
Abstract
Cerebral amyloid angiopathy (CAA) is frequently observed in Alzheimer's disease (AD) and is characterized by deposition of amyloid beta (Aβ) in leptomeningeal and cortical brain vasculature. In 40% of AD cases, Aβ mainly accumulates in cortical capillaries, a phenomenon referred to as capillary CAA (capCAA). The aim of this study was to investigate blood–brain barrier (BBB) alterations in CAA-affected capillaries with the emphasis on tight junction (TJ) changes. First, capCAA brain tissue was analyzed for the distribution of TJs. Here, we show for the first time a dramatic loss of occludin, claudin-5, and ZO-1 in Aβ-laden capillaries surrounded by NADPH oxidase-2 (NOX-2)-positive activated microglia. Importantly, we observed abundant vascular expression of the Aβ transporter receptor for advanced glycation endproducts (RAGE). To unravel the underlying mechanism, a human brain endothelial cell line was stimulated with Aβ1-42 to analyze the effects of Aβ. We observed a dose-dependent cytotoxicity and increased ROS generation, which interestingly was reversed by administration of exogenous antioxidants, NOX-2 inhibitors, and by blocking RAGE. Taken together, our data evidently show that Aβ is toxic to brain endothelial cells via binding to RAGE and induction of ROS production, which ultimately leads to disruption of TJs and loss of BBB integrity. Antioxid. Redox Signal. 15, 1167–1178.
Introduction
The BBB is a tight sealed barrier between the circulating blood and the central nervous system (CNS), consisting of brain microvascular endothelial cells that are surrounded by basement membranes, astrocytic endfeet, and pericytes. The brain microvascular endothelium is characterized by the presence of tight junctions (TJs) and lack of fenestrae, thereby limiting the entry of plasma components, red blood cells, and leukocytes into the CNS, and confer the low paracellular permeability and high electrical resistance of the BBB (16, 50). TJs are complex structures located at the apical region between endothelial cells and are composed of connecting transmembrane proteins (occludin and claudins) that form the primary seal linked via accessory cytoplasmic proteins (zona occludens family members) to the actin cytoskeleton (16).
Occludin is a phosphoprotein with four transmembrane domains and intracellular amino and the carboxyl termini (6a). Occludin expression is associated with increased electrical resistance and decreased paracellular transport. Claudins comprise a multigene family consisting of more than 20 members and contain two extracellular loops and four transmembrane domains and interact in both a homophilic and heterophilic way with claudins of adjacent cells. Claudin-5 is a critical component of the BBB as it prevents the passage of molecules larger than 800 Da (28). Carboxyterminal parts of both occludin and claudins interact with membrane-associated recruiting proteins of the zona occludens (ZO) protein family. ZO proteins are reported to link transmembrane proteins to the actin cytoskeleton and have signaling potential (14).
Leakage of the BBB in AD has been suggested by the detection of plasma proteins associated with amyloid plaques (1, 21, 45) and within AD brain parenchyma (21, 45, 49). Likewise, in CAA, an impaired barrier function was detected associated with cerebrovascular Aβ deposits (45). Opening of the BBB and concomitant altered TJ expression or localization has been attributed to vascular Aβ aggregates (6, 15, 27, 36), which in turn are able to induce reactive oxygen species (ROS) production, mainly generated by NADPH oxidase (NOX), in neuronal and non-neuronal cell cultures (4, 23, 47). Both endogenous and exogenous ROS induce loss of endothelial cell–cell interactions (40) and are able to modulate BBB integrity and disrupt TJs (22, 34). However, to date, the link between Aβ, ROS production and TJs alterations remains elusive.
The involvement of RAGE appears to be very important in the development of the AD and CAA pathology, since RAGE mediates the influx of Aβ into the brain parenchyma and consequently in an unbalanced situation enhances Aβ accumulation. RAGE is also known to be critical regarding the effects exerted by Aβ through its binding to the transporter. Aβ/RAGE interaction has been reported to activate NOX and a cascade of effects such as NF-kB-mediated endothelial activation resulting in secretion of proinflammatory cytokines, the expression of adhesion molecules and suppression of cerebral blood flow (50).
In this study, we combine neuropathological findings in unique brain samples of capCAA patients and show a dramatic loss of TJ proteins in Aβ-laden capillaries. Interestingly, capCAA-affected vessels are surrounded by NOX2-immunopositive activated microglia. We next investigated the link between Aβ toxicity and TJ changes using a human cerebral microvascular endothelial cell line and demonstrated that cytotoxic Aβ via production of ROS, decreased TJ proteins expression which could be rescued by exogenous antioxidants, NOX-2 inhibition, and RAGE blocking antibody.
Materials and Methods
Postmortem tissue
Six patients with extensive capCAA and two age-matched non-demented controls were selected. Human brain specimens were obtained at autopsy with a short postmortem interval (The Netherlands Brain Bank, Amsterdam, The Netherlands and University Medical Centre in Utrecht, The Netherlands). Neuropathological evaluation was performed on frozen tissue and formalin-fixed, paraffin-embedded tissue from occipital pole cortex. CapCAA score was defined as follow: severe (+++), moderate (++), and mild (+). Staging of AD was evaluated according to Braak and colleagues (7). Age, gender, postmortem delay (PMD), Braak, CERAD, and CAA scores and cause of death of all cases used in this study are listed in Table 1.
Immunohistochemistry
For immunohistochemical staining, 5-μm cryosections were air-dried and fixed in acetone for 10 min. Next, sections were preincubated with normal goat serum 1:10, diluted in phosphate buffered saline (PBS) containing 1% bovine serum albumin (Roche Diagnostics, Mannheim, Germany) for 10 min. Sections were incubated O/N with primary antibodies: anti-occludin (mouse, Zymed), anti-ZO1 (rabbit, Zymed) (Table 2) diluted in PBS containing 1% bovine serum albumin. Subsequently, sections were incubated with EnVision goat-anti-mouse horseradish peroxidase (HRP) or EnVision goat-anti-rabbit HRP (Dako, Glostrup, Denmark) for 60 min. Peroxidase labeling was visualized by EnVision 3,3-diaminobenzidine 1:50 (EV-DAB; Dako). Sections were counterstained with hematoxylin. Finally, tissue sections were rinsed with 70% ethanol prior to a 20 min incubation with 50 ml saturated NaCl solution (0.5 M NaCl in 80% ethanol) which was supplemented with 0.5 ml 1% NaOH solution. Then sections were transferred to saturated Congo Red (VWR internationaal, Leuven, the Netherlands) solution supplemented with 0.5 ml 1% NaOH solution for 20 min. Congo Red staining was used to visualize Aβ fibrils. Between all incubation steps, sections were extensively washed with PBS (pH 7.4). PBS served as negative control.
Paraffin sections (5 μm) were mounted on coated glass slides (Menzel Gläzer super frost PLUS, Brainschweig, Germany) and dried O/N at 37°C. Sections were deparaffinized and rehydrated by xylene and a sloping concentration of ethanol (100%, 96%, and 70%). Endogenous peroxidase was blocked by incubating the sections in methanol+0.3% H2O2 for 30 min. Antigen retrieval was established by boiling the sections in 1 mM EDTA buffer for 10 min. Then, sections were O/N incubated with anti-claudin-5 (mouse, Zymed) or anti-NOX2 (mouse) (see Table2) diluted in PBS supplemented with 1% BSA. Then the sections were incubated for 30 min with EnVision anti-rabbit/anti-mouse HRP. Peroxidase labeling was visualized by EVDAB 1:50. Sections were counterstained with hematoxylin. The sections were stained with Congo Red as described above. Finally sections were rinsed twice with 100% ethanol, put in xylene and covered with DePeX mounting medium (Gurr, Germany). Between all incubation steps, sections were extensively washed with PBS (pH 7.4). PBS served as negative control.
Immunofluorescence
For co-localization studies, cryosections were incubated in thioflavin S solution (100 mg/ml) for 5 min to stain Aβ fibrils and washed subsequently three times in ethanol 70%. Sections were preincubated with normal goat serum 1:10 for 10 min and incubated O/N with a mix of primary antibodies: anti occludin/claudin-5 and anti factor VIII or anti ZO-1/RAGE and anti CD31 diluted in PBS containing 1% bovine serum albumin. Sections were then incubated with secondary antibodies: Cy5 labeled goat-anti-rabbit 1:100, diluted in EnVision goat-anti-mouse HRP (Dako) for 30 min. Peroxidase labeling was visualized by reaction with rhodamine-tyramide (1:3000) in presence of 0.01% of H2O2 for 5 min. After washing, slides were covered with Vectashield (Vector laboratories, Burlington, ON, Canada). Between all incubation steps, sections were extensively washed with PBS (pH 7.4). Fluorescent analysis was performed with a Leica TCS SP2 AOBS confocal laser-scanning microscope (Leica Microsystem, Heidelberg, Germany). Quantification of TJ protein-expressing vessel was also performed normal vessels versus Aβ-laden vessels. Four fields per slides were counted and a ratio was calculated (magnification X10).
Cell culture
A human cerebral microvascular endothelial cell line (hCMEC/D3) (44) was maintained in EBM-2 medium (Clonetics, Cambrex BioScience, Wokingham, UK) supplemented with VEGF, IGF-1, EGF, basic FGF, hydrocortisone, ascorbate, gentamycin, and 2.5% fetal bovine serum (FBS) 40. T75 flasks, 96-well plates and 24-well plates were coated with type 1 collagen (Gibco HBSS, Invitrogen, Carlsbad, CA). hCMECs were detached at 37°C with 2 ml trypsin/EDTA in PBS. Cultures were grown to confluence at 37°C in 5% CO2 until the formation of monolayers.
Aβ1-42 preincubation
Synthetic Aβ1-42 (Bachem, Bubendorf, Switzerland) was dissolved in 0.1% ammonium hydroxide and stored in 50 μl, 1 mM aliquots at−80°C. 40 μM Aβ1-42 was preincubated in EBM-2 medium without FBS for 3 days in order to form aggregates. Then Aβ1-42 was further diluted in cell medium to obtain appropriate concentrations.
Electron microscopy
Pre-aggregated Aβ1-42 was applied to formvar carbon-coated copper grids (Stork Veco BV, Eerbeek, The Netherlands) and dried for 10 min. Grids were negatively stained with uranyl acetate for 5 min and examined with a Zeiss EM109 electron microscope to visualize the formation of fibrils.
MTT assay
The cytotoxicity of synthetic Aβ1-42 preparations was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma Aldrich, Germany) assay. hCMECs were cultured in 96-well plates until they reached 100% confluence. Cells were incubated for 24 h with different concentrations of Aβ1-42 (1 nM, 10 nM, 100 nM, 1 μM, 10 μM). Antioxidants and blocking antibody anti-RAGE were applied 2 h prior Aβ incubation and were still present during the entire treatment. Then cells were incubated with MTT (1 mg/ml) for 3 h at 37°C. The formazan-salt generated by mitochondria of viable cells as a result of conversion of MTT was dissolved in glycin/DMSO (ratio 1:6) and the absorbance was measured at 540 nm.
Live/dead assay
Using the LIVE/DEAD Viability/Cytotoxicity Assay Kit (Molecular Probes Inc, Eugene, OR) living and dead cells can be distinguished from each other. hCMECs were cultured in 96-well plates until they reached 100% confluence. Cells were incubated for 24 h with different concentrations of Aβ1-42 (1 μM, 10 μM, 20 μM). Cells were washed gently with warm PBS. Then 1 μl calcein AM and 1.5 μl ethidium homodimer 1 were added to warm EBM-2 medium and 100 μl of this mixture was added per well for 20 minutes. The cell permeable calcein AM is converted into the green fluorescent calcein by intracellular esterase activity (excitation ∼495 nm; emission ∼515 nm). Ethidium homodimer 1 is able to enter cells with damaged membranes. It undergoes a 40-fold enhancement of fluorescence upon binding to nucleic acids and produces a red fluorescent signal in dead cells (excitation ∼495 nm, emission ∼635nm). Four 10 times magnified fields were counted and a ratio was calculated for live and dead cells.
Amplex Red assay
hCMECs were cultured in 96-well plates until they reached 100% confluence. Cells were treated with different concentrations of Aβ1-42 (1 nM, 10 nM, 100 nM, 1 μM, and 10 μM) for 24 h. After incubation, Aβ was removed and H2O2 production was detected using the Amplex red fluorescent dye (Molecular Probes, Breda, the Netherlands) which reacts 1:1 with H2O2 in the presence of horseradish peroxidase, producing highly fluorescent resorufin. Fluorescence was detected at 37°C in a fluorimeter (Galaxy-Fluostar, BMG, Offenburg, Germany). The excitation and emission fluorescent wavelengths were 550 and 590 nm, respectively. The calibration signal was produced by addition of known amounts of H2O2 added to the reaction mix. The velocity whereby H2O2 was produced was calculated using non-linear regression.
mRNA isolation and real-time quantitative PCR
To investigate mRNA expression of TJ proteins cells were grown on a 24-well plate until they reached 100% confluency. Cells were incubated with different concentrations of Aβ1-42 (10 nM, 100 nM, 1 μM) for 24 h. mRNA was isolated by using the mRNA capture kit (Roche Applied Science, Almere, the Netherlands) following the manufacturer's protocol. mRNA was reverse transcribed using the Reverse transcription system kit (Promega, Madison, WI) according to the manufacturer's instructions using GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA). cDNA was diluted three times and quantified for mRNA levels of occludin/claudin-5/ZO-1 relative to the housekeeping gene GAPDH. The accumulation of PCR product is measured using Sybergreen II (Applied Biosystems). Primers were developed using the program Primer Express 2.0 (Applied Biosystems). The sequences of primers are as follows: human occludin: sense 5′-CCCGTTTGGATAAAGAATTGG-3′, antisense 5′-TCAAACAACTTGGCATCAGA-3′; human ZO-1: sense 5′-CCCGAAGGAGTTGAGCAGGAAATC-3′, antisense 5′-CCACAGGCTTCAGGAACTTGAGG-3′. The PCR amplification was performed in triplicate in a 7900 HT Fast Real-Time PCRSystem (Applied Biosystems). Relative expression levels of TJ proteins in relation to the reference GAPDH were calculated using the mathematical model: ΔΔCT. The formula is 2-ΔΔCT where ΔCT=CTtarget - CTreference and ΔΔCT=ΔCTsample - ΔCTcalibrator.
Statistical analysis
Data were analyzed statistically by Student's t-test or analysis of variance (ANOVA) followed by post hoc analysis with Bonferroni's method (*P<0.05, **P<0.01, ***P<0.001).
Results
Reduced tight junction protein expression in CapCAA-affected vessels
Using Congo Red, we observed extensive Aβ deposits throughout the occipital cortex in capillaries and larger vessels. No Aβ was detected in brain vessels of control brains. To investigate the expression of TJ proteins, postmortem tissue of 6 capCAA patients and 2 non-neurological controls was stained for occludin, claudin-5, and ZO-1. We observed a normal vascular expression pattern of TJ proteins in control tissue capillaries and in vessels not affected by Aβ deposition in samples from capCAA patients (Figs.1A, 1C, and 1E). Interestingly, we observed a marked reduction or even complete loss of occludin, claudin-5 and ZO-1 staining in CAA-affected capillaries (Figs.1B, 1D, and 1F). Quantification based on triple fluorescent staining for Aβ, TJs and an endothelium marker confirmed significant loss of TJ proteins expression in Aβ-laden capillaries compared to nonaffected capillaries (Figs. 2d–2f).


Aβ-laden capillaries are surrounded by NOX-2-positive activated microglia
Aβ is known to induce ROS-generating enzymes, including NOX-2 in microglia. NOX-2 is constitutively expressed by microglial cells and under physiological conditions NOX-2 activity is low. However, NOX-2 is strikingly upregulated in response to acute and chronic stimuli, including Aβ (24, 29). We show that NOX-2 is expressed in microglial cells in control brain tissue (Fig. 3A), however NOX-2 is abundantly and widely expressed in microglia throughout capCAA-affected tissue. Cells stained positive for NOX-2 were recognized as microglia based on their morphology (e.g., characteristic long branching processes and a small cellular body). Particularly, Aβ-positive capillaries are engulfed by NOX2-immunoreactive microglia (Fig. 3B), strongly suggesting increased ROS production in close vicinity of Aβ-laden capillaries with TJ changes.

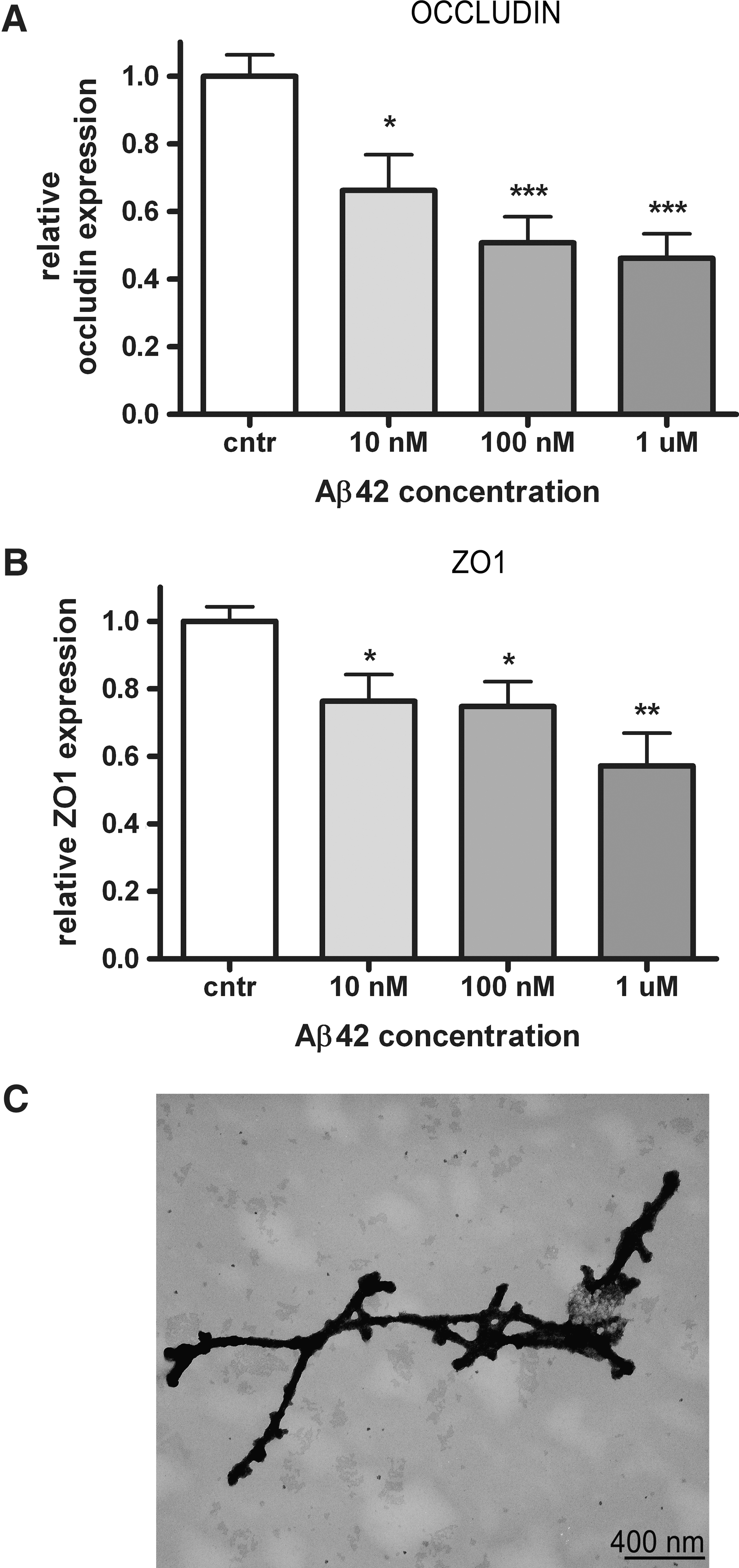
Aβ induces occludin and ZO-1 mRNA downregulation
Immunohistopathological findings showed reduced TJ expression in Aβ-laden capillaries. To investigate the direct effects of Aβ on mRNA expression of TJ proteins, we examined the effects of Aβ1-42 on occludin, claudin-5, and ZO-1, using a human cerebral microvascular endothelial cell line (hCMEC/D3) (44). Endothelial cells treated for 24 h with increasing concentration of Aβ fibrils (Fig. 4C)showed a dose-dependent significant reduction of occludin and ZO-1 transcripts (max reduction of 55% and 45%, respectively) (Figs. 4A and 4B). Remarkably, no changes in claudin-5 mRNA were detected (data not shown). These results are in line with the loss of occludin and ZO-1 in capCAA tissue, and suggest that reduced TJ protein expression might be caused by Aβ deposits in the microvasculature.
Aβ is toxic to brain endothelial cells via enhanced ROS production
In order to test which concentration of Aβ is lethal to brain endothelial cells, we performed a live/dead assay. Hereto, cells were incubated for 4 h and 24 h with different concentrations of Aβ1-42. Cells treated for 4 h with Aβ1-42 did not show any sign of cell death (data not shown), however after 24 h of Aβ treatment, we observed cell death using Aβ concentration of 10 μM and higher. No significant cytotoxicity was detected upon 1 μM Aβ treatment when compared to vehicle-treated cells (Fig. 5A).
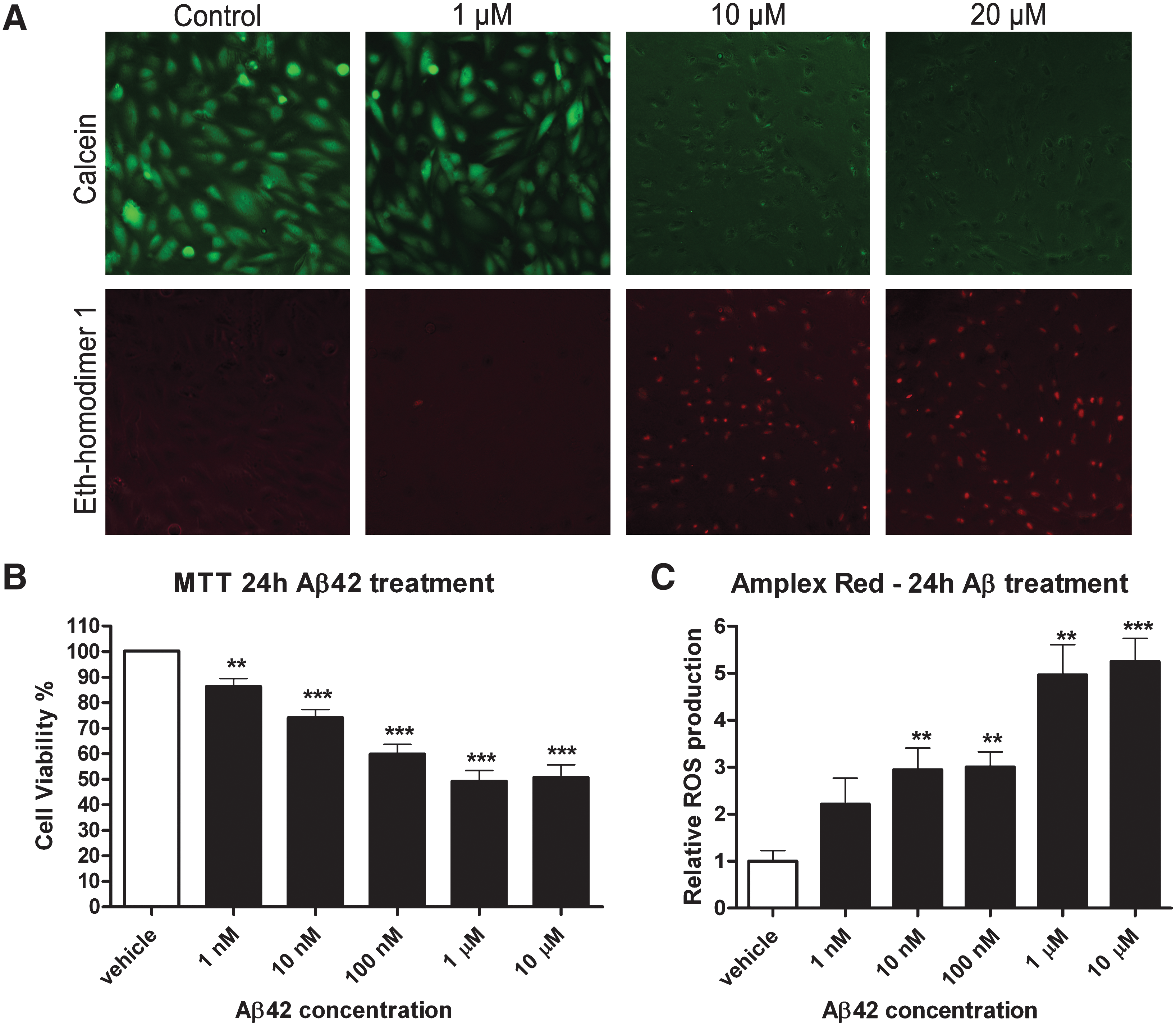
To evaluate the cytotoxic effect of Aβ on hCMECs, we assessed the effect on mitochondrial function as a measure of cell viability. We showed a dose-dependent decrease in brain endothelial cell viability after 24 and 48 h of Aβ1-42 treatment. After 24 h, a dose-dependent effect of Aβ on mitochondrial function was observed with a maximum effect at 1 μM Aβ1-42 with a decline of mitochondrial function around 50% (Fig. 5B). Dose-dependent toxicity was also present at 48 h (data not shown).
We observed cell death and impaired mitochondrial function, which can both be related to ROS production (18, 38). In order to investigate whether Aβ1-42 induces production of ROS, hCMECs were treated with Aβ1-42 for 24 h. Using an Amplex Red assay, we showed that Aβ1-42 induced a dose-dependent increase in H2O2 production compared to vehicle-treated cells (Fig. 5C).
Antioxidants/ROS scavengers rescue endothelial cells from Aβ1-42 toxicity
Since we detected hydrogen peroxide production in response to Aβ incubation, we further elucidated the involvement of NADPH oxidase and xanthine oxidase in Aβ-mediated ROS production using specific inhibitors such as diphenylene iodonium (DPI) and allopurinol. Alpha-lipoic acid (alpha-LA) was used as general ROS scavenger. Endothelial cells preincubated with DPI, allopurinol, and alpha-LA for 2 h were then treated with different concentrations of Aβ1-42 (10 nM and 100 nM) in presence of the different compounds for 24 h. Cell viability was measured by MTT assay. DPI, allopurinol, and LA were all able to rescue hCMECs from Aβ-mediated toxicity (Fig. 6), indicating that Aβ-mediated cytotoxicity is mainly due to Aβ-induced ROS production.

Antioxidants rescue Aβ-dependent downregulation of TJ proteins
We proved that Aβ1-42 fibrils induce ROS generation in our cell system and it has been previously shown that ROS can affect TJs integrity (34, 40). We hypothesized that the downregulation of TJ proteins observed upon Aβ1-42 fibrils treatment is due to the Aβ-dependent ROS production. To elucidate the link between Aβ, ROS, and TJs expression changes, we incubated hCMEC with allopurinol for 2 h prior to Aβ treatment; we then measured the expression of occludin and ZO-1 mRNA levels. As allopurinol was able to rescue cells from Aβ-mediated cytotoxicity, it was also capable of restoring TJ mRNA levels, confirming that indeed Aβ-driven ROS production is responsible for major TJ alterations (Fig. 7).

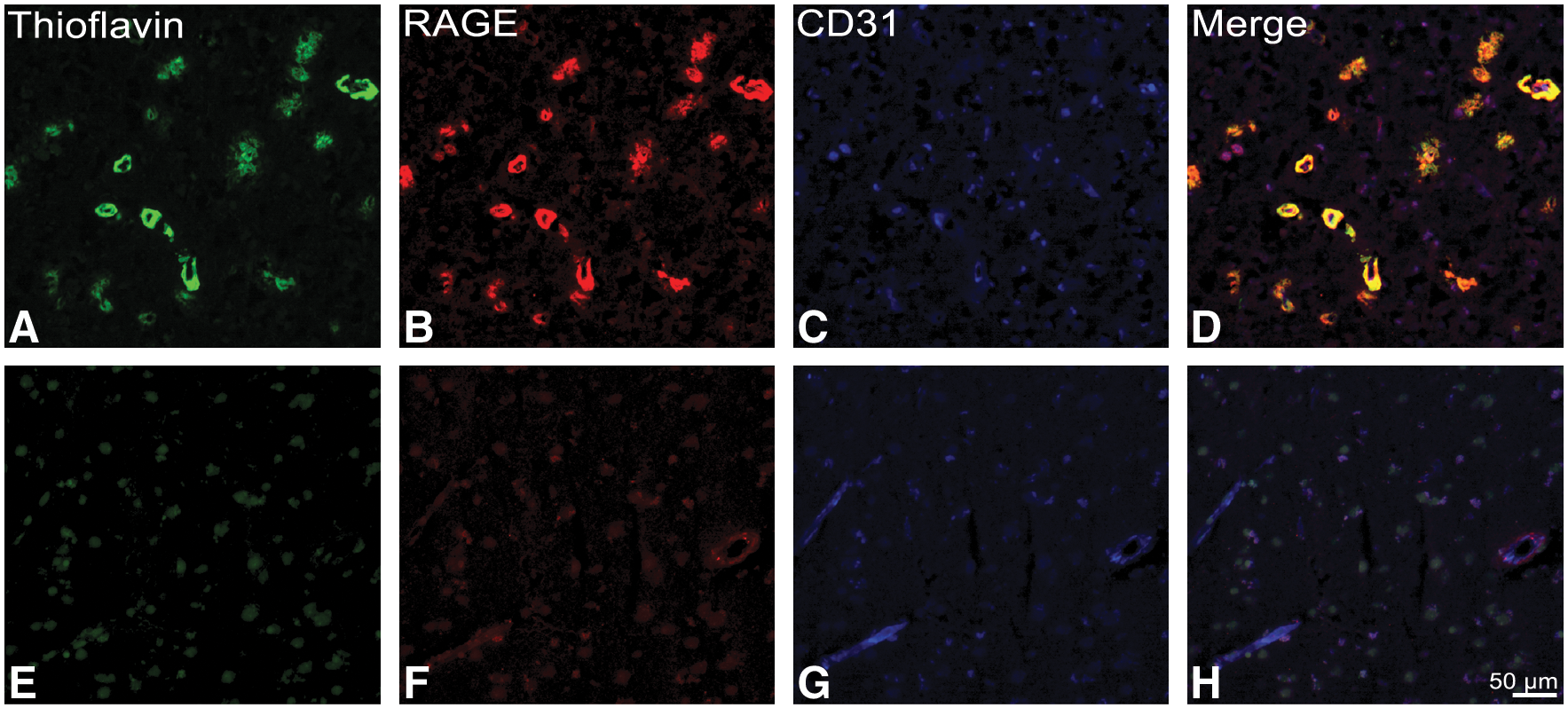
Upregulation of RAGE in capCAA
RAGE is the most important influx transporter for Aβ across the BBB and is expressed at relatively low levels in the microvasculature under physiological conditions. We show by means of immunohistochemical analysis a striking increase in RAGE expression in capCAA-affected capillaries compared to control and nonaffected capillaries. These results confirm, as previously reported (11), that Aβ induces a local upregulation of RAGE (Fig. 8).
RAGE mediates Aβ-induced cytotoxicity
In order to determine the involvement of RAGE in the Aβ cytotoxic effects, we treated hCMEC with a blocking antibody against RAGE. Cells were treated with the blocking antibody for 2 h and for 24 h together with Aβ1-42 fibrils. MTT assay showed that blocking RAGE rescued cells from Aβ-induced toxicity, demonstrating that Aβ effects on endothelial cells are exerted at least partially by its binding to RAGE (Fig. 9).

Discussion
For the first time, we show in this study a dramatic loss of TJ proteins in Aβ-laden capillaries, which are surrounded by NOX2-positive activated microglia. We demonstrated in an in vitro BBB system that Aβ is able to induce ROS formation and decrease TJ mRNA levels, which could be rescued upon pretreatment with NOX inhibitors and lipoic acid. We further demonstrate that blocking RAGE is a way to rescue cells from Aβ-induced toxicity.
Using our unique postmortem tissue, we here provide evidence on the loss of expression of TJ proteins in capCAA. So far, histopathological studies have demonstrated that microvascular alterations can be extensive in AD patients (8, 13). These alterations have been shown associated with vascular Aβ deposition as degeneration of perivascular cells, including pericytes and smooth muscle cells, swollen astrocytic end feet (17, 46), reduced expression of brain endothelial glucose transporter-1 protein, increased pinocytotic vesicles, and decreased numbers of mitochondria. In addition, prominent thickening and local disruption of vascular basement membranes was reported by several research groups analyzing either biopsy tissue or postmortem AD material (8, 26, 30). Vascular abnormalities that are associated with local amyloid accumulation suggest that impaired vascular function and thus impaired BBB integrity represents a common phenomenon in AD pathology. Although there are several studies demonstrating BBB alterations in CAA type II, comprehensive immunohistochemical studies on BBB abnormalities in Aβ-laden capillaries (CAA type I) are limited. In vitro studies support the idea that Aβ deposition affects BBB integrity since different Aβ peptides are able to increase endothelial permeability (6, 36) and induce altered expression and translocation of TJs proteins in human and animal ECs (15, 27). Remarkably, data on putative TJ alterations and the underlying mechanisms in CAA-affected capillaries are lacking.
To provide novel data on the BBB integrity during CAA, we selected a unique cohort of patients with abundant Aβ deposits in cortical capillaries. We observed a striking loss of occludin, claudin-5, and ZO-1 immunostaining in Aβ-laden capillaries, whereas unaffected capillaries showed a normal expression pattern, indicating that loss of TJ expression was predominantly related to microvascular Aβ deposition. Importantly, endothelial cells of capCAA-affected vessels still express endothelial markers, including factor VIII and CD31, excluding brain endothelial cell death as a potential reason for the lack of TJ protein expression. Although leakage of the BBB has been supported by elevated plasma proteins associated with Aβ deposits, we here for the first time provide direct evidence of TJ proteins loss in AD brains linked to Aβ accumulation. The subsequent breakdown of the BBB may in turn disrupt normal transport of nutrients, vitamins, and electrolytes across the BBB, which are essential for proper neuronal functioning.
Interestingly, we detected enhanced expression of the ROS-generating enzyme NADPH oxidase-2 (NOX-2) in microglia surrounding Aβ-laden vessels. Previous data from our group demonstrated that deposition of Aβ throughout the brain parenchyma, especially extravascular deposition of Aβ that from the vessel wall radiates into the neuropil (named dyshoric changes), is able to induce an inflammatory response, consisting in activation of microglia and astrocytes (32, 33). Upon phagocytosis or recognition of Aβ by microglia cells, a series of responses may occur that includes the release of proinflammatory cytokines and ROS. One of the main sources of ROS under neuropathological conditions is enhanced NOX activity, together with the mitochondrial respiratory chain. In particular, NOX-2 is a well-known NADPH oxidase expressed in microglia where it is normally expressed at low levels. However, under pathological conditions, NOX-2 is upregulated and associated with a wide variety of vascular pathologies such as hypertension, diabetes, and hyperlipidemia (35). Enhanced expression of NOX-2 leads to increased ROS levels, particularly superoxide, and induces oxidative stress. In material derived from non-neurological controls, NOX-2 expression is mainly limited to microglia. Notably, the expression pattern of NOX-2 in capCAA tissue was strikingly different; revealing a stronger microglial expression, especially around capCAA-affected vessels, and NOX-2 was clearly expressed in perivascular macrophages associated with Aβ-laden capillaries. From these findings we conclude that Aβ deposition within and surrounding capillaries induces microglial activation and subsequent upregulation of NOX-2 protein expression in activated microglial cells and perivascular macrophages. These results support the idea of increased production of ROS in close proximity of capCAA-affected vessels and are in line with the observation that protein and DNA oxidative damage are increased in AD brains (25, 41).
To unravel pathways involved in BBB damage and Aβ-induced oxidative stress in brain endothelial cells, an in vitro approach was taken using the validated human brain endothelial cell line hCMEC/D3. We first assessed the cytotoxic effects of Aβ1-42, one of the predominant Aβ isoforms accumulating in Aβ-laden capillaries. Aβ1-40 is the predominant form of Aβ in larger CAA-affected vessels, such as arterioles and leptomeningeal vessels. However, the Aβ1-40/Aβ1-42 ratio of capillary Aβ is significantly lower than that of affected arteries and veins but equals that found in senile plaques (32), indicating that Aβ1-42 is a common Aβ isoforms in microvascular CAA. Furthermore, Aβ1-42 is known to be the most toxic form of Aβ to endothelial cells (12). Hence, we used the Aβ1-42 isoform for our in vitro studies. We treated endothelial cells with pre-aggregated Aβ peptide as aggregates were reported to be more toxic to ECs (5). At standardized conditions we could observe cells death after 24 h incubation at concentrations higher than 10 μM. Subtoxic concentrations of Aβ1-42 were then used to assess the effects of amyloid on TJ expression in vitro. Using this set-up, we showed a significant dose-dependent downregulation of TJ proteins occludin and ZO-1 mRNA levels upon Aβ1-42 incubation, but no changes in claudin-5 mRNA level, as also reported by Tai et al. (36), which was in contrast to our postmortem findings. Reduced expression of occludin after 48 h of Aβ1-40 incubation has been previously described in hCMEC, but no changes in claudin-5 or ZO-1 (15). Aβ1-42 has been reported to reduce expression of claudin-5 and occludin in primary rat brain endothelial cells albeit at higher concentrations than the concentration used in this study (27). Our results, together with the previous findings, confirm the significant loss of occludin and ZO-1 in capCAA tissue and directly related these observations to Aβ deposit in the microvasculature. We speculate that claudin-5 protein expression may be altered by Aβ, as our immunohistochemical results strongly suggested. It is conceivable that prolonged exposure to Aβ is needed to decrease claudin-5 expression, as occurs in the patient material, or that loss of claudin-5 reported in capCAA tissue is not due to altered expression at transcriptional level but to a post-translational modification or degradation of the protein.
Our in vitro results showed dose-dependent decrease of mitochondrial function and increased production of ROS upon Aβ1-42 stimulation of hCMEC. One consequence of impaired mitochondrial function is enhanced generation of ROS, and since dysfunctional mitochondria will produce more ROS, a feed-forward loop is set up, resulting in a vicious cycle (42, 48). Aβ is also known to increase ROS production in different cerebral cell types and we showed that both NOX inhibition and exogenous antioxidants were able to counteract the toxic effect of Aβ1-42, confirming that reduced cell viability was indeed caused by ROS.
The cytotoxic effects of vascular Aβ might be caused by Aβ binding to RAGE. We observed for the first time a striking increase of RAGE expression in Aβ-laden capillaries. RAGE is a major influx transporter for Aβ across the BBB and its expression is upregulated in AD and transgenic models of amyloidosis in the affected cerebral vessels, microglia, and neurons (50). RAGE not only imports Aβ into the brain, increasing Aβ accumulation in the cerebral parenchyma, but when bound to its ligands also induces ROS production through NADPH oxidase activation (43). Importantly, we showed that blocking RAGE prevents Aβ-induced decrease of cell viability.
Conclusion
Taken together, we show the occurrence of severe TJ alterations in Aβ-laden capillaries. Interestingly, affected capillaries were surrounded by NOX2-positive activated microglia. In vitro experiments confirmed that Aβ was able to decrease TJ mRNA levels and that both exogenous antioxidants as well as NOX inhibitors limit Aβ-mediated cellular toxicity. We further demonstrated that RAGE is the mediator of the Aβ-cytotoxic effects on endothelial cells. We speculate that due to increased local Aβ-driven ROS production, TJ protein expression is altered in the microvasculature of capCAA brains. It is plausible that extensive microvascular Aβ depositions, enhanced microvascular expression of RAGE, and concomitant loss of TJ protein expression impair BBB function and consequently leads to inefficient transport of nutrients into the brain. Collectively, these pathological processes might hamper Aβ clearance from the brain and thereby contribute to neuronal damage.
Footnotes
Acknowledgments
We thank N. Hahn (Department of Pathology, VU medical center, Amsterdam, the Netherlands) and Prof. Dr. D. Roos (Sanquin Research, Amsterdam, the Netherlands) for providing the NOX-2 antibody.
This work was financially supported by the ‘Internationale Stichting Alzheimer Onderzoek’ (ISAO grants 07517 and 09506) and the European Commission FP6 (ADIT, contract no. LSHB-CT-2005-511977).
Author Disclosure Statement
No competing financial interests exist.