
Abstract
Introduction
Mitochondria are intracellular organelles that play crucial roles, both in the regulation of survival and in the execution of cell death. On one hand, the vital function of mitochondria is explained by the fact that they are the site of energy production within the cell (63). On the other hand, the intrinsic pathway of apoptosis is initiated at the site of mitochondria, which can release multiple intermembrane proteins such as cytochrome c and second mitochondria-derived activator of caspase/direct Inhibitor of apoptosis (IAP) binding protein with low pI into the cytosol (63, 95). This, in turn, results in the activation of caspases that function as downstream death effector molecules (23). Caspases comprise a group of proteases that in steady state conditions are catalytically inactive pro-enzymes, yet become activated during the induction of apoptosis (23). A wide variety of apoptotic stimuli directly or indirectly engage signal transduction processes that result in permeabilization of the mitochondrial outer membrane (63).
Mitochondrial outer membrane permeabilization (MOMP) presents a key event in the signal transduction cascade that links processes within the intra- or intermitochondrial membrane space to the cell death machinery in the cytosol. Therefore, it is not surprising that this step should be tightly controlled to prevent inappropriate initiation of cell death, for example, on accidental leakage of mitochondrial constituents. For example, pro- and antiapoptotic members of the B-cell lymphoma 2 (BCL-2) family can interact with mitochondrial membranes and are intimately involved in the control of the permeability of the outer mitochondrial membrane. Under basal conditions, in nonapoptotic cells, proapoptotic BCL-2 proteins are in an inactive state in the cytosol or within mitochondrial membranes. Moreover, ions and small metabolites are exchanged between the cytosol and the mitochondrial matrix, likewise via the permeability transition pore complex (PTPC), and the mitochondrial inner transmembrane potential is high (2, 12). The PTPC consists of several components including the voltage-dependent anion channel (VDAC), the adenine nucleotide translocase (ANT), cyclophilin D, the peripheral benzodiazepine receptor (PBR), and hexokinase (HK) (63).
Several mechanisms have been identified that can contribute to MOMP (Fig. 1). First, versatile proapoptotic BCL-2 proteins such as BCL2-associated X protein (BAX), which are not associated with mitochondrial membranes under steady state conditions, translocate from the cytosol to the mitochondria, once they become activated. On activation, BAX undergoes a conformational change that exposes its N-terminus and promotes its insertion into mitochondrial membranes (16). By comparison, BCL2-antagonist killer (BAK) changes its conformation locally at the outer mitochondrial membrane on activation, as BAK belongs to the sessile mitochondrial proteins (16). As a result, homo- and/or hetero-oligomers of BAX and BAK are formed that build channels crossing the outer (and perhaps inner) mitochondrial membrane(s) (16). These BAX/BAK channels mediate the release of mitochondrial intermembrane proteins into the cytosol (16).

Second, BCL2 homology 3(BH3)-only proteins, for example, BCL2-interacting mediator of cell death (BIM), the cleaved form of BH3 interacting domain death agonist (tBID), Noxa, and Puma, can trigger MOMP by neutralizing antiapoptotic BCL-2 proteins or by directly activating BAX and BAK (16).
Third, mitochondrial permeability transition (MPT) contributes to MOMP. To this end, the prolonged opening of the PTPC favors the collapse of the mitochondrial membrane potential and leads to the swelling of the mitochondrial matrix secondary to an osmotic imbalance (63). The MPT typically results in the physical rupture of the outer membrane, as the surface area of the outer mitochondrial membrane is smaller than that of the inner mitochondrial membrane (63).
Fourth, uncoupling of the respiratory chain leads to the dissipation of the high mitochondrial transmembrane potential and the failure to generate ATP followed by osmotic swelling of the mitochondrial matrix and eventually MOMP (16).
Since mitochondria exert a key regulatory function in the control of cell death and since they are frequently altered in human malignancies (43), there is much interest in developing agents that directly target mitochondria with the scope of eradicating tumor cells.
Targeting Mitochondrial Apoptosis in Cancer
Targeting antiapoptotic BCL-2 family proteins
In an attempt to antagonize antiapoptotic BCL-2 proteins, BH3 mimetics were developed (Table 1). The BH3 mimetics are small molecules that closely resemble endogenous BH3-only proteins, both in their structure and in their function. The BH3 mimetics are considered to exert their proapoptotic function by binding to and antagonizing the pro-survival members of the BCL-2 family of proteins. In addition, there is recent evidence that obatoclax, a natural BCL-2 inhibitor, can directly activate BAX (109).
2DG, 2-deoxy-D-glucose; 2ME, 2-methoxyestradiol; α-TOS, α-tocopheryl succinate; ACL, ATP citrate lyase; AML, acute myeloid leukemia; ANT, adenine nucleotide translocase; ATN-224, tetrathiomolybdate; ATRA, all-trans-retinoic acid; BCL-2, B-cell lymphoma 2; BSO, buthionine sulphoximine; CD437, 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid; CLL, chronic lymphocytic leukemia; DCA, dichloroacetate; GSAO, 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide; GX15-070, obatoclax; GPX, glutathione peroxidase; GSH, reduced glutathione; HK, hexokinase; MCL, myeloid cell factor; MJ, methyl jasmonate; NSCLC, non small-cell lung cancer; PBR, peripheral benzodiazepine receptor; PDK, pyruvate dehydrogenase kinase; PEITCs, phenyl ethyl isothiocyanates; PTPC, permeability transition pore complex; ROS, reactive oxygen species; SCLC, small-cell lung cancer; SOD, superoxide dismutase; VDAC, voltage-dependent anion channel.
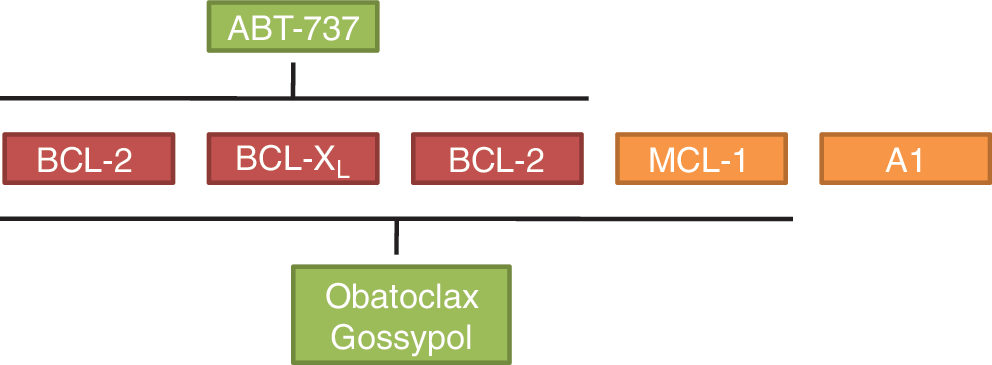
One of the most advanced and best-characterized BH3 mimetic is ABT-737 (Fig. 2), which was developed through rational nuclear magnetic resonance-based screening and structure-based design (90). Similar to BCL2 antagonist of cell death, a BH3-only protein, ABT-737 binds to BCL-2, BCL-XL, and BCL-W, but not to myeloid cell factor (MCL)-1 or A1 (90) (Fig. 3).

In fact, ABT-737 exerts antitumor activity as a single agent in a manner that depends on expression levels of BCL-2 family proteins. Accordingly, cancer cells with high levels of endogenous BCL-2 protein, such as chronic lymphocytic leukemia (CLL) and small-cell lung carcinoma cells, are particularly sensitive to ABT-737 (61, 79, 90). Also, ABT-737 proved to be effective as a monotherapy against primary childhood acute lymphoblastic leukemia (ALL) xenografts implanted into immunodeficient mice (51). In addition, ABT-737 enhanced the antileukemic effects of various antileukemic drugs including L-asparaginase, topotecan, vincristine, and etoposide against drug-resistant cancer xenografts (51). Similarly, the structurally related compound ABT-263 demonstrated significant in vivo activity as a single agent against ALL xenografts, thus resulting in complete remissions in three out of six evaluable leukemias (71).
The MCL-1 expression levels were linked to the resistance of some malignancies to ABT-737, based on the fact that MCL-1 is one of the few antiapoptotic BCL-2 family proteins that are not neutralized by ABT-737 (61, 118). Acquired drug resistance to ABT-737 was also studied in a model of lymphoma cell lines, which were selected by long-term exposure to ABT-737 (129). Interestingly, ABT-737 resistance involved transcriptional upregulation of MCL-1 and BCL-2 related protein A1, which are two antiapoptotic BCL-2 proteins that are not targeted by ABT-737 (129). Accordingly, suppression of MCL-1 levels by RNA interference-mediated silencing or by pharmacological intervention with cyclin-dependent kinase 9 inhibitors, such as flavopiridol or phytohemagglutinin 767491, also restored sensitivity to ABT-737 (129). It is interesting to note that no relationship between MCL-1 expression and the response to ABT-737 was found in vivo in ALL xenografts in immunodeficient mice (51), pointing to additional factors that may regulate sensitivity to ABT-737 in vivo. There is indeed evidence that hypoxia sensitizes cancer cells to BH-3 mimetic-induced apoptosis by suppressing expression levels of MCL-1 (48).
Recently, USP9×was identified as the deubiquitinase that stabilizes MCL-1 protein by removing the polyubiquitin chains that usually target MCL-1 for degradation by the proteasome (105). High expression levels of USP9×in B- and mantle-cell lymphomas, chronic myeloid leukemia, and multiple myeloma were associated with cancer cell survival (105). Accordingly, silencing of USP9×expression resulted in increased susceptibility to ABT-737-triggered apoptosis (105).
To potentiate the antitumor activity of ABT-737, a variety of combination therapies have been designed. For example, ABT-737 has been shown to cooperate with conventional chemo- and radiotherapy in hematological neoplasias and also in several types of solid tumors (47, 56, 66, 78, 79, 112, and 118). Actually, ABT-737 was also demonstrated to cooperate with some targeted agents, for example, the death receptor ligand tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL) (54, 110), proteasome inhibitors (82, 94), histone deacetylases (HDAC) inhibitors (124), and several kinase inhibitors (e.g., inhibitors of break point cluster region-Abelson murine leukemia, FMS-like tyrosine kinase 3, epidermal growth factor receptor, and MAPK/ERK kinase 1/2) (19, 20, 45, 60, 64, and 65).
Mechanistic studies revealed that HDAC inhibitors such as suberoyl bis-hydroxamic acid (SBHA) potentiated ABT-737-induced cell death by upregulating BIM (15). In fact, SBHA-induced BIM was sequestered by BCL-2 and BCL-x(L) unless these two antiapoptotic BCL-2 proteins were inhibited by ABT-737, thereby resulting in cell synergistic killing (15).
In several preclinical mouse models of human cancers, for example, in SCLC and acute leukemia, ABT-737 or its derivative, ABT-263 has demonstrated potent antitumor activity (47, 61, 78, and 79), both as stand-alone agent and within combination regimens (1, 47, 61, 78, and 79).
In reality, ABT-263, an orally available derivative of ABT-737, is currently under clinical evaluation in early-phase clinical trials for SCLC and leukemia/lymphoma, both as a monotherapy and in combination with anticancer drugs or with monoclonal antibodies (i.e., rituximab) (117). In an attempt to identify predictive markers, gene expression profiles of BCL-2 family genes were performed. Sensitivity to ABT-263 correlated with elevated expression of BCL-2 and Noxa, whereas resistance was linked to high MCL-1 expression (113). In addition, global expression profiles revealed patterns of gene expression that differed in sensitive versus resistant SCLC lines and normal lung tissue (113). Such gene expression profiles may provide a rational basis for the selection of patients in future clinical trials. Clinical studies also indicate that thrombocytopenia, the major mechanism-based side effect of ABT-263 due to its negative impact on the BCL-XL-mediated survival of thrombocytes, can be avoided by appropriate dosing schedules (77, 100, 132).
Obatoclax (GX15-070, from Gemin X) is another small-molecule inhibitor of antiapoptotic BCL-2 proteins. Obatoclax, an indole bipyrrole compound, antagonizes BCL-2, BCL-XL, BCL-W, and MCL-1 (87, 96, and 116) (Fig. 3). Since obatoclax also binds to MCL-1, it is active even in cells that are resistant to ABT-737 owing to high MCL-1 levels (87). In cholangiocarcinoma cells, obatoclax was shown to trigger apoptosis as a single agent by direct activation of BAX (109), suggesting that the neutralization of antiapoptotic BCL-2 proteins is not the sole mechanism by which obatoclax kills cancer cells. Further, obatoclax increased TRAIL-induced apoptosis in combination studies in human pancreatic cancer cells (53).
Unexpectedly, obatoclax was reported to reverse resistance of multidrug-resistant childhood ALL cells to glucocorticoids and other anticancer drugs by inducing autophagy-dependent necroptosis (10). Induction of cell death required the expression of receptor-interacting protein (RIP-1) kinase and cylindromatosis (turban tumor syndrome; cylindromatosis) (10), which have both been involved in programmed necrosis (also called necroptosis) (119). Also, obatoclax was recently reported to bypass glucocorticoid resistance in acute leukemia by triggering so-called “autophagic cell death” in addition to apoptosis (50). Obatoclax demonstrated single-agent activity in a phase I clinical study in CLL (89) and is currently under evaluation in hematological malignancies and solid tumors, both as a monotherapy and in combinations (92, 104).
Gossypol (AT-101) is a natural compound from cotton plants (74). Similar to obatoclax, gossypol also inhibits MCL-1 besides BCL-2, BCL-XL, and BLC-W (5). Apogossypol, a derivative of gossypol, has improved antitumor activity and reduced toxicity as compared with gossypol (57). Gossypol was shown to trigger apoptosis in prostate cancer cells by causing DNA damage and p53 activation, suggesting that it may have off-target effects (121). Recently, the Apogossypol derivative BI-97C1 (Sabutoclax) was reported to enhance mda-7/IL-24-induced apoptosis of prostate cancer cells and to reduce tumor growth in a xenograft model in vivo (21). In clinical trials, gossypol demonstrated single-agent activity in a phase I trial in prostate cancer (70). Currently, gossypol is under evaluation both alone and in combination therapy in several malignancies.
Targeting mitochondrial metabolism
Cancer cells characteristically present alterations in their metabolism (Fig. 4). One of the best known examples of cancer-associated metabolic perturbations is the “Warburg effect,” that is, increased glycolytic flow despite high oxygen tension, which was first described by Otto Warburg (122). By producing ATP by aerobic glycolysis, cancer cells gain the ability to shuttle glycolytic intermediates toward various anabolic processes, for example, to glycogen and ribose 5-phosphate synthesis (i.e., glucose 6-phosphate), to lipid synthesis (i.e., dihydroxyacetone phosphate), and to the synthesis of alanine and malate (i.e., pyruvate) (41). Since mitochondria regulate critical steps at the crossroad between metabolism, survival, and cell death, they may present the Achilles' heel to overcome cell death resistance in human cancers (Table 1). Targeting the hyperglycolytic state of cancers may also preferentially affect malignant cells compared with their normal counterparts.
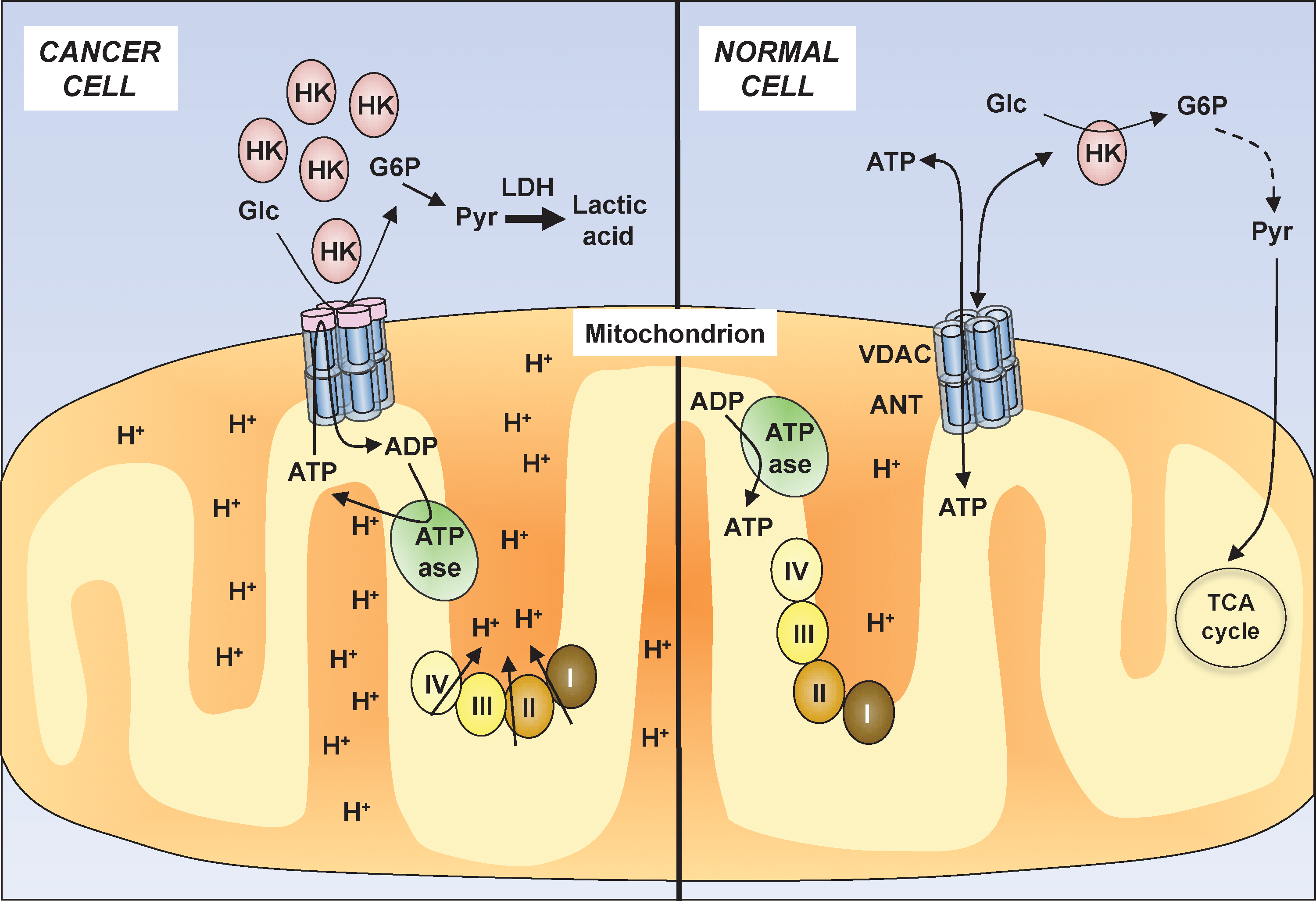
The 2-deoxy-D-glucose (2DG) is an inhibitor of glycolysis that decreases the association between HK and VDAC, thereby enabling enhanced binding of BAX to VDAC. The 2DG has been reported to enhance the sensitivity of cancer cells to conventional chemotherapeutics through the production of reactive oxygen radicals, for example, in head and neck cancer cells (108). Recently, 2DG has been shown to trigger apoptosis in a p53-dependent manner when combined with the antidiabetic drug metformin (7). The 2DG is currently evaluated in early clinical trials in patients with advanced solid tumors or prostate cancer.
Methyl jasmonate (Fig. 2) is a plant hormone that at comparatively high concentrations has the ability to detach HK from mitochondria, thereby initiating mitochondrial apoptosis (44, 101). This mechanism may selectively trigger cell death in cancer cells, as HK is frequently overexpressed in human cancers and is tightly bound to VDAC, especially in cancer cells.
Inhibition of mitochondrial pyruvate dehydrogenase kinase by dichloroacetate (DCA) provides a means to interfere with metabolic reprogramming of tumor cells by switching the abnormally high rate of glycolysis to glucose oxidation. The DCA has been shown to reduce the increased mitochondrial membrane potential of cancer cells, to upregulate the production of mitochondrial reactive oxygen species (ROS), and to activate K+ channels predominately in malignant versus normal cells (11). Reversal of the glycolytic phenotype by DCA has been shown to suppress tumor growth in several types of cancer, including glioblastoma, breast, endometrial, and prostate cancer (13, 81, 111, 126). Currently, DCA is evaluated as a monotherapy in a phase I study in patients with advanced solid tumors.
In fact, SB-204990, an inhibitor of ATP citrate lyase (ACL), can inhibit cell proliferation and growth of various tumor cell lines in vitro and in an in vivo mouse model of lung cancer (49). Actually, ACL is the key enzyme that catalyzes the conversion of citrate to cytosolic acetyl-CoA, thereby linking glucose metabolism to lipid synthesis.
Pharmacologic inhibitors of fatty acid oxidation, that is, etomoxir or ranolazine, were shown to inhibit proliferation and to enhance ABT-737-induced apoptosis of leukemia cell lines, primary human acute myeloid leukemia samples, and quiescent leukemia progenitor cells (103). In a murine model of leukemia, etomoxir demonstrated cooperative antileukemic activity in combination with ABT-737 or cytotoxic drugs such as cytosine arabinoside (103). Similarly, orlistat, an inhibitior of fatty acid synthase (FASN), increased the sensitivity of leukemia cells to ABT-737 (103). Orlistat reduced tumor growth and induced apoptosis in several cancer models in vitro as well as in vivo (14, 31, 59). Moreover, overexpression of FASN was found in a drug-resistant breast cancer cell line (69). Altering FASN expression also modulated drug sensitivity in breast cancer cell lines, thereby causing drug resistance by ectopic overexpression of FASN and vice versa increasing drug sensitivity by reducing FASN expression (69). Similarly, orlistat sensitized breast cancer cells with FASN overexpression to chemotherapeutics (69). Mechanistically, FASN-imposed drug resistance was linked to overproduction of palmitic acid by FASN and subsequent suppression of drug-induced apoptosis (69).
PTPC-targeting agents
There are multiple opportunities to target components of the PTPC (Table 1). For example, 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO), a glutathione-coupled trivalent arsenical compound, blocks the ATP/ADP antiporter activity of ANT by crosslinking a pair of key cysteine residues of ANT at the matrix interface (26). This causes generation of ROS, ATP depletion, and permeabilization of the outer mitochondrial membrane, eventually leading to apoptosis (26). Of note, GSAO was found to trigger apoptosis, in particular in proliferating endothelial cells, whereas it was nontoxic to growth-quiescent, endothelial cells, indicating that it might preferentially target tumor angiogenesis (26). An analog of p-GSAO with the arsenical moiety at the ortho position on the phenyl ring, that is, o-GSAO, was reported to exhibit increased antiproliferative activity compared with p-GSAO, as it accumulated at a higher rate within the cell because of reduced export by the multidrug resistance-associated protein 1 (25).
Lonidamine, an indazole carboxylate compound, is another ANT ligand that triggers mitochondrial apoptosis (6). In early clinical trials in patients with recurrent glioblastoma multiforme, lonidamine was well tolerated and suppressed tumor growth (91).
Clodronate is a bisphosphonate that has been identified as a competitive ANT inhibitor (67). Adjuvant treatment with clodronate has recently been reported to improve the overall survival of women with primary breast cancer who present with micrometastases to the bone marrow (24).
Interestingly, retinoid-related compounds, such as 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid (CD437) and all-trans-retinoic acid (ATRA), can induce MPT in an ANT-dependent manner independently of binding to nuclear receptors (6, 76, 88). Retinoid-related compounds such as CD437 and ATRA have been shown to have remarkable clinical efficacy, especially in patients with acute promyelocytic leukemia (83).
The PBR presents another target of the PTPC. Actually, PBR ligands such as PK11195, RO5-4864, and diazepam demonstrated antitumor activity in vitro and in vivo, including resistant forms of cancer, for example, due to BCL-2 overexpression (22). Diazepam in combination with lonidamine demonstrated a cytostatic effect on tumor growth in a phase II trial in glioblastoma multiforme (91).
ROS regulators
The mitochondrial respiratory chain transfers electrons via several coupled acceptor systems, eventually leading to their reaction with oxygen. When this chain is inhibited or overloaded at any site, ROS are produced. Chemical compounds that produce ROS damage mitochondria and initiate permeabilization of the outer mitochondrial membrane, subsequently leading to apoptosis (Table 1). For example, diazenedicarboxylic acid bis- 5N,N-dimethylamide (diamide), bismaleimido-hexane, and dithiodipyridine crosslink thiol groups and cause thiol-oxidation of ANT (18, 93, 98). By comparison, 2-methyl-1,4-naphthoquinone (menadione) and β-lapachone (ARQ 501) cause oxidative damage to mitochondria by undergoing futile redox cycles on the respiratory chain. Further, inhibition of antioxidant systems can result in ROS production. For example, buthionine sulphoximine inhibits reduced glutathione (GSH) synthesis (75), and imexon depletes the GSH pool due to its thiol-binding activity (32). Phenyl ethyl isothiocyanates (PEITCs) are dietary compounds that inhibit the GSH antioxidant system both by conjugating GSH and by inhibiting glutathione peroxidase, thus resulting in ROS generation and mitochondrial apoptosis (115, 127). Arsenic trioxide's cytotoxicity has also been linked to the generation of oxidative stress secondary to irreversible inhibition of thioredoxin reductase (62, 73). It has been observed that 2-methoxyestradiol, an estrogen derivative, and tetrathiomolybdate (ATN-224), an intracellular copper-chelating agent, were reported to kill cancer cells via inhibition of antioxidant enzymes such as superoxide dismutase (SOD), thereby causing elevated ROS production (52, 55).
Natural compounds and their derivatives
There are several natural compounds that primarily exert their cytotoxic activity by engaging the mitochondrial pathway of apoptosis (Table 1).
Resveratrol, a polyphenolic phytoalexin (Fig. 2), naturally occurs in various fruits and beverages, for example, grapes and red wine (97). Among its multiple functions, resveratrol has been shown to directly affect mitochondria by binding to the γ-subunit of F1-ATPase and, thus, inhibiting mitochondrial ATP synthesis (42). In addition, the resveratrol derivative 3,3′,4,4′,5,5′-hexahydroxystilbene has been reported to trigger downregulation of mitochondrial SOD (MnSOD), an antioxidative enzyme in mitochondria that is involved in the defense against oxidative stress (84). Coupling of resveratrol to the lipophilic, membrane-permeant triphenylphosphonium (TPP) cation further enhanced its targeting to mitochondria (9). Indeed, mitochondria-targeted derivatives of resveratrol, for example, 4-triphenylphosphoniumbutyl-4′-O-resveratrol iodide, accumulated in mitochondria (9).
Another example of mitochondriotoxic drugs is betulinic acid (3b, hydroxy-lup-20(29)-en-28-oic acid), a naturally occuring pentacyclic triterpenoid (37). Betulinic acid has been demonstrated to directly engage MOMP and the release of cytochrome c from mitochondria into the cytosol (38), when incubated with isolated mitochondria in a cell-free system (38). Permeabilization of the mitochondrial outer membrane was blocked by inhibitors of the PTPC or by overexpression of BCL-2 or BCL-XL, but not by caspase inhibitors, pointing to a direct effect on mitochondria (4, 36, 39). Expression levels of pro- and antiapoptotic proteins of the BCL-2 family changed on treatment with betulinic acid in a context-dependent fashion. Accordingly, increased MCL-1 levels were found on exposure to betulinic acid in melanoma cells, whereas no changes in MCL-1 expression were detected in squamous-cell carcinoma cells (106, 107, 114). Betulinic acid was repeatedly shown to induce apoptosis in a p53-independent manner (34, 36, 80, 102, 106, 125, 133). From a therapeutic perspective, it is interesting to note that betulinic acid can trigger mitochondrial apoptosis preferentially in cancer cells compared with normal cells, pointing to a therapeutic window (36, 38, 68).
Vitamin E analogs such as α-tocopheryl succinate (α-TOS) have also been shown to induce apoptosis in cancer cells via mitochondrial perturbations (17). Interaction sites for α-TOS were identified in the proximal and distal ubiquinone-binding sites of the respiratory complex II, responsible for the displacement of ubiquinone from complex II and ROS generation (29). Downstream of ROS production, the preferential formation of BAK channels was recently identified as a key event that mediates apoptosis (99). Importantly, α-TOS also targets endothelial cells besides cancer cells (30). Endothelial cells that had been depleted of mtDNA were also resistant to α-TOS, underlying the key role of the intrinsic apoptosis pathway in α-TOS-induced endothelial cell death (30). Recently, the mitochondrially targeted delivery of vitamin E succinate was improved by the addition of a TPP group (TPP(+)), which resulted in a considerable increase in its antitumor activity (27, 28).
Mitochondrially Targeted Drug Delivery
Different strategies have been developed to specifically target drugs to mitochondria (Table 2). For example, the electrochemical gradient that is built across the inner membrane by the respiratory chain complexes, that is, the mitochondrial transmembrane potential (Δψm), can be exploited to drive drug accumulation into the negatively charged mitochondrial matrix for diagnostic or therapeutic purposes. Delocalized lipophilic cations (DLCs) were reported to readily cross mitochondrial membranes and to accumulate within mitochondria (85). Various DLCs-based fluorescent probes have been developed that are widely used to visualize mitochondria and to assess their functions, for example, chloromethyl-X-rosamine (MitoTracker red) (40, 131). For therapeutic targeting of mitochondria, the addition of the positively charged group TPP (TPP(+)) was shown to increase drug concentration within mitochondria and to potentiate the anticancer cytotoxicity of vitamin E derivatives (27, 28).
Another property that can be exploited for the development of mitochondrially targeting drugs is the presence of a mitochondrial targeting sequence (MTS) of 20–40 amino acids in mitochondrial proteins which are encoded by the nuclear genome. This MTS ensures the translocation of these proteins into the mitochondria after their translation in the cytosol (86, 130). The MTS-containing polypeptides have been used for the delivery of proteins or nucleic acids into mitochondria (120).
Further, vesicle-based carriers can serve for mitochondrial delivery, especially that of large cargos. For example, the MITO-Porter system uses macropinocytosis that is stimulated by specific surface modifications for mitochondrial entry (128). The DQAsomes are vesicles that are based on the mitochondriotropic properties of the cationic bolaamphiphile dequalinium and which are especially useful for the transport of DNA to mitochondria (123).
Conclusions
Mitochondria-targeted drugs offer the possibility to overcome primary or acquired resistance, which often limits the efficacy of conventional chemotherapeutics, thus leading to treatment failure. In contrast to many classical anticancer drugs, which depend on intact signaling cascades upstream of mitochondrial signaling, mitochondria-targeting drugs can also act independently of these processes. Thus, mitochondriotoxic compounds may be effective against refractory cancers. Rational exploitation of the current knowledge on mitochondrial signaling events will be crucial to generate novel and improved therapeutics. In the long run, this approach may offer novel treatment options for patients with cancer.
Footnotes
Acknowledgments
The SF is supported by grants from the Deutsche Forschungsgemeinschaft, Deutsche Krebshilfe, Bundesministerium für Bildung und Forschung, Wilhelm-Sander-Stiftung, the Novartis Stiftung für therapeutische Forschung, the Else Kröner Fresenius-Stiftung, the European Community (ApopTrain, APO-SYS), and IAP6/18. The GK is supported by the Ligue Nationale contre le Cancer (Equipes labellisée), Agence Nationale pour la Recherche (ANR), European Commission (Apo-Sys, ChemoRes, ApopTrain, ArtForce), Fondation pour la Recherche Médicale (FRM), Institut National du Cancer (INCa), Cancéropôle Ile-de-France, and Fondation Bettencourt-Schueller.