
Abstract
Redox-dependent processes influence most cellular functions, such as differentiation, proliferation, and apoptosis. Mitochondria are at the center of these processes, as mitochondria both generate reactive oxygen species (ROS) that drive redox-sensitive events and respond to ROS-mediated changes in the cellular redox state. In this review, we examine the regulation of cellular ROS, their modes of production and removal, and the redox-sensitive targets that are modified by their flux. In particular, we focus on the actions of redox-sensitive targets that alter mitochondrial function and the role of these redox modifications on metabolism, mitochondrial biogenesis, receptor-mediated signaling, and apoptotic pathways. We also consider the role of mitochondria in modulating these pathways, and discuss how redox-dependent events may contribute to pathobiology by altering mitochondrial function. Antioxid. Redox Signal. 16, 1323–1367.
I. Introduction
It is well established that mitochondria produce ROS as a consequence of electron leak during respiration (223, 349). Often this leak is referred to as a byproduct of respiration; however, lack of mitochondrial oxidant production leads to diminished cell signaling in response to growth factors, such as insulin, and decreases the formation of necessary protein disulfides (169, 264, 284, 450, 464). Although some increases in ROS can stimulate these essential pathways, there is a threshold range of concentrations beyond which ROS become harmful (83, 206, 349). Additionally, ROS-damaged mitochondria tend to produce more ROS, thereby activating mitochondrial-mediated apoptotic or necrotic pathways (120, 416). Adaptive antioxidant systems are essential for maintaining the critical redox balance in cells, as are mitochondrial adaptations to changes in ROS flux. Thus, in this review, we highlight the role of key intracellular antioxidant systems and discuss how they may modulate mitochondrial function. We also discuss redox modifications that are essential for normal cellular function and survival, such as the reversible protein thiol oxidation (thiol redox switches) that mediates cell signaling, and discuss the role of mitochondria in these pathways. Additionally, we consider the role of mitochondria in maladaptive responses that lead to cell death, many of which involve mitochondrial injury or dysfunction leading to mitochondrial-dependent apoptosis and necrosis.
II. Cellular ROS, Redox, and Antioxidant Systems
ROS are crucial modulators of cellular function. At low concentrations, ROS are essential participants in cell signaling (136, 323, 450), whereas excess ROS can disrupt normal cellular function and promote damage to cellular lipids, nucleic acids, and proteins. Thus, a balance between ROS production and their removal allows for normal cellular function, whereas an imbalance causes oxidative stress with pathobiological consequences.
A. Sources of cellular ROS
Most cellular ROS are partially reduced forms of molecular oxygen and their derivatives and originate from the one-electron reduction reaction yielding superoxide anion (
Owing to its anionic charge,
B. Mitochondrial generation of ROS
As discussed in more detail below, mitochondrial responses to ROS can increase mitochondrial production of ROS, in part, by increasing electron leak. Within mitochondria, the components of the ETC reside on the inner membrane (Fig. 1). During respiration, electrons are transferred to molecular oxygen via the four complexes of the ETC to generate water while pumping protons (H+) into the intermembrane space at complex I (NADH dehydrogenase), complex III (cytochrome c reductase), and complex IV (cytochrome c oxidase). This gradient of H+ is the major contributor to the mitochondrial inner membrane potential (ΔΨ); the return flux of protons into the matrix through the ATP synthase complex (proton motive force) powers the synthesis of ATP from ADP and inorganic phosphate. During respiration, ∼1%–2% of molecular oxygen is converted to
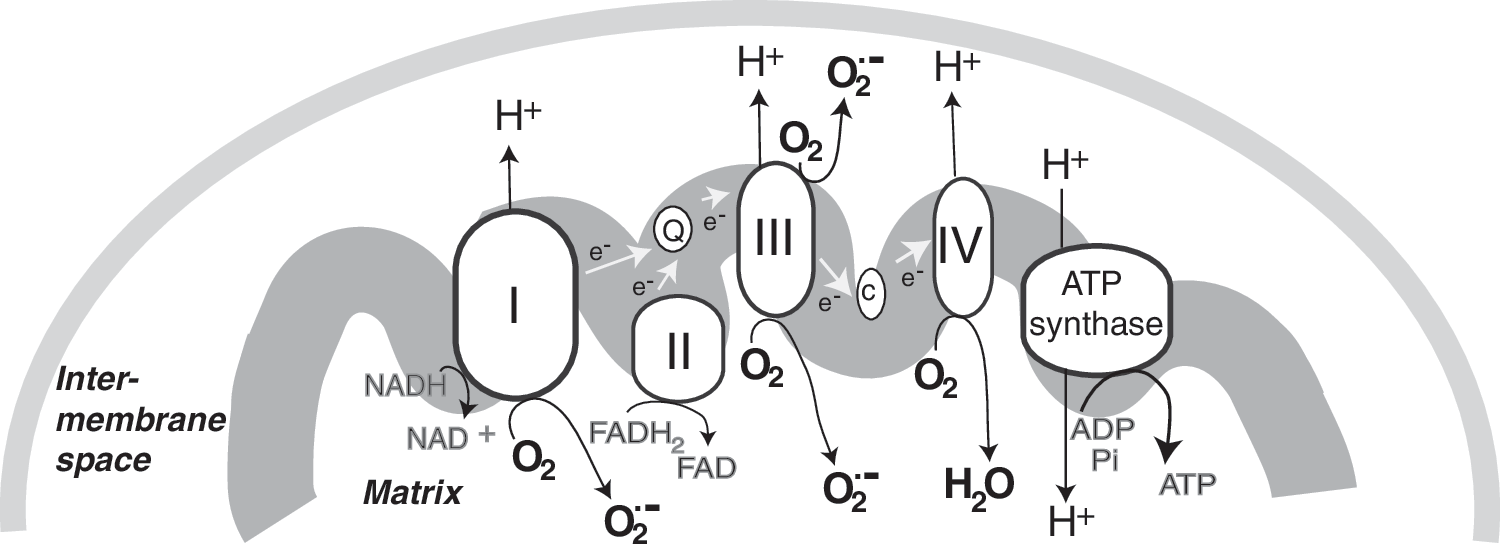
As discussed throughout this review, post-translational modification of mitochondrial subunits, such as complex I, can either promote or attenuate the generation of
ANT, adenine nucleotide transporter; ASK1, apoptosis signal-regulated kinase 1; Cys, cysteine; Drp1, dynamin-related protein-1; Fe-S, iron–sulfur; GPx, glutathione peroxidase; GR, glutathione reductase; Grx, glutaredoxin; ICDH, isocitrate dehydrogenase; JNK, c-jun N-terminal kinase; MDH, malate dehydrogenase; MPT, mitochondrial permeability transition;
C. Redox and antioxidant systems
Owing to the necessity of keeping ROS within a range compatible with normal cellular function, organisms have evolved a number of modulating antioxidant mechanisms. As mentioned in the previous section, the SOD family catalyzes the conversion of

1. SODs
SOD1 or the copper/zinc-dependent SOD (Cu/ZnSOD) is an intracellular enzyme, widely distributed throughout the cell cytoplasm, nucleus, and microsomes; it has even been detected in the mitochondrial intermembrane space of rat liver mitochondria (318). Missense mutations of the SOD1 coding region are associated with some familial cases of amyotrophic lateral sclerosis (ALS), suggesting a role for oxidative stress in this disorder (27). It is not yet resolved, however, as to how these genetic alterations contribute to this degenerative disease, as SOD1-deficient mice are viable with no clear ALS phenotype. Other findings suggest that ALS-associated mutant forms of SOD1 aggregate in the cytoplasm and intermembrane space of the mitochondria and suggest that this aggregation contributes to cell death (75). Recent findings implicate redox mechanisms in the toxicity of these aggregating mutant SOD1 enzymes, as overexpression of the mitochondrial glutaredoxin 2 (Grx2), a redox-active protein that catalyzes disulfide exchange reactions and the reduction of protein-mixed disulfides (177), preserves mitochondrial metabolism and prevents cell death caused by coexpressed mutant SOD1 variants, at least in cell culture models (130). Although SOD1 deficiency in a knockout mouse may not cause ALS symptoms, these mice do exhibit oxidative stress in various tissues (39, 190, 442). Some of the redox changes caused by SOD1 deficiency may be secondary to oxidative inactivation of glutathione peroxidase 1 (GPx-1), a key intracellular enzyme that reduces hydrogen peroxide to water. As discussed further below, GPx-1 plays an essential role in modulating intracellular hydrogen peroxide with consequences for growth factor-mediated signaling and mitochondrial ATP production (169). The extracellular SOD (ecSOD) is a secreted glycoprotein that can associate with cell surface glycosaminoglycans to mitigate the effects of extracellular
As the only mitochondrial matrix SOD, manganese-dependent SOD (MnSOD) has a crucial role in the inactivation of mitochondrial (matrix)
2. Catalase and NADPH
Catalase is a heme-containing tetramer that reduces hydrogen peroxide to water. Although NADPH is not a true cofactor, as the catalytic reduction of hydrogen peroxide by catalase does not require NADPH, NADPH binds to each monomeric subunit of catalase and provides electrons to prevent catalase inactivation by hydrogen peroxide (216); in addition, NADPH binding facilitates the assembly of catalase tetramers, stabilizing the molecule and optimizing its activity (166). Thus, maintenance of the NADPH pool is essential for preserving catalase activity during oxidant stress. Although catalase is highly efficient at reducing hydrogen peroxide, it may not play a central role in modulating hydrogen peroxide responses following many stimuli as it is localized mainly in peroxisomes. Rat liver and heart mitochondria, however, depend, in part, on catalase to reduce endogenous or exogenous hydrogen peroxide (339, 369). These and other findings suggest that in some tissues and cells, catalase is important in maintaining overall redox homeostasis. In particular, in erythrocytes, catalase, together with GPx-1, significantly contributes to the detoxification of hydrogen peroxide (146) as acatalasemic erythrocytes are less able to detoxify hydrogen peroxide. Interestingly, more recent studies with Prx1- and Prx2-deficient mice suggest that these antioxidant enzymes may also significantly contribute to antioxidant protection of erythrocytes.
3. GPx, reduced glutathione, Grxs, and glutathione reductase
GPxs are a family of selenocysteine (Sec)-containing enzymes (with one exception, Gpx-5). GPx-1, the first-identified of these, was initially found in erythrocytes (298). GPx-1 and some of the other members of this family rely on reduced glutathione (GSH) as a cofactor in the reductive inactivation of hydrogen peroxide and/or lipid hydroperoxides (Fig. 3). (Other members of the GPx family rely on other thiol reductants, such as thioredoxin [Trx], which is also used as a source of reducing equivalents by many Prx family members.) During the reduction of peroxide targets, the GSH electron donor is oxidized to oxidized glutathione (GSSG), which is then recycled by glutathione reductase (GR) and relies on NADPH for its reducing activity (358, 359). Thus, the maintenance of GSH for optimal GPx enzyme activity is dependent on maintaining the pool of NADPH stores.
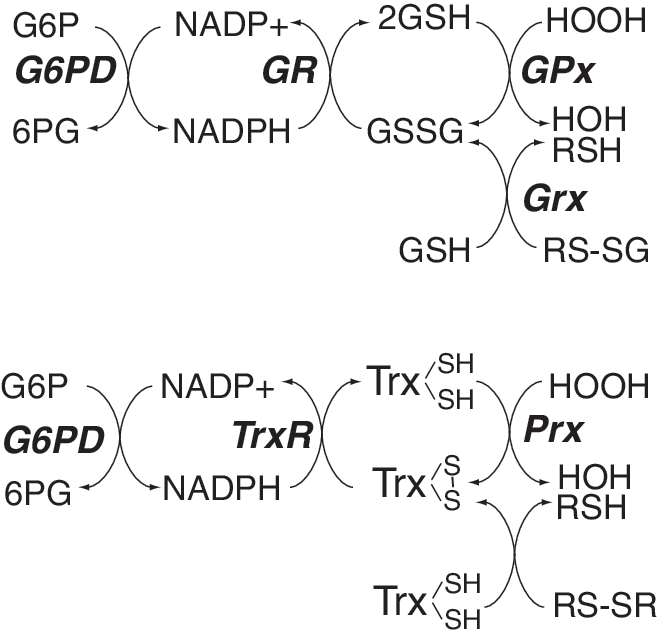
GPx-1 is the most widely studied member of this family [reviewed in Ref. (265)]. In mammalian cells, it is a selenoprotein with Sec at its active site; it is ubiquitously expressed, found in cytoplasm and mitochondrial compartments; and its deficiency is associated with oxidative stress. GPx-1-deficient mice manifest endothelial dysfunction, characterized by a lack of normal responses to endothelium-dependent vasoactive compounds (134, 135). GPx-1 was also found to play a role in ischemia-reperfusion injury in cardiac and neuronal models, and atherosclerosis in susceptible mouse models (103, 248, 256, 418, 452, 459). In human subjects, lower levels of GPx-1 activity are associated with atherogenesis and an increased risk for cardiovascular events (42, 377). Furthermore, GPx-1 modulates mitochondrial function as its overexpression decreases mitochondrial electrochemical potential (decreases ΔΨ), ROS generation, and ATP synthesis (169). Consistent with a role for GPx-1 in modulating mitochondrial oxidants, its deficiency in mice augments release of oxidants from respiring mitochondria (126).
All but one of the members of the mammalian GPx (GPx-1 through GPx-6) family are selenoproteins, with the amino acid Sec. GPx-5, which has a cysteine (Cys) substituted for the active site Sec, is only expressed in the epididymis, and appears to be important in providing antioxidant protection to spermatozoa (90). GPx-2 is highly similar to GPx-1 except that its expression is limited to epithelial cells, especially those of the intestinal tract, where it may play an essential role in preventing tumorigenesis (29). GPx-6 is also limited in its expression to adult olfactory epithelium (344), but has not been widely studied. GPx-3 is a secreted glycoprotein, and its in vivo cofactor is a subject of debate. Deficiency of GPx-3 in humans or knockout mice potentiates platelet activation and thrombosis (139, 193, 212). Furthermore, in GPx-3 knockout mice, deficiency of this extracellular antioxidant causes endothelial dysfunction and increased sensitivity to neuronal injury in a brain model of ischemia reperfusion injury by mechanisms related to its key functions as an antioxidant enzyme and regulator of platelet-dependent thrombosis (193).
GPx-4 is unique in that, unlike the other GPx family members, it exists as a monomer and, intracellularly, it primarily targets membrane lipid hydroperoxides. In addition, unlike the other GPx-family members, GPx-4 knockout is lethal in mice (465). GPx-4 is expressed as multiple isoforms generated from the same gene: one of these targets the mitochondria where it has a vital role in protecting against membrane lipid peroxidation; in its absence, mitochondria accumulate damaging lipid peroxides, leading to apoptogen release (253, 383). Targeted overexpression of the mitochondrial form of GPx-4 in transgenic mice attenuates cardiac ischemia-reperfusion injury, lowers mitochondrial lipid peroxidation, and preserves ETC complex activities after ischemia-reperfusion injury, confirming a vital role of redox alterations in modulating mitochondrial function (106).
Importantly, the Sec functionality in the mammalian GPxs is a redox-active site (Se-H), similar to Cys (S-H). During the enzymatic reduction of hydrogen peroxide, Sec undergoes sequential oxidation by hydrogen peroxide to form a selenenic acid, Se-OH, followed by reaction with GSH to form a Se-SG intermediate before a second GSH combines to form GSSG, resolving the active site to Se-H (133, 278). Upon exposure to oxidant stress, there is accordingly an increase in the oxidation of GSH and a concomitant decrease in the intracellular ratio of GSH/GSSG that appears to be related to the action of GPx-1 as in the absence of GPx-1 there is less detectable change in GSSG (140, 254). Interestingly, the cellular pool of GSH is also necessary to prevent oxidative inactivation of GPx-1 by excess oxidants (433). In vitro, GPx-1 has been found to be highly susceptible to oxidative inactivation by
Intracellularly, glutathione is predominantly reduced (GSH), with the cytosolic content of GSH maintained via the cyclic reduction of GSSG by GR, de novo biosynthesis, and cellular uptake of extracellular glutathione (disulfide) (288). Accurate measurements of GSSG are difficult, especially in isolated intracellular compartments, as GSH is easily oxidized to GSSG during sample isolation. Nonetheless, GSH has a low redox potential in cytosol (E 0=−260 to −200 mV) (374) and measurements from mitochondria suggest an even lower glutathione redox potential (−330 to −300 mV) in this intracellular compartment (156), consistent with ratios of GSH:GSSG of over 100:1 (374). This ratio is in contrast to the endoplasmic reticulum where GSH:GSSG ratios between 1:1 to 1:3 (and corresponding redox state of −180 mV) have been reported (182, 374).
Recent studies have utilized redox-sensitive biosensors to monitor real-time measurements of intracellular GSH-redox state. In yeast cells, this has been accomplished by using a redox-sensitive yellow fluorescent protein (rxYFP) that forms disulfide bonds in proportion to intracellular GSH redox potential (319). Using a series of genetically deficient yeast lines, it was shown that equilibration of the sensor with intracellular GSH:GSSG relies on intracellular Grx. Grx proteins are small (12–16 kDa) redox active proteins that catalyze thiol-disulfide exchange reactions of oxidized protein disulfides and mixed disulfides, as well as those of low-molecular-weight disulfides such as GSSG (205) [Grx are discussed further below]. Other investigators have further modified the rxYFP protein to target it to mitochondrial compartments (178). Despite the advantage of the real-time values that can be tracked by these intracellular sensors, there may be other drawbacks of these protein-based probes due, in part, on their reliance on cellular Grxs and their lag-time to equilibrate with cellular GSSG (178, 319). In addition, these redox probes are highly sensitive to changes in intracellular pH. Furthermore, it is unclear whether other intracellular components can alter their redox status, although, to date, no other intracellular redox component has been found to influence their function (178, 319). A similar biodetector, engineered by combining human Grx with a redox-sensitive GFP, was used to image GSSG-redox changes within human cells (163). This recombinant sensor apparently equilibrates more quickly with cellular GSSG than redox-sensitive probes that lack the Grx domain in cis with the redox-sensor domain. Overall, relative measurements calculated with these biosensors suggest even lower redox potentials for intracellular GSH (−289 mV) and higher GSH:GSSH ratios than those calculated by measuring GSH and GSSG concentrations with biochemical methods (163, 178, 319). These biosensors hold great promise in furthering our understanding of GSH redox status in normal cellular processes and those associated with redox stress.
It has been estimated that mitochondria may contain ∼10%–15% of cellular GSH, but according to a recent review on mitochondrial GSH, the overall concentration of GSH is similar in cytosolic and mitochondrial compartments (measured between 1 and 14 mM) (275). In mitochondria, GSH stores are distinct from that in the cytosol, and maintained by active transport through the dicarboxylate or 2-oxoglutarate carriers (287, 393) and by enzyme-mediated reduction of GSSG by mitochondrially targeted GR (414). Deficiency of cellular glutathione leads to mitochondrial damage and apoptosis (287), in part, by loss of GPx-mediated protection against mitochondrial oxidants. GSH, however, has other cellular functions, including the formation of mixed disulfides with redox-active protein thiols. These interactions can modulate the reactivity and activity of a variety of cellular targets, as discussed further below.
Glutaredoxins (Grx) are important determinants of protein-mixed disulfides. In yeast, five isoforms of Grx have been described, whereas in mammalian cells there are three isoforms, Grx1, Grx2, and Grx5. These proteins have some conserved features, including a Trx fold. Grx1 is a cytosolic protein with a Cys-Pro-Tyr-Cys active site motif. Alternative splicing of the Grx2 transcript results in the production of one enzyme isoform that targets to the nucleus and one that is found in the mitochondrial matrix (267). Grx2 contains a different active site motif, Cys-Ser-Tyr-Cys, and reduction of the oxidized form of Grx2 can be mediated by GSH or NADH and thioredoxin reductase (TrxR) (195). Grx1 and Grx2 catalyze protein disulfide reduction (R-S-S-R) via a dithiol mechanism and protein-mixed disulfide (R-S-S-G) reduction via a monothiol mechanism (177). Notably, Grx2 added to isolated mitochondrial membranes mediates the reversible glutathiolation of proteins in a manner dependent on the ratio of GSH/GSSG (36). One of the targets of glutathiolation, mitochondrial complex I, was also found to have decreased activity under oxidative conditions favoring glutathiolation, suggesting an important role for Grx2 in modulating mitochondrial ROS production by reducing complex I glutathiolation. Grx2 also differs from Grx1 in that it is associated with 2Fe-2S clusters (194). The Grx2/Fe-S complex, which has mostly been found in mitochondria, consists of 2 Grx2 linked to the Fe-S cluster through the N-terminal Cys of the Grx2 active site. Two GSH molecules were also found to associate with this Grx2/Fe-S complex, which has been suggested to function as a redox sensor. Thus, Fe-S sequesters active sites of Grx2 in the setting of high GSH:GSSG ratios and releases active monomers following oxidative shifts in the redox state (194). Grx2 is essential for the normal cellular responses to oxidant stress. In cultured human cells, depletion of mitochondrial Grx2 was found to increase cell sensitivity to apoptosis in response to agents causing oxidant stress (255). Other studies have found that overexpression of Grx2 decreased apoptosis in response to similar agents, confirming a role for Grx2 in protecting against redox-mediated apoptosis (124). Neither of these studies on the role of Grx2 in apoptosis reported changes in protein glutathiolation; however, the latter study, which focused on apoptotic signaling, found that Grx overexpression reduced cardiolipin oxidation to prevent cytochrome c release and caspase activation. Grx5 also protects against oxidative stress and apoptosis; Grx5 overexpression attenuates hydrogen peroxide-induced apoptosis and cardiolipin oxidation in osteoblasts, whereas its deficiency augments cell death (259). Similar to Grx2, the eukaryotic Grx5 is also primarily found in mitochondria. Unlike Grx1 and Grx2, Grx5 has only a single thiol in its active site: Cys-Gly-Phe-Ser (in human Grx5) (205). Grx5 has also been found to play an important role in Fe-S cluster formation in yeast, as well as in higher eukaryotic organisms (355, 436). Thus, a deficiency of Grx5 has been found to alter cellular iron metabolism, the production of heme, and the activity of Fe-S cluster-dependent enzymes, such as aconitase (259, 299, 448). The role of Fe-S clusters in modulating the activities of mitochondrial ETC complexes and enzymes of the tricarboxylic acid (TCA) cycle is discussed further in section VI.
GR is essential for the recycling of GSSG to GSH, thereby playing an important role in maintaining Grx and GPx enzyme activities. GR enters the mitochondria via an N-terminal targeting sequence but is also found in the cytosol (186). Its activity has been shown to regulate Akt and endothelial NOS (eNOS) phosphorylation in response to flow in bovine aortic endothelial cells, in part, by maintaining Grx activity (441). Interestingly, the function of GR can be oxidatively inactivated at a Cys residue in the active site (372, 428). In studies of renal nephrotoxicity, oxidative inactivation of GR and GPx by alkylating agents was found to contribute to oxidative stress, characterized by increased lipid peroxidation and cell death in renal proximal tubules that involve mitochondrial disruption and the production of excess oxidants by mitochondria (428). In RAW264.7 cells, activation by a mixture of endotoxin and interferon-γ, a condition that increases endogenous NO production through the stimulation of inducible NOS (iNOS) in this macrophage-like cell line, results in S-nitrosation of GR. Loss of GR function was also correlated with decreased cellular GSH and increased oxidant stress in these cells (59). Other studies similarly suggest that conditions that inhibit GR as well as Grx disrupt the cellular GSH:GSSG redox state and contribute to cell death; for example, suppression of GR and Grx appears to be responsible for oxidative damage caused by exposure of human monocyte-derived macrophages to oxidized low-density lipoprotein (443). Mitochondrially localized GR has also been shown to be instrumental in maintaining cellular growth and the activities of some Fe-S proteins, at least in fission yeast Schizosaccharomyces pombe (395); at the current time, it is unclear whether GR in higher eukaryotes similarly influences the function of Fe-S containing proteins.
4. Prxs, Trx, and TrxR
First identified in yeast, the mammalian peroxiredoxins (Prx) comprise a large family of enzymes that also catalyze the reductive metabolism of hydrogen peroxide or lipid peroxides (346). Most peroxiredoxins utilize Trx as an electron donor (Fig. 3), although Prx6 apparently uses GSH (132). These enzymes have been subgrouped according to the number of Cys involved in their enzymatic action: thus, Prx1–4 are typical 2-Cys forms, Prx5 is an atypical 2-Cys form, and Prx6 is a 1-Cys enzyme. As homodimers, the typical 2-Cys Prxs reduce hydrogen peroxide by sequentially forming a sulfenic acid at one Cys residue that then forms an intersubunit disulfide, which is resolved by Trx (66). In the atypical 2-Cys Prx5, which is a monomer, the disulfide (from vicinal dithiols) forms intramolecularly after sulfenic acid formation. The Prx6 mechanism is similar to that of the GPxs where an S-OH (sulfenic acid) formed at the active site is reduced by GSH after the formation of an intermediate mixed disulfide (S-SG). Recent findings suggest a necessary role for glutathione-S-transferase π in the glutathiolation of Prx6, as Prx6 function is attenuated in the absence and stimulated by the presence of this glutathione-S-transferase (273). Prx6 is also unique in that it can reduce phospholipid hydroperoxides similar to GPx-4 (132); however, of these two phospholipids peroxidases, only GPx-4 targets to mitochondria.
Theoretically, Prx are less efficient at catalyzing hydrogen peroxide reduction than catalase or GPx-1; however, some of the Prx isoforms (Prx1 and Prx2) are highly abundant, and Prxs have a comparatively low K m (<20 μM) for hydrogen peroxide (65). Thus, it seems likely that Prxs regulate normal hydrogen peroxide flux within cells. Experimental evidence supports this concept, as deficiency of various Prxs augments cellular responses to oxidant-inducing stimuli, whereas their overexpression enhances antioxidant protection. Thus, cells isolated from mice with deficiencies in Prx1 or Prx2 show increased production of hydrogen peroxide and increased sensitivity to oxidant damage (88, 239, 313). In particular, erythrocytes from either Prx1- or Prx2-deficient mice have decreased lifespan and excess accumulation of oxidative proteins. Similarly, deficiency of the mitochondrially specific Prx3 in cells increases mitochondrial production of ROS and promotes apoptotic changes in mitochondria, whereas its overexpression decreases ROS and attenuates apoptotic signaling (73). Furthermore, Prx6-deficient mice have increased cardiac ischemia-reperfusion injury with an increase in oxidative stress (308).
Thus, as shown in many studies, Prxs protect against the damaging consequences of excess oxidants in cell culture and in vivo systems; however, many of the mammalian 2-Cys forms are susceptible to overoxidation, a process in which the –S-OH of the oxidized Cys-active site is further oxidized to −SO2H (sulfinic acid) (338, 462). As discussed further in section II.D.1, in most proteins, sulfinic acid formation is an irreversible oxidation state. The intriguing aspect of sulfinic acid formation in Prxs is the existence of an enzyme, sulfiredoxin (Srx) (74), that specifically targets and reduces sulfinic acid in 2-Cys Prxs. Srx is only found in eukaryotes, suggesting that it evolved as a compensatory mechanism to restore function of eukaryotic 2-Cys Prxs, which, unlike their prokaryotic counterparts, are highly sensitive to overoxidation. The importance of this overoxidation/reduction mechanism is uncertain at this time. Recent work using an Srx1-deficient mouse indicates that Srx protects mice from lipopolysaccharide (LPS)-induced sepsis as deficient mice have increased sensitivity to LPS (333), a stimulus known to produce oxidant stress in vivo. Although the in vivo presence of overoxidized Prxs was not shown in this study, in mouse embryonic fibroblasts, Srx deficiency resulted in excess accumulation of intracellular oxidants in untreated cells and increased oxidation of Prx1/2 in response to exogenous hydrogen peroxide, implicating Srx-mediated preservation of the redox state of Prx1/2 in the antioxidant mechanisms of Srx.
The floodgate theory suggests that peroxiredoxins act as sensors of hydrogen peroxide concentration such that their oxidative inactivation allows downstream hydrogen peroxide-mediated signaling to proceed (454). Sulfiredoxin restoration of Prxs would be one mechanism of reestablishing the gateway. Many arguments can be made for or against this theory; however, experimental evidence indicates that the multitude of systems regulating hydrogen peroxide flux are not redundant, suggesting that each system (catalase, GPxs and Prxs) may contribute to overall cellular peroxide tone. In models of hydrogen peroxide-dependent hydrogen peroxide flux in Jurkat cells, the relative contributions of GPxs and Prxs to hydrogen peroxide reduction were about 100-fold greater than that of catalase, with GPx-1 responsible for 44%–57% of the flux and Prxs 32%–42% of the flux (3). Although these findings may not account for all of the molecular interactions involved in cellular hydrogen peroxide flux and may not hold true in all cell types, this model also predicted a significant effect of hydrogen peroxide on the protein disulfide pool. As discussed later, the formation of many disulfide bonds is modulated via the flux of oxidants produced [especially hydrogen peroxide (464)], in part, via mitochondrial respiration (169, 464).
The activity of the 2-Cys peroxiredoxins is also regulated by the availability of reduced Trx, a low-molecular-weight redox-active protein (137). Two major forms of Trx, cytosolic (Trx1) and mitochondrial (Trx2), play an important role in reducing protein disulfides, including those of the Prxs. Recent findings indicate that mitochondrial Prx3 can also be functionally reduced by mitochondrial Grx2; this was shown in both in vitro kinetic assays as well as in situ in HeLa cells (170). In HeLa cells, Trx2 deficiency (or Grx2 deficiency) alone had little effect on the Prx3 oxidation; however, simultaneous knockdown of both Trx2 and Grx2 substantially increased Prx3 oxidation. Trx2 is, however, important in maintaining cellular redox status and, similar to the effects of Prx3 overexpression in attenuating apoptosis, cells with excess Trx2 are more resistant to oxidant-induced apoptosis (79). The mitochondrial (Trx2) and cytosolic (Trx1) forms of Trx have two active Cys residues that are oxidized to form an intramolecular disulfide after the reduction of protein disulfides. Trx-disulfides are reduced by the cytosolic or mitochondrial NADPH-dependent selenoproteins, the TrxR, TrxR1 or TrxR2, respectively. A third TrxR subtype is expressed solely in testes. These proteins are discussed further below. Trx1 and related family members that are targeted to the cytoplasm and nucleus also modulate the activity of other redox-dependent proteins, including enzymes and transcription factors, to control processes ranging from apoptosis and DNA synthesis to DNA binding by transcription factors. Both Trx1 and Trx2 appear to be essential, as complete deficiency of either subtype causes early embryonic lethality in mice (280, 317). Most of Trx exists in the reduced form in cells and mitochondria, but these proteins can become oxidized under certain apoptotic stresses. Excess oxidation of Trx2 is associated with activation of the mitochondrial transition pore during apoptosis (348), illustrating the importance of Trx-redox in maintaining normal mitochondrial function.
The mammalian TrxR proteins are Sec-containing proteins with the Sec amino acid in the pentultimate site of the protein as part of the C-terminal active site (Gly-Cys-Sec-Gly-COOH). Homodimers form the functional TrxR protein in which the monomers are arranged in a head to tail (Ying-Yang) configuration. The mechanisms of TrxR enzymatic reduction of Trx is reviewed in Ref. (18). Interestingly, selenoproteins are essential for modulating both the mammalian Trx system (through TrxRs) and the GSH system (through GPx enzymes). Deficiency of selenium has been associated with cardiomyopathy (Keshan disease) in regions of China with nutritional inadequacy of this micronutrient stemming from its low soil content. Decreased activity of antioxidant enzymes, such as GPx-1 and TrxRs, has been suggested to contribute to the cardiomyopathy associated with selenium deficiency (82, 100, 145, 241). Treatment with dietary selenium supplements limits the disease phenotype, consistent with a primary causative role of selenium deficiency in the development of this disease.
Both TrxR1, which is primarily cytosolic, and TrxR2, which is primarily mitochondrial, are essential, as knockout of either gene is embryonically lethal with distinct phenotypes. Loss of TrxR1 causes severe growth retardation and deficits in mesoderm formation, whereas loss of TrxR2 has profound effects on hematopoiesis, liver development, and heart development, with profound anemia and reduced myocardial cell proliferation in TrxR2-deficient embryos (18, 100). Conditional knockout solely in cardiac cells implicated TrxR2, but not TrxR1, in heart development. Loss of cardiac-specific TrxR2 was found to result in a dilated cardiomyopathy that caused neonatal death (100). In mouse embryonic fibroblasts with TrxR2 deficiency, there was no evidence of excess ROS by RedoxSensor Red CC-1 staining compared to wild-type fibroblasts, suggesting that in the absence of TrxR2, other antioxidant systems could substitute under basal conditions. However, after cellular GSH depletion, these deficient fibroblasts showed enhanced sensitivity to apoptosis compared to wild-type cells and had enhanced ROS production in mitochondria, conditions that could be attenuated by N-acetylcysteine treatments (100). Consistent with the importance of TrxR2 in modulating mitochondrial ROS, this study also found that hearts from the cardiac-specific TrxR knockout mice had severe mitochondrial malformation and mitochondrial swelling (100). Taken together, these findings show the essential nature of the TrxR proteins in development and the importance of mitochondrial antioxidant enzymes such as TrxR2 in cardiac development.
5. Cytosolic and mitochondrial NADPH
NADPH plays an important role in maintaining the activity of all of the hydrogen peroxide-targeting antioxidants (Fig. 3). It is essential to maintain the active state of catalase and is used as a source of reducing equivalents in the enzymatic recycling of the essential cofactors of these enzymes, GSH and Trx. Glucose-6-phosphate dehydrogenase (G6PD) is essential for the production and maintenance of the NADPH pool by catalyzing the reduction of NADP+ during the first and rate-limiting step of the pentose-phosphate pathway. This enzyme is ubiquitously expressed, and is essential in maintaining cellular redox balance: its deficiency is associated with oxidant stress, whereas its overexpression protects cells against ROS accumulation (63, 244, 245). In humans, G6PD deficiency, due to the presence of lower activity variants, is common worldwide, but especially in regions with endemic malaria. Deficiency of G6PD actually confers protection against severe forms of malaria, possibly due to the early removal of erythrocytes infected with the malarial parasites owing to their accumulation of oxidative damage (63); however, G6PD deficiency may promote susceptibility to other diseases, such as diabetes and hypertension, owing to redox imbalance and oxidant stress (243). For example, acquired deficiency of G6PD caused by high glucose-mediated inhibition of G6PD expression and activity increases ROS generation in pancreatic islet cells (476). Specifically, increased oxidant stress caused by deficiency of G6PD leads to increased apoptosis, decreased proliferation, and impaired insulin secretion in islet cells. Furthermore, G6PD-deficient mice that contain a hypomorphic G6PD gene (335) have impaired glucose tolerance and smaller islets than wild-type mice (476), further illustrating how G6PD deficiency contributes to diabetes. Curiously, the naturally occurring adrenal steroid hormone dehydroepiandrosterone (DHEA), which is a noncompetitive inhibitor of G6PD, improves insulin sensitivity and obesity in cell systems as well as in human and animal models (461). Although this may be due to the effects of DHEA on enzymes other than G6PD, it is possible that a decrease in NADPH-reducing equivalents may promote increased ROS in response to insulin to enhance insulin-mediated signaling and lessen insulin resistance found in obesity or the metabolic syndrome. Other studies find that G6PD deficiency leads to renal oxidant stress, with decreased NADPH and GSH and increased albuminuria (460), and that deficiency of G6PD promotes enhanced cytokine responses to endotoxemic stress with reduced survival (447). Thus, the protective or harmful effect of G6PD suppression may depend on the context: the level of stress, the degree of G6PD inhibition, and whether other systems are simultaneously altered (as mentioned above in the discussion of androgenic steroid inhibition of G6PD). Consistent with a protective role of G6PD, targeted overexpression of G6PD to dopaminergic neurons decreases the toxic effect of 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine in a mouse Parkinson disease model (289).
Although complete deficiency of G6PD is incompatible with life, it is not the only enzyme that contributes to cellular NADPH biosynthesis. In mitochondria, the enzymatic actions of nicotinamide nucleotide transhydrogenase (NNT), which relies on the mitochondrial electrochemical proton gradient for its activity, regenerates NADPH from NADP+ utilizing NADH produced in the TCA cycle (361). Thus, mitochondrial deficiencies that decrease matrix NADH or mitochondrial membrane potential will theoretically decrease mitochondrial NADPH regeneration, contributing to oxidative stress (226). NADPH is an essential source of reducing equivalents for GSH- and Trx-dependent antioxidant enzymes, including those in mitochondria. In addition to transhydrogenase, mitochondrial NADPH stores can also be restored by certain dehydrogenases present in the mitochondria, such as isocitrate dehydrogenase (ICDH) and malate dehydrogenase (MDH) (468). NAD+ can also be a precursor for NADP+ via NAD kinase found in the cytosol. In addition to G6PD, cytosolic isoforms of ICDH and MDH may also play important roles in the regeneration of NADPH to prevent oxidative stress (270), at least in some cell types.
D. Redox-active Cys
Along with GSH, Cys, and other low-molecular-weight thiols, protein cysteinyl residues represent a large pool of available thiols that can serve as a buffer for oxidant stress. As discussed further below, however, not all protein thiols are affected equally by oxidants, thus allowing for some specificity with regard to the signaling effects of ROS and resulting in reproducible cellular targets for thiol modification by GSH or nitrosating species. Here we examine thiol modifications, their potential functional consequences, and the role of mitochondria in these processes. (See Table 1 for examples that affect mitochondrial function.)
1. Cys oxidation states and functional modification of protein thiols
Hydrogen peroxide promotes cell signaling, in part, via the reversible oxidation of protein thiols. Protein thiols can exist in a number of oxidative states, including sulfenic (S-OH), sulfinic (S-O2H), and sulfonic (S-O3H) acids, as well as disulfide (R-S-S-R1) forms (Fig. 4). Thus, key to its usefulness as a signaling mechanism, thiol oxidation is targeted preferentially at critical thiols that are redox active, such as those that exist as thiolate anion (RS−) under physiological conditions due to the local environment that fosters a decreased pKa at this redox-active site (368). Although a simple motif for identifying redox-active thiols is not available, analysis of known modifiable sites suggests a role for nearby noncharged polar residues in the tertiary structure and a possible role for His residues N-terminal to the redox-active Cys (368). With the exceptions of some Prx in which sulfinic acid is reversed enzymatically by Srxs (74), formation of sulfinic acid and the more highly oxidized sulfonic acid are considered overoxidations that are irreversible. In terms of signaling, formation of sulfenic acid or disulfides may result from levels of hydrogen peroxide consistent with receptor-mediated signaling events. Formation of sulfenic acid, which is unstable, is widely believed to precede disulfide formation (345), which can form between protein thiols or between protein thiols and GSH (S-glutathiolation) or homocysteine (S-homocysteinylation). Essentially, the electrophilic nature of the sulfenic acid increases its susceptibility to nucleophilic attack by thiols or thiolates anions (345). Disulfide bonds are more stable than sulfenic acid and amenable to reduction by Trx (protein disulfides) or Grx (protein disulfides and protein glutathiolations). Recent work suggests that thiols involved in reversible disulfide formation comprise two groups: those with structural and those with functional (thiol-disulfide switch) consequences (464).
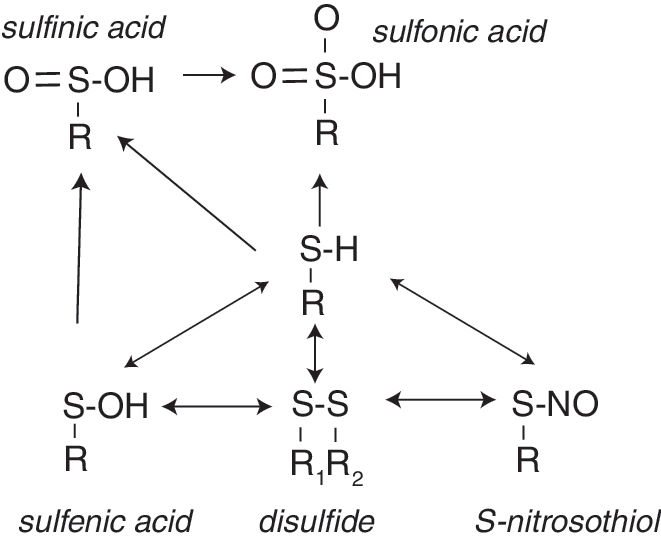
The functional consequences of redox-active modifications are only partially understood. Thus, some changes may modify active sites of enzymes, whereas other redox-sensitive changes may occur in sites remote from the active or ligand binding sites that modify enzyme function (allosteric redox modulation) (197, 464). The role of thiol oxidation on signal transduction has been studied due to the susceptibility of many protein tyrosine phosphatases to oxidative inactivation by this mechanism. Examples include the phosphatase and tensin homolog (PTEN), which antagonizes growth factor-mediated phosphatidylinositol 3 kinase (PI3K) action to attenuate Akt signaling; PTP1B, which modulates insulin signaling; and mitogen-activated protein kinase (MAPK) phosphatases (MKPs or dual specificity phosphatase [DUSPs]), which target phosphotyrosine as well as phosphoserine/phosphothreonine residues within MAPK (114). All of these pathways are regulated to some extent by ROS produced as a result of growth factor-mediated receptor activation. Oxidative modification of these phosphatases prolongs kinase action, whereas diminished ROS attenuates the oxidative inactivation of these phosphatases to suppress kinase-mediated signaling pathways and lessen sensitivity to receptor-activation.
Mitochondria can be the source of the inactivating ROS as well as the downstream target of ROS-mediated signaling. Thus, in some cells, the growth factor-mediated signal transduction that activates Akt requires mitochondrial output of ROS, as, for example, suppression of mitochondrial ROS attenuates growth factor-mediated phosphorylation of Akt (78, 169). In addition, Akt-activation downstream of growth factor receptor or direct oxidative stimuli appears to be important in promoting mitochondrial biogenesis as inhibition of Akt-signaling can suppress mitochondrial biogenic responses downstream of ROS (330).
Other redox-active proteins are susceptible to sulfenic acid or disulfide formation due to the flux of cellular oxidants (50, 160, 234, 381, 464). Some of these modifications have been found under basal conditions due, in part, to oxidants produced during normal respiration in mitochondria, whereas other protein oxidation events are induced after oxidative stress. Within mitochondria, complex I is especially sensitive to redox damage, and thiol oxidation of complex I promotes
2. Protein S-glutathiolation
Similar to protein disulfides, mixed protein disulfides with glutathione most likely form through a sulfenic acid intermediate whereby formation of R-SO− under physiological conditions promotes a reaction with GSH (136). Other mechanisms, such as thiol-disulfide exchange with GSSG, can mediate some S-glutathiolation (-S-SG) reactions [reviewed in Ref. (296)]. Protein S-nitrosation can also precede S-glutathiolation, with glutathione replacing the nitroxyl anion (180). Glutathiolation, as a targeted and reversible change, has been suggested to be an adaptive mechanisms that preserves protein function; for example, in the sarcoplasmic-endoplasmic reticulum calcium ATPase (SERCA) pump, loss of glutathiolation of the Cys674 thiol leads to overoxidation of SERCA (421). By contrast, S-SG formation may represent an oxidation reaction that modulates protein function either by reversibly inactivating active sites or by promoting protein activity: Ras family members are activated by –S-SG in vascular smooth muscle cells and other cell types, a process augmented by oxidative stress (2, 95). In mitochondria, glutathiolation of the 51-kDa peptide of complex I is associated with a decrease in complex I activity and an increase in
The intracellular reduction of mixed protein disulfides is primarily achieved through the action of the Grx enzymes, which, similar to Trxs, can also reduce protein disulfides. Grx requires GSH for the reduction of disulfides and is, therefore, dependent on NADPH and GR to maintain GSH stores (Fig. 3). Of the major forms of Grx, Grx2 targets the mitochondria, although deficiency of either Grx2 or Grx1 decreases mitochondrial complex I activity presumably by redox-dependent mechanisms (120, 209). Other studies have similarly found that lack of Grx1 leads to a loss of mitochondrial membrane potential, in part, due to the overoxidation of the VDAC, an outer mitochondrial membrane protein (363) that facilitates flux of
3. Protein S-nitrosation
Redox-active thiols can also combine with RNS to form S-nitrosothiols (-S-NO). Similar to –S-SG, –S-NO formation is reversible, targeted to certain redox-active thiols, and capable of altering protein function. For example, S-nitrosation of caspase 3, a proapoptotic enzyme, inhibits its activity to attenuate apoptotic signaling (356). Importantly, many of these modifications can be detected in response to physiological and pathophysiological production of NO·. Thus, in endothelial cells the normal tonic production of NO· by eNOS is sufficient to cause –S-NO formation (463). Overall, there are three genes encoding NOS isoforms (8): eNOS, expressed primarily in endothelial cells; iNOS, which can be induced in a variety of cell types, including macrophages, during the response to cytokines; and neuronal NOS (nNOS), which was first characterized as an encoding neuronal enzyme. nNOS and eNOS isoforms require Ca2+ and calmodulin for their regulation and are constitutively expressed (1, 58, 366). In contrast, iNOS is transcriptionally induced by cytokines and is regulated by substrate (L-arg) availability (456).
There are several studies that suggest the presence of an NOS isoform in mitochondria (mtNOS); however, the mitochondrial localization of NOS and its nature are highly controversial, with little consensus among different laboratories. Rat liver was first shown to contain an NOS isoform with characteristics similar to those of nNOS and eNOS, as this NOS was found to be Ca2+-dependent and inhibited by L-Arg analogues, such as NG-monomethyl-L-arginine (152). In contrast, another report suggested that liver mitochondrial NOS is most similar to iNOS, although it was found to be constitutively produced (415) rather than induced under stress. Another study countered that although mitochondria contain some eNOS, the majority of mtNOS was a unique form of NOS, as NOS activity was measured in mitochondria from eNOS-deficient mice (231). To add yet another layer to this controversy, other investigators suggested rat liver mitochondria did not contain any NOS isoforms, as neither calmodulin, which is required for NOS activity, nor NOS isoforms were identified in a proteomic analysis of trypsin-digested proteins from purified mitochondria (431). In this study, calmodulin or eNOS was readily identified by mass spectrometry when their recombinant forms were added postisolation to purified mitochondria, suggesting an ability to identify trypsin fragments from these proteins by mass spectrometry (431). However, it is impossible to determine whether the lack of m/z peaks that can be assigned to calmodulin or NOS was caused by the method of sample processing, or a matter of abundance, or, as reported, evidence for the absence of these proteins. To support their claim that NOS was not in the mitochondrial compartment, these authors also showed negligible NOS activity in isolated liver mitochondria (431). Although recombinant NOS added to these mitochondria had detectable activity (suggesting that there was no inhibitory substance in these preparations), an alternative explanation for the failure to detect NOS activity is that the isolation procedures inactivated the endogenous enzyme. Still, these thorough studies cast serious doubts on the presence of NOS in liver mitochondria. A subsequent study by this group reported that one of the commonly used NOS activity assays that measures radiolabeled [14C]-L-Arg catabolism to L-citrulline, a byproduct of NO production, can also detect arginase-mediated urea production if insufficient levels of arginase inhibitors are used (432). Briefly, in this NOS activity assay, urea can be coeluted with L-citrulline and thereby provide a false-positive signal for NOS activity. In cardiomyocytes, several reports also propose a role for mitochondrial NOS. In one such study, microsensors were used to monitor the release of NO from single isolated mitochondria, providing some evidence for NO production in mitochondria (208). Although it was reported that this mtNOS originated from the nNOS gene, other studies of cardiomyocytes suggest that mtNOS is a mixture of iNOS and eNOS (471). A more recent study of cardiac mitochondria similarly suggests that mtNOS is a Ca2+-sensitive enzyme that can be inhibited by the presence of Mg2+ (472). Many of these arguments for or against mtNOS rely on the purity and integrity of the mitochondrial preparations. Although NO is capable of diffusing into mitochondria, the presence of NOS isoforms within mitochondria has significance for mitochondrial functions owing to the ability of NO to nitrosate mitochondrial targets and its ability to combine with superoxide, a byproduct of electron transport, to form peroxynitrite.
Nitric oxide does not directly react with protein thiols; rather, higher oxides of NO· that are more reactive than NO· are believed to be the S-nitrosating agents, such as N2O3 (199, 213, 312, 449). Furthermore, nitric oxide can be transferred to protein thiols by transnitrosation. Essentially, low-molecular-weight thiols, such as GSH or Cys, or susceptible protein thiols readily become S-nitrosated in vivo and can transfer NO to additional thiol targets (263). Mitochondria play a central role in protein S-nitrosation: many S-nitrosoproteins target to the mitochondria or perimitochondrial regions in cells, and mitochondrial function is necessary for their formation (463). Furthermore, S-nitrosation of mitochondrial targets can modify mitochondrial function (97, 98, 474).
4. Mitochondrial function and the thiol redox state
Mitochondria are a major source of cellular oxidants, and the basal production of oxidants by mitochondria are essential for maintaining the protein thiol redox state of cells, affecting both global S-nitrosation and disulfide formation of proteins (463, 464). Thus, in a staining-based method based on the biotin-switch procedure first developed by Jaffrey and colleagues (191), we detected S-nitrosoproteins in the mitochondrial or perimitochondrial compartments of cells (463). Pretreatment of cells to remove their functional mitochondria (pseudoRho0 cells) eliminated most of the detectable S-nitrosoproteins. Use of mitochondrial inhibitors (rotenone, myzothiazol, or uncouplers) that reduce mitochondrial ROS production diminished S-nitrosoprotein detection, whereas use of antimycin A, which is known to increase ROS production, had little effect on the detection of the S-nitrosoproteins, confirming a role for mitochondrial ROS in modification of redox-reactive thiols with NO·. Although the specific modified thiol was not identified within these proteins, proteomic methods allowed for the identification of several S-nitrosated target proteins, many of which have previously been shown to have redox-active thiols.
Other investigators have similarly used a biotin-switch-based method to identify susceptible protein thiols that can become S-nitrosated in whole cells (232) or isolated cardiac mitochondria (305). In these latter studies, following biotin labeling of –S-NO-labeled thiols, proteins were digested with proteolytic enzymes before streptavidin capture and liquid chromatography (LC)/tandem mass spectrometry (MS/MS) analysis of isolated peptides. One advantage of peptide-based purification is that it allows for an identification of the modified site, at least in peptides with a single Cys. The disadvantage is that not every peptide may be amenable to LC-based methods and MS/MS identification. Consistent with our findings, only a subset of available Cys were S-nitrosated in these studies. Although the functional consequences of –S-NO modifications have not been determined in these mapping studies, these data provide useful information regarding possible targets that may mediate the protective and harmful effects of NO·.
In other studies, we found that mitochondrial ROS are also essential for global protein disulfide formation, as inhibitors of mitochondrial ROS significantly reduce disulfide bond formation in cells, as did a mitochondrially targeted catalase (464). From this study, it is not clear whether the disulfides analyzed were due to interprotein, intraprotein, or mixed disulfide (such as with GSH) formation; however, these findings identify a subset of proteins with redox-active thiols that form disulfides under basal conditions. In support of a role for basal respiration in protein disulfide bond formation, studies in yeast, for example, found that anaerobic growth substantially decreased detection of proteins with sulfenic acid or disulfide bonds (234). In yeast, inhibition of the Trx-mediated pathways augmented disulfide formation, suggesting a tonic effect of Trx in modulating the extent of protein oxidation under normal metabolic fluxes. Inhibition of GSH synthesis pathways, in contrast, suppressed overall detectable protein disulfide formation, possibly due, in part, to a lack of protein glutathiolation and/or overoxidation of many redox-active thiols (to sulfinic and sulfonic acid) in an environment lacking GSH. In addition, other researchers have used proteomic methods to study the effects of oxidant stress on protein thiols, finding a subgroup of proteins with increased sensitivity to thiol oxidation (32, 50, 51, 104), including susceptible thiols within the mitochondrial complexes I, II, and IV (257). Although none of these studies correlated thiol oxidation with mitochondrially produced ROS, they establish a requirement of ROS for their formation and indicate that these redox-dependent events only occur in a subset of the thiol proteome. Our more recent study showed that decreasing cellular ROS through the overexpression of the antioxidant GPx-1 disrupts normal cellular disulfide formation with consequences for mitochondrial function (169). Thus, excess GPx-1, in both the cytosol and the mitochondria, decreases mitochondrial potential and ATP production. Furthermore, this suppression of mitochondrial function contributes to diminished responses in growth factor-mediated Akt activation and cellular proliferation (169). These studies highlight the essential nature of mitochondrial ROS for normal cellular and mitochondrial function.
5. Mitochondrial disulfide-relay system
Similar to the endoplasmic reticulum, enzymes within the mitochondrial intermembrane space catalyze the formation of disulfide bonds in mitochondrially targeted proteins (347). This process has been most widely studied in yeast strains where a disulfide-relay system has been shown to be linked to the import of proteins into the intermembrane space of mitochondria (292). A crucial component of this system is the FAD-containing sulfhydryl oxidase Erv1, which maintains the intermembrane protein Mia40 in an oxidized form (40, 391). Mia40 then passes the disulfides to reduced proteins, after their entry into the intermembrane space. Erv1 serves to shuttle electrons from Mia40 to cytochrome c of the ETC in order to maintain its oxidized state (10, 40, 105, 292). GSH plays an important role in this process by preventing the overoxidation of redox-active thiols during disulfide bond formation within the intermembrane space (40, 292). The function of this system is less well studied in mammals; however, a mutation in GFER, the human homolog of Erv1, alters the protein content of the mitochondrial intermembrane space, and decreases mitochondrial complex I, II, and III activity (116). This rare human mutation was found to cause a progressive mitochondrial myopathy and congenital cataracts, hearing loss, and developmental delay in affected individuals.
E. Metabolism, NADH/NAD+, and NADPH/NADP+
1. Regulation of glycolysis
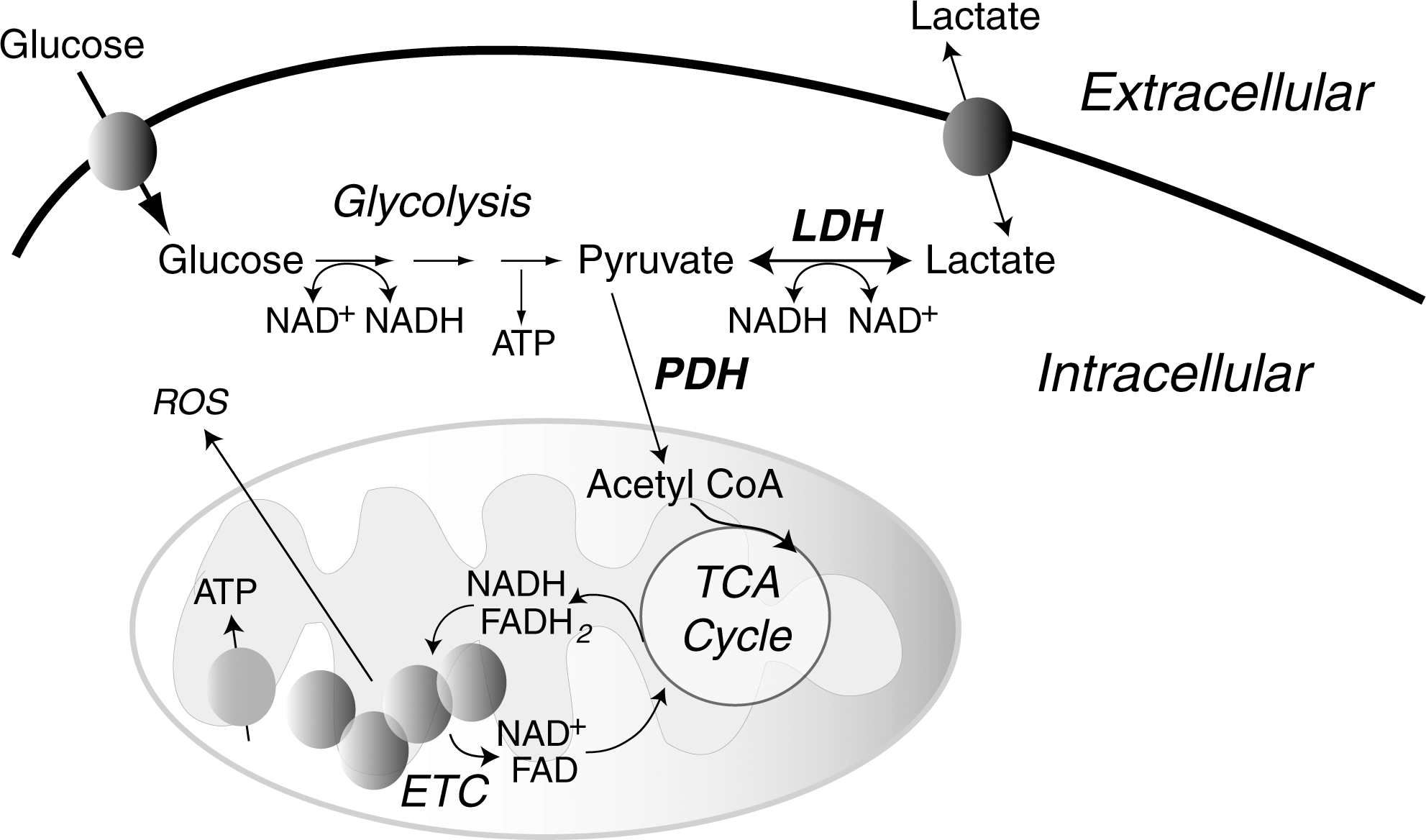
Within the mitochondria, the enzymes of the TCA cycle supply reducing equivalents (NADH and FADH2) for ETC-mediated oxidative phosphorylation and production of ATP. Cellular metabolism of glucose or lipids feeds into the TCA cycle after the formation of acetyl CoA (Fig. 5). The entry of carbons from glucose into the TCA cycle is regulated, in part, by isoforms of lactate dehydrogenase (LDH) that either favor lactate production from pyruvate (lactate dehydrogenase A [LDHA]) or pyruvate formation from lactate (lactate dehydrogenase B [LDHB]) (276). The LDHA production of lactate results in oxidation of NADH (pyruvate+NADH→lactate+NAD+) whereas the LDHB production of pyruvate causes the reduction of NAD+. Certain glycolytic cells may also secrete lactate that is taken up by other neighboring cells. For example, in brain, lactate secreted by astrocytes is taken up by the monocarboxylate transporter into neurons where it is used as a fuel source (117). Other cells can similarly utilize lactate as an energy source following its conversion to pyruvate. Pyruvate dehydrogenase (PDH) is another enzyme that regulates the utilization of pyruvate; it is itself regulated by PDH kinase (PDK), becoming inactivated by phosphorylation. It is well established that high rates of fatty acid oxidation inhibit glucose oxidation by inhibition of PDK, in part, due to high NADH/NAD+; the reverse is also true: low rates of fatty acid oxidation promote glucose oxidation (341, 400). These mechanisms suggest mitochondrial adaptation to available substrates (Randle cycle); however, alterations in the normal balance between glucose and fatty acid oxidation has been suggested to contribute to excessive cellular proliferation, as is seen in some cancers and in pulmonary hypertension (408, 429). Redox activation of c-jun N-terminal kinase (JNK) also fosters a decrease in PDH activity by increasing its phosphorylation (480) to limit TCA-mediated NADH production and subsequent ETC activity. Loss of pyruvate-mediated, high-yield energy production due to deficiencies in the TCA cycle and oxidative phosphorylation determinants is found in aging, neurodegenerative diseases, and also in cancer and during hypoxia. In type 2 diabetics, disruption of normal ETC activity in skeletal muscle may also contribute to a reduction of ATP generation (351). Although some cells are more glycolytic in nature (ending glycolysis with the production of lactate, rather than the complete oxidation of pyruvate in the TCA cycle), hypoxia mediates a glycolytic shift to anaerobic glycolysis caused by the coordinate upregulation of proteins such as LDHA and PDH kinase by the hypoxia-inducible transcription factors, especially hypoxia-inducible factor-1 (HIF-1), resulting in lactate production and an NAD+ increase (385, 386). The corresponding increase in glucose uptake by the upregulation of the glucose transporter-1 (Glut1) helps to maintain the cellular NADH/NAD+ ratio and provides an energy source for this anaerobic glycolysis. (During anaerobic glycolysis, cytosolic NADH produced by glyceraldehyde-3-phosphate dehydrogenase is recycled into NAD+ by LDHA production of lactate from pyruvate, whereas during the complete oxidation of glucose, pyruvate entry into the TCA cycle results in NADH production that is oxidized by respiratory complex I.) Importantly, recent work with selective genetic sensors for cytosolic and mitochondrial NADH suggests that, in contrast to the cytosolic compartment, cells have a strong tendency to maintain mitochondrial NADH homeostasis in response to a variety of oxidant stresses (477). The coordinate metabolic changes caused by HIF-1 that alter mitochondrial function are attributable, in part, to the HIF-1-mediated transcriptional upregulation of microRNA 210 (miR210), which specifically inhibits Fe-S complex formation to alter mitochondrial bioenergetics and ROS production (Pasteur effect) (69). These pathways are discussed further below.
2. SIRT, NAD+, and metabolic regulation
Although there are a number of NAD+-requiring enzymes in mammalian cells (219), the SIRT, a family of NAD+-dependent protein deacetylases that target lysine residues on histones, nuclear factors, and other proteins, have the greatest effect on coordinating metabolism and mitochondrial function. In mammals, this family has several members of which SIRT1, SIRT6, and SIRT7 are nuclear proteins; SIRT1 is found mainly in the cytoplasm; and the SIRT3, SIRT4, and SIRT5 are found in the mitochondria (165, 295, 389). SIRT1 is known to be activated in response to metabolic changes, such as low glucose or exercise, that increase cellular NAD+ (62, 354). SIRT1 can also be activated by AMP kinase (AMPK), which is a metabolic sensor of low ATP. SIRT1 has been shown to target the deacetylation of PGC-1α, which may influence mitochondrial biogenesis and increase mitochondrial respiration (33), in part, by enhancing fatty acid oxidation (150) (Fig. 6). Similarly, other SIRT family members have been found to affect metabolism. SIRT6 activation coordinately regulates the expression of glycolytic genes (479), and mice deficient in SIRT6 have severe metabolic defects, signs of accelerated aging, and die prematurely. The mitochondrial SIRT3 has been found to modulate mitochondrial function, possibly by targeting the deacetylation of mitochondrial complex I or other mitochondrial proteins, including ICDH-2, a potential source for NADPH (6, 389, 394). Thus, SIRT3 activity may decrease oxidative damage by increasing mitochondrial NADPH to allow for GR and TrxR reduction of GSH and oxidized Trx, respectively (394). In mice, SIRT3 is normally upregulated during fasting in liver and brown fat where it plays a role in fatty-acid oxidation by maintaining the deacetylation of long-chain acyl coenzyme A dehydrogenase in mitochondria (175). In its absence, SIRT3-deficient mice show reduced utilization of lipid stores during fasting: they have decreased fatty acid oxidation, accumulate triglycerides, and have diminished production of hepatic ATP.

Owing to their growing importance as metabolic regulators and the findings that SIRT homologs in Caenorhabditis elegans and Drosophila melanogaster may extend lifespan (201, 419), there is an interest in developing pharmaceutical mediators of SIRT function. There is, however, some recent controversy regarding the role of SIRT in extending lifespan, in that recent reports fail to show robust effects of SIRT on lifespan in these same model systems (56). Nonetheless, these NAD+-responsive genes serve to coordinately regulate metabolism and mitochondrial function. AMPK mechanisms apparently upregulate SIRT function (62), as does resveratrol (33, 44). A more recent study used pharmacological methods to induce the oxidation of NADH by the cytosolic NADH:quinone oxidoreductase 1 (NQ1) (183). NQI is an obligate two-electron reductase that participates in cellular antioxidant protection by reducing reactive quinones to hydroquinones avoiding the formation of free radical semiquinones. In this study, β-lapachone supplementation was found to increase intracellular NAD+ and, in vivo, this treatment decreased adiposity, dyslipidemia, glucose intolerance, and fatty liver in diet-induced obesity as well as the ob/ob obese mouse. Furthermore, these metabolic improvements correlated with increased gene expression of some SIRT family members and with upregulation of PGC-1α and nuclear respiratory factor-1 (NRF1), two factors involved in mitochondrial biogenesis (see Table 1 for examples of acetylation that modulate mitochondrial function). Consistent with the upregulation of these biogenic factors, β-lapachone supplementation also increased skeletal muscle mitochondrial content as well as oxygen consumption. Although the increase in NAD+ indirectly implicates SIRT-mediated activation, protein acetylation and SIRT activity were not assessed in this study. Nonetheless, these findings suggest a dramatic metabolic shift caused by modification of intracellular NADH/NAD+ ratio. It should be mentioned, however, that β-lapachone and other similar quinones are used chemotherapeutically for their ability to cause tumor cell death, which has been attributed to oxidative stress caused, in part, to the decrease in cellular NADH/NAD(P)H pools and/or the production of toxic forms of hydroquinones that generate ROS (332).
III. Functional Consequences of Redox Modifiers
A. NO·
Physiological, pathophysiological, and pharmacological levels of NO· can mediate protein –S-NO formation. In addition, NO· may affect cellular functions via activation of soluble guanylyl cyclase (sGC) and the latter's activation of cGMP-mediated protein kinase G pathways that involve protein phosphorylation rather than protein S-nitrosation. Nitric oxide may also promote protein modification through nitration of tyrosines to form the stable 3-nitrotyrosine. Accumulation of 3-nitrotyrosine in proteins is indicative of ROS and RNS stress (188). In this section we next consider evidence for protein –S-NO modifications that specifically alter mitochondrial function (Fig. 7).

1. Apoptosis and NO·
At low concentrations, NO· is considered protective against apoptosis, whereas at higher concentrations, such as those that can be generated during inflammatory states, it can mediate apoptotic responses due, in part, to the formation of peroxynitrite (281, 439). The beneficial or harmful effects of NO· may also depend on the cell type, duration and intracellular location of its production, and redox status of the cell. We found that bovine aortic endothelial cells grown in the presence of an NO· donor at 1 mM causes a decrease in mitochondrial membrane potential, with the release of cytochrome c and activation of caspase cascades (439). Furthermore, hypoxia augments these effects, whereas uric acid, a peroxynitrite scavenger, attenuates these responses. In an in vivo model of neuronal apoptosis due to target deprivation (a model which replicates changes thought to occur in development, or following brain injury or degeneration in which loss of a neuron's target fosters neuronal death due, in part, to loss of retrograde stimulation from the innervated target), intracellular generation of ROS and NO· promotes protein modifications like S-NO and 3-nitrotyrosine formation in mouse brain (277). In this model, apoptosis was absent in mice deficient in nNOS or those treated with NOS inhibitors (277), implicating NO· in the apoptotic mechanisms. Although these studies did not identify –S-NO targets specifically, there are a number of studies that correlate the pro- and antiapoptotic mechanisms of NO· with S-nitrosation of protein targets that directly or indirectly affect mitochondrial function. Effector caspases mediate the pathways that lead to cell death, including chromatin condensation and DNA fragmentation, and are activated in both the extrinsic (death-receptor mediated) and intrinsic (mitochondrial) pathways of apoptosis (54). Caspase 3-S-nitrosation is antiapoptotic as it inhibits the activation of this effector caspase (424). In the intrinsic pathway of apoptosis, Bcl-2 prevents the mitochondrial release of pro-apoptotic factors, such as cytochrome c, by forming heterodimers with pro-apoptotic proteins, such as Bax. S-nitrosation of Bcl-2 prevents the proteasomal degradation of this antiapoptotic protein after apoptotic stimuli to enhance its antiapoptotic function (21). The mitochondrial inner membrane adenine nucleotide transporter (ANT) may also be a target for S-nitrosation (305). Although the functional consequences of ANT S-nitrosation in cells has not been demonstrated, it is known that modification of redox-sensitive thiols in this mitochondrial protein regulates the formation of the mitochondrial permeability transition (MPT) pore and cell death (101). Redox regulation of ANT and MPT pore formation is discussed further below.
2. NO· and mitochondrial fission and fusion
Nitric oxide has also been found to alter intracellular mitochondrial networks. Briefly, mitochondria exist as a dynamic network, the nature of which may be regulated by several factors, including interaction with the cytoskeleton and fusion and fission (93, 138). It is believed that alterations in mitochondrial fusion and fission may serve functional purposes to respond to oxygen and metabolic fluctuations (24, 446). The significance of these rearrangements in mitochondrial architecture are not entirely clear; studies suggest that ROS may promote mitochondrial fission (457), whereas other studies suggest that fission may promote mitochondrial generation of ROS (387). Possibly, changes in mitochondrial network structure provide an example of ROS-mediated ROS generation where ROS plays a role in mitochondrial fission to augment ROS generation from restructured mitochondria. The relationship between mitochondrial network formation and bioenergetics is not well understood. In cell culture systems, inhibition of oxidative phosphorylation is known to promote mitochondrial fission; yet, not all genetic defects in oxidative phosphorylation alter mitochondrial network formation, suggesting a more complex relationship between bioenergetics and mitochondrial dynamics (371). Furthermore, the effects of mitochondrial dynamics on bioenergetics are unresolved. In the process of myogenic differentiation, NO·-induced mitochondrial elongation (fusion) is a necessary step in myogenesis that appears to require repression of the pro-fission GTPase dynamin-related protein-1 (Drp1) (113). Blocking these necessary NO·-mediated pathways in differentiating myoblasts significantly alters mitochondrial function and reduces oxidative phosphorylation. Drp1 was also proposed as a direct target for S-nitrosation to promote the toxic effects of β-amyloid in Alzheimer disease due to an NO-mediated enhancement of Drp1 activity that promotes mitochondrial fission and subsequent fragmentation (86). A later study, however, challenged the role of Drp1 S-nitrosation in Alzheimer disease, suggesting, instead, that NO· promotes activation of Drp1 by phosphorylation rather than –S-NO modification as –S-NO forms of Drp1 are detectable in normal brains as well as those from Parkinson and Alzheimer diseases (45). Thus, this study supports a role for sGC/cGMP signaling pathways rather than S-nitrosation in modulating mitochondrial fission. Nonetheless, in neurons, NO· production from nNOS is increased in response to β-amyloid oligomers, and upregulation of NO· production and S-NO formation are common features of other neurodegenerative diseases, including Parkinson disease, Huntington disease, ALS, and stroke (218). In addition, excess NO· production has been shown to promote mitochondrial fission in neurons (28), an effect opposite of its role as a factor that promotes elongation of mitochondria during myogenesis (113). Further analysis to elucidate the pathogenic role of NO· in neurodegeneration may lead to useful development of drugs to modulate these processes, perhaps by targeting nNOS or its neuronal associated pathways. Additional research indicates that GSH is an important mediator of NO· cytotoxicity in neurons as depletion of GSH enhances NO·-mediated apoptosis by enhancing mitochondrial apoptosis-inducing factor (AIF) release (15). This study further finds that enhanced SOD1 overexpression is protective against these changes, suggesting the usefulness of (targeted) antioxidant agents in protecting against the damaging effects of excess neuronal NO·.
3. NO· and mitochondrial respiration
As mentioned above, NO· can target and modify subunits of the ETC complexes to reduce their activities. Mapping of –S-NO sites in mitochondria identified seven potential sites within six subunits of complex I and as well as other sites of S-nitrosation in complexes I and II (305). Earlier studies had previously provided evidence that exogenous NO· donors could inhibit complex I in whole cells by a mechanism that can be attenuated by low-molecular-weight thiol antioxidants, such as dithiothreitol or GSH, or by light of the appropriate action spectrum for –S-NO species, suggesting a role for –S-NO formation in this inhibitory effect (98). Using similar methods, other studies have found a role for NO· in the inhibition of complex IV (cytochrome c oxidase) (97, 474) or in the ATPase complex (405). Studies of the effects of NO· in endothelial cells identified two Cys of porcine complex IV as targets of inactivating –S-NO modification (474). Other studies of isolated mitochondria suggest that S-nitrosothiols such as GSNO are better donors of NO· for transfer to redox-active thiols than other forms of NO· donors (108). Many of the above studies were performed using exogenous NO· donors; however, other studies have found that some of these –S-NO modifications can be detected in isolated whole heart models (Langendorff preparations) of cardiac function. Using Langendorff perfused hearts, endogenously produced NO· was found to cause S-nitrosation of complex I during ischemic preconditioning (57), a process in which hearts are subjected to intervals of brief ischemia and reperfusion to stimulate innate protective mechanisms against injury resulting from more prolonged ischemia/reperfusion. It is generally believed that mitochondrial dysfunction and release of oxidants contribute to the damage accompanying reperfusion injury. Thus, it has been hypothesized that the –S-NO-mediated inhibition of mitochondrial respiration may be cardioprotective (57). Based on this premise, mitochondrially targeted S-nitrosothiols have been used as protective agents against ischemia-reperfusion injury to slow the burst of respiration (and oxidants) produced during reperfusion. In separate studies, pre-ischemic (306) or postischemic (336) use of these reagents lessened ischemia-reperfusion injury, suggesting these methods may be promising in terms of limiting cardiac reperfusion injury after myocardial infarction. One possible advantage of these treatments is the labile nature of –S-NO, thus suggesting that any effects are temporary. These S-nitrosating compounds, however, target other mitochondrial proteins as well (91), and may have other cellular effects neither completely understood nor well characterized. Therefore, there is a need to explore in more detail the consequences of modifying ETC and other mitochondrial targets in vivo. Earlier studies, for example, linked S-nitrosation of complex I with an increase in production of
B. Reactive oxygen species
Under some conditions, upregulation of cellular ROS production has been found to increase further ROS production (ROS-mediated ROS generation) by augmenting mitochondrial production of ROS. There are many contributors to this effect, some of which are further addressed in this section. Additionally, we examine other modulators of mitochondrial ROS, including growth factor receptor signaling and uncoupling proteins (UCPs), which serve to lessen mitochondrial ROS through their induction of a proton leak against the mitochondrial proton gradient.
1. NADPH activation of mitochondrial ROS
Several studies have implicated ROS and mitochondrial damage in the pathogenic effects of AII (111, 112, 300). AII is known to stimulate NOX activation, but a role for mitochondrially derived oxidants, downstream of AII-mediated receptor activation, is also emerging (121, 215). In fact, AII-activated NOXs were found to cause mitochondrial dysfunction and upregulate mitochondrial release of oxidants, in part, by stimulating ETC-mediated production of ROS (121). Interestingly, peroxynitrite formation was shown to be involved in these pathways as uric acid, a peroxynitrite scavenger, blocked mitochondrial ROS generation and mitochondrial dysfunction, attenuating AII-mediated reduction of ΔΨ, and decreased the respiratory control ratio, a measure of coupling between respiration and phosphorylation. Mitochondrial potassium channels were also implicated in enhanced mitochondrial ROS generation and increased uncoupling. Furthermore, in these studies, there was a concurrent decrease in GSH levels in the mitochondria and cytosol, indicating overall redox stress caused by the increase in cellular ROS. In support of a role for GSH-mediated oxidation in these events, other studies examining the effects of GSH/GSSG redox status on mitochondrial function found that a more oxidative ratio correlated with mitochondrial uncoupling and a decrease in oxidative phosphorylation-mediated ATP generation (13, 174).
Of the NOX isoforms, NOX4 has been uniquely found to target to mitochondria in some cells, although there is controversy regarding the intracellular compartment in which NOX4 is localized. In human endothelial cells, NOX4 was found in the endoplasmic reticulum, but this study did not specifically examine mitochondrial localization by protein-based methods. Rather, the investigators used in silico analysis (PSORTII) to suggest that NOX4 lacks a mitochondrial localization sequence and is more likely to be found in endoplasmic reticulum fractions than other subcellular compartments (77). In mouse heart, however, endogenous NOX4 has been found in both mitochondria and microsomal fractions (4). This report of a cardiac mitochondrial NOX4 also provided experimental evidence that the N-terminal region of NOX4 is capable of targeting proteins to the mitochondria. To date, it is unclear whether NOX4 has the same distribution in all cell types. In mouse hearts, chronic treatments with pressor agents (AII, phenylephrine) and transverse aortic constriction (TAC) were found to increase NOX4 expression. In addition, cardiac expression of NOX4 was found to increase during normal aging (4). Notably, the increase in cardiac mitochondrial ROS following these hypertrophic stimuli was found to be due to both increases in NOX4 expression as well as enhanced mitochondrial-dependent ROS production, suggesting a relationship between the enhanced expression of NOX4 and mitochondrial ROS production and dysfunction. In support of a role for NOX4 in altering mitochondrial ROS production and function, forced upregulation of mitochondrial NOX4 in the heart, by using a targeted transgene, increased thiol oxidation of many mitochondrial proteins including the ETC complex I, aconitase, and other TCA enzymes (4). Some of these targets were also found to be oxidized after TAC-induced hypertrophy. Interestingly, cardiac overexpression of NOX4 also caused a decrease in the mitochondrial biogenic signal in the heart, resulting in decreased levels of mitochondrial DNA (mtDNA) in cardiomyocytes from these overexpressing mice. Several studies suggest that ROS may be involved in retrograde signaling from mitochondria to the nucleus to stimulate the biogenic response, possibly as a means to replace oxidized mitochondrial proteins or DNA. Retrograde signaling may also help to coordinate growth factor responses to augment mitochondrial production of energy (350). Possibly, in the case of chronically overexpressed NOX4, excess ROS may reach a threshold at which it promotes apoptotic rather than biogenic responses. As discussed further below, the extent of redox imbalance and oxidant damage may also dictate the choice between the normal replacement of mitochondria, which may additionally include the targeted removal of damaged mitochondria by mitophagy, and cell death via apoptosis.
2. Other ROS-mediated ROS generators
The above studies suggest the utility of suppressing AII signaling and/or NOX upregulation as means of protecting against ROS-induced mitochondrial dysfunction. Other studies indicate that mitochondrial complex I is especially sensitive to redox damage, and that thiol oxidation of complex I promotes
Excess oxidants may additionally augment mitochondrial ROS by upregulating the expression of the lifespan regulator p66shc to the mitochondrial intermembrane space (297). Within the mitochondria, p66Shc increases oxidant production by oxidizing cytochrome c to produce hydrogen peroxide and activate the mitochondrial transition pore (153). Furthermore, knockout of p66Shc increases lifespan, reduces hydrogen peroxide generation, and enhances survival to oxidant stress (297). Crucial to the apoptotic function of this protein are two redox centers in the N-terminus: one center plays a role in the copper-dependent catalytic site involved in ROS formation, and the other center is important for tetramer formation, which appears to be essential for the apoptotic function of this protein (151). Intracellular antioxidants such as Trx or GSH are thought to maintain the mitochondrial form of p66Shc in an inactive state.
Mitochondrial dysfunction and excess production of oxidants have been considered contributors to the development of the metabolic syndrome and diabetes. Remarkably, p66Shc-deficient mice have been found to be resistant to many complications of experimental diabetes, including diabetic nephropathy, diabetic cardiomyopathy, cardiac stem cell death, hyperglycemia-induced endothelial dysfunction and enhanced lipid peroxidation, and impaired wound healing (61, 128, 291, 357). Furthermore, in high glucose or AII-mediated models of renal tubular injury, p66Shc was found to cause injury by oxidative mechanisms involving mitochondrial dysfunction (increased ROS, decreased mitochondrial potential, decreased mtDNA) and stimulation of apoptotic pathways involving increased cytochrome c release, caspase 9 activation, and mtDNA damage (406).
3. Extracellular redox state and mitochondrial function
In the intracellular environment GSH/GSSG is the major redox couple but, extracellularly, in human plasma, Cys and cystine (CySS) are the predominant low-molecular-weight thiol redox determinants (155). In the oxidized extracellular environment, there is more CySS than Cys. The Cys/CySS ratio tends to become even more oxidized with aging, smoking, diabetes, and obesity (198, 302). Recent findings also suggest that modifying the Cys/CySS ratio in cell culture systems can enhance inflammatory responses and modulate cell growth (192). Apparently, the Cys/CySS ratio can affect the intracellular redox balance, altering the oxidation of mitochondrial Trx2, leading to the increased production of mitochondrial ROS (157, 184). Redox-active thiols accessible at the plasma membrane were found to mediate these effects through mechanisms that may involve receptor proteins and subsequent signaling-dependent oxidation of actin and other intracellular protein targets (157). A role for redox mechanisms in receptor-mediated activation has previously been shown (89, 207); however, more research is needed to understand these mechanisms and their role in modulating cellular function.
4. Mitochondrial ROS and receptor-mediated signaling
The mitochondrial generation of ROS downstream of receptor signaling is an essential step in signal transduction (Fig. 8). As discussed above and in Figure 8, ROS generated by mitochondria contribute to phosphatase inactivation and promote cell signaling. Furthermore, mitochondrially produced ROS can activate insulin receptor autophosphorylation after insulin stimulation to promote receptor-mediated actions (402). Suppression of mitochondrial ROS, either by inhibiting mitochondrial function with uncouplers or by decreasing ROS release with scavengers or antioxidants, significantly reduces receptor-mediated signaling. In support of the notion that ROS modulates receptor signaling, siRNA knockdown of GPx-1 in cultured cells significantly potentiates epidermal growth factor (EGF)-stimulated Akt phosphorylation, whereas overexpression of GPx-1 or catalase suppresses these pathways (169). Furthermore, in mouse in vivo models, lack of GPx-1 was found to augment insulin-mediated Akt activation in skeletal muscle (264), whereas overexpression of GPx-1 attenuated these responses in transgenic mice (284). Mitochondrial dysfunction is believed to play a role in type 2 diabetes, which is characterized by insulin resistance and dysregulation of glucose and lipid metabolism. Thus, a lack of response to insulin may ensue by a loss of beneficial ROS production in response to receptor-mediated events. One thought is that mitochondrial insufficiency may result in a blunted production of essential ROS necessary for signal transduction in diabetics (210, 325, 434). Even though such a concept is supported by some experimental data, this view is controversial. Many studies implicate, instead, mitochondrial dysregulation of metabolism in diabetes that may coincide with mitochondrial generation of excess ROS, which exceeds a threshold compatible with normal responses to insulin [insulin signaling, metabolism and mitochondria are reviewed in Ref. (83)].
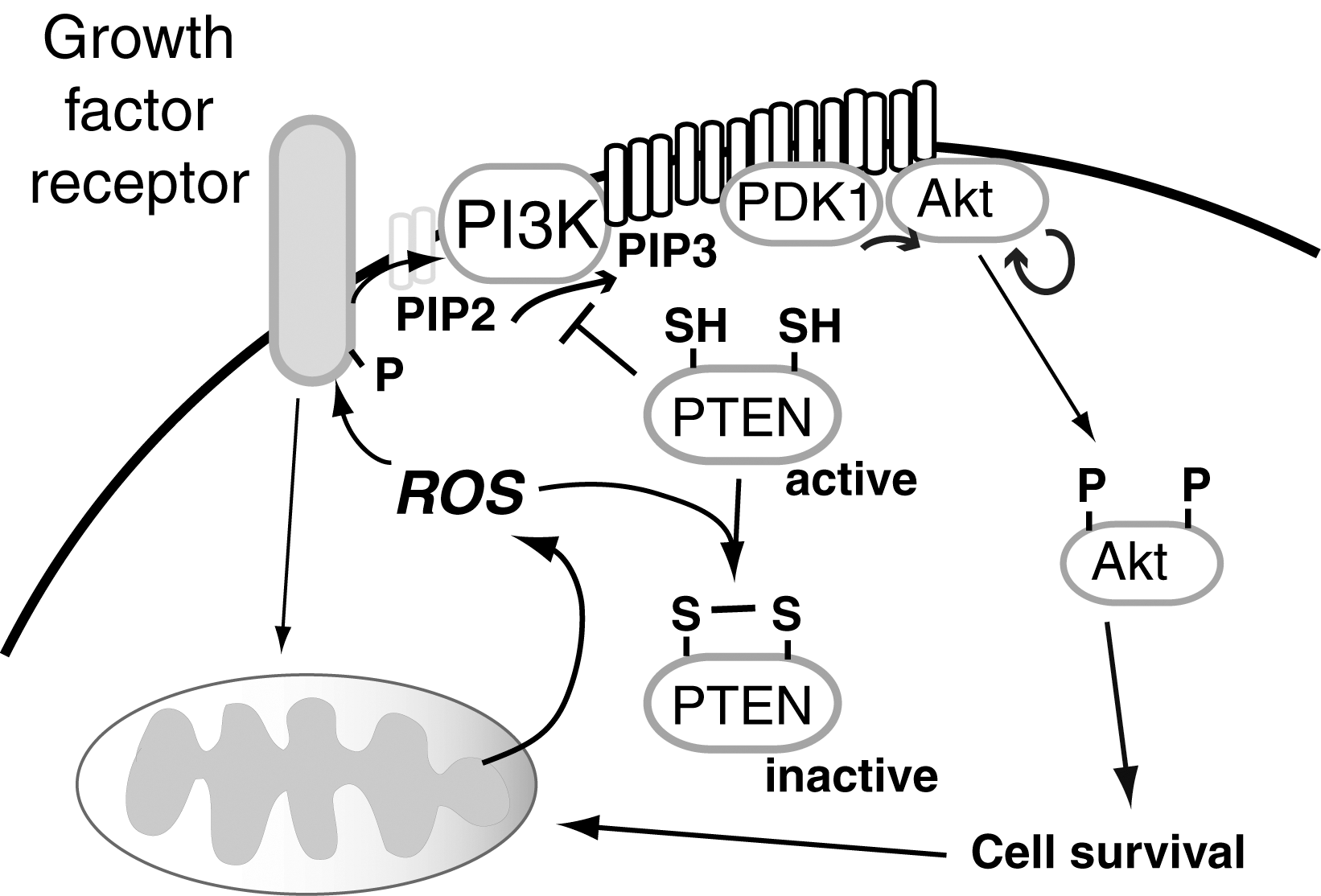
5. Mitochondrial respiration, inner mitochondrial membrane potential, and ROS
The outer membrane of the mitochondria is freely permeable to molecules <5000 kDa, in contrast to the inner mitochondrial membrane, which is highly impermeable. As discussed in section II.B, during respiration, protons are pumped into the intermembrane space to create a gradient of H+, which is the major contributor to the mitochondrial inner membrane potential (ΔΨ); concurrently,
6. UCPs and mitochondrial function
UCP1 plays a role in adaptive thermogenesis in mammalian brown adipose tissue to produce heat (314), whereas the actions of UCP2 and 3 are upregulated in response to oxidant stress caused by nutrient deprivation, glucose sensing, fatty acid metabolism, or other metabolic changes in order to lessen mitochondrial ROS production (49). These anion carriers lessen mitochondrial ROS formation by lowering the mitochondrial membrane potential by increasing the leak of protons from the intermembrane space back into the matrix, a process that also occurs to some extent through the ANT (22). The end result of this mild uncoupling of oxidative phosphorylation from ATP synthesis is a decrease in electron leak in the ETC, which attenuates ROS production (301).
It is unclear precisely how UCPs modulate proton leak; however, experimental evidence implicates fatty acids in the transport of protons by these inner mitochondrial membrane proteins (23). These processes have been most widely studied for the UCP1. One theory suggests an indirect role of UCPs in mediating proton transport (147). In this model, fatty acids are the carriers of protons from the intermembrane space to the matrix: protonated fatty acids diffuse across the inner mitochondrial membrane. After the release of protons from the fatty acids to the matrix, UCP1 transports the fatty acid anion to the intermembrane space (147). Another theory suggests that fatty acids directly associate with UCP1, and then facilitate proton transport across the membrane (217). Overall, studies suggest that fatty acid concentrations may be important in activating proton transport and may also regulate the expression and function of UCP2 and UCP3, as well as UCP1 (68).
Recent studies suggest that UCP2 and UCP3 may be modulated by S-glutathiolation, which maintains the carrier in an inactive state. In response to hydrogen peroxide, GR promotes the removal of –SG to activate UCP function (271). Further analysis is needed to understand fully the significance of this regulation, as these UCPs are also upregulated by transcriptional and translational mechanisms and have a short half-life in many tissues (22). Several studies have found an importance of these UCPs in nutrient regulation, fatty acid metabolism, and glucose-mediated insulin release. Modulation of UCPs has been considered as a strategy to prevent damage during ischemia/reperfusion injury in various models of cardiac and neuronal injury, as upregulation of proton leak due to UCP- or ANT-mediated mechanisms has been found to contribute to ischemic preconditioning in the heart (285, 307). In addition, overexpression of UCP2 or UCP1 has been found to be protective against hypoxia/reoxygenation death in isolated cells or in whole heart models of ischemia (41, 417); however, other studies implicate the metabolic changes (lower ATP and acidosis) caused by UCP2 overexpression with reduced survival in cardiomyocytes (43). Consistent with these latter findings, knockout of UCP2 was found to be protective in a model of cerebral ischemic injury (110). Some of this protection, however, may not be due to the lack of UCP2; rather, protection from oxidative injury may result from the compensatory upregulation of SOD2 and preservation of GSH in UCP2 knockout mice after ischemia compared to the wild-type mice.
Other studies, however, suggest that UCP2 helps to preserve dopamine neurons in a mouse model of Parkinson disease due, in part, to its ability to reduce ROS production: in this model deficient mice had more ROS and increased cell loss compared to wild-type controls, whereas UCP2 overexpressing mice were more protected with less ROS and less cell death (12). Possibly, differences in levels of expression and disease models account for some of these apparently contradictory findings regarding the beneficial role of UCP2. Overall, it seems that chronic changes in the expression of UCPs (or use of pharmacological uncouplers) may not provide a beneficial effect in all cases that involve excess production of oxidants, possibly due to long-term effects on cellular bioenergetics and substrate utilization that need further exploration. UCP2 may modulate insulin sensitivity, in part, by regulating oxidants in pancreatic β-cells, although there is contradictory evidence as to the beneficial role of UCP2 in mediating these processes. Early reports suggested the utility of decreasing UCP2 expression to enhance glucose-mediated insulin release from pancreatic β-cells (473); however, subsequent findings suggest that excess oxidants generated by lack of UCP2 may promote pancreatic islet dysfunction, at least in some congenic strains of mice (328). These later studies propose that compensatory upregulation of antioxidant enzymes in the context of UCP2 deficiency decreases oxidative stress but also attenuates glucose-mediated insulin release. As discussed in other sections of this review, excess expression of antioxidant enzymes, such as GPx-1, can substantially reduce growth factor-mediated signaling due to the loss of essential ROS (169) and these alterations can contribute to insulin insensitivity (284). Studies in human populations support a role for UCP2 in regulating metabolism and diabetes as variants of the UCP2 gene have been associated with obesity, altered metabolism, and diabetes (68). Similarly, variants of UCP3, which is expressed in skeletal muscle, are associated with abdominal obesity, a known risk factor for type 2 diabetes (367), and expression of UCP3 is suppressed in diabetic muscle (68).
IV. Redox Regulation of Mitochondrial Turnover
As discussed above, we now understand mitochondria as a network of structures that are constantly undergoing fission and fusion in order to adapt to cellular metabolic demands. Each mitochondrion may contain a different number of mitochondrial genomes, and the content of cellular mitochondria differs according to cell type. Cellular mitochondrial content is also in flux due to responses to growth factors and nutrients that may promote the biogenesis of new mitochondria or the selective removal of damaged mitochondria during mitophagy, a process that may serve to avoid apoptosis. In this section, we highlight the redox-dependent mechanisms that contribute to these processes.
A. Mitochondrial biogenesis
1. Overview of biogenesis
Mitochondrial biogenesis is a complex process that involves the coordinated expression of genes from both nuclear and mitochondrial genomes, as well as replication of mtDNA (176). The compact mtDNA genome encodes 13 essential subunits of the oxidative phosphorylation complexes; the remainder of the oxidative phosphorylation components and other mitochondrial proteins are encoded in nuclear DNA. Coordinated upregulation of mitochondrial and nuclear genes is accomplished, in part, via PGC-1α and to some extent PGC-1β (337, 458). These coactivators activate the transcription factors NRF1 and NRF2 to increase the transcription of oxidative phosphorylation genes, antioxidant genes, and other mitochondrial-specific genes, including those involved in mtDNA transcription and replication, such as transcription factor A (TFAM) (458). These transcription factors regulate ETC function by binding to distinct DNA target sequences in overlapping sets of genes (154, 373). NRF1 may also coordinately upregulate genes that regulate cell cycle and proliferation (60), and NRF2, which has also been called GA-binding protein (GABP), uniquely targets the cytochrome oxidase subunit 4 promoter (154, 373). The coordinated upregulation of antioxidant genes along with ETC proteins also provides protection against cellular ROS (398, 427). Other nuclear factors, such as the estrogen-related receptors (ERR), ERRα, ERRβ, and ERRγ, may similarly induce the biogenic process in conjunction with PGC-1-coactivators (380). Changes in cellular AMPK, growth factor receptor-mediated signaling, NO· and cGC/cGMP signaling, or oxidant stress may all influence mitochondrial biogenesis, in part through of PGC-1-induced pathways. For example, in skeletal muscle cells, the increase in intracellular Ca2+ caused by muscle contraction leads to activation of calcium/calmodulin and CREB activation to promote PGC-1α transcription, while the consumption of ATP during contraction causes accumulation of AMP, which activates AMPK. AMPK upregulates PGC-1α by transcriptional pathways and also by directly phosphorylating PGC-1α to release it from a repressor protein. Indirectly, release from the repressor increases PGC-1α accessibility to SIRT deacetylases, to enhance PGC-1α activity. Also, indirectly, AMPK can promote the activation of other biogenic pathways, such as the activation of MEF-2 [see Ref. (294), for a review of biogenic signaling mechanisms]. In the next sections we focus on the role of redox regulation in biogenesis.
2. ROS and biogenesis
In several model systems, acute stimulation with exogenous oxidants was shown to activate mitochondrial biogenesis, as measured by increases in mitochondrial mass, mtDNA, and the expression of biogenic regulators, such as PGC-1α, NRF1, and TFAM (237, 238). In other models, acute injury with oxidants was found to lead to early mitochondrial dysfunction followed by recovery of mitochondrial function caused by subsequent upregulation of PGC-1α. In renal proximal tubule cells, this PGC-1α-dependent biogenic response was found to involve activation of p38-MAPK to promote EGF receptor (EGFR)-induced signaling (342). Although this study reported no effect of Akt-mediated pathways downstream of EGFR in PGC-1α activation, other studies have implicated PI3K/Akt-mediated pathways in the regulation of biogenic responses that are promoted by oxidants, such as tert-butylperoxide (330). One difference between these studies is that the latter used a cell model in which NRF1 was relatively overexpressed, possibly allowing for identification of Akt-mediated NRF1 phosphorylation and activation pathways that may not be essential for oxidant-induced biogenesis in all cell types. Redox inactivation of PTEN phosphatase that antagonizes PI3K signaling was proposed to play an essential role in this pathway of Akt-mediated biogenesis (330). It is well known that redox-inactivation of PTEN plays an important role in allowing phosphatidylinositol-3,4,5-triphosphate (PIP3) accumulation in the cell membrane to recruit PDK1 and Akt as part of the PDK1-mediated phosphorylation of Akt (269). In some cell types, Akt3, rather than Akt1, may play a role in growth factor-mediated stimulation of biogenic pathways (455), and other studies identify a role for Akt pathways in the heme oxygenase–mediated modulation of biogenesis, which involves oxidant activation of NRF1 transcription via antioxidant response elements in the NRF1 promoter (329).
One functional consequence of mitochondrial biogenesis following overexpression of PGC-1α is the reduction of mitochondrial ROS and prevention of apoptosis in response to stressors such as high glucose (427). It is well known that mtDNA mutations, oxidative modifications of mitochondrial proteins, and deficiencies in functional mitochondrial complex subunits contribute to excess production of ROS in mitochondria. Thus, PGC-1α-mediated mitochondrial biogenesis may promote the replacement of dysfunctional mitochondria with increased electron leak, with those that perform more efficiently with less electron leak to reduce effectively mitochondrial ROS production. However, PGC-1 proteins may additionally decrease ROS due to the coordinate upregulation of antioxidant proteins downstream of PGC-1α. Thus, PGC-1α upregulation is associated with increased expression of antioxidants, such as UCP2, Prx3, Prx5, Trx2, TrxR2, catalase, SOD2, and GPx-1, in various experimental models (398, 427). There has been keen interest in using PGC-1α to improve in vivo metabolic profiles in type 2 diabetes, for instance, as overexpression of PGC-1α improves insulin sensitivity in cultured skeletal myocytes (224, 293). Contrary to predictions, however, transgenic PGC-1α overexpression in skeletal muscle did not improve the in vivo metabolic profile in mice (87). Rather, although the skeletal muscle of these mice had increased mitochondria, they were also more sensitive to high-fat-induced insulin resistance. Thus, the effects of PGC-1α on other genes, such as those involved in fatty acid transport/utilization, may have increased fatty acid oxidation and intramuscular lipids to decrease insulin signaling. Alternatively, as mentioned above, PGC-1α-mediated upregulation of antioxidant genes may contribute to a loss of insulin-mediated signaling responses by decreasing necessary ROS flux under certain conditions. In other studies, electrotransfection of PGC-1α constructs into rat skeletal muscle appeared to improve lipid metabolism and insulin signaling (37). It is unclear whether the lack of deleterious effects on metabolism in this latter study was due to the lower levels of overexpression achieved in the muscle or the relatively short time frame of overexpression compared to the transgene model. Furthermore, in the rat in vivo transfection model, fatty acid transporters were upregulated to some extent. Interestingly, PGC-1α overexpression reduced triacylglycerol, diacylglycerol, and ceramide in muscle of treated obese Zucker rats, with some increases in levels of triacylglycerol in lean rats. Other treatments that more modestly increase PGC-1α-expression also have biogenic effects that may be beneficial. Thus, drugs such as thiazolidinediones (TZDs), that are used to improve insulin sensitivity in diabetics by PPARγ-mediated mechanisms, were found to improve mitochondrial function in a hyperglycemic model through a PGC-1-dependent mechanism that increases mitochondrial content, reduces ROS generation, and upregulates antioxidant enzymes (142). Some TZDs, however, have been associated with an increased risk of myocardial infarction (315), possibly caused by other adverse metabolic actions of these drugs. Drugs that stimulate AMPK may also activate these biogenic pathways to lower mitochondrial dysfunction and ROS production (228), suggesting another mechanism by which these agents may improve cellular metabolism and reduce oxidative damage.
3. Telomere dysfunction and mitochondrial biogenesis
Aging and diseases of aging, such as diabetes and cardiomyopathy, are associated with decreased mitochondrial function. Telomeres, the nucleoprotein complexes at the ends of chromosomes that serve to protect the ends of the chromosome, also undergo an age-related decrease in length that is linked to cellular senescence (322). The telomerase holoenzyme, which maintains telomeres, includes an RNA subunit and a telomerase reverse transcriptase (TERT). Recent findings correlate deficiency of TERT (and subsequent loss of telomeres) with deficiencies in mitochondrial function attributed to decreases in PGC-1α and PGC-1β expression and suppressed mitochondrial biogenesis (364). In this study, concurrent with decreased cellular mitochondrial content, cellular respiration was suppressed and ROS production increased. Furthermore, mice deficient in telomeres developed cardiomyopathy and had altered metabolism (364). One role of telomeres is to suppress p53 activation (84). p53 activation was found to suppress expression of the PGC-1α and PGC-1β (364), thus linking telomeres to mitochondrial biogenesis. Other studies similarly report that overexpression of TERT can decrease ROS production and improve mitochondrial function (185), possibly via mitochondrial biogenesis. Additionally, increased expression of TERT may also improve antioxidant status by stimulating PGC-1α and PGC-1β, which are known to promote transcriptional upregulation of many antioxidant genes. TERT has also been found within mitochondria, although its role here is unclear. There have been reports that its mitochondrial localization can promote apoptosis, whereas other reports suggest that mitochondrial TERT may improve mitochondrial function or attenuate apoptotic activation (322). Although these noncanonical effects of TERT are not well understood, its translocation out of the nucleus following ROS-mediated phosphorylation by src-kinases has been associated with apoptosis.
B. ROS and mitophagy versus apoptosis
Autophagy is the intracellular removal of cellular macromolecules and organelles through a catabolic process involving lysosomal degradation (469). An initial step in this process is the formation of a double-membrane isolation membrane around the cellular components, called an autophagosome, that later fuses with the lysomal compartment. This process can occur nonselectively during nutrient deprivation as a means of providing molecular sources of energy and building blocks for cell survival, or it can also occur in a more directed fashion to remove damaged or excess organelles. Some of the autophagy component proteins involved in starvation-induced autophagy, such as the Atg4 family of enzymes, are regulated at redox-active Cys (375). It has been suggested that increased ROS production in starvation provides a necessary control mechanism in coordinating the activity of Atg4 during autophagy.
The selective engulfment of mitochondria and removal of mitochondria is referred to as mitophagy. Mitophagy can remove mitochondria damaged by oxidants and those producing excess oxidants. Recognition and removal of these mitochondria by mitophagy can avoid apoptotic changes by preventing the release of mitochondrial apoptogens, such as cytochrome c or AIF (242). Some of the specific components involved in mitophagy include the outer mitochondrial membrane protein PTEN-induced kinase 1 (PINK1) and parkin, an E3 ubiquitin ligase. Mutations and/or dysregulation of these genes have been implicated in neurodegeneration found in Parkinson disease (148, 426). Also crucial to these processes is mitochondrial fission. In damaged mitochondria, accumulated PINK1 fosters the translocation of parkin to mitochondria, the subsequent ubiquitination of mitochondrial proteins, and engulfment of mitochondria in autophagosomes (311, 437, 469). Interestingly, complete loss of PINK1 can promote mitochondrial oxidant production and subsequent mitophagy (107), possibly due to mitochondrial membrane damage and loss of mitochondrial potential, which may promote the association of parkin with mitochondria (311). Importantly, mitophagy serves a protective function; loss of essential mitophagy pathways is thought to promote mitochondrial dysfunction and cell death.
C. Heme oxygenase, carbon monoxide, and mitochondrial function
Heme oxygenase-1 (HO-1) is rapidly induced by inflammatory cytokines. It catalyzes the degradation of heme to carbon monoxide (CO), biliverdin, and ferrous iron (467), and contributes to cellular protection against damage caused by ischemia-reperfusion injury (467), hypoxia (92), inflammatory cytokines (240), and other cellular insults. The mechanisms by which HO-1 promotes protection are not entirely clear, but some of these effects appear to be mediated via the production of CO and its heme-binding properties (272). Activation of the HO system, however, has also been associated with detrimental effects in diseases, such as Alzheimer disease or in aging, due, in part, to increases in mitochondrial iron accumulation and disruption in mitochondrial complex IV function, that correlate with increased HO-1 expression (376). One issue may be the duration and extent of HO-1 expression and function. Thus, both CO and heme degradation productions have been shown to mediate protective and harmful effects, depending on the context of their production (115, 272). It is well known that excess CO is damaging due, in part, to its high affinity for heme moieties involved in oxygen transport and mitochondrial respiration. CO, however, has also been shown to promote mitochondrial biogenesis and may in some instances be protective against mitochondrial dysfunction and cell death. In mice, brief (1 h) periods of exposure to CO were found to attenuate cardiac mitochondrial dysfunction, and lessen cardiac mitochondrial loss, oxidative stress, and apoptosis caused by doxorubicin (404). Doxorubicin, a chemotherapeutic agent, is well known to cause cardiomyopathy that contributes to comorbidity in cancer patients (409). Interestingly, exposure to CO in the doxorubicin studies correlated with increased HO-1 production in heart, suggesting that other heme degradation products could also contribute to the cardioprotective effects of inhaled CO. In cell culture studies, overexpression of HO-1 similarly attenuated doxorubicin-induced apoptosis and loss of mitochondrial content (404). These findings are consistent with other studies suggesting a role for HO-1 in upregulating mitochondrial biogenesis, by a pathway that implicates mitochondrial ROS as an essential contributor to biogenesis (329). Earlier studies by these and other investigators have shown that CO binds to cytochrome oxidase a3 heme to increase mitochondrial leak of hydrogen peroxide (470, 475). Apparently, upregulation of these mitochondrial sources of ROS production are essential to promote both PGC-1 and NRF1 gene expression leading to increased mitochondrial biogenesis. This mechanism relies, in part, on ROS-mediated activation of Akt and its subsequent activation of NRF1 (329). This is another example of the signal-transduction role of moderate levels of excess ROS that serve to maintain (or reset, in this case) cellular homeostasis. In vivo, overexpression of cardiomyocyte-targeted HO-1 improved survival after myocardial infarction by reducing myocyte-specific apoptosis and postinfarction left ventricular remodeling, in part, by preserving mitochondrial function (440). In particular, analysis of isolated cardiomyocytes and mitochondria from HO-1-overexpressing and control mice indicated that excess HO-1 decreased mitochondrial respiration and attenuated Ca2+- or phenylarsine oxide-induced mitochondrial swelling and mitochondrial membrane permeability transition. Furthermore, in this study, use of the CO donor tricarbonylchloro(glycinato)ruthenium (II) similarly improved mitochondrial function and protected the heart from left ventricular remodeling and apoptosis following infarct in an in vivo model of heart failure (440). Unlike the previous studies, this later study did not examine the effects of HO-1 overexpression or CO donors on mitochondrial biogenesis; however, these findings are consistent with a mechanism involving CO-induced protection of mitochondrial function. Another recent study (382) suggested that ROS induced by CO exposure regulate the function of an L-type Ca2+ channel by a redox mechanism to enhance cardioprotection. Thus, in isolated rat myocytes, exposure to CO or a CO donor causes reversible inhibition of L-type Ca2+ channels that could be replicated in HEK293 cells transfected with the α1C subunit of the human cardiac L-type Ca2+ channels containing a C-terminal splice variant. Apparently, CO-induced ROS derived from mitochondria inhibit channel activity by a mechanism involving three Cys residues in the splice insertion region of this channel: mutation of any one of these Cys was sufficient to block CO's inhibitory effect. These studies do not show how CO-induced ROS affect the L-type Ca2+ channel, and only suggest Cys oxidation as a possible mechanism. In cardiomyocytes, however, Ca2+ overload can cause cell death, in part, via mechanisms involving uptake of excess Ca2+ into mitochondria, subsequent ROS production, and activation of the mitochondrial permeability pore transition (as discussed further in the next sections). Thus, an effect of CO on ROS and L-type Ca2+ channels could potentially modulate mitochondrial dysfunction, swelling, formation of the MPT pore, and apoptosis. Further analysis, however, is necessary to determine whether CO-mediated L-type Ca2+ channel regulation contributes to the in vivo effect of CO on cardiomyocytes.
V. Redox Regulation of Apoptosis
As discussed above, redox-sensitive thiols play a role in many aspects of apoptotic signaling. In this section we discuss redox mechanisms that effect apoptosis, analyzing some known pathways related to protein and phospholipid oxidation that drive apoptotic changes (159, 225).
A. ASK1 and Trx-mediated regulation of JNK
JNK1 and JNK2 are members of one of three major subfamilies of MAPK that mediate cell signaling in response to receptor and other stimuli. Each of these MAPK are activated by upstream kinases. The extracellular signal-related kinases, or ERK, are part of growth factor-mediated signal transduction scheme that promotes cell proliferation, differentiation, and survival pathways. In contrast, p38 MAPK and JNKs, which have collectively been referred to as stress-activated MAPK (SAPK), mediate the cellular responses to cytokines, oxidant stress, and other agents (230). Although the SAPKs can also mediate proliferative responses, prolonged activation of these SAPK can lead to apoptosis (80, 162). Oxidants downstream of cytokines and other stress activators are involved in JNK activation, as antioxidants can block JNK activation. This redox effect is due, in part, to regulation of the JNK activating kinase, apoptosis signal-regulated kinase 1 (ASK1) (Fig. 9). ASK1 is inhibited by Trx due to their protein–protein interactions that maintain ASK1 in an inactive state (365). Trx redox state dictates, in part, ASK1 activity: Trx only binds to ASK1 when it is reduced, oxidized Trx dissociates from ASK1, leading to ASK1 activation (141). Once dissociated from Trx, ASK1 may also associate with other proteins such as TNF-receptor-associated adaptor proteins (TRAFs) in a signalosome that may be essential for ASK1 activation (282, 283). Downstream of ASK1, JNK can modulate nuclear factor targets to upregulate the transcription of pro-apoptotic factors and it can also migrate to the mitochondria. The absence of JNK was previously shown to limit uv stress-induced mitochondrial release of cytochrome c (422), and other studies have shown that translocation of JNK to the mitochondria was essential for cytochrome c release and apoptosis downstream of other stress activators (67, 189). Furthermore, targeting of JNK to the mitochondria resulted in a significant increase in oxidants generated by complex I (67). One possible mediator of JNK's effects on mitochondria is p66shc, which apparently is a target of JNK (235, 250). Phosphorylation of p66shc may enhance its apoptotic effect in mitochondria (297). Prolonged JNK activation can also be caused by oxidative inactivation of DUSPs that dephosphorylate JNK (206). Thus, in some cell systems, increased expression of antioxidant proteins or other antioxidant treatments can lessen DUSP inactivation to reduce JNK activation and inhibit apoptosis.
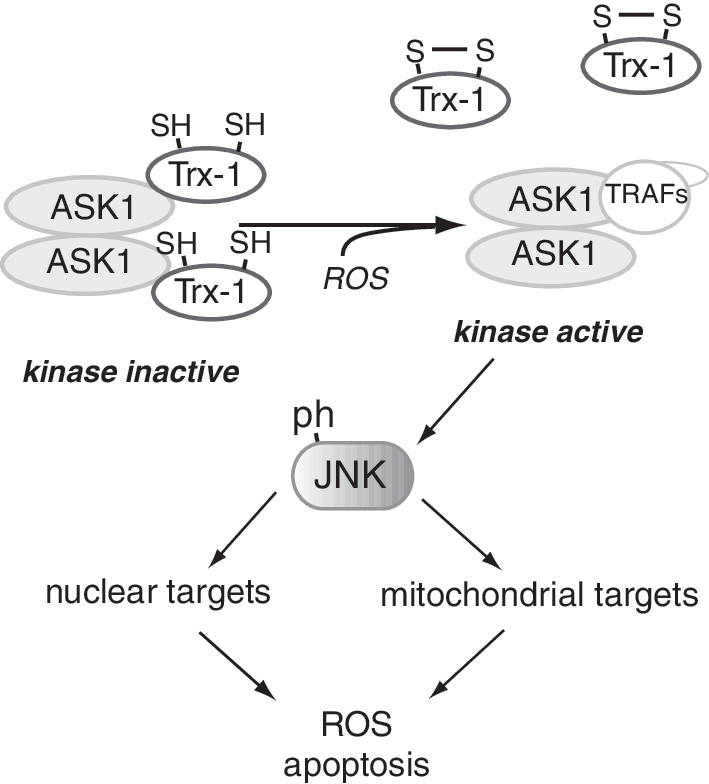
B. MPT pore and cell death
1. Mitochondrial permeability transition
The MPT involves loss of inner mitochondrial membrane integrity due to the opening of a nonspecific pore (MPT pore) that allows for flux of small molecules, <1500 Da, and protons, leading to mitochondrial swelling, loss of mitochondrial membrane potential, outer membrane rupture, and apoptotic or necrotic death. Formation of the pore can occur in response to many stimuli, including Ca2+ overload and oxidant stress, but MPT does not play a role in all forms of apoptosis (46, 125, 309, 438). In fact, some pathways lead to apoptosis and mitochondrial release of apoptogens in the absence of MPT induction.
The composition of the pore responsible for the enhanced inner membrane permeability is not clear. Implicated in MPT pore formation are the inner membrane ANT; the outer membrane VDAC; cyclophilin D, a matrix protein that is a peptidyl prolyl-cis, trans isomerase; and, possibly, the mitochondrial phosphate carrier (247). Although much experimental data exist for a role of ANT in the MPT (e.g., the ANT inhibitor bongkrekic acid, 20-carboxymethyl-6-methoxy-2,5,17-trimethyldocosa-2,4,8, 10,14,18,20-hepataenedioic acid, blocks MPT), liver mitochondria that lack the major forms of ANT (ANT1 and ANT2) can still exhibit MPT depolarization (222), suggesting that ANT may be a modulator of MPT, rather than an essential component of the MPT pore [reviewed in Ref. (247)]. Similarly, absence of all known subtypes of VDAC does not impair MPT pore formation (26). Cyclosporin A, an inhibitor of cyclophilin D, is known to inhibit MPT responses to some stimuli. Further evidence for the involvement of cyclophilin D in MPT formation comes from studies with cyclophilin D knockout mice (25, 309). Consistent with a role of cyclophilin in some, but not all, pathways involving mitochondrial-dependent cell death, lack of cyclophilin D does not lessen cytochrome c release or apoptotic death in response to Bcl-2-mediated mechanisms, such as those activating Bax or tBid. Accordingly, some researchers consider that cyclophilin D-dependent (i.e., cyclosporin A-inhibitable) forms of MPT lead to necrosis rather than apoptosis. Nonetheless, cells from cyclophilin D-deficient mice show resistance to MPT and death from Ca2+ overload and oxidant stress. In addition, in the absence of cyclophilin D, mice are more protected from cardiac damage after ischemia/reperfusion (25, 309); however, lack of cyclophilin D in cardiac cells may be detrimental under certain conditions, as this deficiency compromises Ca2+ handling in mitochondria, decreasing the ability of mitochondria to efflux Ca2+ after alterations in myocardial workload due to lack of beneficial (perhaps transient) MPT pore formation (123). Consequently, in cyclophilin D-depleted cells, faulty Ca2+ handling altered mitochondrial substrate utilization, decreasing fatty acid utilization in response to stressors (such as pressure overload) and promoting a reliance on glucose instead. This metabolic shift was found to contribute to cardiac hypertrophy and increased sensitivity to heart failure after pressure overload (123). A reliance on glucose rather than fatty acid metabolism is characteristic of heart failure in humans, as well (425). Interestingly, at very high levels of Ca2+, cyclophilin D-deficient cells or cells treated with cyclosporin A still have an MPT response (25, 31, 167, 309). These findings have led to the suggestion that other proteins, such as the phosphate carrier mentioned above, may be essential components of the MPT pore (247). Interestingly, in addition to cyclosporine A or bongkrekic, the MPT response can be blocked by polyamines, such as spermine or spermidine, possibly due to their antioxidant effects (5).
Both ANT and cyclophilin D may be targets for redox regulation. One study identified possible redox-active Cys in cyclophilin D. In this study, reducing and oxidizing conditions were used to study the role of each of the 4-Cys in cyclophilin D on its activity, intrinsic Trp fluorescence (a comparative measure of conformation), and ability to form intramolecular disulfides (258). Based on a series of experiments with single Ser for Cys substitutions in the protein, it was determined that two of these Cys, Cys203 and Cys157, may be involved in redox-mediated disulfide bond formation (258). These experiments, however, were mostly performed on isolated proteins and there is not a clear role for these reactive thiols within the mitochondria. Further study is needed to determine the functional significance of these thiols in cyclophilin D-mediated MPT pore opening. In contrast, the redox-sensitive thiols of ANT have been well studied as targets for modification by nitric oxide, peroxynitrite, thiol crosslinkers, and other thiol-reactive reagents (101, 435). ANT has three thiols that are implicated in its function that are located in three distinct hydrophilic loops (102, 167, 171, 286). ANT has two different conformational states depending on the location of the substrate binding site in the matrix (m-state) or cytosolic (c-state) side. Redox-sensitive disulfides form between Cys160 and Cys257, to enhance the “c” conformation of ANT, which is associated with diminished binding to ADP. The third thiol, Cys57, can be induced to form an intermolecular disulfide between ANT subunits only when it is in the “m” state but not the “c” state, confirming the proximity of these residues in the functional homodimer; however, these dimers also block transport function (171).
2. Mitochondrial Ca2+, redox, and mitochondrial dysfunction
As discussed above, overload of mitochondrial matrix Ca2+ can lead to MPT and cell death. Uniporters regulate mitochondrial matrix uptake of Ca2+ when its intracellular concentration is in the micromolar range (161). At nanomolar concentrations of intracellular Ca2+, its uptake into mitochondria has been proposed to be regulated by a Ca2+/H+ exchanger that would favor import when mitochondria are hyperpolarized: the function of this exchanger is still highly speculative, but it may have bidirectional properties (370). The uptake of Ca2+ into mitochondria is blocked by ruthenium red, which actually blocks VDAC-mediated transport of Ca2+ into the intermembrane space (362). Ca2+ release from mitochondria is known to be driven by cell-specific Na+-dependent and Na+-independent pathways. In cardiomyocytes, for example, Na+-dependent pathways predominate. Mitochondrial uptake of Ca2+ is regulated, in part, by the release of intracellular stores from compartments such as the endoplasmic reticulum or the sarcoplasmic reticulum in muscle cells (261, 352). Mitochondrial uptake of Ca2+ can also buffer intracellular Ca2+ that enters cells from extracellular pools through Ca2+ channels. In contracting cardiomyocytes, normal release of Ca2+ from intracellular stores allows for uptake by mitochondria and the stimulation of Ca2+-sensitive enzymes in the TCA cycle (PDH, 2-oxoglutarate dehydrogenase, and NAD+-linked ICDHs) to coordinate energy production and demand, increasing NADH production and mitochondrial respiration (261). In other cells, mitochondrial buffering of Ca2+-channel uptake can maintain Ca2+ channel activity by decreasing local concentrations of Ca2+. Alternatively, Ca2+ uptake by mitochondria can serve to regulate intracellular Ca2+ transport (370). Excess Ca2+, however, can cause oxidative stress, in part, by its effects on mitochondrial phospholipid disposition and oxidation. In particular, Ca2+ affects cardiolipin, a membrane phospholipid that is found primarily in mitochondria. The role of oxidized phospholipids, including cardiolipin, in apoptosis is discussed further in the next section; however, it is important to mention briefly here the role of cardiolipin in Ca2+-mediated mitochondrial ROS production and MPT activation.
Both Ca2+ overload and cardiolipin oxidation enhance mitochondrial ROS production (321). The mechanisms by which Ca2+ promotes excess ROS generation are not well understood: it is thought that excess Ca2+ enhances ROS production, in part, due to its effects on TCA cycle enzymes, but possibly, the association of Ca2+ with cardiolipin contributes to ROS generation by disrupting the flow of electrons in complex III. One consequence of mitochondrial Ca2+ overload is the formation of the MPT pore. Antioxidant treatments have been found to attenuate Ca2+-induced MPT, whereas coexposure to oxidants augments MPT pore formation (64, 225, 268). As discussed above, transient MPT pore formation may be protective; however, prolonged MPT pore opening is associated with the release of apoptogens to promote apoptotic cell death.
3. STAT3 and mitochondrial function
STAT3, initially characterized as a nuclear factor that regulates gene expression in response to IL-6 and other cytokines, has been recently found to have a specific role in modulating mitochondrial function through mechanisms independent of its function as a nuclear factor, but dependent on its phosphorylation at a serine residue (412, 445). The molecular mechanism by which STAT3 modulates ETC activity is not well understood, but deficiency of STAT3 has been shown to decrease complex I and complex II activity and ATP production while increasing mitochondrial ROS at complex I (412). Interestingly, these changes do not involve alterations in the expression of the mitochondrial subunits. Instead, it has been proposed that a direct interaction between mitochondrial STAT3 and complex I mediates the effects of STAT3 on mitochondrial respiration (412). Excess mitochondrially targeted STAT3 increases electron transport and oxidative phosphorylation, while decreasing ROS to protect against MPT pore formation in response to Ca2+ overload. Thus, cardiac-specific deletion of STAT3 in mice result in cardiac inflammatory fibrosis, dilated cardiomyopathy, and heart failure, and lack of STAT3 eliminates the protective effects of ischemic preconditioning (173). Confirming a role of STAT3 in cardioprotection, studies have found that overexpression of STAT3 in cardiomyocytes protects mice against doxorubicin toxicity, which is known to involve mitochondrial dysfunction. To study specifically the mitochondrial function of STAT3, independent of its function as a nuclear factor, a recombinant form of STAT3 that targets to the mitochondria and lacks the DNA binding domain was engineered (MLS-STAT3E). Expression of this mutant form of STAT3 to cardiomyocytes was found to protect hearts from ischemic damage by decreasing mitochondrial generation of ROS and attenuating cytochrome c release in a stop flow model of ischemia (411). Similar to STAT3-deficient mitochondria, overexpression of MLS-STAT3E decreased complex I and complex II activities; however, unlike the STAT3 deficiency, the MLS-STAT3E did not increase ROS generation at complex I. The reason for diminished complex I and II activities caused by overexpression of the DNA-binding domain-deficient construct is not clear, nor are the means by which endogenous STAT3 modulates mitochondrial function. These intriguing findings suggest the need for further analysis of the role of proteins that modify mitochondrial electron transport.
C. Lipid oxidation and apoptotic signaling
1. Cardiolipin oxidation and apoptosis
Cardiolipin is a membrane phospholipid found primarily in mitochondrial membranes. It has a unique structure with four fatty acyl side chains arranged in a symmetrical fashion on 2 (joined) diacyl phosposphatidyl acids. In the heart, cardiolipin is noted to have a high content of linoleic acid that is essential for its ability to promote ETC complex III and IV activity (396). Cardiolipin also fosters the formation of supercomplexes within the inner mitochondrial membrane that have enhanced activity (327, 353). The presence of cardiolipin within the inner mitochondrial membrane may also modulate the activity of other resident proteins, such as ANT (96). Cardiolipin is thought to play an essential role in maintaining cytochrome c in the inner membrane where it constitutes nearly 20% of membrane lipids. Oxidation of cardiolipin disrupts its interaction with cytochrome c to foster the release of cytochrome c from mitochondria, which promotes apoptosis. Compared to the other major phospholipids of mitochondria, phosphatidylethanolamine and phosphatidylcholine, cardiolipin was found to be more sensitive to oxidative stress (451). Interestingly, cardiolipin may be oxidized, in part, through the peroxidase activity of cytochrome c in response to increased production of hydrogen peroxide after disruption in electron transport (34, 310). It has been proposed that cytochrome c association with cardiolipin is key to its transition from an electron acceptor/donor in ETC to a peroxidase (30, 204). Cytochrome c only functions as a peroxidase in association with cardiolipin, resulting in the oxidation of cardiolipin, which subsequently potentiates cytochrome c release from the mitochondria. Once in the cytosol, cytochrome c promotes the formation of the apoptosome, which involves the oligomerization of apoptotic releasing factor-1 (Apa-f1). Apoptosome recruitment of caspase 9, an initiator caspase, begins the cascade that activates caspase 3, leading to apoptotic cell death (251).
The preservation of cardiolipin helps to maintain mitochondrial function and prevent apoptosis. Thus, there is an interest in targeting cardiolipin/cytochrome c interactions to modulate cell survival or apoptosis. Melatonin, a lipophylic hormone that has been shown to have properties as a free radical scavenger (9), was found to reduce lipid peroxidation in cardiac mitochondria after ischemia/reperfusion injury while preserving complex I and III activity (326). In addition, in vitro studies on isolated mitochondria indicated that melatonin had a similar protective effect on complex I and III function and preserved mitochondrial cardiolipin content, suggesting that melatonin protected mitochondria directly, perhaps via an antioxidant effect rather than an indirect effect activating other protective cellular mechanisms (326). Similarly, in studies of intact hearts, inhibition of complex I (with rotenone) during ischemia preserved mitochondrial cardiolipin and cytochrome c content (246), suggesting a role for ETC leak in cardiolipin oxidation and a possible target to prevent cardiolipin oxidation. Other therapeutic means to regulate ETC leak are being developed, including S-nitrosating compounds, that were discussed previously (306, 336), and cationic plastoquinone derivatives (392). The function of GPx-4 to limit specifically mitochondrial phospholipids oxidation has also been explored, as discussed next.
2. Membrane phospholipids and AIF-mediated cell death
Of the GPxs, GPx-4 is somewhat unique because it will reduce membrane phospholipid hydroperoxides, as well as soluble hydroperoxides. Recent findings, however, suggest that GPx-4 primarily targets oxidized membrane phospholipids (383). A role for GPx-4 in regulating cardiolipin oxidation in particular has not been examined, but one of the isoforms of GPx-4 targets to the mitochondria, where possibly it may also serve to reduce mitochondrial phospholipids. In fact, overexpression of mitochondrial targeted GPx-4, but not the nonmitochondrial form, suppressed apoptosis, and reduced mitochondrial hydroperoxide content (316, 340) in response to a variety of exogenous agents, including 2-deoxyglucose, etoposide, t-butylhydroperoxide, and diquat. Complete absence of GPx-4 is known to be lethal (465), and studies show that deficiency of GPx-4 promotes cellular apoptosis via the accumulation of lipid peroxidation in the absence of exogenous oxidants or stimuli (383). Interestingly, a deficiency of GPx-4 appears to potentiate 12/15-lipoxygenase-mediated lipid peroxidation to increase cell death via the release of AIF, an apoptogen from mitochondria (383). Consistent with previous reports (407), AIF migration from the mitochondria to nucleus was found to mediate apoptosis in a manner that could not be blocked by overexpression of the antiapoptotic Bcl-2 or by caspase inhibition. The absence of GPx-4 was suggested to play a role in the activation of 12/15-lipoxygenase and to promote lipid peroxidation. Lipid peroxidation and cell death could also be potentiated in GPx-4-replete cells by inhibiting GSH synthesis. This finding is consistent with the role of GPx-4 in modulating lipoxygenase activity (378). The role of lipid peroxides in promoting apoptosis was proven by the finding that the lipid-soluble α-tocopherol could prevent apoptosis, whereas N-acetyl Cys or other water-soluble antioxidants could not block cell death in GPx-4-deficient cells (383). In models of GPx-4 overexpression, excess GPx-4 also blocked cytochrome c release from mitochondria and caspase activation in models where oxidative stress was induced by exogenous agents (316, 340), although it did not prevent apoptosis from FasL (extrinsic) pathway activation. Taken together, these findings suggest a crucial role for antioxidant mechanisms in preserving mitochondrial membrane redox potential to prevent apoptosis.
VI. Mitochondrial Responses to Hypoxia
HIF family transcription factors are master regulators of hypoxic responses. These factors are heterodimers that consist of a common β chain that is constitutively expressed and not responsive to oxygen levels, and one of three α chains (HIF-1α, HIF-2α, and HIF-3α), which are primarily regulated at the level of protein stability, according to oxygen tension. Of these HIF-1α is most well studied for its role in modulating metabolic responses to hypoxia in most tissues. HIF-2α may upregulate some unique targets, such as erythropoietin, and, in some cells, may also contribute to the hypoxia-mediated increase in other genes, like vascular endothelial growth factor or plasminogen activator inhibitor-1 (343). Here, we will focus on HIF-1α, due to its effects on mitochondrial respiration and ROS (Fig. 10). Under normal oxygen tension, HIF-1α is targeted for proteasomal degradation through the concerted action of prolyl hydroxylase (PHD) enzymes that modify two conserved prolines in the HIF-1α protein (279, 397). PHD are O2-dependent enzymes that utilize ferrous iron and α-ketoglutarate (16, 320). PHDs are inactive at low O2 and can be inactivated by the accumulation of TCA metabolites fumarate and succinate, which may occur from mutations in the tumor suppressor genes succinate dehydrogenase subunit B or fumarate hydratase (187, 334, 384). [Succinate is a product of the PHD hydroxylation reaction; other TCA metabolites, such as fumarate as well as oxaloacetate, have been found to inhibit PHDs (221).] Prolyl hydroxylation allows for the interactions of HIF-1α with von Hippel-Lindau tumor suppressor (VHL), which fosters the ubiquitination and degradation of prolyl hydroxylated-HIF-1α (203). Thus, under hypoxia, prolyl hydroxylation is inhibited, and HIF-1α escapes degradation, accumulates in the nucleus, and initiates gene transcription. Alterations in normal PHD/VHL/HIF-mediated pathways is thought to contribute to the metabolic adaptation found in Warburg tumors, that is, those that manifest enhanced glycolysis in the presence of O2 (35, 444).
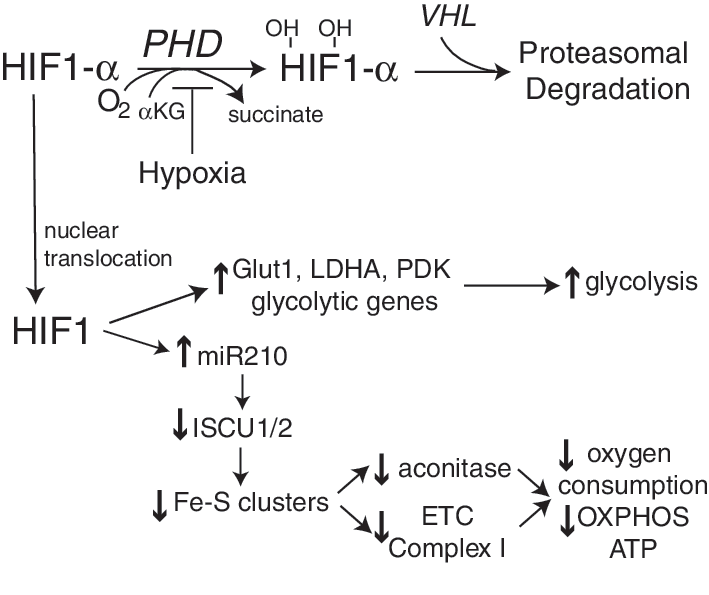
There is an ongoing debate as to the role of mitochondrial oxidants in regulating HIF-1 activation and the hypoxic response (72). Experimental evidence implicates a hypoxia-induced burst of mitochondrial ROS as necessary for HIF-1α and HIF-2α stabilization, as cells deficient in mitochondrial ROS, due to genetic or pharmacological treatments, have decreased HIF activation (53, 164, 274). One possible explanation for the potentiating effect of an ROS burst on HIF-α signaling is that an oxidant burst may promote oxidation of the Fe2+ required for the function of PHD enzymes, thereby reducing HIF-α degradation (202). Alternatively, these enzymes also are inhibited by succinate, and fluctuations in the TCA cycle could similarly diminish their ability to hydroxylate HIF-α. Interestingly, lack of mitochondrial ROS does not prevent activation of HIF-α subunits in anoxia, thus suggesting that some oxygen is necessary for mitochondria to promote HIF-α signaling. Although mitochondrial complex III has been implicated in the oxidant burst by mitochondria, to date, the mechanism by which reduced oxygen tension mediates ROS generation is not well understood. Nonetheless, experimental evidence suggests that enhanced ROS production is not a sustained event in hypoxia (69, 385).
HIF-1 coordinately upregulates many genes to regulate metabolism, energy production, and mitochondrial function (385). Thus, under hypoxia, cells switch to oxygen-independent glycolysis (the Pasteur effect) through activation of HIF-mediated transcriptional responses. HIF-1 coordinately upregulates PDH kinase, LDHA, and other glycolytic genes to increase glycolysis; decreases the activity of the TCA cycle; and reduces electron flux through mitochondrial complexes I, III, and IV (214, 386). To compensate for the increased reliance on glycolysis, HIF-1 also upregulates the expression of the Glut1, a glucose transporter. This transcription factor also promotes a subunit switch in cytochrome c oxidase 4 or COX4 from 4-1 to 4-2 during hypoxia, which improves the efficiency of electron transport at complex IV (143). The result of these expression changes optimizes the control of energy production and minimizes the generation of potentially toxic ROS from increasingly inefficient mitochondrial electron transport under conditions of hypoxia.
Our recent findings suggest yet another mechanism by which HIF-1α modulates electron transport and metabolism by ultimately suppressing the formation of Fe-S clusters, which are integral to the function of the respiratory chain. This HIF-1α-mediated response relies specifically on upregulation of a microRNA, miR210, that decreases the expression of iron–sulfur cluster assembly proteins (ISCU1/2) by inhibiting their translation. Subsequent studies by other investigators confirm the role of miR210 in suppressing ISCU1/2 and as well as other factors (cytochrome c oxidase assembly protein) essential for ETC assembly (81, 129). ISCU1/2 arise from splice variants of the same transcript. ISCU1 protein is found in the cytosol, whereas ISCU2 targets to the mitochondria (420); we refer to them as ISCU1/2 here. These proteins facilitate the assembly of [4Fe-4S] and [2Fe-2S] Fe-S clusters, which are essential for the function of some apoproteins. These Fe-S prosthetic groups promote electron transport and redox reactions integral to many cellular processes ranging from electron transport and oxidative phosphorylation, ribosome biogenesis, purine catabolism, heme biosynthesis, DNA repair, to iron metabolism, among others; mutations in ISCU have been associated with a metabolic myopathy (360). Fe-S clusters are found in key mitochondrial respiratory complexes (complexes I, II, and III) where they facilitate electron transport (360) and in aconitase, a TCA enzyme.
We found that the miR210-induced decrease in ISCU1/2 has functional consequences that directly alter the metabolic phenotype of endothelial cells by decreasing complex I function, mitochondrial respiration, and ATP synthesis (69). Similarly, in other studies, miR210-mediated suppression of ETC and TCA enzymes were found to correlate with enhanced anaerobic glycolysis and increases in lactate production (81, 129). In our studies, we found that we could restore the ETC and TCA enzyme activities in the presence of elevated miR210 by using ISCU1/2 gene constructs that lack the miR210 recognition sequence in their 3′ untranslated region, suggesting that these miR210-induced deficiencies were primarily due to lack of Fe-S clusters. We also established a link between ICSU1/2 and suppression of miR210 expression by showing that siRNA knockdown of miR210 during hypoxia augments ROS production, illustrating a role for miR210 in modulating compensatory mechanisms to protect cells from ROS during hypoxia. Interestingly, during normoxia, forced expression of miR210 caused an increase in ROS generation (69, 81), possibly due to deficiencies in complex I that may promote electron leak in the presence of molecular oxygen. We suggest, however, that miR210-mediated inhibition of Fe-S clusters promotes cell survival, in part, by its suppression of mitochondrial respiration; possibly, miR210 upregulation may also promote cell survival by lessening ROS generation during the hypoxia to normoxia transition (Pasteur effect). miR210-enhanced cell survival, however, may not always be beneficial. Thus, in cancer or in pulmonary hypertension, metabolic shifts promoted by HIF and miR210 may increase cell growth and proliferation to contribute to tumor survival or vascular remodeling, respectively (122, 131, 390). In addition, in a chronic setting, suppression of Fe-S cluster formation may have consequences for the synthesis of heme, the uptake and intracellular metabolism of iron, and genomic stability (430, 466). Forced expression of miR210 in cancer cell lines slowed initiation of tumor growth after implantation of these cells in nude mice; however, miR210 overexpression did not appear to increase cell death (179). Thus, these findings may suggest in the absence of other HIF-regulated genes miR210 may not produce the same results as when it is upregulated downstream of HIF. Other considerations that need further study are the additional targets that may be suppressed by miR210 in hypoxia, which may contribute to the adaptive responses to hypoxia.
In addition, the hypoxia-induced suppression of Fe-S protein activity can be contrasted with the suppression of Fe-S protein activity found in Friedreich's ataxia, a hereditary neurogenerative disease that results from decreased production/function of the frataxin protein (17, 220). Frataxin is a mitochondrial iron chaperone; its precise function is still a matter of debate but its deficiency leads to an increase in mitochondrial iron, mitochondrial dysfunction, and decreased function of Fe-S cluster containing proteins, including complex I and complex III, succinate dehydrogenase (which is both complex II in the ETC and part of the TCA cycle), and aconitase. Decreased expression of frataxin also increases the Fenton reaction due to excess free iron (17, 220). Studies indicate that loss of normal frataxin leads to excess mitochondrial production of hydrogen peroxide. Increased production of mitochondrial oxidants was found to correlate with a decrease in both total GSH as well as a decrease in the GSH/GSSG ratio in cells with frataxin deficiency; these changes may lead to the increased accumulation of protein S-glutathiolation in this disease (17). Interestingly, as mentioned above, forced miR210 overexpression under normoxic conditions is also associated with excess production of cellular oxidants (69, 81), although less is known about the species produced and the consequence of this production on GSH redox. Cells with frataxin deficiency show an inability to upregulate mitochondrial MnSOD, leading to some controversy regarding the importance of MnSOD and superoxide in the disease phenotype (17, 220); however, other studies confirm a role for hydrogen peroxide rather than superoxide in the damaging effects of this deficiency. For example, in a Drosophila model of Friedreich's ataxia, superoxide scavengers had no protective effect on the phenotype, whereas mitochondrial targeted catalase or Prx were protective (11).
VII. Conclusion
Redox signaling allows for the rapid adaptation to a variety of environmental stimuli, such as hormones, nutrients, and O2 tension. The mitochondrion plays a central role in cellular adaptation: modulating metabolic, energetic, and ROS responses to promote cell growth and survival or cell death in response to a range of stimuli (Fig. 11). The overall redox status is kept in balance by a host of antioxidant enzyme systems, notably GPxs/GSH/GR/Grx and Prx/Trx/TrxR, that modulate cellular ROS flux and lipid oxidation. Grx/GSH/GR and Trx/TrxR also reduce protein disulfides. Notably, each of these enzyme systems require NADPH to regenerate the reducing cosubstrate (GSH or Trx), highlighting the importance of G6PD to regenerate NADPH. In mitochondria, the NNT enzyme, which reduces NADP+ utilizing the proton gradient and NADH from the TCA cycle, may also have a crucial role in maintaining mitochondrial NADPH.
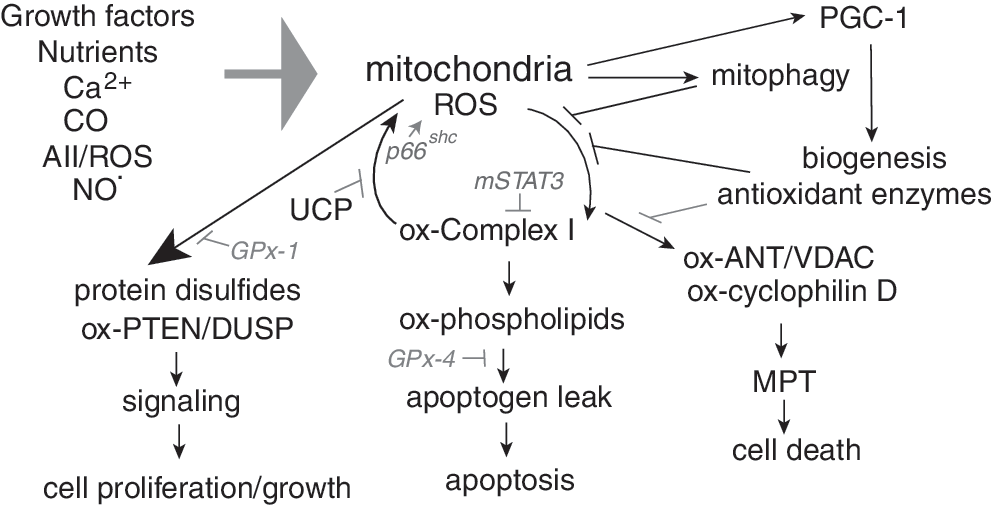
Proteins offer unique redox switches that are reversibly modified by ROS to alter their function. Protein localization, thiol accessibility, pKa, and protein–protein interactions may all contribute to the specificity of these responses. Mitochondrial function can be modulated directly by protein thiol oxidation or by lipid peroxidation (Fig. 12), and indirectly following the redox-dependent activation of phosphatases (Fig. 8), HIF (Fig. 10), ASK1 (Fig. 9), or other redox-sensitive proteins. Nutrient changes can also lead to NAD+ accumulation and activation of SIRT-dependent pathways that can include mitochondrial biogenesis (Fig. 6). Sites within the mitochondria can be regulated by SIRT deacetylation, or by direct oxidative modification. Complex I, for example, is a target for S-NO and other forms of thiol oxidation, which may inhibit its function, or possibly, promote electron leak (Fig. 7). Other proteins, such as mitochondrial STAT3, may protect complex I from oxidative inactivation by mechanisms not well understood that involve preserving ETC function and decreasing ROS production after oxidative stress (Fig. 11). In contrast, oxidative mechanisms that activate p66shc to promote its mitochondrial localization can augment ROS generation, contributing to redox modifications to proteins and phospholipids. Downstream of ROS, other modifications of mitochondrial proteins can lead to mitophagy or activation of the MTP. Mitophagy and biogenesis are seen as two complementary processes that help to maintain metabolic homeostasis and reduce ROS accumulation. Brief openings of MTP pore may serve a protective function in mitochondria, but more prolonged MTP pore formation can lead to cell death. Excess mitochondrial ROS can also lead to detrimental cardiolipin oxidation. Such alterations in phospholipid oxidation promote mitochondrial-dependent apoptosis. Downstream of HIF activation, inhibition of Fe-S complex formation reduces ETC and TCA cycle enzymes to decrease ROS generation in hypoxia. The examples discussed in this review provide some insights into mechanisms by which redox events regulate mitochondrial function to alter cellular energetics and metabolism.

Footnotes
Acknowledgments
The authors wish to thank Stephanie Tribuna for her expert editorial assistance. This work was supported by grants HL61795, HL48743, HL70819, HL107192, and HL108630.