
Abstract
Introduction
The discovery and characterization of genes associated with familial PD, together with epidemiological and pathological investigation of sporadic PD cases, has yielded much insight into the pathways of PD. Mitochondrial dysfunction, oxidative stress, and abnormal protein aggregation have been strongly implicated in PD pathology, converging on dopaminergic neurons, impinging on normal cell function and contributing to cytotoxicity.
In general, mitochondria are key regulators of cellular bioenergetics and can also be effectors of cell death. Mutations in mitochondrial DNA (mtDNA) and the release of reactive oxygen species (ROS) from mitochondria are believed to contribute to the processes of aging; importantly, aging is the greatest risk factor associated with PD and many other neurodegenerative diseases. The brain is acutely sensitive to oxidative damage, due in part to its high content of easily oxidized unsaturated fatty acids, a high oxygen consumption rate, and a relative paucity of antioxidant enzymes compared to other organs (73), and DA neurons are believed to have heightened sensitivity to oxidative damage due to the generation of ROS secondary to dopamine metabolism (32). Disrupted mitochondrial energy metabolism leads to impaired Ca2+ homeostasis, increased generation of ROS, and decreased adenosine triphosphate (ATP) production, which in turn, may compromise function of the ubiquitin proteasome system (UPS), further contributing to abnormal protein aggregation in a deleterious feed-forward cycle. The role of protein aggregation in DA neurotoxicity in PD is supported by the presence of aggregated α-synuclein protein within Lewy bodies, which are the histopathological hallmark of PD. Alpha-synuclein, which is encoded by the gene SNCA, was the first protein to be linked to genetic PD (76). Further connecting mitochondrial function and genetic PD, are observations that a remarkable number of proteins that are associated with familial PD, including α-synuclein, parkin, PINK1, and DJ-1 localize in or interact with mitochondria (59, 115).
Additionally, the selective degeneration of dopaminergic neurons following systemic exposure to toxins strongly supports a role for mitochondrial dysfunction in sporadic PD. NADH-CoQ dehydrogenase (complex I) is the first of a series of multimeric enzyme proteins of the mitochondrial respiratory chain. Uncoupling of mitochondrial respiration by inhibition of complex I of the electron transport chain (ETC) has been shown to be a primary mechanism of toxin-induced mitochondrial dysfunction (90). Environmental pesticides and toxins that are complex I inhibitors thus directly impair mitochondrial energy production, consequently leading to oxidative stress and deficits in ATP. Overall, mounting experimental evidence from studies in both genetic and toxin models of experimental PD suggest that mitochondrial dysfunction occurs early and acts causally in PD pathogenesis (59). In this review, we will focus on the role of mitochondrial dysfunction in toxin models of parkinsonism.
Mitochondrial Dysregulation in Parkinson's Disease
Impaired mitochondrial dynamics in Parkinson's disease
Mitochondria function in a variety of biochemical pathways, including calcium buffering, carbohydrate, amino acid, lipid, and steroid metabolism, energy production by oxidative phosphorylation, and the provision of ATP for cellular energy requirements, and act as transducers of intracellular signaling pathways for apoptosis. Neurons derive the vast majority of their energy by oxidative phosphorylation and have a very high density of mitochondria. Mitochondria are the main sites of ROS generation within the cell because oxidants, such as H2O2 and superoxide radicals, are produced as natural byproducts of oxidative phosphorylation. Thus, a normal homeostatic function of mitochondria is to offset basal levels of ROS with a battery of endogenous antioxidants. However, under certain pathological conditions where acute or sustained mitochondrial respiration defects occur, the amount of generated ROS can overwhelm the neutralizing capacity of these endogenous antioxidants, leading to overall mitochondrial dysfunction.
Additionally, mitochondria are highly dynamic organelles; emerging evidence suggests that disruption of the fundamental balance between mitochondrial fission and fusion (collectively designated ‘mitochondrial dynamics') contributes to neurodegeneration as mitochondrial dynamics are indispensable for neurotransmission, synaptic maintenance, mitochondrial biogenesis, and overall neuronal homeostasis and survival (97). Disruption of mitochondrial dynamics and resulting mitochondrial dysfunction can emanate from mutations in the genes that encode fission- or fusion-related proteins or from post-translational modifications that regulate their stability and activity. The PD-related proteins parkin, PINK1, α-synuclein, and HTRA2/Omi, have been shown to potentially affect mitochondrial dynamics (11). Moreover, toxins used to model experimental PD, such as 6-hydroxydopamine (6-OHDA) and rotenone, have likewise been shown to impinge on mitochondrial dynamics by inducing mitochondrial fission (3, 38). Figure 1 is a summarized depiction of mitochondrial fission and fusion (Fig. 1).

Mitochondrial DNA mutation and deletion in Parkinson's disease
Systemic defects in mitochondrial respiration have been recognized in PD patients for some time, initially based on experiments using cybrid cells (99). In these experiments, Swerdlow and colleagues used platelet-derived mitochondria from PD patients to repopulate neuroblastoma cells in which mitochondria were absent. The cybrid cells that resulted from these experiments were found to have increased levels of oxidative stress and decreased basal mitochondrial complex I activity with concomitant enhanced susceptibility to cell death by complex I inhibition. These findings suggested that the mitochondrial genome may contribute to the complex I deficiency observed in PD patients.
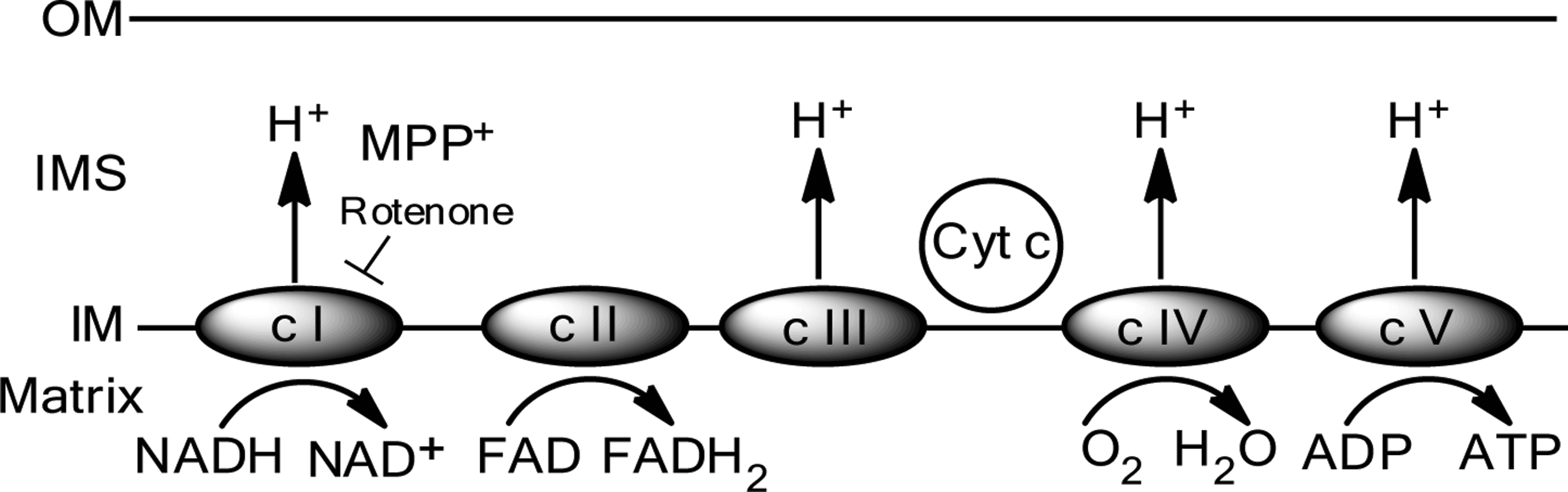
Most mitochondrial proteins are encoded by genomic DNA. However, the mitochondrial genome encodes 13 subunits of the ETC, including seven subunits for complex I, one for complex III, three subunits for complex IV, and two complex V subunits. Additionally, mtDNA is thought to be especially vulnerable to oxidative damage because it is hypomethylated, employs a relatively inefficient repair mechanism, and is proximal to the ETC in the inner mitochondrial membrane, which is a potent source of ROS due to its role in oxidative phosphorylation; all of these factors are believed to contribute to a high rate of mtDNA mutation and deletion (59). Corroborating the notion that mtDNA deletions may contribute to neurodegeneration in PD, increased levels of mtDNA deletions have been observed in the striatum and in substantia nigra (SN) neurons of PD patients; however, the role of single base pair mutations, polymorphisms and haplogroups in human PD remains controversial (118). Importantly, previous methods of measuring mtDNA mutations have recently been called into question, supporting a more conservative interpretation of the data associating accumulation of mtDNA mutations and age (113). Overall, studies of mtDNA deletions and mutations in the context of PD suggest that the mitochondrial genome may be involved in the pathogenesis of PD, but additional genetic and functional studies are necessary to further elucidate this proposed connection.
Mitochondrial complex I deficit in sporadic Parkinson's disease
In addition to a proposed systemic defect in mtDNA and overall compromised mitochondrial respiration in PD, compelling evidence implicating mitochondrial dysfunction in PD originates from postmortem biochemical studies wherein a disease-specific and drug-dependent defect of the mitochondrial respiratory complex I was observed in SN tissue from patients with idiopathic PD (43, 86). Moreover, complex I deficiency has also been observed in the frontal cortex, in platelets, and though somewhat inconsistently, in skeletal muscle and lymphocytes of PD patients (reviewed by (11)). The detection of complex I deficiency in multiple brain regions and tissues suggests that sporadic PD is characterized by a systemic defect in mitochondrial complex I activity.
Environmental Toxins and PD: Mitochondrial Dysfunction in Toxin-Induced Dopamine Neuron Death
The ideal animal model for PD, whether induced by environmental neurotoxins or genetic manipulations, would successfully recapitulate the characteristics of human PD, including behavioral abnormalities (motor deficits), pathological features (selective, progressive nigrostriatal degeneration with the presence of Lewy bodies), and molecular dysfunctions affecting brain regions other than the SN. Unfortunately, all current animal models of experimental PD fail to completely mimic the etiology, progression, and pathology of human PD. Many genetic mouse models of parkinsonism, both those with autosomal dominant and autosomal recessive inheritance patterns, fail to produce neurodegeneration of DA neurons and all but one of the genetic mouse models of PD fail to exhibit cytoplasmic inclusions reminiscent of Lewy bodies (105). There are limitations to toxin-induced animal models of parkinsonism as well, including adverse systemic toxicity, challenges in reproducibility, variability in response to dopamine or dopamine agonist therapy, and a tendency to fail to model early stage parkinsonism and extra-nigral pathology (111). Despite these limitations, toxin-induced models of parkinsonism have been widely used, continue to evolve, and have yielded a wealth of insight into PD neuropathogenesis while also providing disease models in which to define putative pharmacological targets and to test potential therapies.
Toxin-induced animal models have been used to study PD for more than half a century. Introduced in the late 1950s, the 6-hydroxydopamine (6-OHDA) model of parkinsonism was the first toxin model to be developed. The structure of 6-OHDA is very similar to that of dopamine, but an additional hydroxyl group on 6-OHDA renders it specifically toxic to DA neurons. Much of the biochemical, physiological, and behavioral effects of nigral DA neuron loss and striatal DA depletion has been gleaned from the 6-OHDA model of parkinsonism. While recent data implicate a potential role for mitochondrial dysfunction and apoptotic signaling in 6-OHDA-induced cell death (93), and report a gender-specific role for mitochondria in the vulnerability of mesencephalic neurons to 6-OHDA treatment (67), oxidative stress is widely regarded to be the primary mechanism of DA cell death in the 6-OHDA models of parkinsonism. However, toxins used to model parkinsonism subsequent to the 6-OHDA model, such as MPTP and various environmental pesticides, have been shown to directly involve mitochondrial dysfunction in cell death. Figure 2 (Fig. 2) depicts various mitochondrial toxins that have been used to model experimental PD.

MPTP Models of Parkinsonism
Overview of MPTP animal models
The meperidine analog 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was first recognized as a neurotoxin when a group of intravenous heroin users developed an acute version of parkinsonism that was symptomatically indistinguishable from sporadic PD (54). MPTP was discovered to be a contaminant in the synthetic heroin that poisoned the drug users, and after it was recognized to function as a DA neurotoxicant, animal models were quickly developed, initially through repeated injections of MPTP in monkeys, thereby providing the first effective nonhuman primate model of parkinsonism (12). MPTP-treated nonhuman primates exhibit the major symptoms of human PD (with the exception of tremor) and respond to all drugs that are currently used in the treatment of PD (50). The MPTP model in nonhuman primates is basically a model of nigrostriatal degeneration that occurs in late-stage human PD; it is not progressive, does not affect other brain regions that are affected in human PD patients, and Lewy bodies are not observed (49, 55).
Shortly after the MPTP nonhuman primate model was introduced, a protocol to induce MPTP-dependent parkinsonism in mice was developed (44). The extent, as well as the timecourse of DA neuron loss in the MPTP mouse model can vary depending on numerous factors, including the cumulative MPTP dose, the number of doses, and the frequency of administration (87). An important caveat to the MPTP model of parkinsonism is the salient fact that MPTP is not toxic to nigral DA neurons in many other nonprimate species for reasons that remain incompletely understood. Rats especially are resistant to MPTP-induced neurotoxicity; therefore, an active metabolite of MPTP must be administered stereotaxically or directly infused into the central nervous system (CNS) to achieve nigral DA neurodegeneration (94, 119).
Thousands of articles describing the relationship between MPTP and PD have been published and the MPTP model remains the best-characterized model of experimental PD; despite this vast history, the MPTP model continues to evolve. New routes of MPTP exposure, including repeated intranasal delivery, have been explored (83) and this mechanism of MPTP delivery may more readily mimic human exposure to pesticides and other environmental toxins. Additionally, the MPTP mouse model has recently been shown to include degeneration of DA neurons in the enteric nervous system, recapitulating some of the aspects of gastrointestinal dysfunction observed in human PD patients (2, 70), suggesting that the MPTP mouse model may be appropriate for experiments designed to investigate extra-nigral PD pathophysiology. Importantly, impaired colonic motility due to dysfunction of the enteric nervous system within the gastrointestinal tract is a nonmotor and dopa-unresponsive symptom of human PD, and the extent of enteric Lewy neurite burden has been shown to correlate with motor symptoms of human PD (57).
Interestingly, Fornai and colleagues report that wild-type mice chronically administered MPTP via osmotic pump developed progressive behavioral deficits, nigral DA neuron loss, and nigral inclusions immunoreactive for α-synuclein and ubiquitin (reminiscent of Lewy bodies), whereas wild-type mice sporadically administered MPTP via single or multiple intraperitoneal bolus injections exhibited nigral DA neuron loss but did not manifest with nigral α-synuclein/ubiquitin positive inclusions (34). In these studies, the ubiquitin proteasome system was more inhibited in terms of magnitude and duration in the chronic MPTP administration model relative to the sporadic MPTP administration model, and was α-synuclein dependent, as the effects of chronic MPTP administration were significantly attenuated in α-synuclein-deficient mice (34). While mitochondrial dysfunction was not examined in this study, the results of Fornai and colleagues demonstrate that the route of MPTP administration is highly relevant in MPTP animal models.
MPTP metabolism and mitochondrial mechanisms of neurotoxicity
Following systemic injection, MPTP readily crosses the blood-brain barrier (BBB). The MPTP pro-toxin is then metabolized by the enzymatic action of monoamine oxidase B (MAO-B), which is enriched in endothelial cells of the BBB microvasculature and in glial cells, into the metabolite 1-methyl-4-phenyl-2,3-dihydropyridium (MPDP+), which is then deprotonated (likely due to spontaneous oxidation) to the active neurotoxin 1-methyl-4-phenylpyridinium (MPP+) (19). The actively toxic species MPP+ is then released into the extracellular milieu where it enters DA neurons via selective uptake by the dopamine transporter (DAT) (79). Figure 3 (Fig. 3) depicts MPTP metabolism within DA neurons and the proposed mechanism of MPP+-induced DA neurotoxicity. Midbrain neurons have been shown to have the highest concentration of DAT per cell (15), perhaps partially explaining the selective sensitivity of midbrain DA neurons to MPTP. DAT is requisite for MPTP-induced toxicity; mouse strains known to be sensitive to MPTP but which harbored null mutations for DAT were completely protected from MPTP-induced cell death (36).
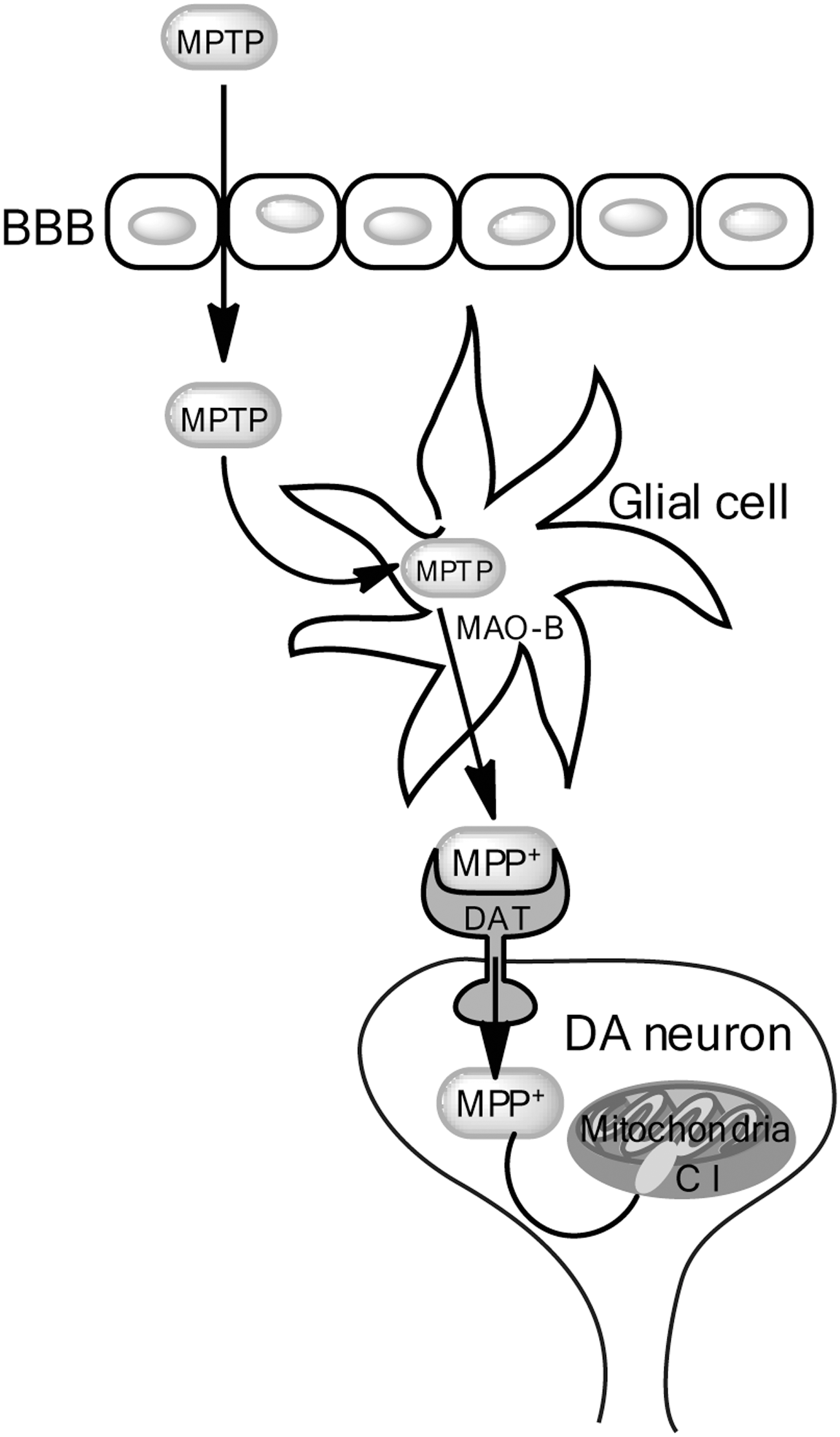
In mice, MPTP intoxication causes acute DA neuron dysfunction and death within days, as well as a prolonged sequelae of events that contribute to DA neurodegeneration for months to years (48). Once inside the neuron and during the acute phase of MPTP-induced death, there are a few routes that MPP+ can take: it can enter the vesicular pathway, bind to vesicular monoamine transporters and translocate into synaptosomal vesicles (61), it can remain in the cytosol and interact with various cytosolic enzymes (53), or it can be concentrated within mitochondria via a mechanism dependent on mitochondrial transmembrane potential (80, 84). Within the mitochondria, MPP+ binds to complex 1, uncoupling the oxidation of NADH-linked substrates and consequently disrupting the flow of electrons along the ETC, resulting in decreased ATP production and increased generation of ROS (72). Furthermore, in addition to inhibition of complex I, MPTP has been suggested to inhibit mitochondrial complexes III and IV (but not II) in vitro (28).
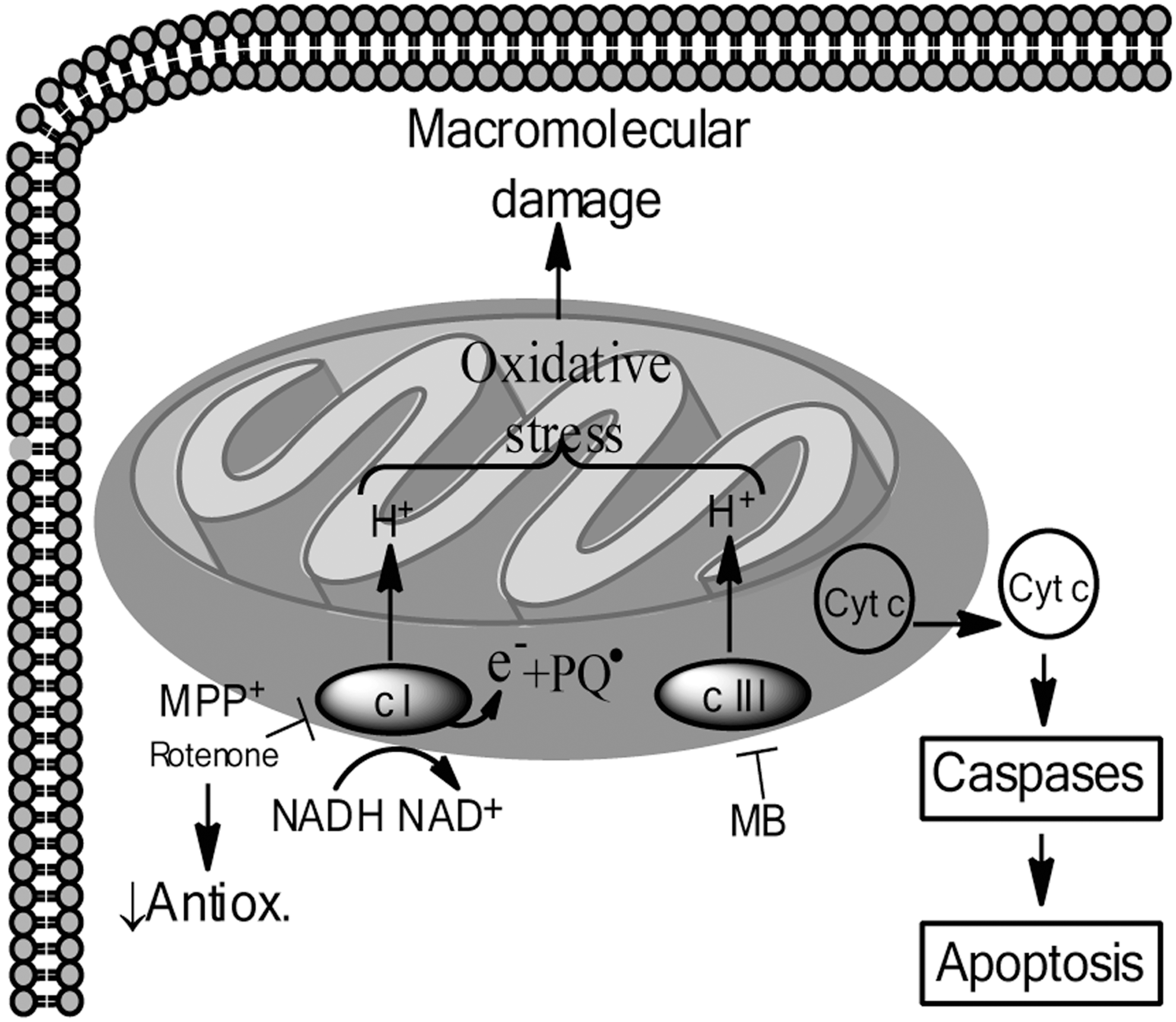
Excessive oxidative stress is one proposed mechanism of DA neurotoxicity (Fig. 4) and it is hypothesized that MPTP-induced toxicity resulting from MPP+ inhibition of mitochondrial complex I is primarily dependent on oxidative stress. Support for this hypothesis is provided by the observation that MPP+-related ATP depletion causes only an approximately 20% reduction in ATP levels in the striatum and midbrain, which is not likely to be a severe enough depletion of ATP to be the primary cause of DA neuron death (17, 25). Further support for the oxidative stress-mediated mechanism of mitochondrial dysfunction by MPTP is evidence that transgenic mice overexpressing the antioxidant Cu/Zn-superoxide dismutase enzyme have attenuated MPTP-induced DA neuron loss (78). Conversely, mice deficient in CuZn-superoxide dismutase (Cu/Zn SOD) or glutathione peroxidase exhibit enhanced DA neurotoxicity in response to MPTP (122). Additionally, increased nitric oxide (NO) signaling has been specifically implicated in MPTP-induced toxicity, both through pharmacologic inhibition of nitric oxide synthase (NOS) enzymes and through the use of transgenic animals (27, 88).
Recent experiments conducted in cell cultures and in an MPTP mouse model have further elucidated MPTP-induced mechanisms of mitochondrial dysfunction. Petit-Paitel and colleagues showed that MPTP/MPP+ induces specific activation of the GSK-3β isoform of glycogen synthase kinase (GSK)-3 by phosphorylation of Tyr216. Interestingly, MPTP/MPP+ treatment increased the cytosolic pool of active GSK-3β, while simultaneously depleting the mitochondrial pool of active GSK-3β, leading to depolarization of the mitochondrial membrane and subsequent caspase activation and cell death. Blockade of GSK-3β activity with specific inhibitors or via siRNA-mediated GSK-3β knockdown abolished these effects, suggesting that inactive or diminished mitochondrial levels of GSK-3β contributes to MPTP/MPP+-induced cell death (75).
Additionally, a recent in vivo proteomic analysis of changes in protein abundance in an MPTP mouse model identified 58 proteins associated with mitochondrial function (including subunits of all five mitochondrial complexes) that exhibited significant brain region-dependent changes in abundance following MPTP treatment (123). This proteomic study supports the general notion of MPTP-induced mitochondrial dysfunction, as many of the protein ‘hits' identified have been previously validated in other studies. However, several proteins known to play a role in mitochondrial function but previously unstudied in the MPTP model were observed to change abundance in response to MPTP treatment. Secondary validation of the function of these proteins in the paradigm of MPTP-treatment may further elucidate mechanisms of mitochondrial dysfunction in PD.
In summary, despite its limitations, such as its acute nature, the absence of clearly-defined Lewy bodies and Lewy neurites, and the lack of effects in other brain regions affected in human PD, the MPTP model is one of the most widely used models of experimental PD. The MPTP model provided proof of principle for the involvement of environmental toxins in sporadic PD, and supplied the initial experimental evidence to strongly implicate mitochondrial dysfunction in the pathogenesis of PD. Additionally, the MPTP primate model has been indispensable in the development and testing of novel PD therapeutics as well as in studies examining side-effects of current PD therapies, such as dyskinesias in response to levodopa (49). Furthermore, recent data reporting gastrointestinal dysfunction in the MPTP mouse model (2, 70) support the notion that MPTP-treated mice may be an appropriate model in which to study extranigral PD pathophysiology.
Environmental Toxins, Pesticides, and Parkinson's Disease
Almost immediately after the discovery of the neurotoxicity of MPTP, the potential involvement between environmental exposure to pesticides/herbicides and PD pathology became an area of intense research. Epidemiological studies over the years have consistently proposed an association between PD and exposure to pesticides (4). The types of pesticides that have been prospectively correlated with PD are numerous and varied, including: insecticides (e.g., organophosphates, carbamates, rotenone); herbicides (e.g., bipridyls and paraquat); fungicides (e.g., maneb); and organochlorines (e.g., DDT and dieldran) (10). Some of these pesticides are overtly neurotoxic both in vivo and in vitro (6, 7) and others induce or inhibit enzymes that function in xenobiotic pathways (56) and may therefore impact metabolism of other endogenous or exogenous neurotoxins. These and similar studies exploring the proposed connection between pesticides and PD have implicated numerous mechanisms of pesticide-induced dopaminergic neurotoxicity, including mitochondrial toxicity, oxidative stress, and aberrant interactions with α-synuclein. Further associating pesticide exposure with increased susceptibility to PD is the structural resemblence between MPTP and many pesticides/herbicides (Fig. 2), which (like MPTP) are capable of inhibiting mitochondrial complex I, thereby enhancing hydrogen peroxide and superoxide generation, and leading to sustained mitochondrial dysfunction (39). From an epidemiological standpoint, it is likely that environmental toxins that inhibit mitochondrial complex I contribute to PD pathogenesis in concert with genetic predispositions and other environmental factors.
Rotenone Models of Parkinsonism
Overview of rotenone animal models of parkinsonism
Rotenone is a naturally occurring organic agent historically used as a pesticide and to control nuisance fish populations in lakes and reservoirs. Rotenone is a specific mitochondrial complex I inhibitor and is highly lipophilic, readily crossing the BBB and biological membranes independent of a receptor or transporter; therefore, rotenone functions as a systemic complex I inhibitor. In spite of inhibiting complex I systemically, rotenone has been shown to selectively lead to neurodegeneration of nigral DA neurons and is therefore used to model experimental parkinsonism. Very recently, rotenone has been associated with PD in humans (odds ratio 2.5, 95% confidence interval 1.3, 4.7) (102).
Because rotenone is poorly absorbed by the gastrointestinal (GI) tract, the initial studies using rotenone treatment in rats utilized stereotaxic infusion of the toxin into the parenchyma (45). While robust decreases in striatal dopamine and serotonin levels were observed, these early versions of the rat rotenone model involved high concentrations of rotenone and were not very specific for dopamine neurons and terminals. However, as rotenone is a systemic complex I inhibitor and systemic complex I deficiency was observed in sporadic PD patients, systemic administration of lower concentrations of rotenone were then used to model parkinsonism in the Lewis rat strain. Chronic intravenous infusion of rotenone in rats caused a 75% reduction in complex I activity and selective degeneration of nigrostriatal DA neurons (6). In these studies, rotenone-induced pathology closely recapitulated that observed in human sporadic PD, in that the neurodegeneration began in nerve terminals within the striatum and retrogradely progressed to the cell bodies in the SN, involved oxidative protein damage, elicited motor symptoms of bradykinesia and rigidity, and for the first time in an animal model of experimental PD, cytoplasmic α-synuclein and ubiquitin immunoreactive inclusions reminiscent of Lewy bodies were observed in surviving neurons within the SN and striatum (6, 89). However, these studies were limited by variability in the percentage of animals that exhibited a defined nigrostriatal lesion following rotenone administration; while this variability in the extent of nigrostriatal lesion in the rotenone rat model can be frustrating in terms of consistent reproducibility, it may reflect a complex interaction between variable genetic susceptibility factors and inherent differences in xenobiotic metabolism within rats. Nonetheless, subsequent development of chronic intraperitoneal administration models of rotenone has been shown to elicit more consistent and reproducible behavior and neurochemical deficits in rats (13).
Recently, chronic rotenone exposure in rats was reported to reproduce GI neuropathology similar to that observed in human PD. Drolet and colleagues showed that the chronic intraperitoneal model of administering low-dose rotenone (1.0 mg/kg or 2.0 mg/kg of rotenone 5 times weekly for 6 months) which is subthreshold to induce overt nigrostriatal lesions and concomitant akinetic motor deficit, was sufficient to elicit loss of TH-positive enteric neurons and to slow GI transit time (31), mimicking symptoms of constipation and enteric pathology observed in human PD (57). Additionally, formic acid-resistant, α-synuclein immunoreactive aggregates resembling Lewy bodies were also observed in the small intestine of rotenone-treated rats. These observations indicate that this low-dose, chronic, and systemic rotenone paradigm may provide a good experimental model in which to study extranigral PD pathology in the GI system and may provide a “pre-clinical” model of parkinsonism, possibly enabling the study and delineation of early DA neurotoxic events, and thus providing opportunities to test novel therapeutics and to potentially develop PD biomarkers that may enable early diagnosis.
Also recently, a chronic rotenone mouse model of parkinsonism was developed. Inden and colleagues showed that chronic oral administration of rotenone at 30 mg/kg for 56 days in C57BL/6 mice caused a significant and selective loss of nigral DA neurons and induced behavioral impairment. In addition, α-synuclein immunoreactive cytoplasmic inclusions similar to Lewy bodies were observed in surviving DA neurons in a time-dependent manner (47). This recently optimized mouse rotenone model may thus provide a powerful model system in which to study gene–environment interactions if the chronic rotenone regimen is used in mice that are transgenic for genes linked to human PD.
Rotenone metabolism and mitochondrial mechanisms of neurotoxicity
The mitochondrial complex I enzyme oxidizes NADH, transferring electrons to coenzyme Q10 (also referred to as ubiquinone), which is a lipid-soluble electron carrier. Additionally, complex I translocates protons from the mitochondrial matrix to the intermembrane space, thus assisting in establishment of the electrochemical gradient that is used to drive ATP synthesis (42). Rotenone is a classical, high affinity inhibitor of mitochondrial complex I (81) that binds at the acceptor end of the complex, increasing its reduction state and thus precipitating the leakage of electrons which combine with O2 to generate superoxide (71).
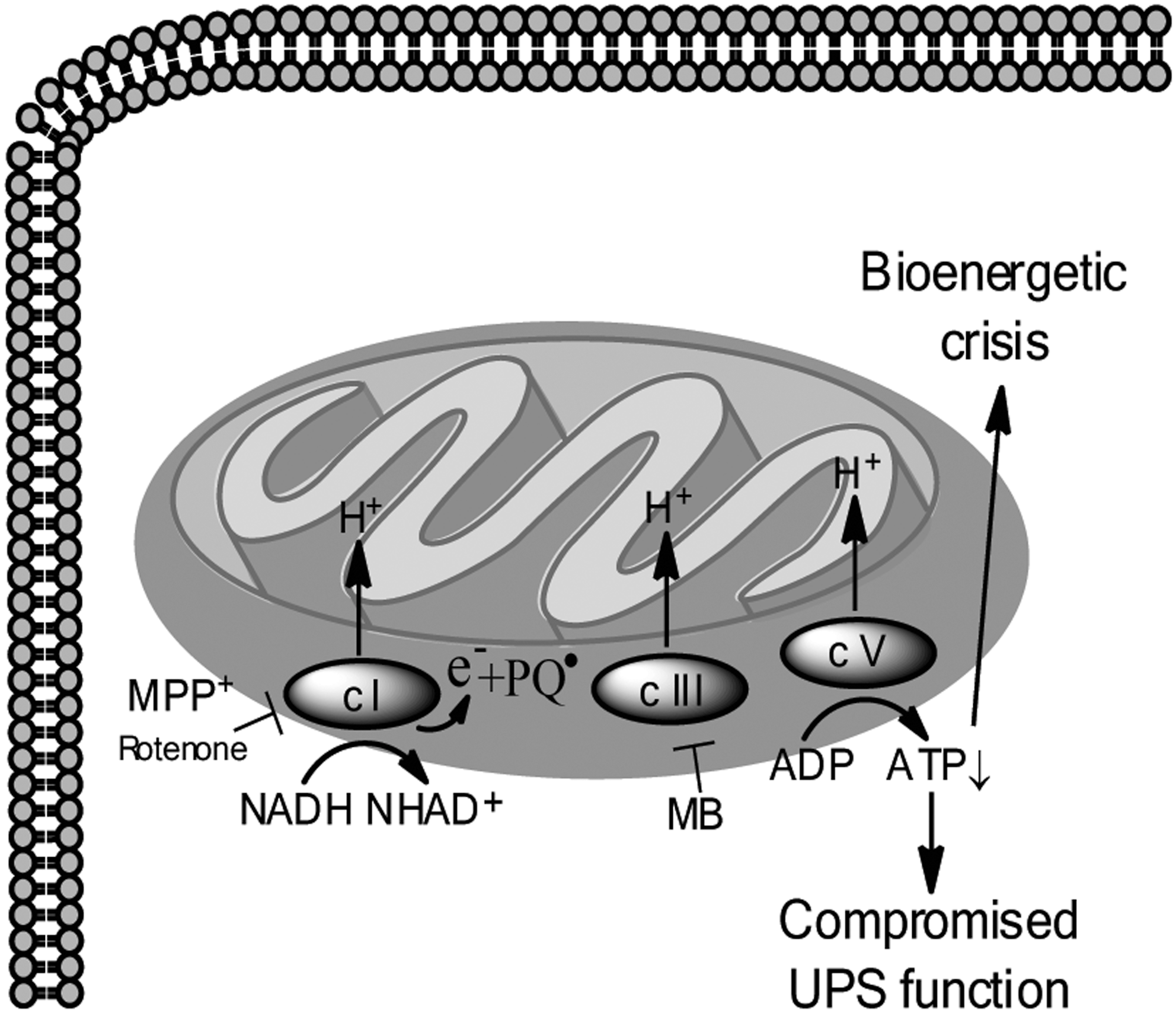
Therefore, the consequences of inhibition of mitochondrial complex I by rotenone are multifarious (Fig. 5). Generation of ROS, which drives further oxidative stress, is thought to be a result of the accumulation of electrons that occurs upon complex I inhibition, and indeed, rotenone-induced toxicity secondary to oxidative stress has been extensively studied and is attenuated by antioxidants in vitro (89) (Fig. 4). Inhibition of complex I can also sensitize DA neurons to an NMDA receptor-mediated bioenergetic crisis as levels of ATP become diminished (71). Figure 6 (Fig. 6) depicts the ATP crisis model of DA neurotoxicity. Additionally, inhibition of complex I may lead to aberrant opening of the mitochondrial permeability transition pore, which can cause depolarization of mitochondria, and facilitate mitochondria-dependent apoptotic or necrotic cell death (41). In fact, nanomolar concentrations of rotenone have been shown to selectively cause caspase-3 activation in DA neurons (other neuron populations were not similarly affected) and the caspase-3 inhibitor DEVD-fmk can attenuate rotenone-induced death of DA neurons in vitro (1). Likewise, overexpression of Bcl-2, a mitochondria-resident anti-apoptotic protein that regulates the permeability of the outer mitochondrial membrane, was shown to dramatically inhibit rotenone-induced changes in mitochondrial potential, further confirming involvement of mitochondrial dysfunction in rotenone-induced apoptotic cell death (100).
Additionally, mitochondrial Ca2+ overload as a result of dysregulated Ca2+ homeostasis and glutamatergic excitotoxicity have been proposed as another model of DA neurotoxicity and is depicted in Figure 7 (Fig. 7). High levels of Ca2+ are acutely cytotoxic, therefore intracellular Ca2+ levels are tightly regulated; in most neurons, the opening of Ca2+ channels is a rare event, usually occurring during action potentials, but in the case of DA neurons in the SN, Cav 1.3 Ca2+ channels are open much of the time to facilitate autonomous pacemaking functions (96). Glutamatergic synaptic input to SN DA neurons results in Ca2+ entry into the neuron via the NMDA receptor, which combined with Ca2+ entry through Cav 1.3 Ca2+ channels, may synergistically cause increased cytosolic Ca2+ levels that can be transiently buffered by mitochondrial uptake of Ca2+(98, 114). Sustained elevations in mitochondrial Ca2+ can however, lead to permeabilization of the mitochondrial membrane due to opening of the mitochondrial permeability transition pore, causing Ca2+ diffusion back into the cytoplasm. Interestingly, the vulnerability of nigral DA neurons to the toxins MPTP, 6-OHDA, and rotenone is increased by calcium influx (16). Moreover, low nanomolar concentrations of rotenone have been shown to significantly increase NMDA receptor-mediated Ca2+ influx (71, 116). Thus, the mechanisms of ATP depletion and Ca2+ overload may converge as a result of rotenone-induced inhibition of complex I; in the context of mild to moderate ATP depletion and increased Ca2+ influx via the NMDA receptor, rotenone has been shown to increase glutamate release from isolated terminals, thereby sensitizing DA neurons to glutamatergic excitotoxic cell death (52, 71). Further supporting the Ca2+-overload model of DA neurotoxicity is the observed inverse correlation between expression of calbindin, a mobile calcium buffering protein, and susceptibility to DA neurodegeneration (5).
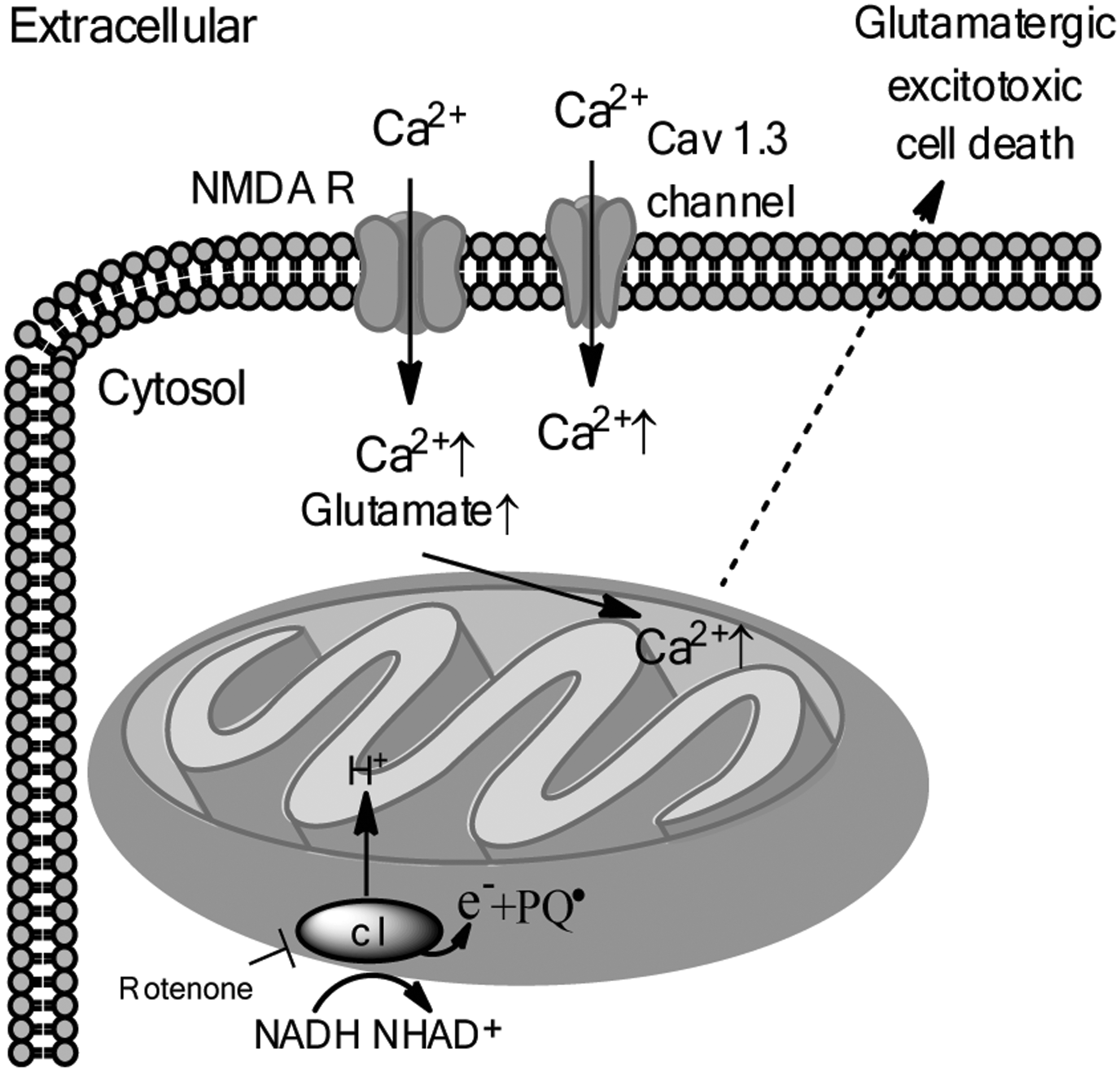
Moreover, rotenone-induced neurodegeneration may involve Ca2+-independent glutamate-mediated excitotoxicity. Kilbride and colleagues report that a partial inhibition (∼40%) of complex I was sufficient to induce Ca2+-independent release of glutamate in synaptosomes isolated from rats. The increase in glutamate correlated with a significant decrease in ATP and led to depolarization of the mitochondrial membrane potential, essentially implicating both the mechanisms of ATP energy crisis and glutamatergic excitotoxicity in the demise of DA neurons (52).
As with any animal model of experimental PD, the rotenone model has limitations, including potential systemic toxicity. Inconsistency in symptomatic phenotype has been observed among different strains of rats, and striatal degeneration in addition to nigral neuron loss has also been reported (20). However, much valuable insight into the pathophysiological mechanisms of DA neurotoxicity has been gained as a result of experiments conducted in rotenone animal models. The rotenone model of parkinsonism lends excellent support to the environmental hypothesis of PD, especially in regard to pesticide exposure and PD, and was the first model to provide experimental evidence that modest systemic defects (such as systemic inhibition of complex I) may lead to specific and robust neuropathology, effectively modeling parkinsonism, while simultaneously demonstrating that nigral DA neurons have an exquisite sensitivity to mitochondrial complex I inhibition (40).
Paraquat and Maneb Models of Parkinsonism
Overview of paraquat animal models of parkinsonism
Paraquat (1,1’-dimethyl-4,4’-bipyridinium dichloride; PQ) is structurally similar to MPTP (Fig. 2.) and is a widely used herbicide that has very recently been associated with human PD (odds ratio=2.5, 95% confidence interval 1.4, 4.7) (102). PQ treatment elicits a selective and dose-dependent loss of nigral DA neurons with associated modest decreases in striatal dopamine nerve terminal density and mildly decreased motor behavior (9, 64). Following sustained systemic intraperitoneal administration, PQ was found to have a very long half-life in the mouse brain (up to 28 days) and was associated with ongoing lipid peroxidation (77). Moreover, in a chronic rat PQ model, minor decreases in DA neurons and slight increases in DA neurotransmission were observed at 4 weeks; by 24 weeks significant DA neuron loss and DA neurotransmitter depletion were observed, indicating a potentially chronic neurodegenerative process induced by PQ treatment (74). These results support the consideration that PQ treatment in rodents may provide a “pre-clinical” PD model that may be useful in studying early events in PD pathology.
Paraquat metabolism and mitochondrial mechanisms of neurotoxicity
Though structurally similar to MPTP, PQ crosses the BBB (via a neutral amino acid transporter) much less efficiently than MPTP although detectable levels of PQ have been measured in the CNS after its systemic injection into rodents (23, 63). Interestingly, despite its structural similarity to MPTP and an apparent interaction with mitochondrial complex I, the deleterious effects of PQ on DA neurons are distinct from the toxic mechanisms of MPTP and rotenone, both of which potently inhibit complex I, while PQ is a rather weak inhibitor of mitochondrial complex I activity and does not require DAT for neurotoxicity (82).
However, PQ (but not MPTP or rotenone) is a potent redox cycler, readily converting to a free radical that interacts with molecular O2 to generate superoxide anions and other ROS within mitochondria (51, 120) (Fig. 8). Redox cycling is a primary cause of PQ-induced toxicity as evidenced by protection from adverse PQ effects by overexpression of superoxide dismutase (SOD) (108) or via administration of SOD mimetics (68). Conversely, deficiency in antioxidant enzymes causes hypersensitivity to PQ treatment in vivo (112). In vitro in SH-SY5Y cells, PQ has been shown to increase generation of ROS and to concomitantly decrease antioxidants (117). Importantly, mitochondria are the principal cellular sites of PQ-induced ROS production and complex III of the ETC has been shown to prominently contribute to this process as a novel role for complex III-dependent mitochondrial ROS production by redox cycling herbicides such as PQ has recently been described (14, 30).
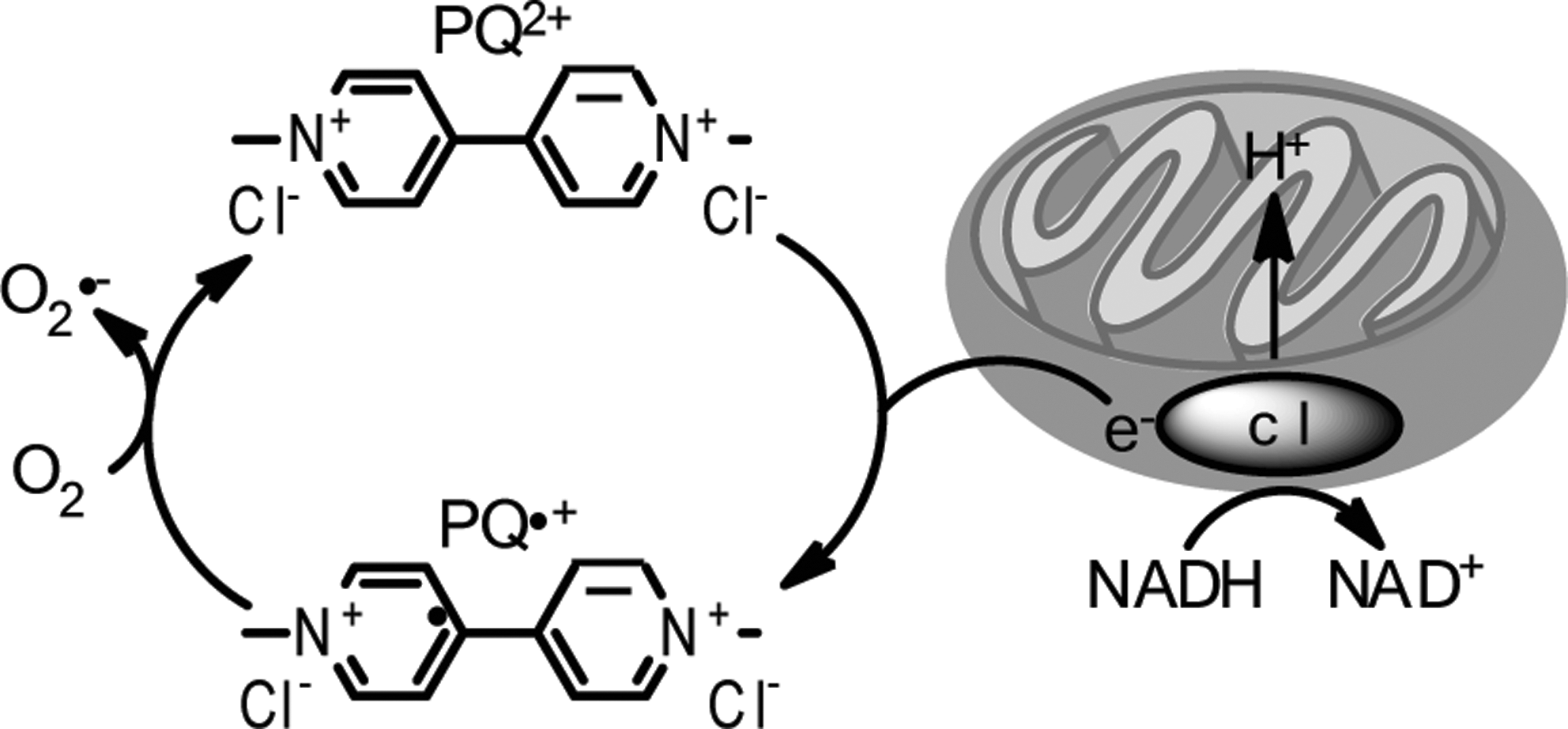
Like MPP+, PQ accumulates in mitochondria, but by a distinct mechanism. MPP+ directly crosses the inner mitochondrial membrane driven by the membrane potential (26), whereas PQ seems to be taken up into mitochondria via a low-affinity carrier-mediated process (21). Once inside the mitochondria, both MPP+ and PQ stimulate mitochondrial ROS production from complex I, but again by divergent processes; PQ accepts electrons from complex I for redox cycling (21) whereas MPP+ directly inhibits complex I activity.
Another mechanism of PQ-mediated ROS generation within mitochondria is believed to arise from interactions between PQ and glutamate. Subcutaneous administration of PQ to rats elicited glutamate-mediated excitotoxicity via Ca2+ efflux into the cell by depolarization of NMDA receptor channels and by activation of non-NMDA receptor channels (91). This influx of Ca2+ is believed to stimulate production of NOS. Nitric oxide may then diffuse into DA terminals, further inducing mitochondrial dysfunction, and leading to continuous and prolonged dopamine overflow in a DAT-dependent manner (91). Corroborating these findings, PQ-mediated DA neuron loss is attenuated by inhibitors of NMDA and NOS as well as by inhibition of caspase cascades (92).
Recently, an additional mechanism of PQ-induced DA neurotoxicity has been proposed to involve the mitochondrial pro-apoptotic protein Bak. Bak protein function has been implicated in mitochondrial outer membrane permeabilization, and Fei and colleagues report that Bak-deficient mice are resistant to the effects of a PQ treatment paradigm that selectively depleted nigral DA neurons in wild-type mice, suggesting that PQ neurotoxicity occurs in a Bak-dependent manner. These studies also showed that, unlike MPTP-induced cell death, Bax (another pro-apoptotic protein involved in mitochondrial outer membrane permeabilization) is not required for PQ-induced cell death; rather, the PQ-dependence on Bak is downstream of BNip3 and Noxa activation (33).
Overview of the maneb model of parkinsonism and mitochondrial mechanisms of toxicity
Maneb (MB) is an organomanganese belonging to the dithiocarbamate (DTC) family of fungicides; chronic exposure to MB has been linked to development of parkinsonian-like symptoms in humans (66). Additionally, in experimental mouse models, maneb administration causes motor deficit (69) and potentiates the effects of MPTP (101).
Relative to MPTP, rotenone, and paraquat, much less is known about the mechanisms of maneb-induced neurotoxicity. The potently fungicidal ingredient in commercial MB is manganese ethylene-bis-dithiocarbamate (Mn-EBDC), which is thought to be neurotoxic (111). Within the body, Mn-EBDC may be metabolized to manganese and EBDC, either or both of which may be toxic to DA neurons. Indeed manganese is known to be relatively nontoxic to adults except in the brain, where it causes irreversible PD-like neurologic symptoms (58). Furthermore, the EBDC component of maneb is believed to contribute to neurotoxicity based in part on observations that both mancozeb (Mn-Zinc-EBDC) and zineb (Zinc-EBDC) elicit neurotoxicity in vitro (95), EBDC enhances the toxicity of MPTP (65), and Mn-EBDC administered into the lateral ventricle of adult rats caused extensive striatal DA efflux comparable to what is observed in response to MPP+ (121). In a DA neuron cell line, Mn-EBDC treatment elicited proteasomal inhibition, oxidative stress, and cytoplasmic α-synuclein aggregation in vitro, although mitochondrial dysfunction was not examined in this context (124). Interestingly, Mn-EBDC has been shown to preferentially inhibit complex III of the ETC within mitochondria, contributing to increased oxidative stress and overt mitochondrial dysfunction (121) (Fig. 4).
Synergistic neurotoxic effects of paraquat and maneb
Although the overall toxic effects of paraquat and maneb are not as profound as those of MPTP or rotenone, several studies have demonstrated that the neurotoxicity of these two compounds is considerably enhanced when they are used in combination, suggesting a synergistic interaction between PQ and MB in vivo (109, 118). This proposed synergy between PQ and MB is of potential epidemiological importance as the estimated agricultural use of PQ and MB overlaps geographically (106); thus, there is likely environmental exposure of humans to the synergistic neurotoxicity of these two pesticides. Further supporting synergistic DA neurotoxicity between PQ and MB, in a population-based case-controlled study, Costello and colleagues report that combined exposure to PQ+MB nearly doubled the risk of older subjects developing PD compared to exposure to either PQ or MB alone (24). Interestingly, it was recently shown in mice that MB treatment alone induces apoptosis through Bak activation, while combined PQ+MB treatment inhibits the Bak-dependent apoptotic pathway and instead potentiates apoptosis via Bax activation (33).
The PQ+MB mouse model has also provided interesting insight into developmental and age-related effects of environmental pesticide exposure and onset of parkinsonism. Experimentally, postnatal administration of PQ+MB in mice caused a 38% loss of DA neurons in adulthood, whereas postnatal exposure to PQ+MB followed by re-exposure to the toxins during adulthood induced a 70% loss of nigral DA neurons and concomitant locomotor deficit (110). Furthermore, there may be an age-related effect for sensitivity to PQ+MB, as older mice have been reported to be more susceptible to systemic PQ+MB treatment (107) perhaps due to changes in transport across the BBB (22).
As with any animal model of experimental PD, there are limitations to the PQ, MB, and PQ+MB animal models of parkinsonism. As with most toxins, these pesticides tend to be acute, and experimental methods of administering them involve doses and routes of exposure that may not recapitulate human environmental exposure to pesticides. Additionally, while α-synuclein-positive inclusions have been reported after prolonged exposure to PQ (62, 124) classical Lewy bodies are not observed in most models using PQ, MB, or PQ+MB. Moreover, PQ especially, has been associated with acute systemic toxicity. However, much valuable insight has been gained from studies using PQ and MB, as described previously. Perhaps the most important lesson learned from the PQ+MB model is the realization of complex interaction between multiple environmental factors, providing rational support for the “multiple-hit” hypothesis of PD etiology.
Other Pesticides and Complex I Inhibitors in Parkinson's Disease
There is compelling evidence implicating both primary and secondary mitochondrial dysfunction, and complex I deficiency in particular, in PD pathogenesis (85). Significantly, a common feature of the toxin models of parkinsonism is that they involve inhibition of mitochondrial complex I as a central mechanism of dopaminergic neurotoxicity. Several compounds, many naturally occurring, are known to be complex I inhibitors. In experiments analyzing a large number of naturally occurring herbal, microbial, or synthetic complex I inhibitors, Hollerhage and colleagues found that lipophilicity was an important co-factor determining the toxicity of the compounds tested, likely due to enhanced access to a binding site in mitochondrial complex I; interestingly, the specific complex I binding site of the compounds did not seem to affect their biological effects (46). Importantly, Sherer and colleagues demonstrated that complex I inhibition is requisite for the neurotoxic effects of several pesticides (90).
Of the many different complex I inhibitors that have recently been studied in vitro, some have been used to develop animal models of experimental Parkinson's disease. The mitochondrial complex I inhibitor trichloroethylene (TCE) (Fig. 2), which is used extensively in industry as a degreasing agent and is a common environmental contaminant, has recently been epidemiologically linked to Parkinson's disease in humans who worked near a TCE source and who were chronically subjected to inhalation and dermal TCE exposure (37). Subsequently, in two different studies conducted in rats, TCE treatment led to selective mitochondrial complex I impairment, selective loss of DA neuron cell bodies in the SN and cell fibers in the striatum, and motor deficit (37, 60). In one study, intracellular α-synuclein accumulation was observed in nigral neurons and in the dorsal motor nucleus of the vagus nerve (60). In addition to lending further support to the environmental exposure theory of sporadic PD etiology, the TCE animal model of parkinsonism may recapitulate in rodents features of early to middle stages of PD in humans; further studies are thus warranted to better characterize the TCE rodent model of parkinsonism.
Conclusions
Over the past several years, genetic and toxin-based models of parkinsonism have significantly contributed to our understanding of mechanisms involved in the neuropathogenesis of Parkinson's disease. The various toxin models of parkinsonism, as discussed previously, have clearly elucidated a conserved role for mitochondrial dysfunction in the pathophysiology of PD. However, much still remains to be determined in terms of discerning the underlying causes that initiate the pathogenic mechanisms and contribute to their chronic and progressive nature, especially within the contexts of human exposure to multiple environmental toxins and complex gene/environment interactions in the complicated etiology of PD.
As discussed elsewhere and in this review, a large body of evidence strongly supports a role for mitochondrial dysfunction as a common pathogenic mechanism in PD. Thus, mitochondria-targeted therapeutic approaches are an attractive and promising direction of ongoing neuroprotective research. Toxin models of parkinsonism have been used to study the effects of bioenergetic agents that improve mitochondrial function. Of these bioenergetic agents, both creatine, a naturally produced amino acid believed to have antioxidant properties, and Coenzyme Q10 (CoQ10), which is an electron acceptor for complexes I, II, and III of the ETC, attenuated nigral cell loss in MPTP mouse models of parkinsonism and progressed to clinical trials in human PD populations (reviewed by (18)). Interestingly, there is divergence in the neuroptrotective properties of the presumed antioxidant melatonin, as it was found to be neuroprotective in a mouse MPTP model (104) but to potentiate neurodegeneration in the rat rotenone model of parkinsonism (103). Such incongruous results may reveal important differences in pathogenic mechanisms between toxin animal models of parkinsonism, as well as potentially indicate that a combinatorial approach of more than one compound may be necessary to achieve therapeutically relevant neuroprotection in human PD.
Moreover, PD is becoming increasingly appreciated as a disease involving nonmotor symptoms and extranigral pathology. Various nondopaminergic neurotransmitter systems have been implicated in the nonmotor aspects of PD and are disrupted as a consequence of long-term levodopa therapy (35). Additionally, nondopaminergic neurotransmitter systems within the brainstem are likely affected prior to DA neurotransmitter systems; thus, degeneration of non-DA systems may precipitate symptoms such as constipation and sleep disturbances prior to nigral degeneration and overt onset of motor symptoms in PD (35). Interestingly, postmortem histopathological analyses of brain tissue from human PD patients show that the incidence of Lewy bodies occurs in the brainstem prior to observation of Lewy bodies within the SN (8), lending further support to the notion that certain nonmotor aspects of PD may precede nigral DA neurodegeneration and motor symptoms. Therefore, extranigral and nonmotor aspects of PD must be considered both in efforts aimed at developing better animal models of parkinsonism as well as in pursuit of better PD therapeutics.
Despite much recent progress, there still remains a disconnect in terms of translating what has been observed mechanistically in animal models of experimental PD into bona fide disease modifying therapeutics for human PD patients. Good animal models are requisite for such an end; however, there are currently no accepted progressive models of parkinsonism that fully mimic the processes known to occur during cell death and that result in the motor deficits, extranigral pathology, biochemistry, and drug responsiveness as seen in human PD patients. Continued efforts to optimize established animal models of parkinsonism as well as the development and characterization of new animal models of experimental PD are therefore essential.
Footnotes
Acknowledgments
This work was supported by NIH Grants NS059806, ES018058 (JTG) and T32 NS07391 (TNM) and the American Parkinson Disease Association.
We apologize to the many authors who have contributed substantially to the field whose work could not be cited here due to journal format constraints.