
Abstract
Introduction
Mitochondria are a major source of ROS such as superoxide and are also particularly susceptible to damage caused by these species (9, 104). Consequently, mitochondria accumulate oxidative damage that contributes to dysfunction and cell death in a range of diseases, although it should be noted that mitochondria also use ROS such as hydrogen peroxide as a crucial component in redox signaling (9, 97, 101). Therefore, a focus has been on developing small molecules that report on or prevent oxidative damage to mitochondria (38, 106, 123, 142) without disrupting many other vital metabolic processes. A wide variety of mitochondria-targeted pharmacophores and probes could be conceived and developed; however, to date, the mitochondrion has been a neglected drug target (103). Developments to target pharmacophores, probes, and bioactive molecules selectively to mitochondria in vivo by oral, intravenous, or intraperitoneal administration could elevate mitochondria into important drug targets and, in addition, enhance and extend core knowledge about how mitochondria function (Fig. 1). The selective targeting of a pharmacophore to concentrate in mitochondria will decrease the external dose required, while the sequestration within mitochondria may limit toxic side reactions and minimize metabolism of the compound. Extension of this concept is possible, for example, mitochondria-targeted pro-drug compounds may be possible that involve the mitochondrion as an intracellular “reaction chamber” to release pharmacophores or other bioactive molecules selectively to the mitochondrion or to the rest of the cell (Fig. 2).
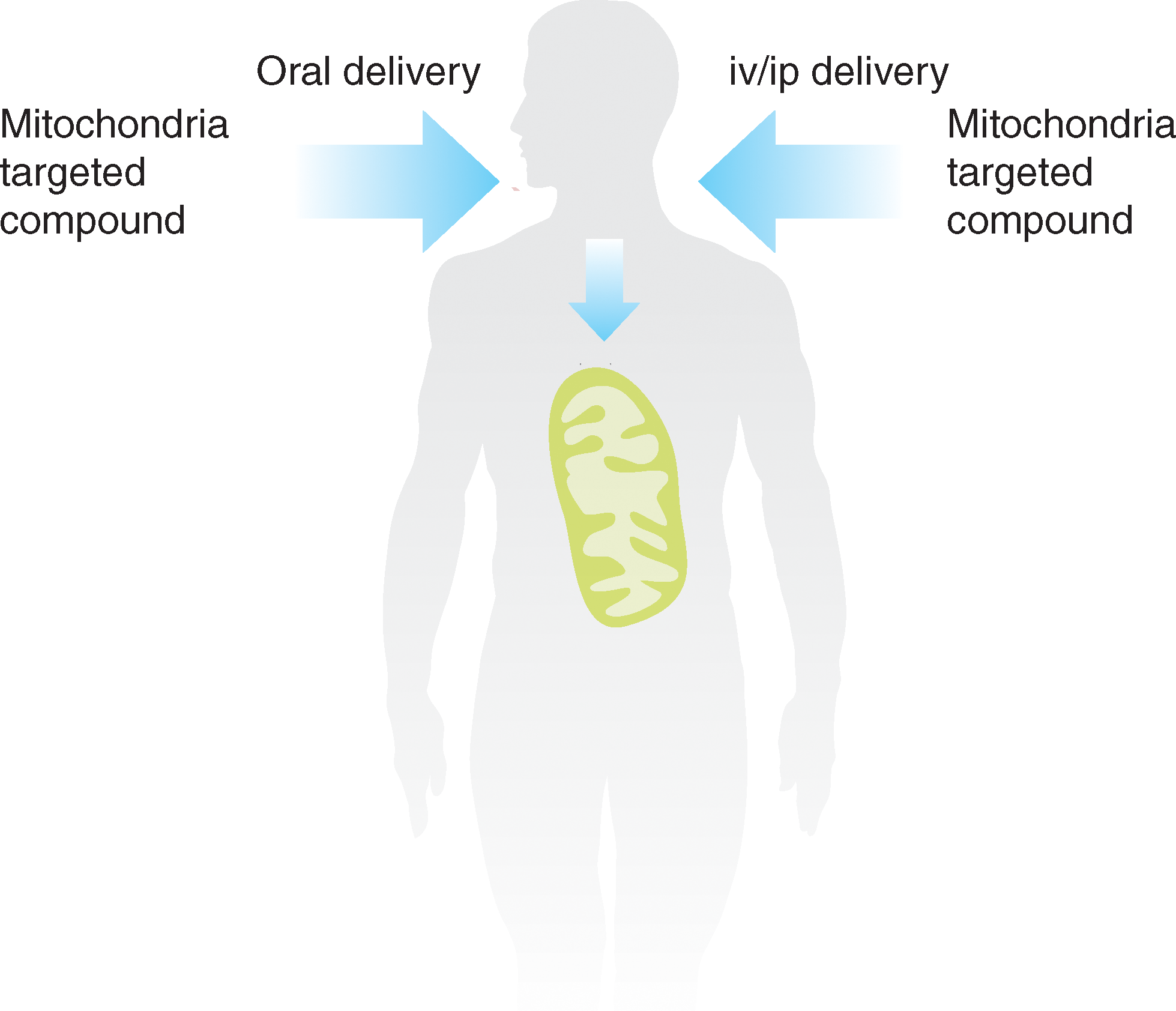
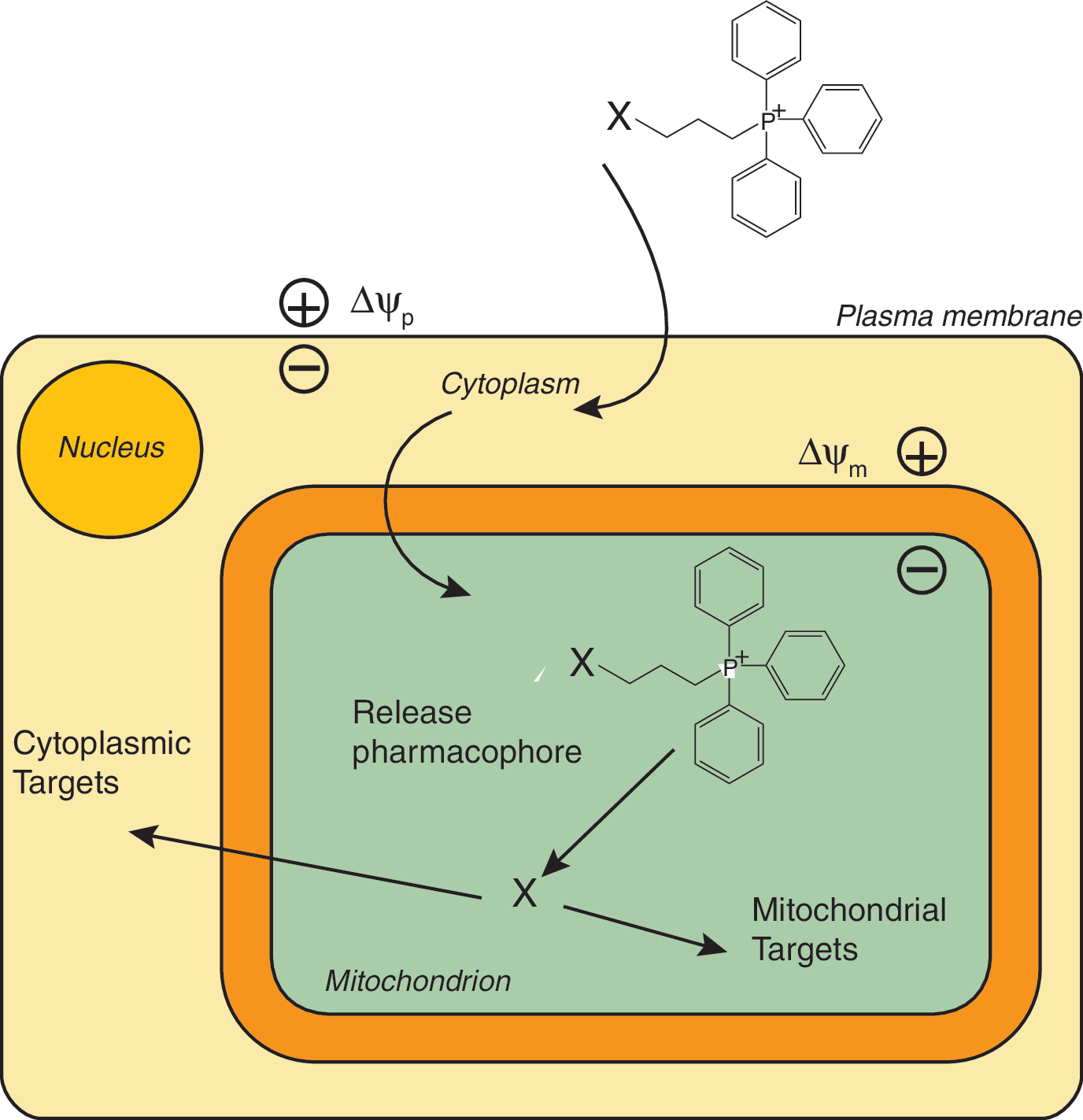
For these reasons, the development of small-molecule mitochondria-targeted compounds and the potential strategies that can be used to deliver therapeutic and probe molecules is important. Here we review the methods that can be used to target small molecules to mitochondria, survey progress to date in the development of probes and therapeutics, and suggest future directions.
Methods of Targeting Small Molecules to Mitochondria
Conjugation to lipophilic cations
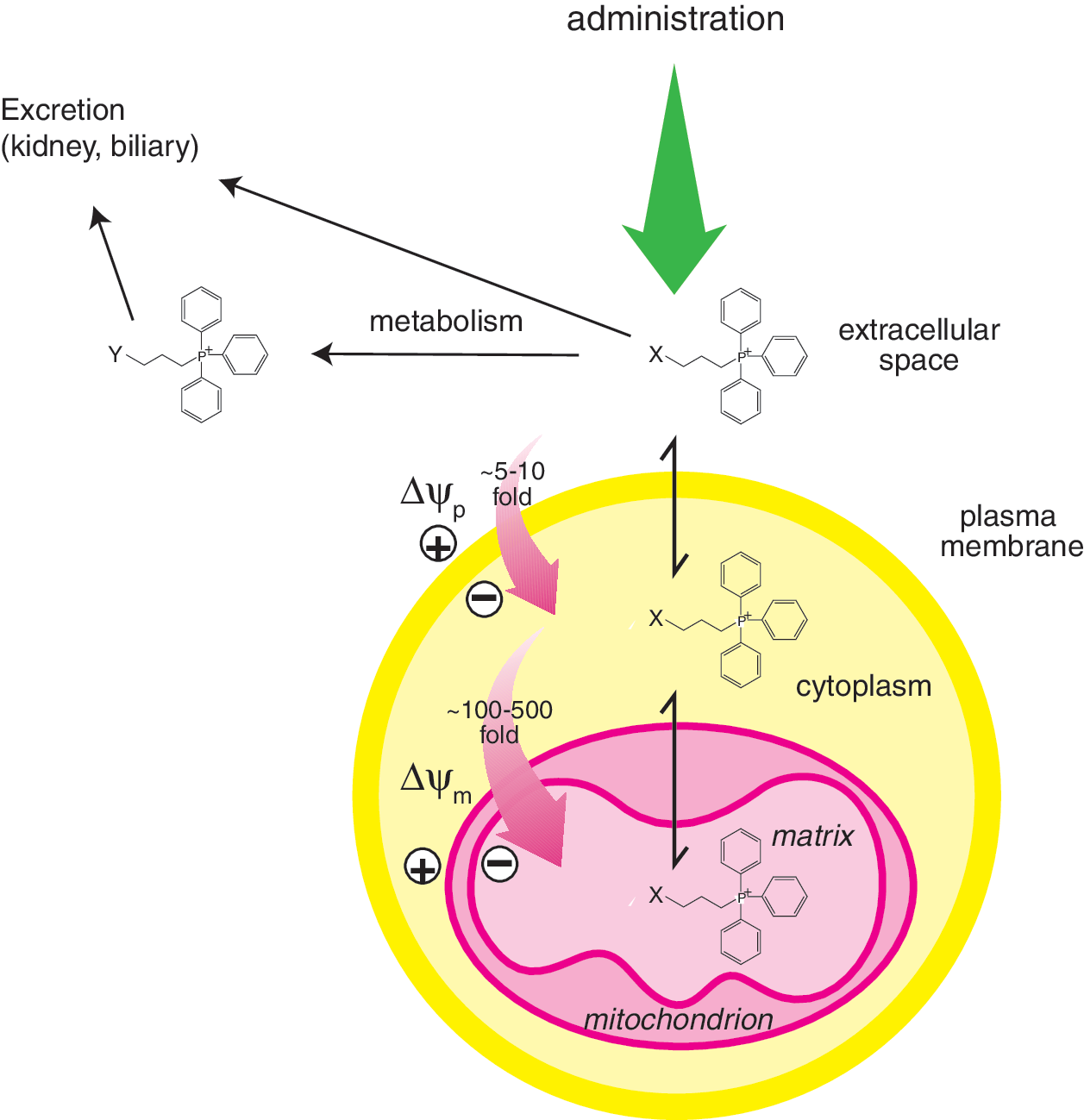
The most generally applicable method to target small neutral molecules to the mitochondrial matrix in vivo is by conjugation to a lipophilic cation, as has been reviewed extensively elsewhere (99, 106, 107, 127). The distinctive feature of lipophilic cations is that their positive charge is delocalized over a large and hydrophobic surface area. Consequently, they can pass easily through phospholipid bilayers, enabling their accumulation into the mitochondrial matrix in response to the membrane potential (51, 73, 127). The uptake of lipophilic cations into mitochondria depends on the very large membrane potential across the mitochondrial inner membrane of up to 150–160 mV (negative inside) which, coupled with the plasma membrane potential of 30–60 mV (negative inside), drives the extensive uptake of cations within the mitochondrial matrix. The Nernst equation indicates that the otherwise unimpeded uptake of singly charged cations increases 10-fold for every 61.5 mV of membrane potential at 37°C (127), and therefore the relative concentration of lipophilic cations in the mitochondrial matrix should then be several hundred- to a few thousand-fold greater than in the extracellular environment. A wide range of lipophilic cations are taken up by mitochondria in this manner. Historically, lipophilic cationic dyes such as Janus green were used as vital stains to visualize mitochondria for light microscopy (81), although the physical basis of the accumulation was not recognized at the time. With the advent of the chemiosmotic coupling hypothesis, it became clear that there should be a large membrane potential across the mitochondrial inner membrane (109), and the demonstration of the distribution of lipophilic cations and anions across mitochondrial membranes was compelling evidence for the central role of the membrane potential in bioenergetics (84, 85). Since then, the uptake of the lipophilic tetraphenylphosphonium and methyltriphenylphosphonium cations have been widely used to measure mitochondrial membrane potential (8, 20). In parallel to these, Chen developed the idea of visualizing mitochondria within cells by the uptake of lipophilic fluorescent cations such as rhodamine 123 and JC-1 (26), and this has led to the routine use of fluorescent lipophilic cations to visualize mitochondria and to assess the mitochondrial membrane potential within cells (34, 65).
Based on the realization that lipophilic cations can be taken up by mitochondria in cells, it was a natural extension to consider linking a lipophilic cation to a moiety that can then be delivered selectively to mitochondria in vivo (99). Chen was the first to achieve this by linking to the fluorescent lipophilic cation rhodamine 123 (26, 33, 147, 148) and using the knowledge that mitochondria in cancer cells have a higher membrane potential than noncancer cells to selectively target anticancer drugs to mitochondria. Subsequently, the idea of taking advantage of the elevated mitochondrial membrane potential in cancer cells to increase the uptake was enhanced to increase the nonspecific toxicity of these cations to cancer cells (139). Selective toxicity to cancer cells was also shown for tetra- and triphenylphosphonium (TPP) lipophilic cations (120) and this was extended by conjugating toxic functionalities such as nonspecific alkylating agents to increase the effectiveness of cancer cell killing (94), or by targeting two distinct lipophilic cations to mitochondria that then react to form a toxic product (112, 119). This represents the first use of lipophilic cations to direct attached components to mitochondria within cells and was aimed at selectively killing cancer cells. Since then, our groups have extended this approach to use lipophilic cations, predominantly the TPP cation, to direct redox probes, antioxidants, spin traps, and other bioactive molecules to mitochondria with the idea of protecting, modifying, or reporting on mitochondrial function (22, 23, 72, 106, 136). In parallel work, the uptake of the TPP cation has been used to construct novel mitochondria-targeted positron emission tomography (PET) probes by conjugation of a TPP cation to a PET-visible nuclide (90, 91).
Use of the TPP cation to target mitochondria
A number of factors support the use of the TPP cation for generating mitochondria-targeted compounds. The biophysics of the movement of TPP cations across phospholipid bilayers has been extensively studied and is well understood (51, 73, 127). In isolated mitochondria and cells, the uptake of TPP compounds is adequately described by the Nernst equation, implying specific molecular transport methods are not necessary and the compounds display extensive binding to the matrix surface of the mitochondrial inner membrane (67, 68, 128). A critical parameter affecting the rate and extent of uptake of TPP cations into mitochondria within cells is their hydrophobicity (128). As the hydrophobicity increases, the activation energy for transport of TPP cations across the plasma membrane is lowered and this greatly enhances the rate of uptake (128). Furthermore, TPP cations have a strong tendency to bind to the surface of phospholipid bilayers in a potential energy well close to the membrane surface, with the “cargo” attached to the TPP cation dipping into the membrane. Increasing the hydrophobicity enhances this tendency to bind to the inner membrane, thereby lowering the amount of unbound compound that equilibrates with the membrane potentials. As a consequence, the overall uptake into mitochondria of more hydrophobic TPP compounds is increased relative to less hydrophobic compounds at the same membrane potential (7, 67, 68, 128). The hydrophobicity of the moiety attached to the TPP is a critical parameter and provides an opportunity to modulate the extent and rate of uptake of these compounds into mitochondria.
Utilizing a TPP moiety to generate mitochondria-targeted lipophilic cations has a number of practical advantages, as it is chemically relatively easy to introduce a TPP into a compound at the end of a synthetic scheme, usually by displacing a leaving group by reaction with a triphenylphosphine (134). This facilitates the production of modified versions of mitochondria-targeted compounds containing a radio- or stable isotopically labeled TPP moiety, widely used for studying the uptake and metabolism of these compounds, conveniently towards the end of the chemical synthesis (134). These advantages have led to a range of mitochondria-targeted compounds based on TPP being widely used by a number of groups to target antioxidants, bioactive molecules, and probes to mitochondria, and a selection is shown in Figures 3 and 4. However, despite the many advantages of using TPP, it should be noted that many different lipophilic cations could be used in principle to target attached bioactives to mitochondria comparable to TPP. This may be particularly useful if it is necessary to modify the targeting cationic moiety to alter its hydrophobicity or to avoid structure-specific problems such as selective excretion or metabolism.
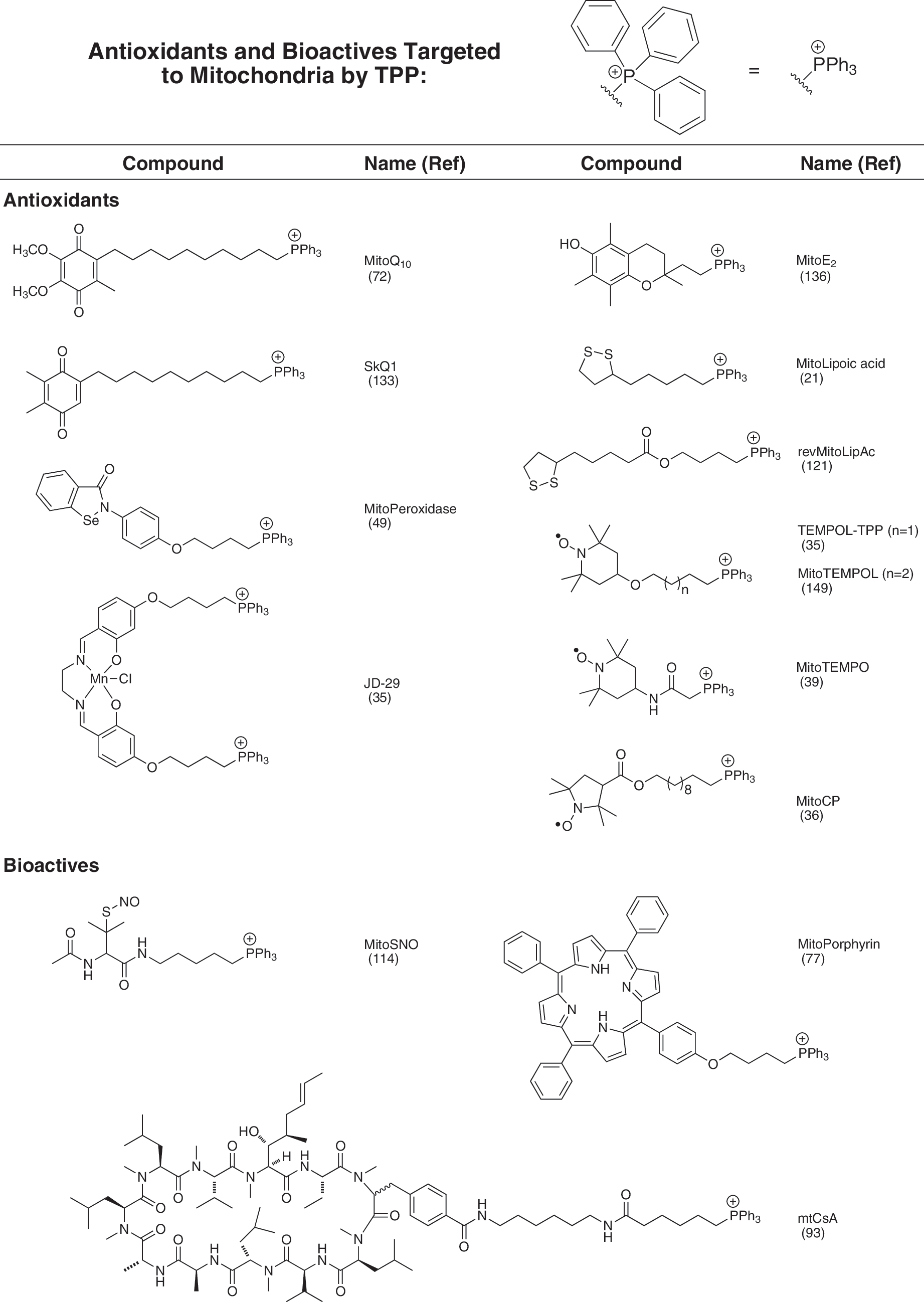
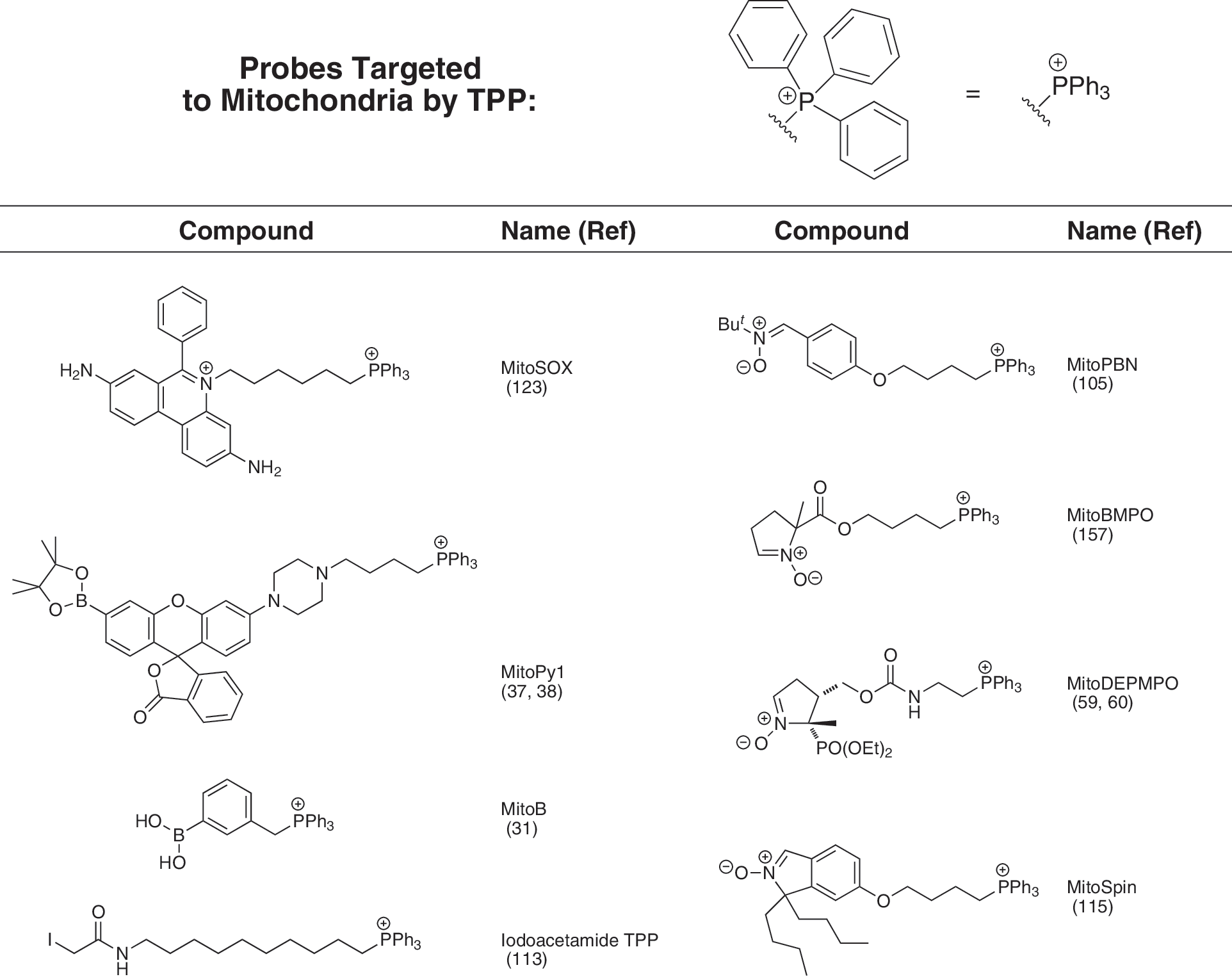
Uptake of TPP cations by mitochondria in vivo
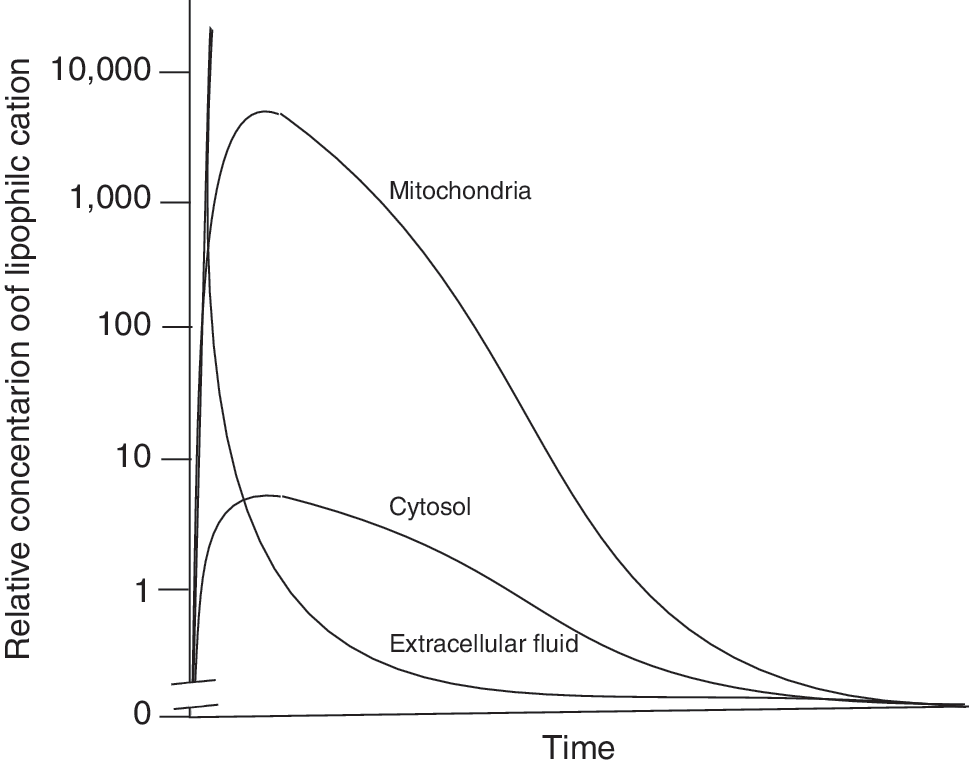
The ability of TPP-based compounds to be taken up by mitochondria in vivo has been assessed in detail (113, 124, 137). TPP cations are taken up rapidly into mitochondria within organs in vivo following oral, intravenous, or intraperitoneal administration with negligible residual amounts present in the blood, consistent with the uptake into the organs from the circulation driven by the plasma and mitochondrial membrane potentials (113, 124, 137). Of particular note is the very rapid uptake of lipophilic TPP compounds into mitochondria within organs in vivo within 5 min following intravenous administration (113), which may facilitate application to acute therapies. The toxicity of TPP compounds is relatively low and they can be safely administered indefinitely to give significant levels in organs such as the heart, liver, and kidneys (113, 124, 137). There is also some uptake of TPP compounds into the brain (124, 137), but it is less than into other organs and the reason for the difference is unclear at this time. Nonetheless the antioxidant MitoQ10 (Fig. 3) is still protective in the brain against the selective damage caused by the 1-methyl-4-phenyltetrahydropyridine (MPTP) neurotoxin (54). The metabolism of TPP compounds in vivo has been investigated and there is no evidence of any significant modification of the TPP functionality (82, 83, 128) although the bioactive group can be altered, for example, the ubiquinol moiety in MitoQ10 is modified by sulfation or glucuronidation followed by excretion by the biliary pathway, or by the kidneys into the urine (82, 83). The metabolism is very dependent on the chemistry of the bioactive component of the TPP compound, for example, a targeted lipoic acid (MitoLipoic acid, Fig. 3) was reduced and the free thiol groups methylated when incubated with cells in culture (21). The loss of TPP compounds from tissues has also been investigated, and after a single bolus intravenous injection into mice, they are taken up rapidly into a range of organs with a maximum concentration obtained within a few minutes and very low amounts remaining in the blood (113). The TPP compounds are then gradually lost from these organs with half lives in the range of 2–15 hours, depending on the compound and organ (113). This is consistent with efflux from the tissues being primarily determined by the Nernstian distribution. Once the level of the compound in the extracellular fluid and blood decreases due to metabolism and excretion through the kidneys and the biliary pathway, then the compounds will redistribute from the mitochondria and the cytosol into the extracellular fluid and be excreted. The factors that govern the uptake, distribution, metabolism, and excretion of lipophilic cations in vivo are shown schematically in Figure 5 and an idealized profile for this process is shown schematically in Figure 6. The Nernst equation governs accumulation of different TPP conjugates independently, but membrane disruption that is possible with very high accumulation will probably depend on the sum of TPP-containing compounds. In principle, the uptake of lipophilic cations is well understood with the relative concentrations of compound taken up by different compartments being determined by the concentration in the blood, the plasma and mitochondrial membrane potentials, the extent of binding in the cytosol and mitochondria, and the relative sizes of those compartments. However, the difference in the extent of uptake of TPP compounds by different tissues is not solely determined by their equilibration between the various pools and is a complicated function of the location of the organ relative to its site of administration, its rate of uptake across the plasma membrane, the magnitude of any selective efflux, and metabolism pathways associated with the payload attached to the TPP, the size of the plasma and mitochondrial membrane potentials, and the cytoplasmic and mitochondrial volumes within the organs. Therefore, the distribution shown in Figure 6 is idealized and the details will vary with organ and compound.
Variations of the TPP targeting system
A number of modifications to the TPP delivery system have been developed that may become widely utilized over time. One of these has been the study of the uptake of TPP dications which, from consideration of the Nernst equation could be taken up 10-fold into mitochondria for every 30 mV of membrane potential as compared with 60 mV for the same effect with monocations (126). This could potentially lead to significantly greater uptake and concentration of mitochondria-targeted dications into mitochondria than monocations. While dications were taken up by isolated mitochondria to a greater extent than monocations, the enhancement was less than predicted by the Nernst equation for reasons that are currently unclear (126). It was also evident that lipophilic dications had to be significantly more hydrophobic than a comparable monocation in order to be taken up across a biological membrane, presumably to help counteract the increase in activation energy for membrane transport associated with the larger charge (126). The use of dications may in time yield compounds that are taken up into mitochondria to a greater extent than current TPP monocations, and uptake of lipophilic dications into mitochondria has yet to be explored in vivo (1, 89, 126).
TPP cations can also be used as delivery systems for the release of bioactive molecules within mitochondria. In this approach, a TPP moiety is attached to its cargo by a labile connection that enables the bioactive molecule to be taken up into mitochondria and then released on cleavage of the linkage to TPP. This has been done for the bioactive molecule nitric oxide (NO) in which a mitochondria-targeted NO-donor MitoSNO (Fig. 3) was taken up rapidly by mitochondria and released NO within the mitochondrial matrix that can diffuse out from the mitochondria to induce vasodilation and also to modulate mitochondrial respiration (114). In addition, MitoSNO selectively S-nitrosates mitochondrial protein thiols and protects the heart from ischemia-reperfusion injury (29, 114). The bioactive molecule lipoic acid was also delivered to mitochondria by conjugation to a TPP delivery system by a labile ester linkage (revMitoLipAc, Fig. 3) that was cleaved by mitochondrial aldehyde dehydrogenase (ALDH2) to release lipoic acid within mitochondria (121). Related approaches have been used to conjugate a TPP moiety to a peptide nucleic acid (PNA) by a disulfide link to deliver it across the plasma membrane (48); however, the relatively slow subsequent uptake across the mitochondrial membrane coupled with the rapid reduction of the disulfide bond in the cytosol meant that this procedure only delivered the PNA effectively to the cytosol. Since then, the methodology for linking a TPP moiety to a compound by a disulfide bond has been optimized (95), so it should now be straightforward to assess if this approach can successfully deliver a molecule to the mitochondrion without excessive loss by reduction of the disulfide bond in the cytosol. In another application, attempts to tag stem cells by using TPP modified polythiophenes was unsuccessful when using molecules containing a cleavable disulfide linkage, whereas more chemically robust linkages gave encouraging results (43). This approach can potentially be further modified to deliver a wide range of molecules to mitochondria in vivo and modulated by altering the linkage between the cargo and the TPP to increase or decrease the rate and selectivity of cleavage. One issue that must be considered with this approach to delivering molecules to mitochondria is that after removal of the TPP cation there may no longer be a thermodynamic driving force to retain the delivered molecule within the mitochondrial matrix and it may rapidly diffuse out of the mitochondrion once released from its delivery module. A final variation on the use of TPP to deliver molecules to mitochondria is by simultaneously targeting two different TPP cations to mitochondria so that both accumulate in the same molecular environment and react due to the high local concentration within the mitochondrial “reaction chamber” to form a new bioactive product (112, 119). There are a number of intriguing possibilities to extend the scope of delivery to mitochondria by conjugation to lipophilic cations that remain to be explored.
Common questions about using lipophilic cations to target mitochondria
There are frequently asked questions about the phenomenon of the uptake of lipophilic TPP cations into mitochondria in vivo and some comment is appropriate at this point. A common concern is whether the several thousand-fold accumulation of lipophilic cations within mitochondria in vivo will result in the continual accumulation of more and more compound within mitochondria upon subsequent dosing and ultimately lead to unacceptable toxicity. This does not occur because, as is illustrated in Figure 6, uptake of lipophilic cations into mitochondria is effectively self-limiting, due to the rapid equilibration of the compound between the circulation and the mitochondria and then depletion in the circulation level due to excretion. Once the level in the circulation decreases, the reversibility of the membrane transfer process will result in loss of the compound from mitochondria by passing back to the circulation to maintain the relative concentrations of the compound as determined by the membrane potential. Exceptions are possible, for example, where the TPP compound is metabolized to a membrane impermeable product, or forms a covalent attachment to a macromolecule. A second question relates to the requirement of a membrane potential for the uptake of TPP cations into mitochondria, which could be compromised by the lack of a mitochondrial membrane potential during pathological conditions. While the complete loss of a mitochondrial membrane potential would prevent uptake, this occurs very rarely as, even under extreme mitochondrial dysfunction, such as in cells entirely lacking mitochondrial DNA, there is still a large membrane potential of about 70 mV (5). In most disorders it is unlikely that the membrane potential will fall much below the normal range in vivo of 130–150 mV, as this is counteracted by reversal of the ATP synthase and it is likely that cells in which the mitochondrial membrane potential is negligible are already damaged beyond repair. Another concern that has been raised is that some mitochondria-targeted antioxidants can also have prooxidant effects because, under some conditions in vitro, many quinols can redox cycle to produce superoxide (69), and this has been demonstrated for mitochondria-targeted quinols in vitro (42, 45, 67, 110, 133). This was assessed in vivo by measuring mitochondrial oxidative damage in mice that were fed MitoQ10 for up to 24 weeks (124). There were no changes in oxidative damage to the phospholipid cardiolipin (CL) (111), the accumulation of protein carbonyls (32, 80), the activity of mitochondrial respiratory complexes, mtDNA copy number, and damage to mtDNA (129). Furthermore, MitoQ10 had no effect on the expression of the manganese superoxide dismutase, MnSOD, which is sensitively upregulated in response to increased mitochondrial superoxide production (64, 76). This indicates that MitoQ10 does not increase oxidative damage or ROS levels in vivo and is supported by the one year's safety data for this compound in humans (138). Thus, while pro-oxidant reactions of some mitochondria- targeted antioxidants can be measured in vitro, they do not occur in vivo with TPP-based antioxidants and this is probably because most of these compounds will be adsorbed to membranes in vivo, thereby preventing these types of reactions (135). A related concern is whether mitochondria-targeted antioxidants might block the useful production of ROS in vivo, such as that used by neutrophils in bacterial killing or in redox signaling. Administration of MitoQ10 for up to 28 weeks in mice and for up to 1 year in humans did not lead to any changes in immune function or gene expression (124, 135), suggesting that the location of these compounds in the mitochondrion minimizes inhibition of the production of “useful” ROS at the cell surface by NADPH oxidases. The mitochondria-targeted antioxidants that have been used in vivo to date such as MitoQ10 do not react with hydrogen peroxide (67), which is likely to be the major ROS involved in mitochondrial redox signaling (101, 102). However, this issue may arise in the future as mitochondria-targeted antioxidants with selectivity for other ROS are used in vivo. A final point that has been raised is the potential toxicity of large amounts of accumulated lipophilic cations within mitochondria. In experimental studies, parallel incubations with otherwise inactive control TPP compounds are carried out to determine any contribution of nonspecific effects (124). It was found that the biological efficacy of compounds such as MitoQ10 occurs at a far lower concentration than that at which control TPP compound toxicity starts (71), giving a therapeutic window of ∼1000 fold in that study. Most importantly, compounds such as MitoQ10 have been given to animals for up to 6 months and to humans for up to 1 year with no toxic side effects at concentrations that are effective against various pathologies (124, 135).
Mitochondria-targeted peptides
The other major approach to targeting small molecules to mitochondria is by using mitochondria-targeted peptides (4, 143, 159, 160). The first such molecules developed were the Szeto–Schiller (SS)-peptides that are taken up by mitochondria within cells and some of which have intrinsic antioxidant activity, due to the incorporation of a dimethyl tyrosine residue (163). The SS-peptides act as mitochondria-targeted antioxidants and have been shown to selectively protect isolated mitochondria and cells from oxidative damage in a number of studies (163). These peptides comprise four alternating aromatic/basic amino acids, some of which are nonstandard, such as dimethyltyrosine, along with a D-amino acid in the first or second position, and amidation of the C-terminus to increase stability (e.g., SS-31, Fig. 7). At physiological pH, these peptides have a positive charge of three and are rapidly bound to isolated mitochondria by a process that is largely independent of the mitochondrial membrane potential and was not significantly affected by physically disrupting the mitochondria (163). These findings suggest that these peptides are bound selectively to the mitochondrial inner membrane rather than being accumulated within the mitochondrial matrix, although the nature of this interaction is uncertain. The SS-peptides protect isolated mitochondria against oxidative damage in vitro which is consistent with some type of selective interaction with mitochondria (163). This class of peptides is also taken up into cells by a nonsaturable process through the plasma membrane, consistent with nonmediated passage directly through the phospholipid bilayer (162). Within the cell, these peptides localize to mitochondria, as demonstrated by confocal microscopy using a peptide tagged with a neutral fluorophore, β-anthraniloyl-L-α,β-diaminopropionic acid in place of one of its lysine residues (145, 163).
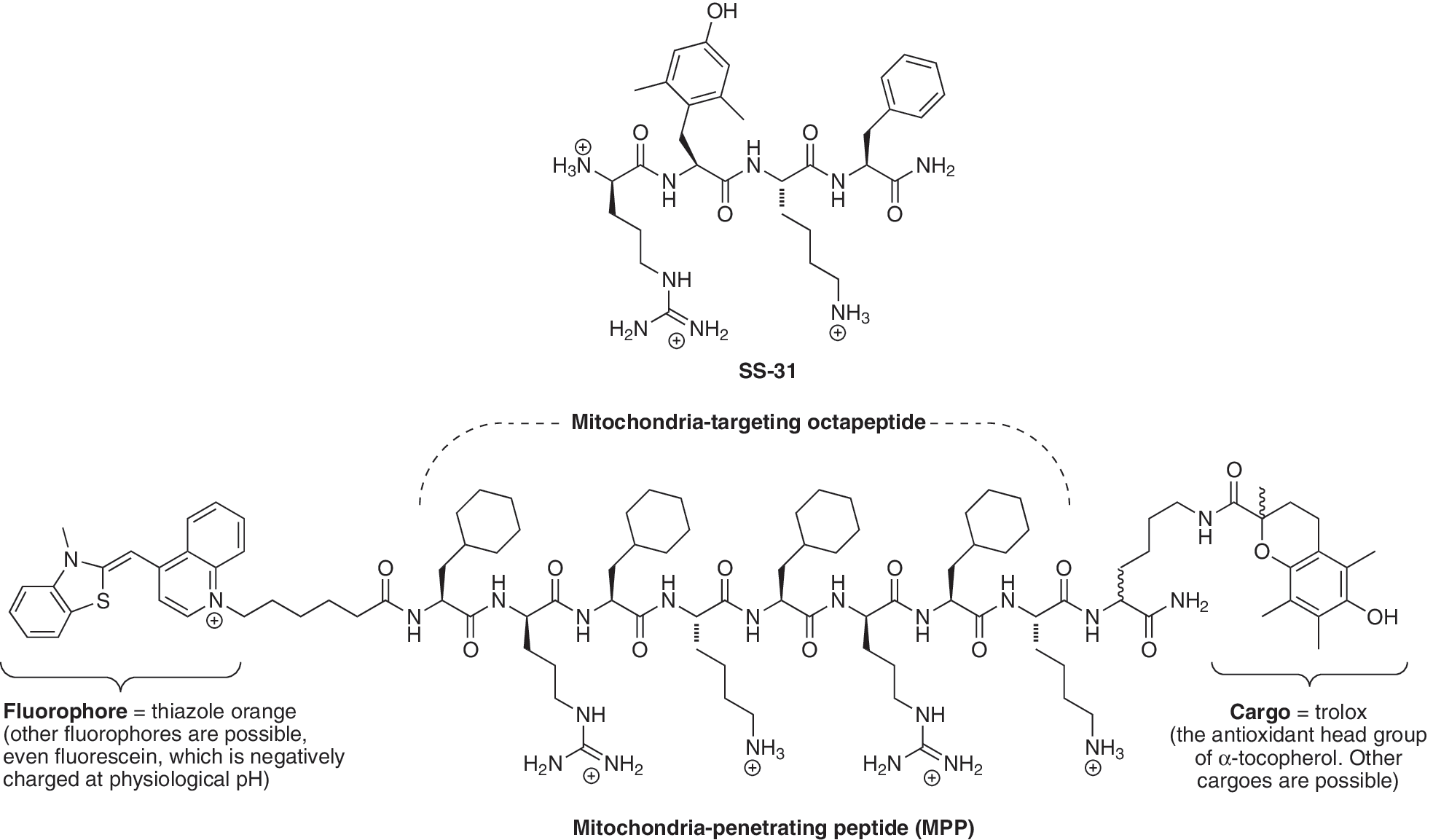
Another class of peptide with similar structure to the SS-peptides has been developed by the Kelley laboratory (63, 160). This series of peptides initially comprised four to eight amino acid residues, with alternating alkyl and basic functionalities, some of which are nonstandard, such as a reduced (cyclohexyl) derivative of phenylalanine, along with D-arginine to increase stability (e.g., MMP; Fig. 7). The alternating arrangement of positive residues does not seem to be essential for mitochondrial uptake, although concentrated blocks of positive charge do impede transport (159). These peptides have net positive charges of three to five and are taken up rapidly by mitochondria within cells. However, in contrast to the SS-peptides, this uptake is driven by the membrane potential and is critically dependent on the balance between the molecular charge and hydrophobicity (63). The profile of the uptake of these peptides has many similarities to that of lipophilic cations (63, 159); however, it is unclear if the charge on the peptides is delocalized and more work is required to better understand the similarities and differences in the mechanism of uptake between lipophilic cations such as TPP and targeted peptides. The peptides can be used to deliver a variety of attached cargo molecules to mitochondria (159). The initial peptides investigated contained a thiazole orange fluorophore to facilitate measurement of its intracellular location (63). As this fluorophore is itself a lipophilic cation connected to the peptide by a hydrophobic methylene bridge, the extent to which this appendage assists the uptake into mitochondria was unclear. However, subsequent studies using an anionic fluoroscein fluorophore also showed mitochondrial localization within cells (159), consistent with the peptides themselves having the intrinsic ability to localize to mitochondria. It would be informative if the mitochondrial and cellular uptake of radiolabeled versions of these peptides, without the additional molecular complexities of the fluorescent tags, could be investigated. This would facilitate quantitation of uptake into isolated mitochondria and cells, allow estimation of the applicability of the Nernst equation to these systems, and reveal the charge level of the species that are actually traversing the membrane. It would also be particularly interesting to see if these compounds can sustain a diffusion potential across a black lipid bilayer and, if so, whether its magnitude indicates the charge of the species crossing the membrane as has been done for TPP cations (126). In addition, there is a growing list of peptides that incorporate positively charged amino acids, particularly those containing the guanidinium group, that also show uptake into mitochondria (16, 47, 55). Therefore, it is likely that many other peptide variants will be designed and developed to optimize delivery to mitochondria. Towards this goal it would be useful to have more detailed information about the mechanism of the uptake of these compounds into mitochondria and their passage through biological membranes.
Mitochondria-targeted peptides can be used as cargo-delivering modules by conjugation to the molecule to be delivered to mitochondria. As mentioned previously, the peptides from the Kelley laboratory have been conjugated to fluorescent tags in order to demonstrate mitochondrial uptake in cells, and these have also been shown to be capable of delivering biotin and antioxidants to mitochondria in cells (63, 92, 159). Similarly, the SS-peptides can be used to deliver attached fluorophores to mitochondria in cells (141). Thus it should be possible to conjugate mitochondria-targeted peptides to functionalities and routinely deliver these to mitochondria. It will be interesting to see if the cargoes that can be delivered by peptides are different from those that can be directed to mitochondria by conjugation to a lipophilic cation, and a particular area of great interest would be to determine if peptides could be used to deliver more polar or negatively charged cargo that may not be possible for singly charged lipophilic cations (159).
The uptake of mitochondria-targeted peptides in vivo has been less extensively investigated than that of lipophilic cations to date. SS-peptides can be safely administered intravenously, intraperitoneally, and subcutaneously to rodents (4, 141), but their efficacy or distribution following oral administration has not been reported. The half life of the SS-peptides in rat and sheep plasma is relatively long (144), consistent with their uptake into the tissues. However, the uptake of these peptides into mitochondria within tissues in vivo and their metabolism has not yet been reported (4, 143, 158, 162). A number of animal studies have shown that SS-peptides that can act as mitochondria-targeted antioxidants, such as SS-31, are protective in a range of animal studies in which mitochondrial oxidative stress is thought to be involved. These include ischemia-reperfusion injury to the isolated heart (163); prevention of insulin resistance in skeletal muscle (4); protection against the MPTP model of selective neuronal injury in the brain (141). These findings are encouraging and support the view that these peptides are taken up by mitochondria within tissues in vivo where their antioxidant activity protects the mitochondria from damage. Even so, it is important that at some stage the uptake of these compounds into mitochondria in vivo is confirmed, quantified, and their metabolism and excretion is better understood to enable the rational optimization of this class of mitochondria targeted compound.
Other strategies to target small molecules to mitochondria
While mitochondria-targeted lipophilic cations or peptides are currently the most recognized, robust, and adaptable modes for targeting small molecules to mitochondria, there are a number of other methods that can in principle be used in this context. Even though these other approaches have not been widely utilized to date, they may prove useful in the future for the targeting of particular compounds to mitochondria. Among these possibilities is the utilization of the many specific transport machineries present in the mitochondrial inner membrane. This has been demonstrated for the protein import pathway where it has been shown in proof of principle experiments with isolated mitochondria that small molecules can be imported by conjugation to a signal peptide (152, 153). Many other specific mitochondrial transport systems could also be possibly used and this might enable the mitochondrial delivery of molecules that are too polar to be transferred by conjugation to lipophilic cations or mitochondria-targeted peptides, but which can be accommodated by the protein machinery of that specific transport process.
Other ways in which compounds could be selectively targeted to mitochondria is by the use of high affinity mitochondria-specific binding sites. This has been suggested for the accumulation of nonylacridine orange which binds to the mitochondria-specific phospholipid cardiolipin (52). A related approach is the relatively selective binding of porphyrins to the mitochondrial benzodiazepine receptor, which is present on the mitochondrial outer membrane (151). This approach may be used in photodynamic therapy to kill cells selectively in cancer therapies, as cancer cells express more of these receptors (151). In addition, there is considerable interest in developing ways of selectively inducing the mitochondrial pathway of cell death by targeting the proteins, such as those of the Bcl-2 family, that are present on the mitochondrial outer membrane and regulate the permeabilization of the mitochondrial outer membrane during the induction of apoptosis (6, 19). The development of molecules that bind to these sites has yet to yield a general strategy to target mitochondria; however, these approaches may prove useful when it is necessary to target to other locations in mitochondria such as the outer membrane or the inter membrane space.
Mitochondria-Targeted Small Molecule Therapeutics
Antioxidants
The most widespread use of mitochondria-targeted compounds as therapies has been with antioxidants designed to ameliorate mitochondrial oxidative damage. This area has already been covered in detail by a number of recent reviews (106, 135, 141) and therefore here it will be discussed concisely. The rationale for the development of mitochondria-targeted antioxidants is that, although oxidative damage to mitochondria contributes to a wide range of pathologies (3, 11, 50, 57, 141), antioxidant therapies have performed poorly in clinical trials (17, 30). One of the reasons for this lack of success may be related to the fact that only a very small proportion of the antioxidant in vivo is actually located in the mitochondria, where it is needed most. The driving force for the development of mitochondria-targeted antioxidants was to overcome this limitation (30).
A number of mitochondria targeted antioxidants have been developed by conjugation to the TPP cation (reviewed in Ref. 106) and the first example was a targeted version of Vitamin E, called MitoE2 (Fig. 3) (136). Since then many other antioxidants have been targeted to mitochondria in this way, including a ubiquinone (72), the peroxidase Ebselen (49), lipoic acid (21), a plastoquinone (133), nitroxides (36, 39, 149), and nitrones (59, 60, 105, 157). Most of these have shown protection against oxidative damage in isolated mitochondria and in cells, although only a few have been used in vivo. In animal studies, the best characterized mitochondria-targeted antioxidant is MitoQ10, which consists of a ubiquinone moiety linked to a TPP cation by a ten-carbon alkyl chain (7, 66, 68, 72, 106, 137). The ubiquinol form of MitoQ10 acts as an antioxidant and in doing so it is oxidized to the ubiquinone form, which is then rapidly re-reduced by complex II in the respiratory chain, restoring its antioxidant efficacy (68). As MitoQ10 is primarily found adsorbed to the mitochondrial inner membrane, and its linker chain enables the active ubiquinol antioxidant component to penetrate deeply into the membrane core, it is an effective antioxidant against lipid peroxidation (7, 72). The oral administration of MitoQ10 to mice and rats for up to at least 24 weeks is safe (124) and a number of in vivo studies have shown that orally administered MitoQ10 can protect against oxidative damage in a number of animal models of pathology, including: cardiac ischemia/reperfusion (I/R) injury (2, 108), damage to endothelial cells in vivo by nitroglycerin (46), hypertension (56), sepsis (88, 140), adriamycin toxicity (25), kidney damage in type I diabetes (24), MPTP toxicity in the brain (54), kidney cold preservation for organ transplantation (98), and cocaine toxicity (150). Other antioxidant moieties such as plastoquinone (133) and the nitroxide TEMPO (149) have also proven effective protective agents in vivo (39). Together these findings suggest that antioxidants targeted to mitochondria in vivo are protective against pathological changes in a number of animal models of mitochondrial oxidative damage that are relevant to human diseases.
The positive results in animal models led to the development of MitoQ10 as a pharmaceutical and it was assessed in phase II trials in humans. It was first used to ascertain if it could slow disease progression in Parkinson's disease (PD) in the PROTECT trial (
Mitochondria-targeted antioxidants based on SS-peptides have also been assessed in animal models of disease (143), most often with the SS-31 peptide that has antioxidant function (141). The peptide SS-31 showed protection in an ex vivo cardiac reperfusion model of I/R injury (163), while the peptides SS-02 and SS-31 were also protective against cardiac I/R injury when added during the reperfusion phase (143). Intraperitoneal injection of SS-31 resulted in uptake into the brain and protection against the neurotoxin MPTP-induced cell damage that mimics that of Parkinson's disease, to the substantia nigra (158). The SS-20 peptide, which does not have antioxidant ability in vitro, was also effective in this disease model, perhaps suggesting that, in this case, the protection may not be simply related to its antioxidant ability (158). Intraperitoneal injection of SS-31 was also protective against insulin resistance in the skeletal muscle in a high fat-fed mouse model (4). While the SS-peptides are effective in a number of animal models of disease involving mitochondrial damage and can be delivered in vivo by intraperitoneal or intravenous administration, they have not been yet shown to be orally active. While human trials have yet to be reported with SS-peptides, these are planned in the near future (H. Szeto, personal communication).
Using mitochondria-targeted compounds as anticancer agents
The application of mitochondria-targeted compounds as anticancer agents is another potentially important aspect and, as this field has been reviewed extensively, it will not be dealt with in detail herein (6, 14, 19, 40). The general strategy employed is to selectively kill the cancer cell by exploiting an aspect of cancer cell mitochondrial metabolism that differs from the situation in normal cells (14, 116). The most straightforward approach is to use the feature that mitochondria in cancer cells have a larger membrane potential than normal cells and consequentially there is increased uptake of lipophilic cations into cancer cells (26, 33, 147, 148). This increased concentration of the lipophilic cation in the cancer cell can then lead to selective toxicity by either the nonspecific effects arising from the accumulation of large amounts of the lipophilic cation in the mitochondria (120, 139), or by conjugation of the lipophilic cation to a suitable toxic agent (41). Functionalities such as an alkylating agent which damages DNA and disrupts the mitochondrion function or leads to toxicity elsewhere within the cancer cell have been utilized (94, 147, 148). An alternative approach involves the simultaneous targeting of two molecules to concentrate within mitochondria that then react to produce a toxin that damages the cancer cell in the neighbourhood of where it was formed (112, 119). Alternative approaches include to direct compounds to mitochondria that can render them more susceptible to other anticancer drugs or to selectively target aspects of mitochondrial metabolism that may contribute to resistance to anticancer drugs. For example, the targeting of gold compounds to mitochondria to inhibit mitochondrial protective antioxidant enzymes may be of use in limiting resistance to some cancer drugs (13, 62). Other approaches have been based on photodynamic therapy (40), involving the use of mitochondria targeted compounds that act as photosensitizers and the selective toxic effect is achieved by targeting only the tumor with light to induce local cell death (77).
Targeting pharmacophores and bioactive molecules to mitochondria
While most therapeutic or bioactive molecules that have been targeted to mitochondria have been either antioxidants or anticancer agents, the development of methodologies to target small molecules to mitochondria opens the way to directing many other pharmacophores and bioactive molecules to mitochondria in vivo (99, 103, 160). At this time there are only a few examples of such mitochondria-targeted molecules as exemplified by the mitochondria-targeted nitric oxide (NO) donor, MitoSNO (Fig. 3). This compound reacts with the intramitochondrial environment and produces NO locally that can diffuse out of the mitochondria and affect vasodilation in adjacent cells. In addition, the NO can also compete with oxygen at cytochrome oxidase to prevent hypoxia (114). Furthermore, MitoSNO can directly S-nitrosate mitochondrial protein thiols and thereby affect the activity of mitochondrial enzymes such as complex I and aconitase which correlates with protection in the heart in vivo during a model of cardiac I/R injury (29, 114). Another example of the selective targeting of bioactive molecules to mitochondria has been the delivery of compounds designed to selectively decrease the mitochondrial membrane potential, which may be of use in decreasing obesity or selectively decreasing the production of superoxide by mitochondria (61). One possible approach involved targeting protonophores to mitochondria by direct conjugation to the TPP cation with the intention of developing self-limiting uncouplers (15); however in one case reported, the TPP cation rendered the protonophore inactive (18). Nonetheless, some mitochondria TPP molecules do seem to interact with the mitochondrial inner membrane in such a way as to stimulate proton movement (87) and others have shown the potential to carry fatty acids across the inner membrane (131). A note of caution in this area is appropriate given that in wild-type mice fed MitoQ10 there was no effect on whole body energetics, suggesting that any such effects in vivo may be minimal (124). Cyclosporin A (CsA) has attracted interest as it inhibits the activity of cyclophilin D, thereby preventing much of the mitochondrial damage that occurs during I/R injury due to induction of the mitochondrial permeability transition (58). In order to improve mitochondrial targeting, CsA has been conjugated to TPP and this modification increased the selectivity of the modified CsA for mitochondrial cyclophilin D over cyclophilin A which is present outside the mitochondria. The modified CsA also showed protection against the mitochondrial permeability transition in cell models, even though conjugation to the TPP decreased the binding affinity to cyclophilins (93). Finally, it should be noted that molecules targeted to mitochondria need not always be protective to give interesting information. Illustration of this point is provided by the example of a photosensitizer targeted to mitochondria that generates singlet oxygen locally within mitochondria when illuminated, enabling the mitochondrial response to oxidative stress to be investigated (92). In summary, there is considerable scope to target a wide range of pharmacophores and bioactive molecules to mitochondria in vivo in order to act as therapies or to modify mitochondrial function.
Mitochondria-Targeted Probes
As well as using mitochondria-targeted molecules to intervene in mitochondrial function, there is also considerable interest in using probes to assess how mitochondria operate within cells and in vivo. There are a number of aspects of mitochondrial function that can be interrogated with small molecule probes.
Probes of the mitochondrial membrane potential
The mitochondrial membrane potential is a critical variable in assessing mitochondrial function and dysfunction (100, 109). The membrane potential, generated by proton pumping across the mitochondrial inner membrane by the respiratory chain, is utilized for ATP synthesis, uptake of molecules into mitochondria, and for thermogenesis (109). Consequently, the magnitude of the potential is one of the most important indicators of mitochondrial functional status. The extent of uptake of lipophilic cations by mitochondria has been widely used to assess the mitochondrial membrane potential in isolated mitochondria and cells (8, 20). The most commonly used methods for assessing the mitochondrial membrane potential in cells and in vivo is through the use of lipophilic fluorescent cations such as tetramethylrhodamine and the MitoTracker series of compounds that are selectively taken up into mitochondria in response to the magnitude of the mitochondrial membrane potential. The extent of uptake into the mitochondria can be assessed by confocal microscopy, measurement of fluorescence or, in tissues, by two photon fluorescence microscopy. These optical approaches have been reviewed extensively (12, 79, 109).
In some circumstances, it would be advantageous to be able to assess mitochondrial membrane potential by nonoptical approaches, for example, in whole animals or patients. One of these approaches is to directly measure the uptake of a lipophilic cation within mitochondria in vivo by positron emission tomography (PET) which enables the location and concentration of the PET nuclide within the body of an animal to be accurately determined. In principle, the local concentration of such a mitochondria-targeted PET probe within the body would reflect the amount of mitochondria in the region and their membrane potential. Therefore, tumors, which have a higher mitochondrial membrane potential, or areas of damaged tissue, which have a lower mitochondrial membrane potential, could be identified precisely and thus aid diagnosis and assessment of treatments in patients. To date, a number of PET probes based on the TPP lipophilic cation containing the 11C (90), 18F (28, 118), and 64Cu (74, 155) PET nuclides have been used in animal models with promising preliminary results (74, 90, 91). It is likely that these and other related direct methods to determine the mitochondrial membrane potential in whole organisms and patients by nonoptical means will be developed.
Mitochondria-specific probes for reactive oxygen species
Mitochondria produce the reactive oxygen species (ROS) superoxide which in turn forms hydrogen peroxide and peroxynitrite and this leads onto oxidative damage and also can be at the center of mitochondrial redox signaling (9, 70, 102). Consequently, there is considerable interest in measuring the levels of ROS such as superoxide and hydrogen peroxide within mitochondria in cells. This can be achieved by using fluorescent probes that are specific for a particular ROS and then modifying the molecule, generally by conjugation to a lipophilic cation, so that it is taken up by mitochondria (37, 38, 123). Among these is MitoSOX (Fig. 4), a mitochondria-targeted version of hydroethidine that reacts with superoxide to form a fluorescent product, thus enabling observation of the changes in fluorescence to indicate changes in mitochondrial superoxide (123). The chemistry of the reaction of MitoSOX with reactive oxygen species is complicated as it yields two fluorescent products, one of which is superoxide specific, while the other is formed in response to general oxidative stress, potentially affecting the interpretation of the fluorescent measurements, although the two products can be separated by HPLC (161, 164). To measure mitochondrial hydrogen peroxide, the TPP lipophilic cation has been linked to a phenylboronate ester (MitoPy1, Fig. 4) which reacts with hydrogen peroxide to generate a fluorescent product, thereby enabling the measurement of hydrogen peroxide within mitochondria (37, 38). The phenylboronate moiety also reacts with peroxynitrite, so these probes will respond to this ROS as well (132). These mitochondria-targeted fluorescent probes can be used to assess the production of various ROS within mitochondria in cells more selectively than untargeted probes and it is likely that many more selective fluorescent probes will be developed with enhanced sensitivity and selectivity for particular ROS species.
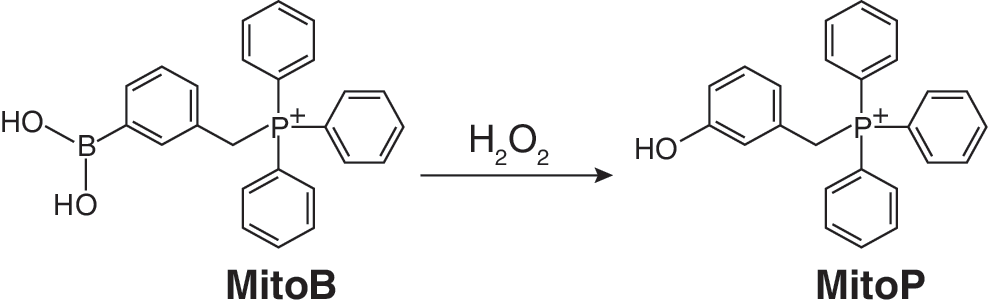
While mitochondria-targeted fluorescent ROS probes can be used effectively in cell systems and to some extent in vivo in transparent organisms such as zebrafish or in the outer layers of cells in tissues by the use of two photon confocal microscopy, there are many experimental systems, particularly in vivo, that are not accessible. Therefore it would be very useful to have alternative modes of ROS measurement using mitochondria-targeted probes that are not optically limited. One possible approach is to conjugate a spin trap to a lipophilic cation in order to direct the spin trap to mitochondria. Spin traps react selectively with free radicals to generate a relatively stable spin-trap adduct molecules containing an unpaired electron that can then be detected by electron paramagnetic resonance (EPR) spectroscopy (125). The advantage of this approach is the inherent selectivity for free radical species as a result of using the EPR spectroscopy detection system, as it is specific for products containing an unpaired electron. However, there are also a number of practical limitations to this approach including the issue that the reaction rate with free radicals is often quite low, so very high concentrations of the spin trap are required. Furthermore, the spin adduct often decomposes or is rapidly metabolised in vivo to form EPR silent products (125). Even so, a number of mitochondria-targeted spin traps have been developed by conjugating the TPP lipophilic cation to known spin traps such as PBN (105), DEPMPO (59, 60), BMPO (157), and cyclic nitrones (115) (Fig. 4). While these molecules are taken up by mitochondria and react with free radicals, to date, the low sensitivity of the probes developed has meant that none has so far been used to report on mitochondrial ROS production directly in vivo. Conversely in some cases, the targeted spin traps have proven useful as antioxidants to selectively decrease particular oxidative reactions within mitochondria (105). An alternative way to retain the selectivity of the reaction of a ROS-selective mitochondria-targeted probe but with greatly increased sensitivity is to use mass spectrometry to detect and quantify the reaction product (31). Our groups have extended this approach by using a ratiometric mass spectrometric mitochondria-targeted ROS probe named MitoBoronic acid (MitoB, Fig. 4) (31). We chose an arylboronic acid, based on the pioneering work of Chang (37, 97), who showed that the reaction of this moiety with H2O2 to form a phenol can be used to assess H2O2 within living cells. This function will also react with peroxynitrite (ONOO−) (132) so the extent of the conversion of MitoB to its phenol product (MitoP) in vivo (Fig. 8), the MitoP/MitoB ratio, will report on mitochondrial H2O2 and ONOO− production. After extracting the compounds from the whole organism, mass spectrometry was used to determine the MitoP/MitoB ratio, and accurate quantitation was achieved by including deuterated internal standards to correct for any variability in extraction and detection (82). The inherent positive charge of the TPP component of the MitoB and MitoP probes facilitates their very sensitive detection by mass spectrometry (10, 156). So far this approach has been used to measure the concentration of hydrogen peroxide within living fruit flies (31), but can be extended to measure many other types of specific ROS.
Other potential mitochondria-targeted probes
Many other probes of mitochondrial function can be developed using similar principles to those outlined above. For example, intramitochondrial thiol status has been examined by using lipophilic compounds containing a TPP function connected to various thiol reactive moieties (e.g., iodoacetamide TPP, Fig. 4). These compounds are taken up by mitochondria, react with protein thiols, and can then subsequently be identified by immunoblotting with antisera against the TPP moiety (86, 113, 128). This approach can be used to infer that protein thiols have been oxidized (86), and can also be used to show that a TPP compound is taken up by mitochondria in vivo (113, 137). A related development is the mitochondria-targeted lipophilic cations that can act as fluorescent probes for levels of mitochondrial iron (117). Thus, it is clear that as a general strategy it should be possible to adapt a wide range of probes to measure the mitochondrial environment by conjugation to a lipophilic cation. It should also be possible to extend this approach to make mitochondria-specific substrates by, for example, measuring the activity of mitochondrial processes in vivo by measuring the formation of a product by LC/MS/MS relative to deuterated standards as has been done for MitoB (31). Thus by appropriate modification and development of the approaches outlined here, it should be possible to generate many new and improved ways to assess mitochondrial function in cells and in vivo that will complement the other currently used methodologies.
Conclusions and Future Perspectives
The effort over the past few years on the development of general methods to deliver molecules to mitochondria in vivo is leading to many new classes of mitochondria-targeted therapies and probes. The delivery by lipophilic cations to mitochondria in vivo is reasonably well understood and it is relatively straightforward to use this approach to deliver small neutral probes, bioactive molecules, or pharmacophores to mitochondria in vivo. The delivery of compounds to mitochondria by conjugation to mitochondria-targeted peptides is also promising, but it will be useful to obtain more fundamental information about the rates, thermodynamic influences which are driving uptake, metabolism in vivo, and oral delivery. It will be particularly interesting to compare the uptake of mitochondria-targeted cations and peptides in vivo to determine whether there are consistent differences in the types of bioactive functionalities that can be delivered or if the uptake into particular tissues can be exploited for therapeutic advantage. Some significant challenges remain for both approaches including discovering how to combine the selective delivery to mitochondria with the specific delivery to particular organs.
It is now established that mitochondria-targeted antioxidants show efficacy in a wide range of animal models. Furthermore, they have been shown to be safe for oral administration to humans for up to one year and have shown promising results in a phase IIA study. Therefore, it is likely that many other mitochondria-targeted antioxidants will be developed as potential therapies for mitochondrial dysfunction for human diseases. This is also likely to lead to the utilization of these delivery strategies to develop many more mitochondria-targeted pharmacophores, bioactive molecules, and probes. To conclude, we are at the beginning of an exciting phase of research, development, and clinical innovation in the mitochondrial field. The important role of mitochondrial function and dysfunction in pathologies is accepted and useful new tools are now becoming available to learn more about these disorders and to treat them effectively.
Footnotes
Acknowledgments
Work in the authors' laboratories is supported by the Medical Research Council, the Biochemical and Biophysical Research Council, the Wellcome Trust, the United Mitochondrial Disease Fund, Antipodean Pharmaceuticals Inc., the Foundation for Research, Science and Technology New Zealand, the Marsden Fund of the Royal Society of New Zealand, the Lloyds TSB Foundation for Scotland and by the Royal Society of Edinburgh.
Author Disclosure Statement
MPM and RAJS hold intellectual property in the area of mitochondria-targeted compounds and act as consultants for Antipodean Pharmaceuticals Inc. RCH holds intellectual property on flavonol-vitamin E hybrid antioxidants and consults for Antoxis.