
Abstract
Junctional adhesion molecules are transmembrane proteins that belong to the immunoglobulin superfamily. In addition to their localization in close proximity to the tight junctions in endothelial and epithelial cells, junctional adhesion molecules are also expressed in circulating cells that do not form junctions, such as leukocytes and platelets. As a consequence, these proteins are associated not only with the permeability-regulating barrier function of the tight junctions, but also with other biologic processes, such as inflammatory reactions, responses to vascular injury, and tumor angiogenesis. Furthermore, because of their transmembrane topology, junctional adhesion molecules are poised both for receiving inputs from the cell interior (their expression, localization, and function being regulated in response to inflammatory cytokines and growth factors) and for translating extracellular adhesive events into functional responses. This review focuses on the different roles of junctional adhesion molecules in normal and pathologic conditions, with emphasis on inflammatory reactions and vascular responses to injury. Antioxid. Redox Signal. 15, 1221—1234.
JAM Proteins: Structure and Molecular Interactions
The members of the JAM family are glycoproteins composed of an extracellular domain, a single transmembrane segment, and a cytoplasmic tail of variable length (Fig. 1). Like other adhesion molecules, the JAM proteins contain immunoglobulin-like folds in their extracellular domain. Specifically, these proteins contain a membrane-distal and a membrane-proximal immunoglobulin-like fold (VH- and C2-type, respectively). Based on this molecular structure, the JAM proteins have been assigned to the larger CTX family, which was named after cortical thymocyte markers for Xenopus (4, 23). In turn, the JAM family comprises two subfamilies, the former composed of the three closely related proteins JAM-A (71, 125), JAM-B (29, 94), and JAM-C (1, 3), the latter composed of JAM-4 (41), JAM-like (JAML) (77), endothelium-selective adhesion molecule (ESAM) (42, 85) and the coxsackie- and adenovirus receptor (CAR) (24).

Several studies have reported on the molecular interactions of the JAM family members, which involve both the extracellular domain and the intracellular tail. The extracellular domain engages in different types of adhesive interactions. As a general rule (with some exceptions), homophilic and heterophilic interactions between the JAM proteins (Fig. 2, solid lines) serve the major purpose of linking adjacent endothelial and epithelial cells at the intercellular junctions. In contrast, heterophilic interactions between JAM proteins and their counterreceptors (mostly of the integrin superfamily; Fig.2, dotted lines) primarily link endothelial (or epithelial) cells with circulating leukocytes, as they occur in trans (i.e., between molecules expressed on different cells), even though other interactions have been reported to occur in cis (i.e., between JAM and integrin molecules expressed at the surface of the same cell). Similarly, platelet JAM proteins interact both with other JAM and with integrins, to adhere to the endothelium and to the leukocytes, respectively. Finally, when localized to the cell–cell contacts, many JAM proteins act as major components of the tight junctions, together with other molecules, such as occludin (14a), claudins (90a), and the zonula occludens proteins (38a).
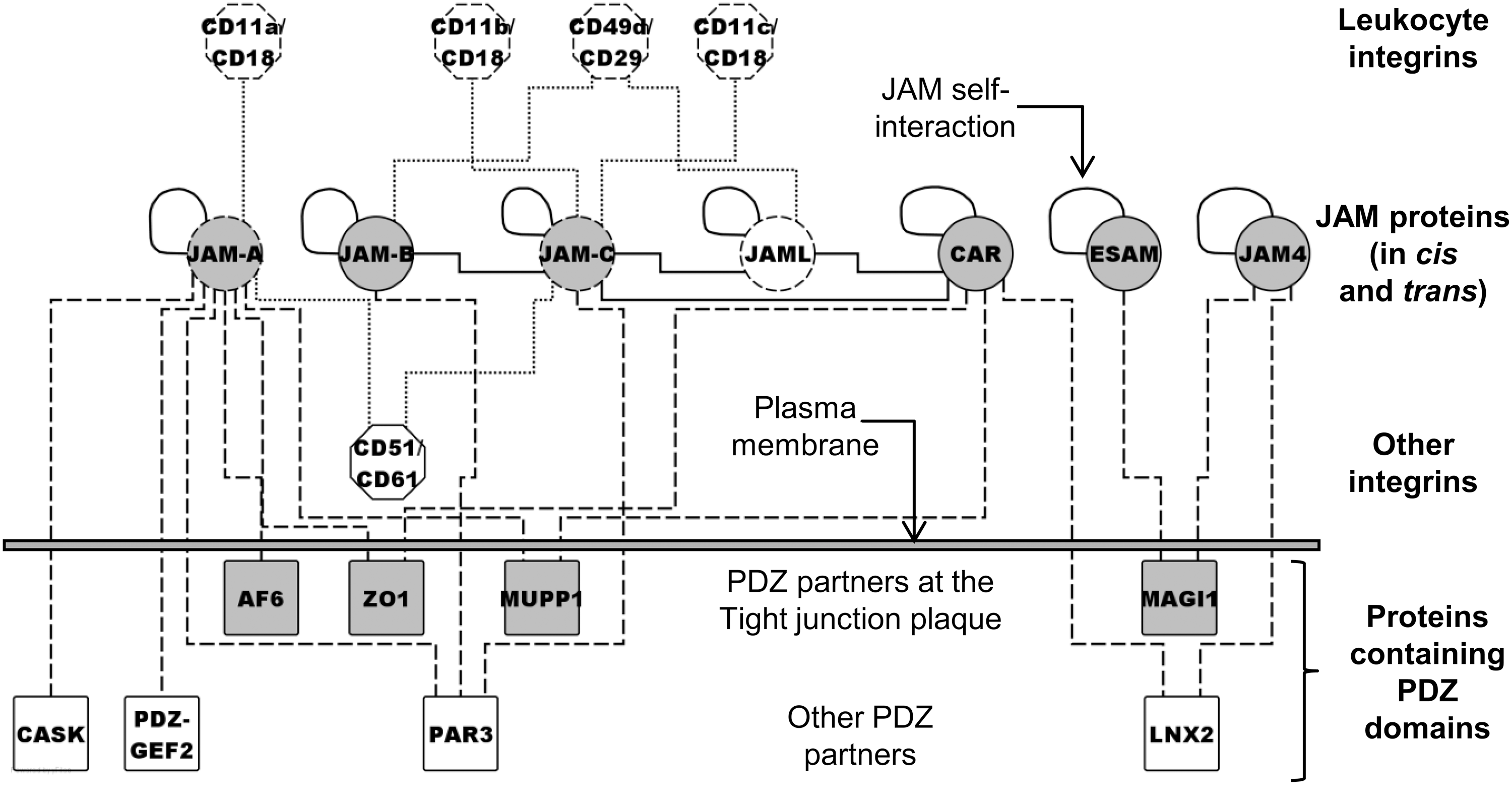
JAM-A (7, 12, 61), JAM-B (29), JAM-C (1), and the other JAM proteins (16) engage in homophilic binding interactions, albeit to different degrees. Concerning the heterophilic interactions, JAM-A (which is ubiquitously expressed on endothelial and epithelial cells, as well as on leukocytes and platelets) binds the leukocyte integrin CD11a/CD18 (αLβ2 or LFA-1; lymphocyte function–associated antigen-1) (90), the endothelial integrin CD51/CD61 (αvβ3) (79), as well as the Reovirus attachment protein σ-1 (8). Hereafter, integrins are designated according to the cluster of differentiation (CD) nomenclature. JAM-B (which is endothelium specific) binds two leukocyte ligands [i.e., JAM-C (1)], as well as the integrin CD49d/CD29 (α4β1 or VLA-4), in a way that requires prior interaction of endothelial JAM-B with leukocyte JAM-C (30). Furthermore, JAM-C (which is expressed rather ubiquitously) binds not only endothelial JAM-B but also the leukocyte integrins CD11b/CD18 (αMβ2 or Mac-1) and (albeit to a lesser extent) CD11c/CD18 (αXβ2 or p150/95) (55, 101), as well as CD51/CD61 (59) and CAR (75). Finally, JAML binds CD49d/CD29 (65), CAR (130), and JAM-C (75).
X-ray crystallographic analysis of JAM-A indicates that a tripeptide linker between the two immunoglobulin folds (Val-Leu-Val) imposes a bent conformation to the extracellular domain, whereas another tripeptide within the membrane-distal fold (Arg-Val-Glu) acts as a dimerization motif, thus suggesting that JAM-A proteins are expressed as homodimers in cis (with the shape of an inverted U) on the cell surface (53, 98). It also is predictable that these U-shaped in cis dimers on one cell self-assemble in trans with similar U-shaped JAM-A homodimers expressed on the surface of an adjoining cell (Fig. 3). The proposed mechanism might represent the molecular basis whereby interactions between JAM-A proteins contribute to homotypic cell–cell adhesion (e.g., between adjacent endothelial cells in a vascular monolayer) together with other classic adhesion molecules (e.g., cadherins). Interestingly, the BV11 antibody, which recognizes JAM-A dimers, blocks JAM-A self-assembly in vitro, thus supporting the hypothesis that formation of in cis dimers precedes the binding of in trans dimers (12).

Finally, a similar dimerization motif is also found in JAM-B (Arg-Leu-Glu) and JAM-C (Arg-Ile-Glu), thus suggesting that JAM-B and JAM-C also might form dimers and exploit dimerization as a means of establishing adhesive interactions.
Although all the JAM proteins studied so far are bona fide capable of homophilic interactions, their ability to seal the pathway of paracellular permeability in endothelial or epithelial cells remains an active area of investigation. In addition, to compound the issue, an individual JAM protein may engage in both homophilic and heterophilic interactions, often with different strengths. For instance, evidence of competition exists between some homophilic and heterophilic (i.e., JAM-C/JAM-C and JAM-C/JAM-B, respectively) interactions of JAM-C (55). Thus, depending on the cell type (and the cell type–specific repertoire of endogenously expressed JAM proteins), transfection of exogenous JAM-C may have different effects on the molecular architecture of the JAM-based junctions and, ultimately, on permeability, as discussed later (3, 70).
Concerning the molecular interactions that involve the intracellular tails, the cytoplasmic carboxyl termini of the JAM proteins represent consensus motifs for binding PDZ domains, modular structures that mediate protein–protein interactions (Fig. 2, dashed lines). PDZ is an acronym for postsynaptic density protein PSD95, Drosophila disc large-tumor suppressor DlgA, and zonula occludens ZO-1, which were among the first proteins to be characterized for the presence of this structural domain. The residues Phe-Leu-Val of JAM-A mediate the interaction with proteins containing type II PDZ domains, such as ZO-1 (13, 33, 37, 46), AF-6/Afadin (33), PDZ-guanine nucleotide exchange factor-2 (PDZ-GEF2) (104), CASK (73), PAR-3 (34, 46), and MUPP-1 (39), as evaluated by in vitro binding assays. Other JAM proteins (except JAML) display motifs for binding additional proteins containing either type II PDZ domains (JAM-B, Phe-Ile-Ile; JAM-C, Phe-Val-Ile) or type I PDZ domains (ESAM, Ser-Leu-Val; CAR, Thr/Ser-Val/Ile-Val; JAM-4, Thr-Leu-Val), and indeed, like JAM-A, also JAM-B and JAM-C bind PAR-3 (32). JAM-4 (41) and ESAM (123) bind MAGI-1, whereas JAM-4 (48) and CAR (76, 113) bind ligand-of-numb protein-X (LNX2). Finally, CAR also binds ZO-1 (24) and MUPP-1 (28).
Notably, the JAM proteins are transmembrane components of the intercellular tight junctions and, as mentioned earlier, they may contribute to the regulation of paracellular permeability by preventing the access of fluids and ions to the spaces between adjacent cells. In addition, some of the intracellular partners of the JAM proteins are structural components of the submembrane plaque of the tight junctions (e.g., ZO-1, AF-6/Afadin, MAGI-1, and MUPP-1), whereas other partners are either regulators of the polarized distribution of the tight junctions (e.g., PAR-3) or mediators of functional interactions between cell–cell and cell–matrix junctions (e.g., PDZ-GEF2), as summarized elsewhere (95) and as discussed later.
JAM Proteins and Leukocyte Extravasation
The cellular and molecular mechanisms that underlie and regulate the extravasation of leukocytes during inflammation have been subject of intense investigation in the past decades (114). Leukocyte ability to engage in adhesive interactions and to migrate across endothelial (and epithelial) barriers (9, 11) is essential to the process of inflammation. As JAM proteins are capable of mediating not only adhesive interactions between circulating leukocytes and vascular endothelial (or epithelial) cells, but also adhesive interactions at the junctions between adjoining endothelial cells [and epithelial cells (62, 68)], they are likely to be important mediators of leukocyte transmigration, as shown in several reports and as reviewed in (22, 86, 122). In addition, the expression levels and subcellular distribution of the JAM proteins are regulated on exposure to soluble mediators of inflammation.
JAM-A and leukocyte extravasation: molecular mechanisms
Concerning JAM-A, early studies provided evidence that JAM-A indeed contributes to leukocyte extravasation. Specifically, the BV11 antibody inhibits monocyte transmigration, both in a chemotaxis in vitro assay and in an in vivo model of skin inflammation (71). In addition, intravenous injection of BV11 reduces leukocyte accumulation in the cerebrospinal fluid, leukocyte infiltration in the brain parenchyma, and blood–brain barrier permeability in a cytokine-induced model of meningitis in mouse (31). However, anti–JAM-A antibodies do not reduce leukocyte accumulation (and may cause endothelial disruption) in a model of infectious meningitis, which occurs after infecting mice with either Listeria monocytogenes or the lymphocytic choriomeningitis virus (58).
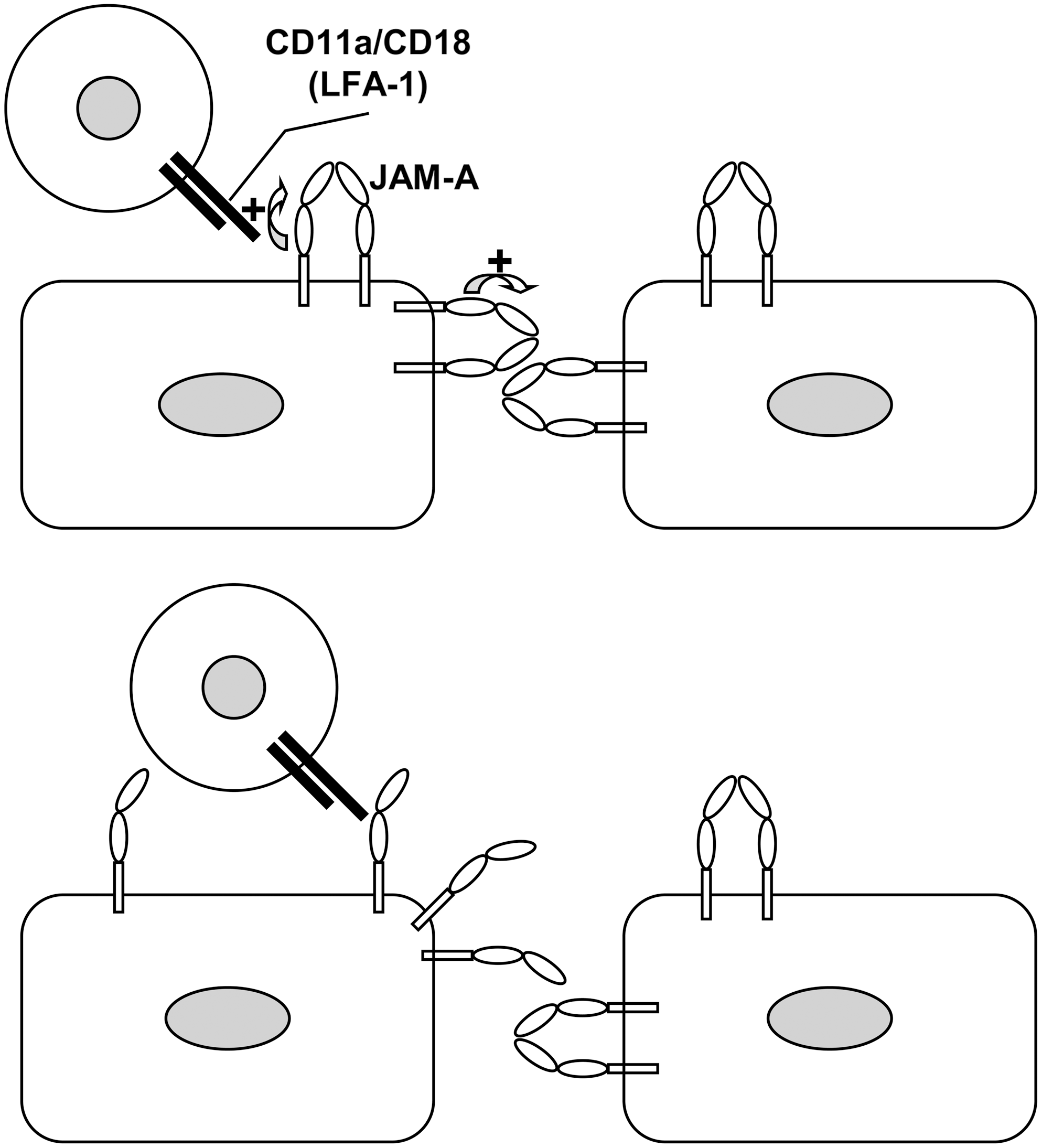
As JAM-A is expressed on both endothelial cells and leukocytes (125) and is capable of homophilic binding (12), the adhesive interaction between vascular endothelial cells and transmigrating leukocytes might rely (among others) on the binding between endothelial JAM-A and leukocyte JAM-A, by analogy with the homophilic binding of JAM-A at the junctions between adjacent endothelial cells. However, endothelial JAM-A can also engage in other adhesive interactions with different leukocyte counterreceptors. As mentioned earlier, JAM-A binds the leukocyte integrin CD11a/CD18 (90), which may account not only for leukocyte transmigration, but also for leukocyte arrest, in particular when cytokines induce redistribution of endothelial JAM-A from the intercellular junctions to the vascular surface, under both static (72, 91) and flow conditions (109). Whereas homodimerization (which may mediate the homophilic interaction of a JAM-A dimer with another JAM-A dimer) requires the membrane-distal immunoglobulin domain of JAM-A (12, 53), the heterophilic interaction of JAM-A with CD11a/CD18 requires the membrane-proximal immunoglobulin domain of JAM-A and the Inserted domain of CD11a (36). In addition, by atomic force microscopy, it has been found that the adhesive forces involved in the homophilic interactions between JAM-A monomers are actually lower than the forces involved in the heterophilic interactions between JAM-A and the integrin CD11a/CD18 (126). The molecular mechanisms underlying these interactions have been analyzed in detail (Fig. 4). In brief, under constitutive conditions, the membrane-proximal immunoglobulin domain of JAM-A stabilizes the homodimeric state and thus possibly the homophilic in trans interactions of the JAM-A homodimers. However, the interaction between the membrane-proximal immunoglobulin domain of JAM-A and the Inserted domain of CD11a removes such stabilizing action. As a consequence, the heterophilic interaction between endothelial JAM-A and leukocyte CD11a/CD18 predictably weakens the homophilic in trans interactions of the endothelial JAM-A dimers. As JAM-A stabilizes the integrity of the junctions (either directly by contributing cell–cell cohesion or indirectly by mediating hypothetically proadhesive signals), it is likely that weakening the homophilic interactions of JAM-A may ultimately facilitate the passage of the migrating leukocyte across the endothelial barrier (126).
JAM-A and leukocyte extravasation: in vivo models of inflammation
The availability of JAM-A–deficient mice (with either ubiquitous or endothelium-restricted defect) has provided proof that endothelial JAM-A is primarily responsible for leukocyte extravasation, even though its contribution depends on the type of stimulus and the type of migration. For instance, in JAM-A–null mice (or in wild-type mice treated with the BV11 antibody), leukocyte transmigration through the cremasteric venules is reduced in response to interleukin 1, but not leukotriene B4 or platelet-activating factor (127). Subsequent studies have shown that, in mediating the interleukin 1–stimulated transmigration, JAM-A acts in concert with other endothelial adhesion receptors, and that a transmigrating neutrophil interacts sequentially (i.e., on its way from the luminal to the abluminal side) with intercellular adhesion molecule-2, JAM-A and platelet endothelial cell adhesion molecule-1. In contrast, the tumor necrosis factor–stimulated transmigration does not depend on these adhesion molecules, as only leukocytes deficient for the tumor necrosis factor-α receptor (but not wild-type leukocytes) fail to migrate across the vessels of mice that do not express intercellular adhesion molecule-2, JAM-A or platelet endothelial cell adhesion molecule-1 (128). In addition, further analysis of tumor necrosis factor-induced transmigration under shear flow has shown that CD11a/CD18 (on the leukocyte), as well as intercellular adhesion molecule-1 and JAM-A (on the endothelial cell), do undergo redistribution and colocalization to a ringlike structure at the interface between leukocyte and endothelial cell. Nevertheless, the role of endothelial JAM-A is less clear when using neutrophils stimulated with phorbol esters, a condition (at variance with endothelial activation with cytokines) associated with enhanced avidity of the leukocyte integrins and not with the redistribution of endothelial JAM-A away from the junctions (108).
Finally, although these studies support an important (albeit stimulus-specific) role for JAM-A in leukocyte transmigration along the paracellular pathway (i.e., across the borders of adjacent endothelial cells), another study indicates that JAM-A is not involved in leukocyte transmigration along the transcellular pathway (i.e., through the interior of individual endothelial cells). In particular, although endothelial JAM-A takes part (together with platelet endothelial cell adhesion molecule-1 and CD99) in a vesicular compartment that surrounds the trans-migrating leukocyte, blocking JAM-A antibodies fail to inhibit this peculiar type of migration (66).
It is worth mentioning that, besides indirectly influencing leukocyte extravasation by regulating junctional dynamics, JAM-A may also regulate cell motility in a direct manner, as shown by the increased trafficking to the lymph nodes of dendritic cells in JAM-A–null mice (19). Along this line, it should be mentioned that JAM-A may also regulate the motility of epithelial (67) and endothelial cells (14, 43). The JAM-A–mediated regulation of epithelial and endothelial motility likely plays an important role in biologic contexts that are unrelated to leukocyte extravasation, such as the repair of wounds in intestinal and vascular monolayers. In particular, in epithelial cells, JAM-A enhances cell motility by virtue of a crosstalk with β1 integrins (106), which requires JAM-A homodimerization (103), the dimerization-dependent co-clustering of the JAM-A–associated (and PDZ domain–containing) proteins AF6/Afadin (33) and PDZ-GEF2 (104), as well as the PDZ-GEF2–dependent activation of the AF6/Afadin-associated GTPase Rap1A (15). In turn, Rap1A acts as an integrin activator and promotes the migration of the epithelial cell (Fig. 5).

JAM-C and leukocyte extravasation: in vitro and in vivo studies
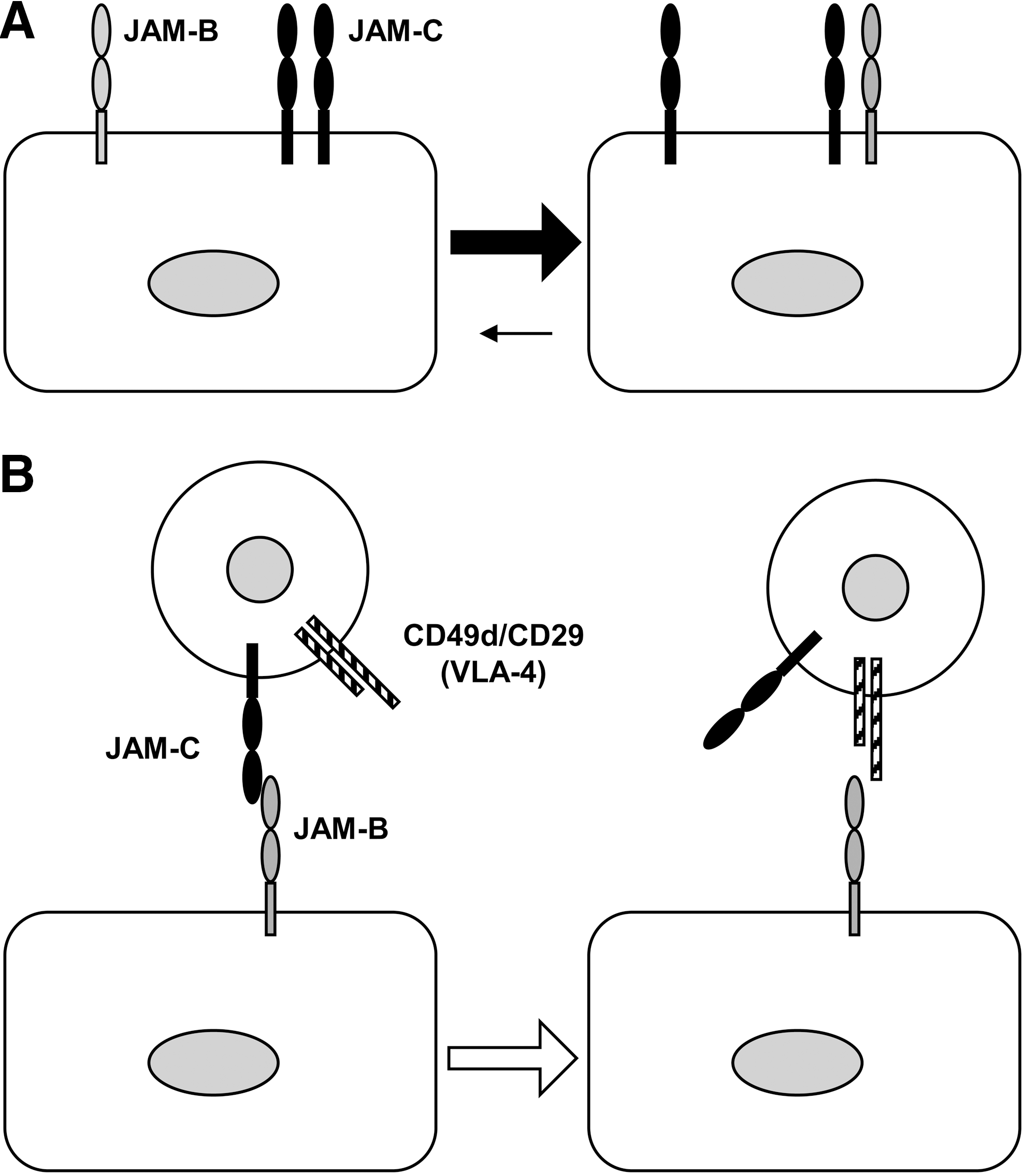
Like JAM-A, JAM-C is involved in the process of leukocyte extravasation. Early studies showed that murine JAM-C localizes to the high endothelial venules within lymph nodes and Peyer patches (3), which are sites of intense leukocyte trafficking. In addition, lymphocytes migrate in greater numbers across monolayers of endothelial cells transfected with JAM-C. Conversely, blocking reagents (i.e., an anti–JAM-C antibody and a soluble JAM-C peptide) reduce the number of leukocytes that transmigrate across JAM-C–expressing endothelial cells (47). The adhesive interactions involved in these responses offer intriguing scenarios. As already mentioned, JAM-C, which is expressed on both endothelium and leukocytes, binds JAM-B, which is restricted to the endothelium (1). An interesting competition exists between stronger heterophilic (JAM-C/JAM-B) and weaker homophilic (JAM-C/JAM-C) interactions (55), which suggests that leukocyte JAM-C may function as a high-affinity counterreceptor for endothelial JAM-B (Fig. 6A). Furthermore, in facilitating the interaction between endothelial JAM-B and the leukocyte integrin CD49d/CD29 (30), leukocyte JAM-C provides an intriguing example of a molecular crosstalk between JAM proteins and integrins that ultimately may facilitate leukocyte transmigration across the endothelium (Fig. 6B). Possibly as a consequence of these molecular dynamics, the interaction between leukocyte JAM-C and endothelial JAM-B was shown to facilitate the adhesion of lymphocytes, natural killer cells, and dendritic cells to the endothelium (60). Conversely, however, endothelial JAM-C also may play an important role, because it can act as counterreceptor for the leukocyte integrins CD11b/CD18 and CD11c/CD18. Actually, the binding of JAM-C with these integrins accounts for the adhesive interactions of leukocytes with different types of JAM-C–expressing cells, which include not only endothelial cells (21), but also epithelial cells (129) and platelets (56, 101). Specifically, blockade of JAM-C (with peptides and antibodies) inhibits neutrophil trans-endothelial migration (with almost complete inhibition in combination with the blockade of platelet endothelial cell adhesion molecule-1) (21), neutrophil trans-epithelial migration (129), and leukocyte–platelet adhesion (101).
More recently, other reports have defined the role of JAM-C in detail. For instance, JAM-C is likely to play a minor role in neutrophil extravasation under flow conditions, because JAM-C overexpression does not increase the response nor does JAM-C blockade inhibit it, even though a combination of blocking antibodies against JAM-C, platelet endothelial cell adhesion molecule-1, intercellular adhesion molecule-1, and CD99 has synergistic effects. Under the same flow conditions, JAM-C is expressed at uniformly low levels at the endothelial surface, sparsely at the junctions, but more abundantly in an intracellular pool (110). In addition, another emerging theme is that endothelial JAM-C (possibly with its endothelial counterreceptor JAM-B) is not primarily involved in facilitating the egress of leukocytes from the vasculature into the inflammatory site, but rather in preventing their reverse transmigration back into the circulation. Thus, it is predictable that the major role of endothelial JAM-C consists of retaining transmigrated leukocytes on the abluminal side (16). This explanation would account for the observation that JAM-C–null mice exhibit an increase in peripheral blood neutrophils during an inflammatory response (44). In other terms, the major effect of disrupting the endothelial JAM-B/JAM-C (or JAM-C/JAM-C) interaction is not just to decrease forward (i.e., luminal to abluminal) transmigration, but rather to increase reverse (i.e., abluminal to luminal) transmigration (17). In this respect, the initial studies that suggested a role for JAM-C in leukocyte extravasation may now find a novel mechanistic explanation. JAM-C absence (or JAM-C blockade) reduces the final number of extravasated leukocytes because of enhanced reverse transmigration. Conversely, JAM-C overexpression enhances the final number of extravasated leukocytes because of reduced reverse transmigration.
Finally, other JAM proteins have been reported to mediate adhesive interactions that may play a role in leukocyte extravasation. Specifically, blockade of JAM-B reduces lymphocyte rolling by preventing the interaction of JAM-B with the integrin CD49d/CD29, as assessed by intravital microscopy (63). In addition, ESAM-null mice show reduced extravasation of neutrophils, possibly by a direct control of endothelial junctions and vascular permeability (124). Furthermore, JAML, which is expressed on neutrophils, likely regulates transepithelial migration through binding interactions with CAR, which is expressed at the epithelial tight junctions, because blocking reagents against JAML and CAR inhibit neutrophil transmigration across epithelial monolayers (130). In contrast, JAML, when expressed on monocytes and lymphocytes (possibly as a monomer), engages in heterophilic interactions in cis with the CD49d/CD29 integrin. Then the interaction of JAML monomers with CD49d/CD29 causes accumulation of JAML dimers and ultimately enhanced adhesion of leukocytes to endothelial cells (65).
JAM Proteins and Inflammation
The ability of the JAM proteins to mediate the transmigration of leukocytes across endothelial and epithelial barriers has led investigators to examine the role of these molecules in both inflammatory models in mice and inflammatory diseases in humans. In addition to the models of skin inflammation (71) and meningitis (31, 58), the contribution of JAM-A to the inflammatory response has been studied with a model of experimental peritonitis in JAM-A–null mice. The study showed that JAM-A is required for the infiltration of neutrophils into the inflamed peritoneum, because, in the absence of JAM-A, neutrophils are impaired in their directional motility and remain entrapped between the endothelial cells of the mesenteric microvessels and the underlying basement membrane (27). Mechanistically, the ability of neutrophil JAM-A to regulate motility (in a cell-autonomous manner) is likely related to the control of integrin internalization (20).
Other studies have focused on the role of murine JAM-A in the intestinal epithelial barrier, which is possibly relevant to inflammatory bowel diseases in humans (46a, 105, 117). One study reported increased infiltration of neutrophils and larger lymphoid aggregates in the colonic mucosa of JAM-A–null mice compared with wild-type controls, as well as compromised barrier function of the intestinal epithelia, with enhanced paracellular fluxes of dextran and reduced trans-epithelial electrical resistance. Also, the JAM-A–defective mice are more susceptible to an experimental colitis induced with dextran sodium sulfate, even though the colonic mucosa shows less injury and increased epithelial proliferation (57). Another study confirmed that JAM-A–null mice display enhanced intestinal permeability and susceptibility to dextran-induced colitis, but also reported on enhanced epithelial apoptosis. Furthermore, the effect of JAM-A is epithelium specific, as Tie-2-Cre-JAM-A–null mice (with selective inactivation of JAM-A expression in endothelial cells) do not display the enhanced susceptibility to the disease (118).
Like JAM-A, JAM-C plays a role in regulating leukocyte transmigration in different tissues, thus possibly accounting for its involvement in diverse experimental models of inflammation. Specifically, administration of blocking antibodies against JAM-C (and JAM-B) before allergen challenge reduces neutrophil infiltration and the extent of allergic contact dermatitis in sensitized mice. Interestingly, the combined treatment with suboptimal concentrations of anti–JAM-C and anti–JAM-B antibodies has a synergistic effect (64). Similarly, in a model of cerulein-induced acute pancreatitis, anti–JAM-C antibodies reduce leukocyte infiltration, local inflammation, and necrosis of the pancreatic acinar cells, whereas endothelium-selective overexpression of JAM-C (in transgenic mice) exacerbates the disease (119). Also, anti–JAM-C antibodies reduce leukocyte accumulation in lung alveoli during acute pulmonary inflammation in mice (5). Furthermore, soluble JAM-C (which blocks the interaction of endothelial JAM-C with leukocyte CD11b/CD18) reduces neutrophil infiltration in a mouse model of thioglycollate-induced peritonitis (21).
JAM-C is also expressed in fibroblasts and endothelial cells of the synovial tissue. Actually, in experimental models of arthritis, a blocking JAM-C antibody reduces neutrophil infiltration into the inflamed synovium, decreases the severity of antigen-induced arthritis, and delays the onset of serum transfer–induced arthritis (93). Furthermore, blocking JAM-C antibodies reduce adhesion of myeloid U937 cells to fibroblasts from the rheumatoid arthritis synovium, but enhance U937 migration through a monolayer of these fibroblasts, thus reinforcing the hypothesis that JAM-C mediates leukocyte adhesion to synovial fibroblasts and retains the migrated leukocytes within the inflamed synovium (99).
As mentioned earlier, emerging evidence supports the involvement of the JAM proteins in human inflammatory diseases as well. To mention just few examples, JAM-A (and the associated PDZ domain–containing protein ZO-1) is abnormally expressed in active white matter lesions (possibly at the endothelial level) of patients with multiple sclerosis (92). Furthermore, JAM-A expression is reduced in specimens from patients with Crohn's disease and ulcerative colitis (118). Also, JAM-B is more abundantly expressed in endothelial cells from chronic bronchopneumonia and chronic nephritis compared with normal tissues (60), whereas JAM-C expression is increased in synovial endothelial cells from patients with rheumatoid arthritis and osteoarthritis (99). Finally, by virtue of their junctional localization, the JAM proteins are likely to be involved in pathologic conditions that affect the blood–brain and blood–retinal barriers, such as brain inflammation (24a) and diabetes (36a), as well as additional conditions characterized by oxidative stress (58a) and amyloid deposition (17a).
JAM Proteins and Vascular Diseases
Atherosclerosis
In addition to the inflammatory conditions described earlier, the JAM proteins also contribute to vascular reactions, such as atherosclerosis and ischemia/reperfusion injury, two different pathologic conditions that share (among other determinants) a well-documented dysfunction of the endothelium, which in turn causes key responses of the dysfunctional cells to oxidative stress. In particular, atherosclerosis is a chronic inflammatory disease of the arterial wall that involves different types of leukocytes, such as lymphocytes, monocytes, and macrophages (40). The recruitment of circulating leukocytes to the atherosclerotic lesions requires several adhesion molecules, including E-selectin, P-selectin, vascular cell adhesion molecule-1 and intercellular adhesion molecule-1, as well as JAM-A and JAM-C. The emerging theme is that reagents capable of blocking the adhesive interactions of the JAM proteins might represent novel tools to prevent and delay the onset of the atherosclerotic lesions.
One of the earliest suggestions that JAM-A might directly contribute to the inflammatory reactions of the dysfunctional vessel wall came from the observation that JAM-A on platelets, where it was originally cloned and named F11 receptor (52, 84, 112), mediates platelet adhesion to surfaces coated with recombinant soluble JAM-A. In addition, platelets adhere to cytokine-activated endothelial cells in a JAM-A–dependent manner, as this type of adhesion can be inhibited with recombinant soluble JAM-A and peptides that correspond to either the amino-terminal region or the membrane-distal immunoglobulin domain of JAM-A (7). Subsequent studies have shown that, besides recruiting platelets to inflamed vessels, endothelial JAM-A also facilitates the attachment of leukocytes to the activated endothelium of atherosclerotic lesions. Actually, JAM-A expression is increased in the atherosclerotic endothelium of carotid arteries from apolipoprotein E–null mice fed an atherogenic diet. Also, a soluble JAM-A-Fc chimera inhibits the CD11a/CD18-mediated binding of mononuclear cells to cytokine-stimulated endothelial cells, the chemotaxis of activated T lymphocytes, as well as the accumulation of mononuclear cells on the endothelium derived from atherosclerotic samples (89). Another study reported high levels of JAM-A (both messenger and protein) in atherosclerotic plaques of cardiovascular patients and apolipoprotein E–null mice, as well as in cytokine-treated endothelial cells from human aortic and venous vessels (6). Similarly, increased expression of JAM-A in blood vessels and infiltrating macrophages from inflammatory sites has been detected with microarray technology (111).
Concerning possible proatherogenic mechanisms, it has been shown that the combined deficiency of JAM-A and apolipoprotein E in mice reduces the extent of neointimal hyperplasia and the content of macrophages (but not smooth muscle and endothelial cells), as well as the arrest and transmigration of monocytes under flow. The arrest effect probably reflects reduced deposition of the platelet-derived chemokine RANTES on the luminal surface of endothelial cells in injured arteries (131). Furthermore, besides favoring the formation of the atherosclerotic lesions, initial evidence suggests that JAM-A may also facilitate the repair of the endothelial lesions by a mechanism involving the recruitment of circulating and JAM-A–positive CD34+ progenitor cells to inflamed endothelial cells and immobilized platelets, as well as the differentiation of these progenitors into mature endothelial cells, thus facilitating re-endothelialization of denuded vessels after injury (115).
Finally, although JAM-A is a transmembrane protein expressed at the surface of vascular endothelial cells, evidence also suggests circulating JAM-A in plasma. In addition, in patients (with known or suspected coronary artery disease) undergoing coronary angiography, the mean plasma levels of circulating JAM-A correlates with the severity of the disease and with the plasma levels of tumor necrosis factor, thus strengthening the association of JAM-A with atherosclerosis in humans (18). Recent evidence obtained in an inflammatory context may provide a cellular basis for this observation, as it indicates that the extracellular domain of JAM-A can be cleaved (with the consequent release of a soluble fragment) on the cytokine-induced activation of metalloproteases of the disintegrin family (51).
JAM-C also might play a role in atherosclerosis based on its ability to recruit inflammatory cells under proatherogenic conditions. Specifically, JAM-C expression is upregulated in atherosclerotic vessels and in spontaneous early lesions of apolipoprotein E–null mice. In addition, oxidized low-density lipoproteins (which is possibly relevant to the clinical condition of hyperlipidemic patients) not only upregulate JAM-C on arterial smooth muscle and endothelial cells but also redistribute JAM-C from the interendothelial junctions to the apical cell surface. As a consequence of the subcellular redistribution, JAM-C becomes capable of mediating the adhesion of circulating leukocytes, thus initiating the inflammatory response of the early atherosclerotic lesions (49). Besides favoring leukocyte infiltration, JAM-C may also favor neointima formation after arterial injury, because anti–JAM-C antibodies reduce neointimal hyperplasia and macrophage content in apolipoprotein E–null mice fed an atherogenic diet. In addition, the JAM-C antibodies inhibit monocyte adhesion to carotid arteries and to coronary artery smooth muscle cells (107). Finally, further to stress the role of the JAM proteins in linking thrombotic and immune responses in the pathogenesis of atherosclerosis, it is noteworthy that platelets, besides mediating adhesion of dendritic cells to injured carotid arteries, induce maturation and activation of the dendritic cells (with enhanced proliferation of lymphocytes). Remarkably, the platelet–dendritic cell interaction can be inhibited with soluble JAM-C, which blocks the interaction of JAM-C (on platelets) with CD11b/CD18 (on dendritic cells) (56).
Ischemia/reperfusion injury
Ischemia/reperfusion injury is a complex response that is due to the acute deprivation and the subsequent restoration of blood flow and oxygen, with production of reactive species, release of inflammatory mediators, enhanced expression of adhesion molecules, and accumulation of leukocytes. It may occur in many clinical conditions (e.g., infarction, trauma, and organ transplantation) and cause damage and dysfunction, at the cell, tissue, and organ levels (97). Evidence now indicates that JAM-A and JAM-C are among the adhesion molecules that are involved in the ischemia/reperfusion injury. Specifically, during the reperfusion of the liver, JAM-A expression is upregulated in the endothelium of the hepatic venules, where it may serve as a receptor for transmigrating leukocytes. Actually, neutrophil transmigration is attenuated in mice lacking JAM-A (both ubiquitously and in the endothelium only), as well as in wild-type mice treated with a blocking anti–JAM-A antibody. However, despite the reduced infiltration of neutrophils, the apoptotic injury of the hepatocytes is increased, possibly because of the retention of neutrophils under the endothelial lining of the hepatic vessels (50). Also, in JAM-A–null mice (or in wild-type mice treated with a blocking antibody), leukocyte transmigration through the cremasteric venules is reduced in response to ischemia/reperfusion injury (127). Whereas these findings point to endothelial JAM-A as mediator of the hepatic or muscle injury, leukocyte JAM-A might play a more important role in other organs. For instance, on ischemia/reperfusion injury in JAM-A–null mice, neutrophil infiltration in the heart is reduced, and high numbers of JAM-A–null neutrophils remain either adherent to the endothelium or entrapped between the endothelium and the basement membrane, which is possibly due to impaired motility of the JAM-A–defective neutrophils (27).
Endothelial JAM-C also mediates leukocyte adhesion and transmigration in response to ischemia/reperfusion injury. Blocking JAM-C with recombinant soluble JAM-C abolishes leukocyte migration across the vessels of kidney and cremaster muscle. Furthermore, both the adhesion and transmigration of leukocytes are suppressed in JAM-C–null mice, whereas they are enhanced in transgenic mice, which overexpress JAM-C in the endothelium. Interestingly, in injured tissues, endothelial JAM-C redistributes from the junctional and intra-cellular pools to the apical plasma membrane, which might facilitate the adhesion of circulating leukocytes (102).
Hypertension
Studies in rats suggest that JAM-A might also be involved in the long-term regulation of arterial pressure and exert a prohypertensive action within the brain stem, even though the proinflammatory action of JAM-A might still represent the underlying mechanism. In brief, in the brain and the peripheral circulation, the levels of JAM-A messenger are higher in spontaneously hypertensive rats compared with normotensive rats. It is unlikely that the enhanced expression is secondary to hypertension, as the levels of JAM-A messenger are higher not only in hypertensive rats but also in young rats at a prehypertensive age. Furthermore, the systolic pressure increases in adult normotensive rats after adenoviral-mediated expression of JAM-A in the nucleus of the solitary tract (121), a brain-stem structure that regulates the set point of arterial pressure. In an attempt at explaining the prohypertensive action of JAM-A, subsequent studies have led to the hypothesis that the enhanced levels of JAM-A in the microvascular endothelial cells of the nucleus of the solitary tract may cause localized adhesion of leukocytes and enhanced release of proinflammatory cytokines (e.g., monocyte chemoattractant protein-1), thus causing cardiovascular autonomic dysfunction and ultimately neurogenic hypertension (120). Notably, studies of JAM-A polymorphisms suggest that JAM-A might also play a role in human hypertension (87).
JAM Proteins and Tumors
Several studies have analyzed the JAM proteins in the context of tumor cells. Thus, although the link between oxidative stress and cancers remains an issue for debate, it is useful to summarize possible functional interactions between JAM expression and oncogenic transformation. In general, changes in the expression and function of several junctional proteins have been documented in transformed cells. Concerning the JAM proteins, work focusing on tumor cells supports the notion that oncogenic transformation is associated with increased expression levels of the JAM proteins and that the enhanced expression is associated with poor prognosis. Possibly, the negative effect is due to the increased ability of tumor cells to migrate onto the matrix and to transmigrate across the vessels wall, during local invasion and metastatic dissemination. For instance, in invasive breast cancer, upregulation of JAM-A (both messenger and protein) associates with poor prognosis. In addition, in MCF7 breast cancer cells, blocking JAM-A reagents decrease cell migration into a wound and reduce the levels of β1 integrin, another molecule essential for migration (74). Nevertheless, the correlation between JAM-A and motility in tumor cells is not straightforward. For instance, another study reported an inverse correlation in different breast cancer cell lines (with the low-motility MCF7 cells expressing high JAM-A levels and the high-motility MDA-MB cells expressing low JAM-A levels) and reported that JAM-A blockade in MDA-MB cells enhances collagen gel invasion (80). As migration is a complex response that relies on a balance between opposing forces, such as attachment to and detachment from the matrix, defining the precise role of any motility regulator requires careful evaluation of the endogenous levels not only of the regulator itself (e.g., JAM-A) but also of the actual effectors of migration (e.g., integrins). Finally, other studies suggest that, in addition to enhancing the motility of the tumor cells, JAM proteins might also facilitate tumor growth by inhibiting immune responses. For instance, the growth and aggressiveness of pancreatic islet cell carcinoma (induced by SV40 T antigen expression in beta cells) are reduced in JAM-A–null mice. Because these tumor cells do not express JAM-A, the effect of JAM-A depends on changes in stroma reactivity and, in particular, on increased transmigration of dendritic cells (the effect being attributable to dendritic or endothelial JAM-A or both) and lymphocytes (78).
Besides JAM-A, other JAM proteins have been involved in tumors. In particular, JAM-B expression is observed in the vessels that are adjacent to carcinomas of colon, lungs, mammary gland, and testis (60). In addition, JAM-C is expressed in cancer cell lines from tumors with high metastatic potential. Small interfering RNA of JAM-C in HT-1080 fibrosarcoma decreases cell adhesion to extracellular substrates and Matrigel invasion, whereas transfection with JAM-C exerts opposite effects in vitro and decreases the life span in mice (38). Furthermore, the levels of JAM-C (together with other cell-surface molecules) are increased in a HT-1080 variant with enhanced ability to intravasate and disseminate (25). Human gliomas express both JAM-C and JAM-B, whereas blocking antibodies against JAM-C and JAM-B inhibit glioma growth and invasion in vivo (116). Finally, in addition to enhancing motility, JAM-C may also facilitate the adhesive interaction between tumor and endothelial cells, which is also central to the metastatic process. For instance, the lung carcinoma cell line NCI-H522, which expresses JAM-C, specifically adheres to immobilized JAM-C, to JAM-C–transfected Chinese hamster ovary cells, and to endothelial cells. The interaction with the endothelial cells is abolished in the presence of soluble JAM-C (100).
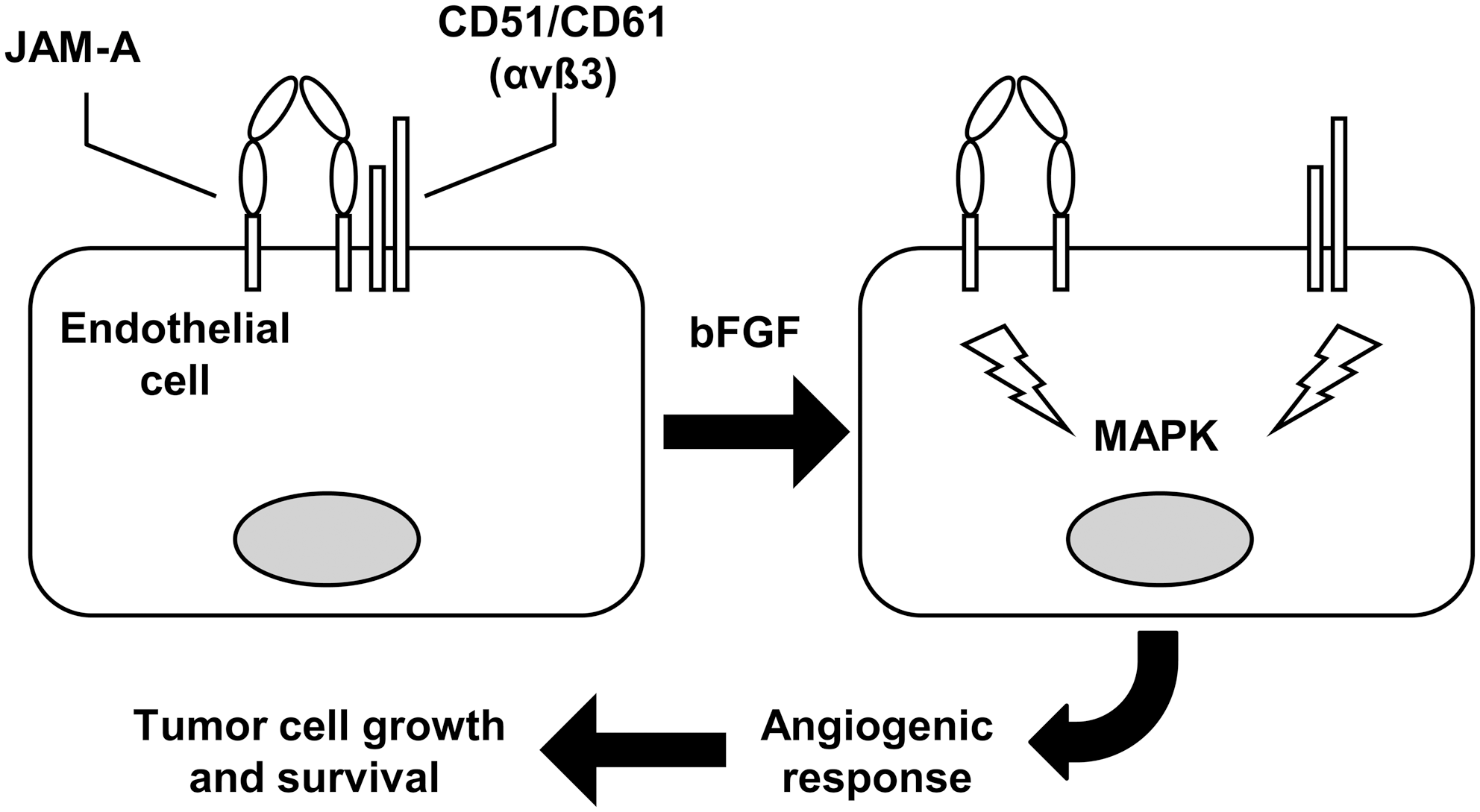
Besides facilitating tumor cell adhesion and migration, the JAM proteins may also mediate other interactions of the tumor cells with the tumor microenvironment. For instance, several studies suggest that JAM-A plays a role in vascular angiogenesis (83, 96), one of the key steps of tumor growth, which requires migration and proliferation of the endothelial cells, as well as interactions between receptors for growth factors and integrins (Fig. 7). Specifically, basic fibroblast growth factor disrupts the association of JAM-A with the endothelial integrin CD51/CD61, which is normally observed in quiescent cells. Then the consequent dissociation of the two adhesion receptors allows the propagation of proangiogenic signals, such as mitogen-activated protein kinase activation (79). JAM-A is also important for the CD51/CD61-dependent migration of endothelial cells onto vitronectin (81, 82). Actually, in JAM-A–null mice, basic fibroblast growth factor-induced angiogenesis is impaired (26).
JAM-C also contributes to angiogenesis, as a monoclonal antibody directed against JAM-C reduces tumor growth and infiltration of macrophages by inhibiting angiogenesis. In addition, the antibody reduces angiogenesis in two experimental models [i.e., hypoxia-induced retinal neovascularization and vessel outgrowth from aortic rings (54)]. Furthermore, JAM-C contributes to permeability, which is essential to angiogenesis, because disruption of JAM-C function decreases basal permeability and prevents increases in microvascular permeability in response to histamine or vascular endothelial growth factor. The loss of JAM-C expression stabilizes VE-cadherin–mediated interendothelial adhesion in a manner dependent on the small GTPase Rap1 (88).
Finally, ESAM plays a role in tumor angiogenesis, as seen in ESAM-null mice (45).
Concluding Remarks
This overview indicates that the JAM proteins are involved in diverse tissue reactions to various conditions of oxidative stress, ranging from acute inflammation to tumor growth. Although evidence comes from different types of experimental models, emerging hypotheses are beginning to integrate in vitro data (at the molecule and cell level) with in vivo findings. Furthermore, initial evidence is accumulating to support a role of this class of molecules in human pathology. Therefore, these findings point to the opportunity to characterize blocking reagents of JAM function as therapeutic targets (2), and it is foreseeable that therapeutic tools of potential interest for several inflammatory and vascular conditions will be developed in the ensuing years.
Furthermore, the involvement of the JAM proteins in the multistep process of leukocyte extravasation illustrates how all the disparate pieces of information gathered in the past decade will require integration into a more general framework that will provide a coherent and mechanistic view. Providing a comprehensive overview of information on cell type–specific expression patterns, hierarchy of interactions between the extracellular domains, coordination between extracellular adhesive events with intracellular signaling responses, and temporal sequence of events will undoubtedly advance our understanding of the JAM-driven responses in biologic settings as complex as the inflammatory and vascular diseases.
Footnotes
Acknowledgment
The generous contribution of the Negri-Weizmann Foundation is gratefully acknowledged.