanide, tert-butyl hydroperoxide, and glucose deprivation) in various mammalian cell lines (65, 72, 105, 106). Nonmammalian systems were no exception as well, for the introduction of human Bcl-2 into Saccharomyces cerevisiae induced resistance against oxidative-stress induced cell death (19). These sets of observations served as the basis for the notion that Bcl-2 was an antioxidant protein. However, this dogmatic view has since been challenged by a number of recent studies. Most, if not all, experimental models employed to establish the antioxidant property of Bcl-2 have either subjected the cells to various ROS-inducing triggers or by exposing the cells to harsh oxidative environment before attaining the antioxidant response conferred by Bcl-2 overexpression. At best, such approaches could only associate Bcl-2 with enhanced intracellular antioxidant capacity, whilst few, if any, of these studies could confirm Bcl-2 as having intrinsic antioxidant ability. Moreover, apoptosis itself could result in the accumulation oxidative damage in dying cells, and proteins such as Bcl-2 could thereby incorrectly be labeled as antioxidant proteins by dint of their anti-apoptotic function. In fact, the initial report by Hockenbery et al. demonstrated that Bcl-2 expression, although able to protect against menadione-induced oxidative damage, could not inhibit peroxide formation in the cells (42), implying that Bcl-2 expression could have enhanced cellular antioxidant capacity, thus providing a shield from oxidative insults. Indeed, later studies demonstrated that Bcl-2 by itself did not possess innate antioxidant capacity (57) and its protective effect against oxidative stress-induced injury/death could be reversed by depleting reduced glutathione (GSH) in the cell (65). These data provide strong evidence that the antioxidant effect of Bcl-2 is indirect, rather than an innate characteristic of the oncoprotein.
It appears that the reason for the reported antioxidant nature of Bcl-2 could be attributed to the fact that most of these studies looked at the effect of Bcl-2 overexpression in cells undergoing overt oxidative stress. However, of late a clearer picture is emerging vis a vis the effect of Bcl-2 on cellular redox status, and to that end, recent studies reveal a pro-oxidant activity of Bcl-2 under normal physiological states (5, 20, 25, 35, 52, 92). Subsequent work further indicated that Bcl-2-mediated protection is invariably associated with an enhanced level of antioxidant defense machineries, with the likes of catalase, glutathione reductase, glutathione peroxidase, GSH, and NAD(P)H being commonly implicated (33, 52, 53, 57, 77). This led to the suggestion that the antioxidant properties purported previously could in fact be an adaptive response to a mild, but chronic, oxidative environment induced by Bcl-2 overexpression, which in turn resulted in the eventual reinforcement of the intracellular antioxidant machinery (33, 35, 57, 65, 77). Bcl-2-mediated prophylactic fortification of the antioxidant armor could therefore serve as a first-line defense against acute oxidative stress, thereby preventing ROS from breaching a threshold deleterious to the cell.
The exact mechanism(s) driving such redox-sensitive response in Bcl-2 overexpressing cells has yet to be conclusively addressed. Eposti et al. reported an increase in NADP+ transhydrogenase activity and hence an increase conversion of NADH to NAD(P)H upon Bcl-2 overexpression (35). Although such enhancement in NAD(P)H levels could account for the elevated antioxidant capacity [by fueling the glutathione reductase and peroxidase system (43)] in Bcl-2 overexpressing cells, the precise mechanism(s) in which Bcl-2-induced pro-oxidant state increases the level of NADP+ transhydrogenase remains a question. Alternatively, it has also been reported that mitochondrial ROS could directly signal the expression of manganese superoxide dismutase (MnSOD) via the protein kinase D (PKD) and NF-κB pathway (93). This is conceivable, as superoxide anions (O2 -) have previously been shown to modify gene expression patterns (30, 45, 104). It is therefore possible that mitochondrial O2 - could leak out to the cytosol via the voltage-dependent anion channels [VDAC (41)] or the translocase of the outer membrane (TOM) complexes (13) and induce upregulation of various antioxidant genes.
Bcl-2 and the pro-oxidant theory of carcinogenesis
The first indication of Bcl-2 being a pro-oxidant came from studies on Escherichia coli whereby the introduction of recombinant Bcl-2 into SOD-deficient E.coli strains resulted in an elevated level of oxidative DNA damage as compared to wild-type strains (92). These findings were subsequently corroborated by studies on mammalian cell lines. Armstrong and Jones demonstrated, via the use of a ROS-sensitive probe (DCFH-DA), that Bcl-2 overexpression resulted in an elevated level of intracellular ROS in HL60 promyelocytic leukemia cells (5). Our group, on the other hand, reported that both HeLa cervical cancer cells and CEM leukemia cells that are stably transfected with Bcl-2 constitutively exhibit a higher MitoSOX Red fluorescence level relative to the mock-transfected counterparts, thus suggesting a higher level of mitochondria O2 - in Bcl-2 overexpressing cells (20, 25).
Despite abundant evidence demonstrating the pro-oxidant effect of Bcl-2, few studies have addressed the mechanism in which Bcl-2 exerts its pro-oxidant activity. Kowaltowski and Fiskum hypothesized that this elevated cellular redox state could be a function of an altered dynamics within the oxidative phosphorylation (OXPHOS) machinery of Bcl-2 overexpressing mitochondria, a phenomenon postulated to be the result of an increase in mitochondrial size as well as mitochondrial NADH levels (51, 52). It was suggested that the observed increase in mitochondrial volume and its associated matrix content is in turn translatable to an increase in electron donors within the mitochondria, thereby increasing the probability in which electrons could be leaked out from the electron transport chain (ETC) for the reduction of molecular oxygen into O2 - (52). However, the precise mechanism by which Bcl-2 expression resulted in an increase in mitochondrial size as well as matrix enzymes was not addressed.
Over the years, our group has been studying the mechanism and translational relevance of redox regulated cell fate signaling in the context of carcinogenesis (26, 82, 83). Despite the dogmatic view of ROS as an agent of detriment, emerging evidence has established that, at nontoxic concentrations, ROS could function as proliferative and/or survival signals (82). To that end, a slight increase in intracellular O2 - has been shown to promote cancer cell survival by inhibiting apoptotic execution, irrespective of the trigger (1, 2, 28, 29, 82, 83). We provided further evidence that the regulation of cell fate by intracellular redox status was a function of a tight balance in the ratio of O2 - to H2O2, which could be impacted by the constitutive expression of antioxidant enzymes, in particular the SODs. Survival signaling was favored in an environment where the ratio tilted in favor of O2 -, whereas a significant increase in H2O2 sensitized cells to apoptotic stimuli (Fig. 2) (27, 28, 82).
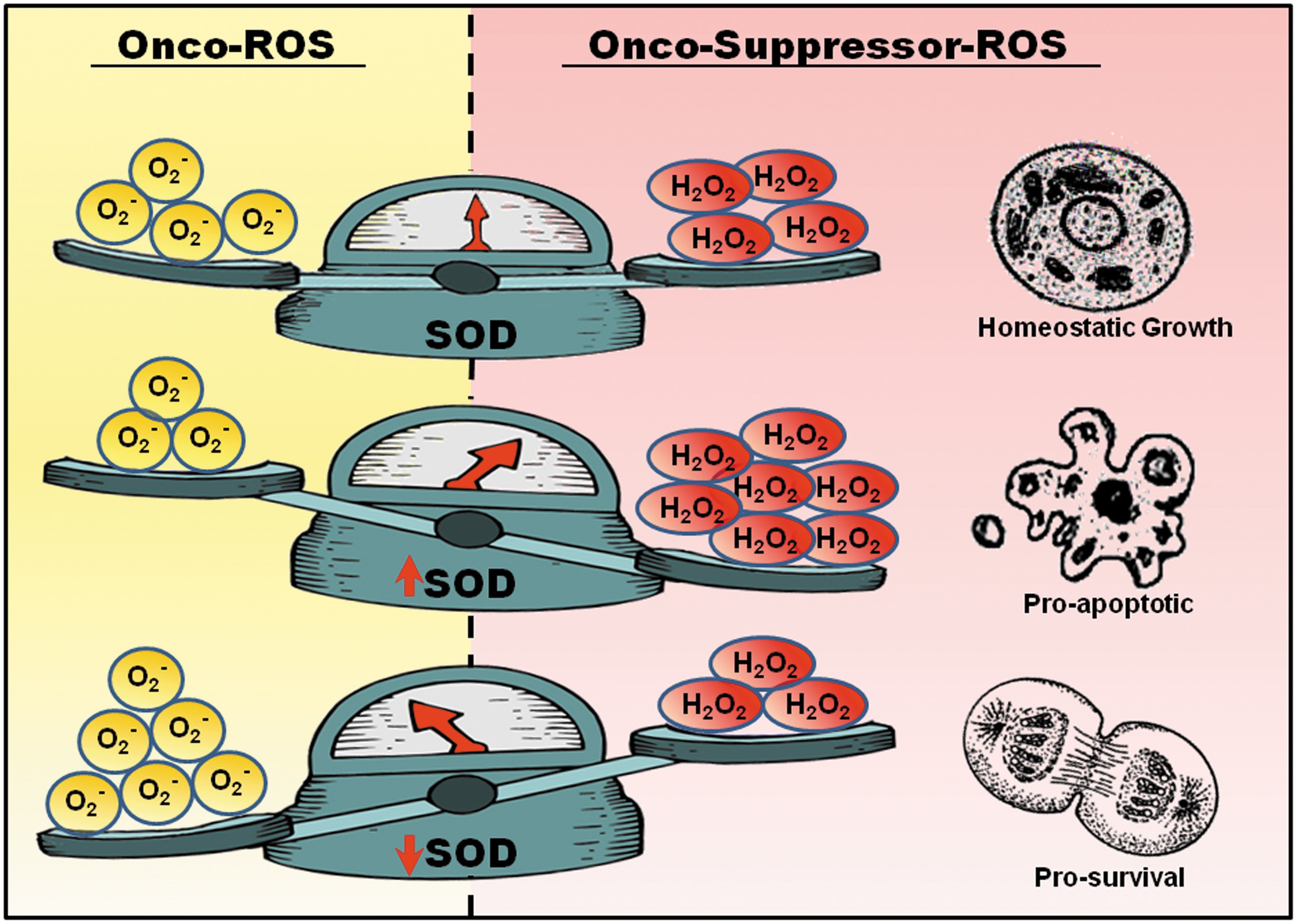
Corroborating the pro-survival effect of a mild pro-oxidant state, the anti-apoptotic activity of Bcl-2 was linked to its noncanonical activity of inducing an increase in intracellular O2
- (25). Using a variety of model systems, such as death receptor- and drug-induced apoptosis, it was demonstrated that resistance to apoptosis in human leukemia CEM cells overexpressing Bcl-2 could be overcome by decreasing overall cellular O2
The source of this pro-oxidant state appears to be the mitochondria ETC, as judged by the elevated fluorescence intensity of the mitochondrial O2 - specific probe, mitoSOX Red, and the increase in mitochondrial oxygen consumption rate observed in Bcl-2 overexpressing cells (20, 25, 61). Nonetheless, the relationship between mitochondrial ETC activity and the rate of O2 - by-production from the mitochondria has been less straightforward than it appears to be. Whereas it has long been established that inhibition of mitochondrial ETC activity by metabolic poisons such as rotenone [complex I inhibitor (14, 60)] and antimycin A [complex III inhibitor (96)] could result in a massive increase in O2 - production from the ETC, our group (20, 22, 61) and others (55, 75, 97), on the other hand, have shown that an augmented mitochondrial respiration rate is associated with an elevated production of mitochondrial ROS. Such discordance could, in part, be due to a different working(s) in which electrons are leaked out to oxygen molecules during different respiratory states. Whereas inhibition of the ETC complexes results in a piling up of reduced electron carriers which increases the propensity of electrons being transferred to oxygen molecules when oxidative phosphorylation is reduced/halted (71), an increase in electron flow across the mitochondrial ETC during normal/uninhibited mitochondrial respiration could also serve to augment the level of mitochondrial O2 -. It is thus plausible that an augmented mitochondrial respiration could increase the probability of mishap during electron shuttling through the ETC with the consequential increase in respiration-dependent O2 - by-production. Indeed, Bcl-2-induced pro-oxidant activity stems from an elevated mitochondrial respiration rate, as evidenced by an increase in mitochondrial oxygen consumption and cytochrome c oxidase (COX; complex IV) activity in Bcl-2 overexpressing cells (20, 22, 61). Corroborating this hypothesis, siRNA-mediated silencing of Bcl-2 in Bcl-2 overexpressing HeLa (HeLa/Bcl-2) and CEM cells (CEM/Bcl-2) resulted in a concurrent reversion of both oxygen consumption and intracellular redox status to a level comparable to the mock-transfected counterparts (20). A similar effect was observed upon functional inhibition of Bcl-2 with the BH3 mimetic, HA14-1 (20). Furthermore, a strong correlation of mitochondrial respiratory rate and O2 - level with Bcl-2 expression was also observed across three distinct tumor cell lines (HK-1, C-666, and HCT-116) with different levels of endogenous Bcl-2 (22), thus lending further support to the effect of Bcl-2 in the regulation of respiration-dependent O2 - production.
Despite suggestions that the slight increase in mitochondrial respiration in Bcl-2 overexpressing cells is insignificant and even negligible (52, 70, 91), we rationalized that any observable increase in mitochondrial oxygen consumption could still be of considerable significance. This is particularly so, as an augmented oxygen consumption rate is directly translatable to an overall increase in electron flux across the ETC, which in turn, would increase the probability of electrons leaking onto molecular oxygen across all complexes (I,II, and III). As such, a small change in mitochondrial oxygen consumption could potentially be amplified into a significant difference in mitochondrial O2 - production. As a matter of fact, a significant increase in mitochondrial respiration would instead be counterproductive, as the amount of ROS generated could easily overwhelm the cells' antioxidant barrier rather than inducing a beneficial adaptive response within the cell.
Bcl-2 as a redox-driven regulator of mitoenergetics
The present theme of tumor metabolism was first proposed by Otto Warburg, whereby cancer cells favor the glycolytic pathway over the OXPHOS system to cater for the majority of their energy needs. For decades, the Warburg effect has been the central dogma of tumor metabolism and progression, with a wide range of tumors shown to exhibit this phenotype; and with the glycolytic phenotype in turn being linked to the aggressiveness of the tumors (36). However, OXPHOS is not always defective in tumors, and energy derived from OXPHOS is seldom, if ever, negligible during the process of carcinogenesis. True to this aspect, OXPHOS has been observed to operate in parallel with the glycolytic pathway in a variety of tumors (34, 59, 62, 109) whilst more importantly, reports have gone on to testify that the OXPHOS system may even predominate in certain tumors, with instances such as lung, breast, ovarian, and cervical carcinomas, as well as bone sarcoma and skin melanoma being implicated before (6, 34, 47, 87, 88, 109). Reinforcing this, it was even suggested that an active OXPHOS system would necessitate tumor progression (88).
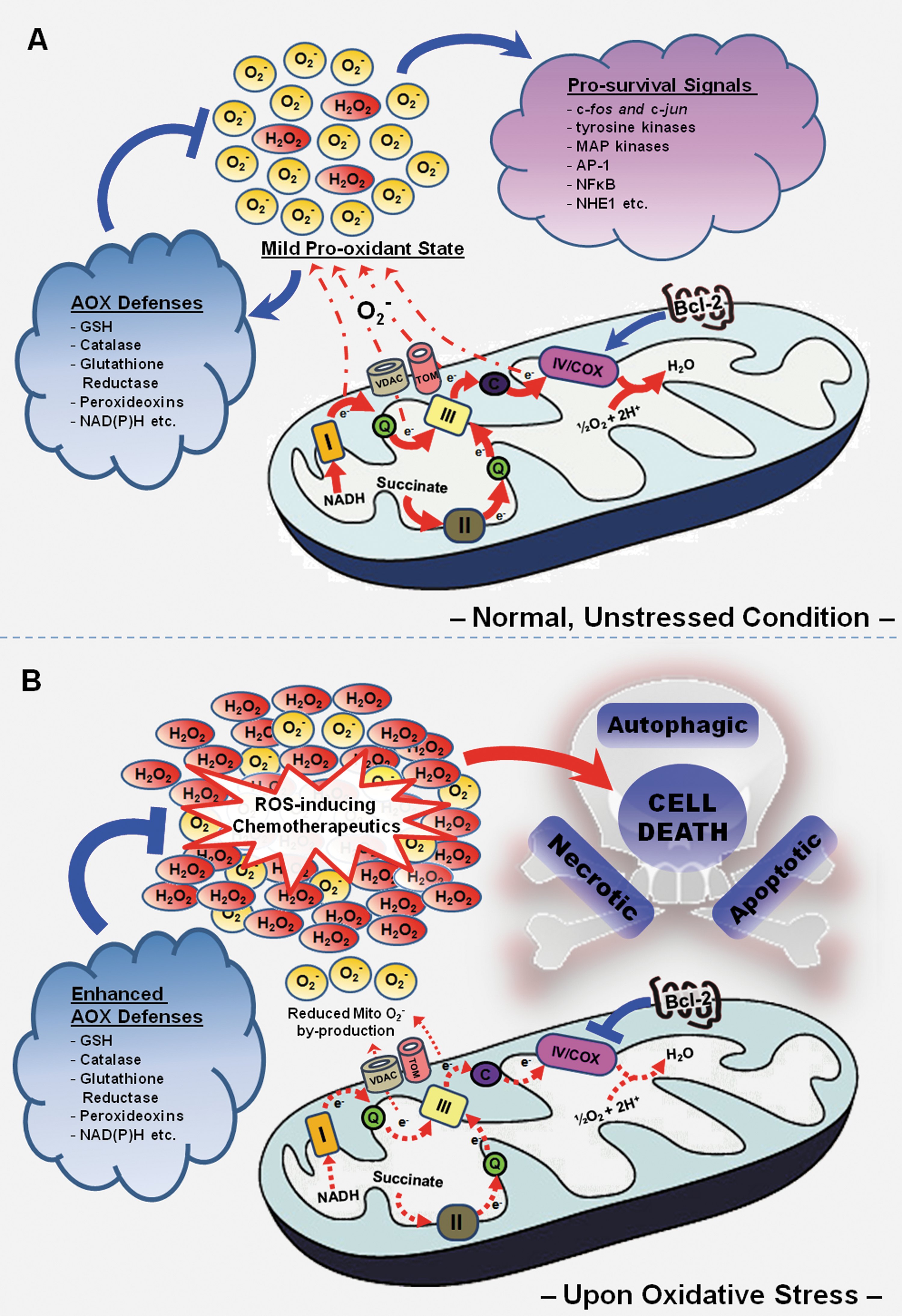
Bearing in mind the importance of the OXPHOS system in tumorigenesis, together with the pivotal role of Bcl-2 in maintaining mitochondrial integrity and function, it is conceivable that Bcl-2 may have a regulatory hand in the workings of the oxidative metabolic machinery. Indeed, our group revealed, for the first time, a novel role of Bcl-2 in the homeostatic regulation of mitochondrial bioenergetics. Using leukemia (CEM) and cervical carcinoma (HeLa) cells as models, we demonstrated that Bcl-2 overexpression is consistently accompanied by a moderately augmented level of mitochondrial respiration and its associated by-production of mitochondrial O2 -, a phenomenon later shown to be a function of an elevated enzymatic activity at the level of the terminal electron transporter, COX (20, 22, 61). This clearly suggests a link between Bcl-2 and COX, though the exact molecular mechanism(s) by which this multi-subunit complex is regulated by Bcl-2 remains a question. Our recent investigations demonstrated that Bcl-2 overexpression promoted mitochondrial localization of two (out of ten) nuclear encoded subunits of the COX complex, COX Va and Vb, suggesting that Bcl-2 may facilitate the assembly of this multi-subunit enzyme complex, so as to form a more efficient COX holoenzyme. Indeed, previous observations associating the level of COX Vb with an increase in COX activity further lends support to our postulation (15). Of note, co-immunoprecipitation studies uncovered a physical association between Bcl-2 and COX Va while mutagenesis studies implicated the BH2 domain and C-terminal region of Bcl-2 in the interaction with COX Va (22). Intriguingly, BH3 mimetic, HA14-1, could also reverse the regulatory effect of Bcl-2 on mitochondrial oxygen consumption and COX activity (22), suggesting that the BH3 domain could also be involved in its interaction with COX Va as well. However, any direct interaction between Bcl-2 and COX Vb could not be detected. Based on these evidence, while not disregarding the fact that both COX Va and Bcl-2 are nuclear encoded, we hypothesize that Bcl-2 may potentially act as a chaperone for COX Va, aiding its targeting to the mitochondria as well as its assembly into the COX subcomplex. On the other hand, the absence of a direct interaction between COX Vb and Bcl-2 suggests that the enhanced presence of COX Vb in the mitochondria upon Bcl-2 overexpression could be secondary to COX Va's inclusion to the mitochondria. This is plausible since the inclusion of COX Va into the COX subcomplex immediately precedes that of COX Vb during the assembly of the holoenzyme, indicating that the integration of COX Va may be vital for the subsequent recruitment of COX Vb into the COX enzyme (74, 103).
Intriguingly, excessive induction of mitochondrial ROS (by antimycin A) resulted in a converse downregulation of COX activity and a consequential reduction in respiration-dependent mitochondrial O2 - production in Bcl-2 overexpressing cells (20). Also, similar observations were obtained even when physiological triggers (serum withdrawal, hypoxia, and glucose deprivation) were employed for oxidative stress induction (22). These unique findings suggest a novel feature of Bcl-2, which steers away from its canonical anti-apoptotic activity, enabling the anti-apoptotic protein to exert metabolic control over the mitochondrial ETC in response to changes in cellular redox status. With this novel characteristic, Bcl-2 overexpression could render cancer cells more resistance to ROS-based chemotherapeutics, maintaining a mild pro-oxidant intracellular milieu which facilitates death evasion while, at the same time, serving as a redox sentry to prevent excessive accumulation of ROS during episodes of oxidative stress by way of downregulating COX activity (Figs. 3A and 3B). With regards to this, we provide evidence that Bcl-2 overexpression protects against the death-inducing effects of resveratrol (RSV), a phytoalexin with promising chemotherapeutic properties (61). Whereas an increase in mitochondrial O2 - was detected in mock-transfected CEM cells upon RSV treatment, CEM/Bcl-2 cells appeared refractory to the death-inducing oxidative burst triggered by RSV (61). This observed protection correlated well with a concurrent decline in COX activity and mitochondrial oxygen consumption, while SiRNA-mediated silencing of Bcl-2 in CEM/Bcl-2 cells restored mitochondrial ROS and COX activity to a level comparable to that of CEM/Neo cells (61). Of note, protection by Bcl-2 against RSV-induced mitochondrial O2 - was not secondary to its inhibitory activity on Bax/Bak-induced mitochondrial ROS production, since similar protection against RSV treatment was observed even in the absence of Bax and Bak in HCT116 cells (61).
Despite promising evidence connoting Bcl-2 as a novel metabolic regulator, the underlying mechanism(s) governing the response of Bcl-2 and COX against impending oxidative distress is still elusive. To address this, CEM/Neo and CEM/Bcl-2 cells were subjected to various modes of physiological stress states (serum withdrawal, hypoxia, and glucose deprivation) and the localization status of the nuclear encoded COX subunits, Va and Vb, was studied by means of mitochondrial fractionation techniques. Interestingly, although no significant change in mitochondria COX Va level was identified, COX Vb localization to the mitochondria was significantly reduced in CEM/Bcl-2 cells, with the reverse being true for CEM/Neo cells (22). While this could explain, at least in part, the stress-activated downregulation of COX activity and mitochondrial ROS production, exactly how Bcl-2 restricts mitochondrial localization of COX Vb during period of oxidative distress remains unclear. In addition, the rapid metabolic response against impending oxidative insults conferred by Bcl-2 overexpression also begs the question of how exactly could the intracellular redox status be sensed and deciphered by the oncoprotein at such rate and precision. Regarding this, the involvement of redox-mediated post-translational modification of Bcl-2 could be possible, especially since Bcl-2 phosphorylation and ubiquitation were shown to be redox-regulated (58), and in addition several Bcl-2 acting kinases and phosphatases are also known targets of intracellular ROS (18, 63, 68).
Bcl-2 regulates mitochondrial GSH
A critical component of cellular antioxidant defenses is the glutathione system. Depending on the oxidation status of the cells, the glutathione content is shuttled between the reduced form (GSH) and the oxidized form (GSSG) via the glutathione oxidase/reductase enzymes. Therefore, GSH to GSSG ratio is a reliable measure of the cellular redox status. Interestingly, recent evidence has demonstrated a novel function of Bcl-2 in regulating the mitochondrial GSH pool. A direct physical interaction between Bcl-2 and GSH-conjugated agarose was demonstrated in vitro, which was disrupted by the Bcl-2 inhibitor, HA14-1, that binds to the BH3 groove of Bcl-2, and by another structurally distinct BH3 mimetic, compound 6. Similarly, a recombinant BH3-only protein, Bim-L was also shown to block the interaction between Bcl-2 and GSH. The availability of BH3 groove is clearly essential for Bcl-2 to interact with GSH and the physical tethering probably serves as a means to sustain the mitochondrial pool of GSH by Bcl-2. Supporting this further, GSH was displaced from mitochondria in intact neurons when its interaction with Bcl-2 was abolished with the use of BH3 mimetics (107). The mitochondrial pool of GSH comes entirely from the more abundant cytosolic pool of GSH (56). Indeed the uptake of [3H] GSH into isolated rat brain mitochondria was reduced upon incubation with HA14-1, which could probably contribute partially to the displaced mitochondrial pool of GSH (107). However, there is no evidence that Bcl-2 has active transport properties for GSH; thus how the physically tethered GSH contributes to the mitochondrial antioxidant capacity remains elusive, but at least physically tethered GSH would likely decrease the susceptibility of oxidation of the proteins at the mitochondrial membrane (100). Nonetheless, these observations provide further evidence that Bcl-2 is able to counteract excessive oxidative stress in the mitochondria through regulation of the GSH pool, thereby serving as a safety valve in the face of overwhelming ROS insults (Fig. 4).
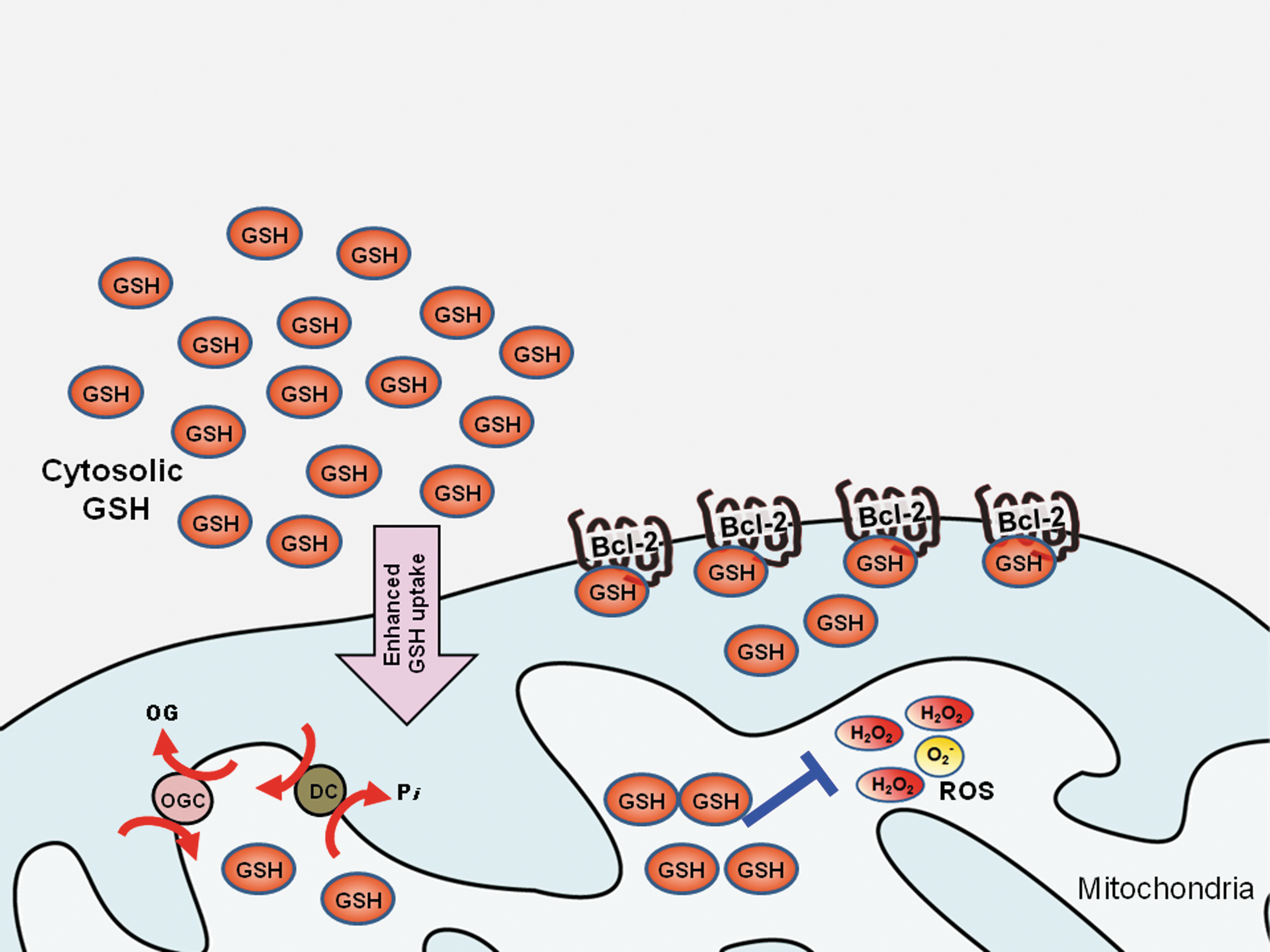
In the preceding sections, we discussed two specific interactions of Bcl-2 with direct relevance to cellular redox signaling in the context of the death inhibitory and oncogenic activity of Bcl-2. These observations have potential implications for the therapeutic management of cancers that are refractory to therapy due to the overexpression of Bcl-2.
Cellular Redox State as a Novel Drug Target in Bcl-2-Induced Chemoresistance
Resistance to apoptosis presents a serious challenge to the therapeutic management of cancer. It is now well established that most cancer cells have acquired mechanisms to resist chemotherapy-induced execution such as the overexpression of Bcl-2, which has been associated with poor disease prognosis. Among hematologic cancers, overexpression of Bcl-2 has been shown to correlate with resistance to chemotherapy in follicular lymphomas, B-cell chronic lymphocytic leukemia (CLL), multiple myeloma, and acute myeloid leukemia (AML) (49). Furthermore, dysregulation of Bcl-2 is increasingly observed in solid tumors such as melanoma (94), prostate (64), breast (9), lung (66), colon (85), pancreatic (37), and head and neck cancers (32). Therefore, some of the promising therapeutic strategies are aimed at overcoming the problem of Bcl-2 overexpression in cancer cells. The new facet in the biology of this remarkable protein could have implications for the design of novel therapeutic strategies aimed at targeting the redox modulating aspect of Bcl-2 in cancers.
Based on recent evidence that a slight increase in intracellular ROS is linked to cell survival, but an overwhelming increase in ROS could tilt the balance in favor of execution, (26, 27, 80), it is tempting to suggest that modulating the levels of intracellular ROS could be a novel strategy to overcome drug resistance in cancer cells. To that end, a combination approach of targeting the pro-oxidant Bcl-2 protein and ROS inducing compounds and/or other functional players involved in the interactome of pro-oxidant Bcl-2 appear as attractive modalities to garner an effective anticancer response (Table 1).
Acivicin: inhibits γ-glutamyltranspeptidase which limits GSH synthesis; Adaphostin and etoposide: ROS-inducing drugs; BSO: L-buthionine (S,R)-sulfoximine, which inhibits GSH synthesis; DEM: diethylmaleate, which conjugates with GSH and rapidly depletes GSH levels; Flavopiridol: cyclin-dependent kinase (CDK) inhibitor with ROS-inducing property; α-TOS: vitamin E analog α-tocopheryl succinate; U0126: 4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio) butadiene, which is a MEK inhibitor; Verapamil: a multidrug resistance protein 1 (MRP1) activator, which induces acceleration of GSH efflux.
Targeting Bcl-2 expression by antisense strategies
Downregulation of Bcl-2 via antisense oligonucleotide-based targeting of the mRNA has been used as a therapeutic modality. One such antisense oligonucleotide, G3139, an 18-mer phosphothiorate oligonucleotide called oblimersen (Genasense™), targets the first six codons of Bcl-2 mRNA (23). Genasense was able to reduce Bcl-2 expression by more than 80% in human breast cancer cells in vitro (23). Antisense-mediated repression of Bcl-2 reduction was associated with a significant reduction in cell viability (80%–95%) due to efficient induction of MOMP and caspase activation (23). Furthermore, in a separate study, Bcl-2 antisense oligodeoxynucleotide sensitized human acute lymphocytic leukemia Jurkat cells to the vitamin E analog α-tocopheryl succinate (α-TOS)-induced apoptosis, which was associated with an early production of ROS (73). In addition, the phosphorylation status of Bcl-2 on the serine 70 residue has been implicated where the loss-of-function mutant S70A was more sensitive to α-TOS-induced apoptosis and the gain-of-function mutant showed the opposite (73).
Interestingly, based on computer modeling studies, α-TOS was shown to adopt a unique hairpin conformation docking onto the hydrophobic groove of Bcl-2, thus acting as a BH3 mimetic (90).
Depletion of antioxidant GSH reservoir maintained by Bcl-2
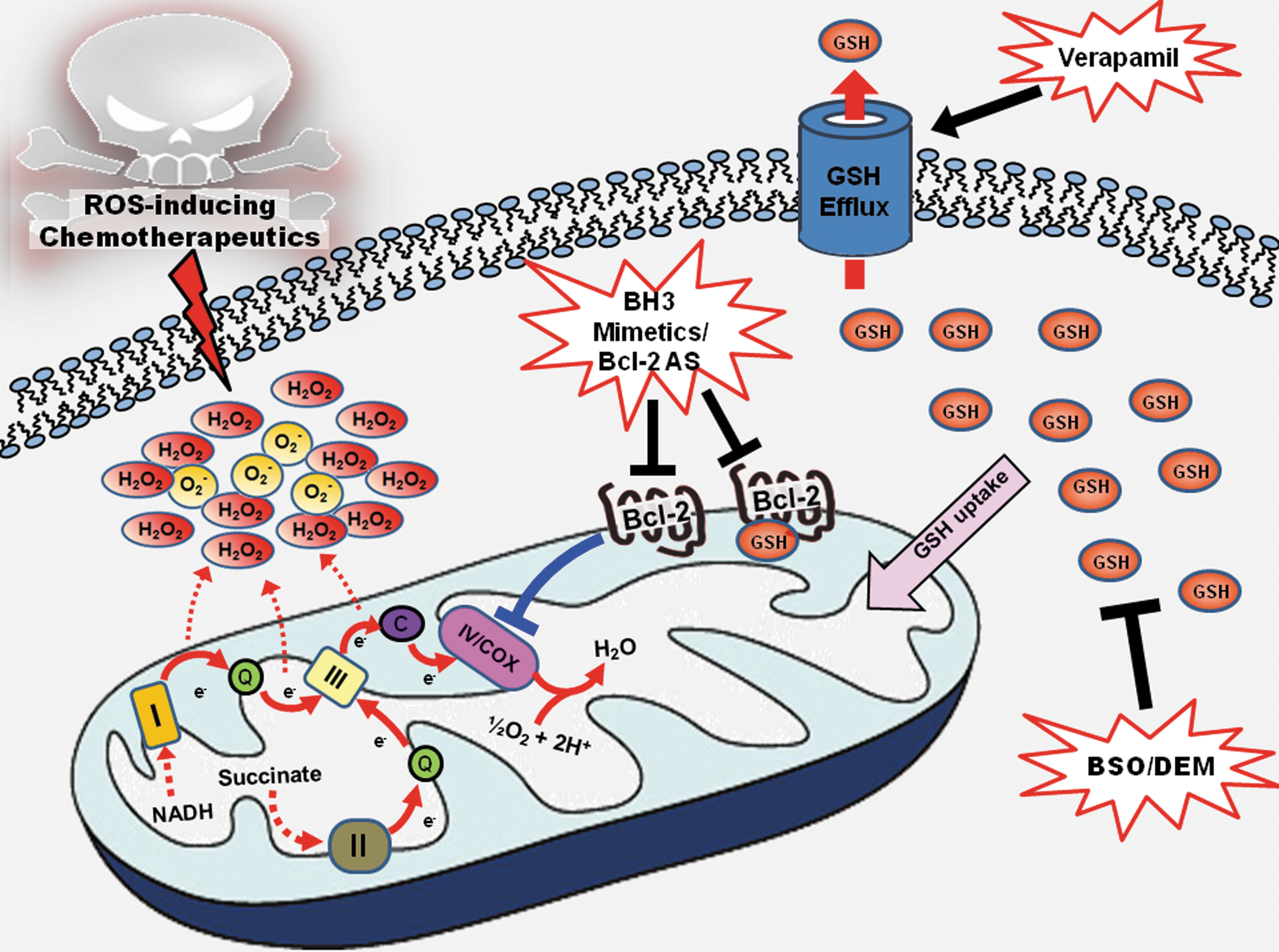
The ability of Bcl-2 to physically tether GSH to the mitochondria reinforces mitochondrial antioxidant capacity and renders Bcl-2-overexpressing cells resistant to oxidative stress. However, this dependence on GSH could be exploited in cells where Bcl-2 overexpression presents a hindrance to chemotherapeutic intervention. Indeed, in line with this prediction, pharmacological inhibition of GSH synthesis with L-buthionine (S,R)-sulfoximine (BSO) sensitized Bcl-2-overexpressing MCF-7 breast cancer and lymphocytic precursor FL5.12 cells to cisplatin-induced apoptosis (89) (Fig. 5). Similarly, depletion of the mitochondrial GSH pool sensitized Bcl-2-overexpressing B16 melanoma cells (B16M) with high (F10) metastatic potential to oxidative stress and tumor necrosis factor-α (TNF-α) in vitro (76). Interestingly, depletion of GSH by itself was sufficient to induce cell death in HL60 human leukemia cells overexpressing Bcl-2, while no significant effect on cell viability was observed in mock-transfected cells (5). Furthermore, GSH depletion by either inhibition of its synthesis or increasing its efflux was combined with Bcl-2 antisense approach to promote cell death within microvessels of metastasized B16M-F10 melanoma cells via endothelium-induced nitrosative and oxidative stress, thus abolishing their invasive capacity (Fig. 5) (11, 76). In yet another study, a sequential targeting therapy was used to deplete, step by step, mitochondrial GSH, Bcl-2, and MnSOD by antisense oligodeoxynucleotide, in combination with TNF-α and chemotherapies (taxol and daunorubicin), resulting in almost complete elimination of the highly resistant metastatic melanoma cells (10). These data provide evidence that targeting cellular redox status could constitute a novel therapeutic approach in combination with conventional chemotherapy agents, particularly in Bcl-2 overexpressing cancers.
Mimicking BH3 ‘only’ proteins-BH3 mimetics
A critical ratio of the anti-apoptotic and pro-apoptotic members of the Bcl-2 family is important in cell fate signaling in cancer cells. Several recent reports have described small molecule compounds that inhibit the anti-apoptotic activity of Bcl-2 by mimicking the actions of the pro-apoptotic BH3 domain, hence the name BH3 mimetics. Functional inhibitors of Bcl-2 were originally discovered based on computational modeling, structure-based design, or high throughput screening. One of the first bona fide BH3 mimetic compounds is TW-37, a benzenesulfonyl derivative, which was designed based on the structure of a naturally occurring Bcl-2 inhibitor, gossypol, derived from cottonseeds (99). TW-37 is a potent inducer of apoptosis in many forms of cancers such as mantle cell lymphoma, diffuse large B-cell lymphoma, acute lymphocytic leukemia (3, 67), and pancreatic cancer (102). A recent report demonstrated synergy between TW-37 and the MEK inhibitor U0126 in melanoma cells through ROS induced-activation of p53, which was highly selective thus reducing secondary toxicity to normal cells (99). The vulnerability of melanoma cells to handle ROS due to their intrinsic dysregulated redox capacity suggests that such kind of combinatorial approach could have an important place in the clinical management of this disease.
Another well-studied BH3 mimetic is ABT-737, developed by Abbott (Abbott Laboratories, Abbott Park, IL), which is derived as an analog of gossypol as well. It inhibits Bcl-2 with affinity two to three orders of magnitude higher than previously reported compounds (98). Interestingly, exposure of human acute lymphocytic leukemia Jurkat cells and human cervical cancer HeLa cells to ABT-737 resulted in a significant depletion in cellular GSH content, resulting in an increase in intracellular ROS production, caspase activation, and subsequent apoptosis (44). ABT-737 also synergized with other ROS-inducing drugs (adaphostin or etoposide) to induce apoptosis in cancer cells, thus suggesting a double hit effect of taking out Bcl-2 and increasing intracellular ROS (Fig. 5). However, ABT-737 is inactive in cells with phosphorylated Bcl-2 and/or overexpression of Mcl-1 due to its weak affinity for Mcl-1. Therefore, inhibition of Bcl-2 phosphorylation (either by using a MEK1 inhibitor or introduction of nonphosphorylatable mutants on all the three sites in the flexible loop region of Bcl-2), as well as suppression of Mcl-1 expression appear necessary to restore the sensitivity of cells with phosphorylated Bcl-2 and Mcl-1 expression (50, 98).
HA14-1 is another small molecule Bcl-2 inhibitor identified by computational modeling (101) and possesses redox altering capacity as well. ROS production was observed following HA14-1 treatment in HL60 human promyelocytic leukemia cells. Therefore, similar to other small BH3 mimetics that work through altering cellular redox status, a combinatorial approach of HA14-1 with ROS inducing agents is likely to have a synergistic effect in drug-resistant cells (Fig. 5). One such example of the ROS inducing agents is the cyclin-dependent kinase (CDK) inhibitor flavopiridol. Although flavopiridol was designed to be an inhibitor of CDK, studies on human leukemia cells showed that it was able to induce mitochondrial injury and ROS production leading to apoptosis (31). Combination of HA14-1 and flavopiridol resulted in apoptosis in human multiple myeloma cells, which are resistant to single agent treatment with HA14-1 or flavopiridol. The apoptotic response was associated with a marked increase in ROS production, and treatment with the antioxidant N-acetyl-L-cysteine attenuated both events. It is highly likely that HA14-1 interferes with Bcl-2's ability to modulate redox status and thus synergized with flavopiridol to induce potent oxidative stress leading to apoptosis (79).
Concluding Remarks
The expression of Bcl-2 functions as a double-edged sword. Overexpression of Bcl-2 has been correlated with chemoresistance in a number of hematologic as well as solid tumors. The increased expression prevents pro-apoptotic family members from targeting mitochondria and triggering MOMP, and at the same time regulates mitochondrial redox metabolism to generate a slight pro-oxidant intracellular milieu that provides cells with a survival advantage. As the death inhibitory activity of Bcl-2 has been linked to its interaction with a host of proteins, such as p53, Bax, GSH, and Cox Va, the conventional wisdom has been to target these interactions in an effort to relieve apoptotic effectors from the clutches of Bcl-2. However, with the new mechanistic insight into the biology of this remarkable protein, there is a strong argument for looking at therapeutic targeting of this protein in a different manner. Perhaps a combination strategy targeting both its mitochondrial stabilizing activity and its ability to modify cellular redox status could have potential for success in the clinical settings. Further understanding of how Bcl-2 acts as a redox “sensor”, in particular, how it regulates COX subunit assembly and activity during oxidative stress, would help greatly in the design of more specific therapeutics to abolish its death inhibitory function.