
Abstract
Introduction
Cells derived from mice carrying gene trap mutations in both Ero1α and Ero1β genes proved to be hypersensitive to depletion of PRDX4. A possible explanation for this unexpected phenomenon is that PRDX4 can substitute for Ero1 in disulfide bond formation. Indeed, depletion of PRDX4 exacerbated the modest defects in disulfide formation seen in the Ero1α–Ero1β double knockout cells (39). The role of PRDX4 as a hydrogen peroxide dependent disulfide catalyst has been confirmed by an independent in vitro study showing the rapid oxidation of PRDX4 by hydrogen peroxide and a consecutive relatively slower transfer of its disulfide to PDI family proteins (35). In accordance with these observations, accumulation of high molecular weight IgM complexes was detected in lymphocytes isolated from PRDX4 knockout mice and differentiated to plasma cells (4).
Innovation
Hydrogen peroxide was long thought to be a harmful byproduct of oxidative protein folding. Currently, however, different pathways were hypothesized to take a role in the recycling of H2O2 to disulfide bond formation. Here, evidence is provided by using in vivo models that hydrogen peroxide can promote disulfide bond formation in the ER.
In wild-type cells, the cooperation between Ero1s and PRDX4 on one hand improves the efficacy of oxidative protein folding by generating two disulfide bonds at the expense of one molecule of oxygen; on the other hand, it would solve the problem of hydrogen peroxide toxicity. However, under the experimental conditions applied by Zito and co-workers (39), hydrogen peroxide sources other than Ero1s should be supposed. The identification of these sources requires further studies.
PRDX4 is presumably not a single candidate as PDI oxidant. A recent study showed that two human glutathione peroxidases (GPx7 and GPx8) are ER-resident PDI peroxidases. They were able to catalyze an efficient protein refolding in the presence of PDI and peroxide. They were shown to interact with Ero1α, suggesting that they can represent a novel pathway for the productive reutilization of hydrogen peroxide produced by Ero1s during disulfide bond formation (28).
The aim of the present study was to demonstrate in vivo the role of hydrogen peroxide in disulfide bond formation. To this end, two experimental approaches were used. First, ER-targeted hydrogen peroxide production was provoked in mice, which express an ER membrane flavoprotein gulonolactone oxidase. The enzyme is responsible for the last step of ascorbate synthesis, and the in vivo administration of its substrate gulonolactone results in ER luminal ascorbate and—as a byproduct—hydrogen peroxide formation (20, 31). In our second model, ER luminal hydrogen peroxide was downregulated by the ER-targeted overexpression of catalase in cellular models where disulfide bond formation is inevitable for proper cellular function. The results confirm and extend the notion that ER luminal hydrogen peroxide can be efficiently reutilized in the electron transfer chain of oxidative protein folding.
Results
ER-targeted hydrogen peroxide production promotes local thiol oxidation
In a set of in vivo experiments, the effect of the ER luminal hydrogen peroxide formation on protein thiol oxidation was studied in the liver of mice. To provoke an oxidative folding-independent hydrogen peroxide production in the ER lumen, gulonolactone was administered intraperitoneally. Gulonolactone oxidase metabolizes gulonolactone to ascorbate with the concomitant production of hydrogen peroxide. In accordance with its predicted membrane topology, the catalytic site is positioned in the lumen, thus the enzyme produces hydrogen peroxide (and ascorbate) intraluminally. The presumed effect on glutathione or protein thiol oxidation was mitigated by the coadministration of dithiothreitol (DTT).
Acute and chronic effects of gulonolactone were investigated. The effective dose of gulonolactone was established in preliminary experiments, which showed that 3.5 mg/kg (approx. 20 mM/kg) body weight gulonolactone was necessary to obtain a well-measurable stimulation of ascorbate production (Supplementary Fig. S1; supplementary data are available online at
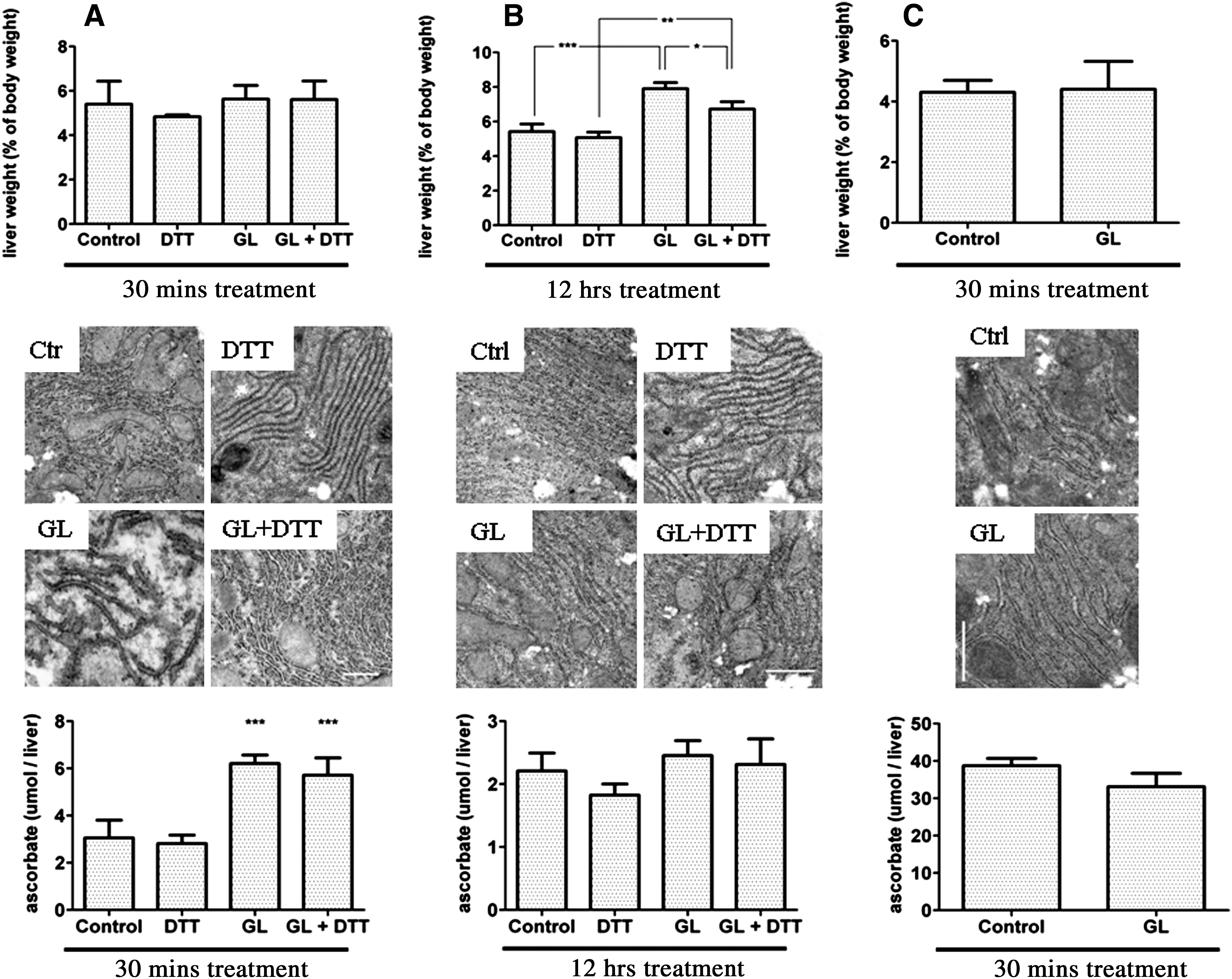
The preventive effect of DTT was not due to the inhibition of gulonolactone oxidase. Gulonolactone addition produced a significant increase in hepatic ascorbate content (expressed as micromoles/liver; concentration would have been misleading due to the swelling), which was not affected by DTT coadministration (Fig. 1A). After 12 hours, the ascorbate level returned to the control value, presumably due to the complete consumption of the added gulonolactone and/or the excretion of gulonolactone and ascorbate (Fig. 1B).
To prove that liver swelling and altered ER morphology were indeed due to the metabolism of gulonolactone (and not to its osmotic or other effect), the treatment was also performed in guinea pigs who lack gulonolactone oxidase activity. As it was expected, gulonolactone administration did not increase hepatic ascorbate levels; moreover, liver swelling and the morphological changes were not observed in guinea pigs (Fig. 1C).
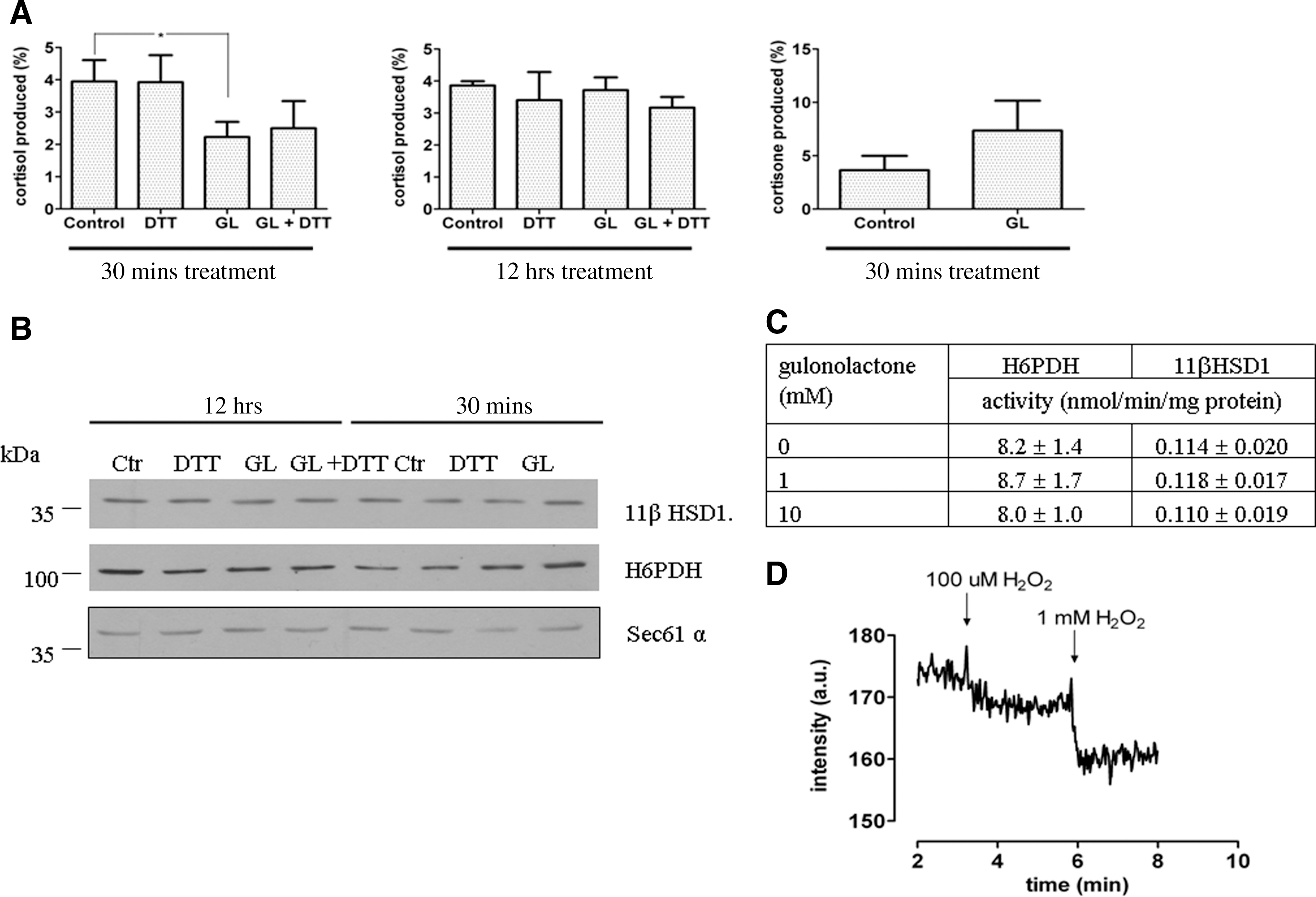
Hydrogen peroxide elimination usually occurs at the expense of NADPH as electron donor in cellular compartments other than the ER. Thus, although ER luminal hydrogen peroxide metabolizing pathways have not been fully understood, we wanted to further characterize the effects of ER-targeted hydrogen peroxide generation on the redox conditions in the lumen of the organelle. The redox state of luminal pyridine nucleotides was shifted towards oxidized direction, as it was estimated by measuring microsomal cortisone–cortisol interconversion (Fig. 2A). This reaction is catalyzed by the luminal enzyme 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) (2). The reaction is reversible and its direction depends on the availability of reduced and oxidized pyridine nucleotides (i.e., on the [NADPH]/[NADP+] ratio) (10). We observed a marked decrease in cortisol production from cortisone in liver microsomes from gulonolactone-treated mice (Fig. 2A, left panel), while cortisone generation from cortisol was increased (Fig. 2A, right panel). DTT treatment did not change the interconversion and failed to prevent the effects of gulonolactone administration (Fig. 2A). The observed changes were not due to the altered expression of either 11βHSD1 or the main ER luminal NADPH-producing enzyme hexose-6-phosphate dehydrogenase (Fig. 2B) (34). We excluded the direct effect of gulonolactone on these enzymes: in microsomal measurements gulonolactone up to 10 mM concentration did not cause any inhibition (Fig. 2C). The alterations in pyridine nucleotide pool were seen only in the 30-min samples and not in the chronically- treated mice (Fig. 2A, middle panel). In accordance with these results, addition of hydrogen peroxide to liver microsomes resulted in a prompt decrease of the fluorescent signal of reduced pyridine nucleotides (Fig. 2D) indicating a direct oxidizing effect of H2O2 on the NADPH/NADH pool.
The redox state of the thiol–disulfide system was also changed in 30-min treated animals. In accordance with our previous results (3, 25, 31), luminally produced H2O2 preferentially oxidizes microsomal glutathione. Gulonolactone-treated mice presented a marked decrease in microsomal GSH content, while GSSG content remained unaffected (Fig. 3A). DTT coadministration could not reverse the effect. At the same time, total hepatic GSH content, measured in liver homogenates, was only moderately decreased (Fig. 3B). The alteration of glutathione levels were reversible; 12 h after the treatment glutathione levels returned to the control values both in microsomal fractions (Fig. 3C) and in the homogenates (Fig. 3D).

Here we found that microsomal protein thiol levels also decreased upon gulonolactone addition; DTT reversed the effect (Fig. 4A). A decrease of protein thiol level in total liver homogenate was not observed (data not shown). The oxidation of the intraluminal thioredoxin-motif containing oxidoreductases could be observed on immunoblots by using AMS labeling. The slower migrating reduced and the faster migrating oxidized form of ERp72 are clearly distinguishable on the blot; gulonolactone treatment resulted in a marked decrease in the reduced form (Fig. 4B). ERp72 might also be present in higher molecular weight disulfides with other proteins; however, they could not be visualized on the blots. In case of ERp46 a redox shift could be observed due to their oxidation upon gulonolactone treatment (Fig. 4C). DTT coadministration counteracted the effect of gulonolactone treatment on the redox state of these proteins. Immunoblotting of ERp72, ERp46, and ERp5 on nonreducing gels showed a faster migration of ERp46 in samples from gulonolactone-treated mice, while in case of the other two proteins the nonreducing gel did not reveal the difference in their redox state (Fig. 4D).
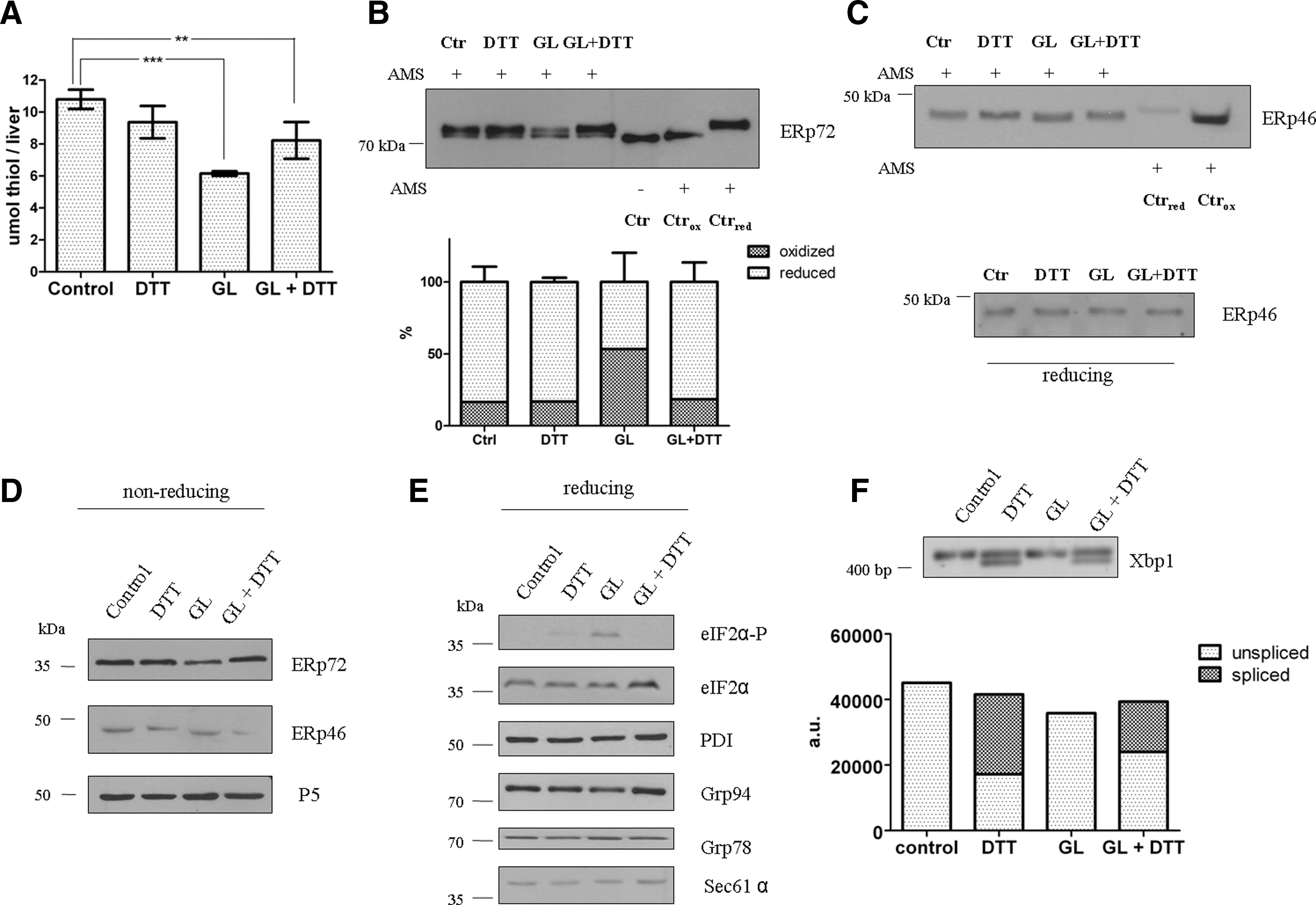
These acute alterations could not be detected in 12-h samples (Supplementary Fig. S3.), in accordance with our previous findings in 24-h samples (25).
The altered redox state of the ER lumen was accompanied by an early sign of ER stress (i.e., the phosphorylation of eIF2α) (Fig. 4E). XBP1 splicing was instead observed in DTT-treated mice (Fig. 4F). Both changes were fully or partially reverted by the coadministration of the two substances (DTT and gulonolactone), indicating the ability of one to antagonize the effect of the other. No signs of ER stress were observed in the 12-h samples (Fig. S3.); moreover, the expression of ER chaperones PDI, Grp78, and Grp94 was not changed by any treatments, measured in either 30-min or 12-h samples (Figs. 4E and S3B).
ER-targeted hydrogen peroxide elimination interferes with oxidative folding
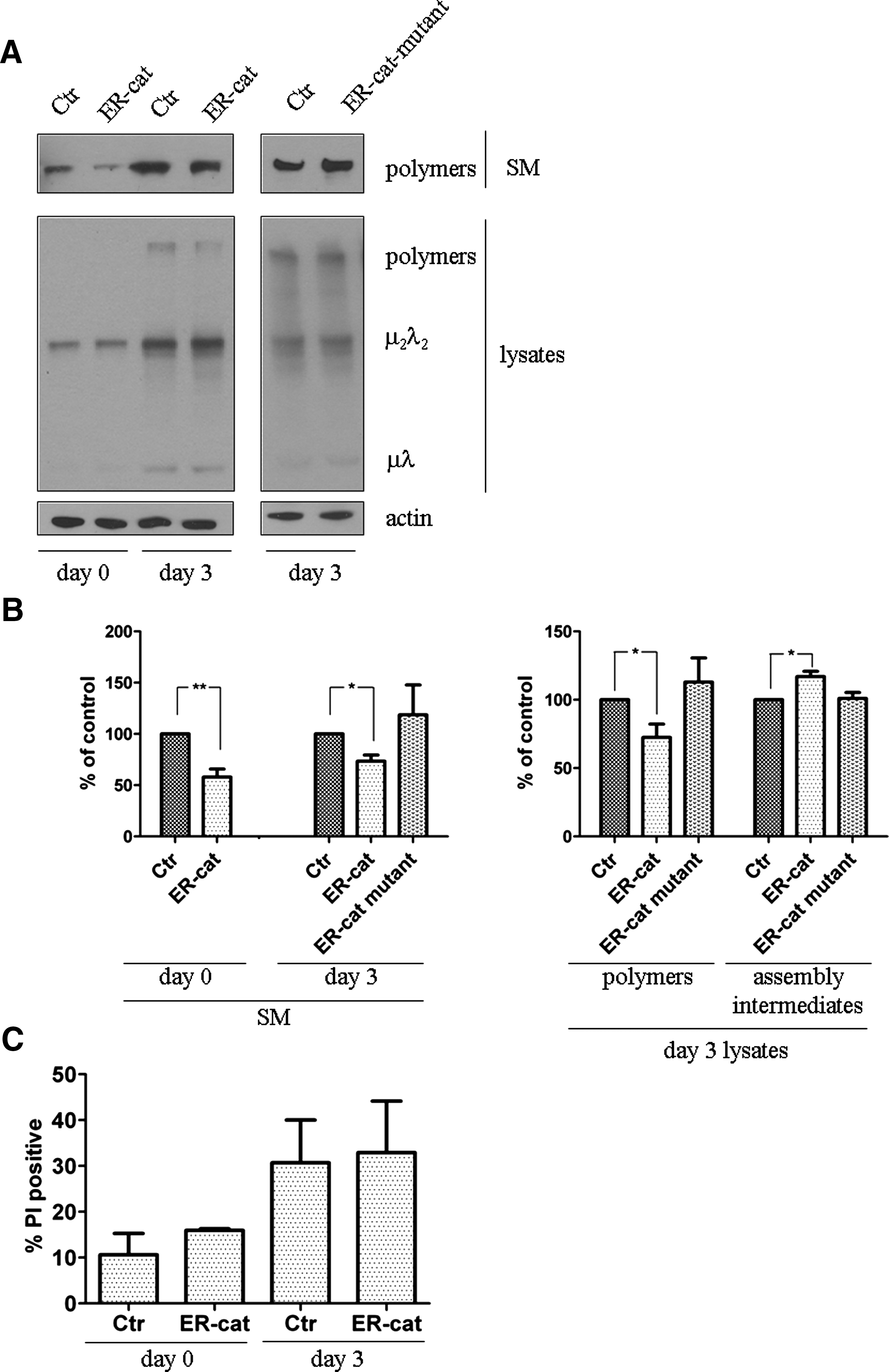
Cell lines devoted to secreting abundant amounts of disulfide-containing proteins (27) were prevented from reutilizing the intraluminal H2O2 produced during active oxidative folding with the expression of ER-targeted antioxidant enzyme catalase. As a control, a catalase mutant (Y358F-catalase) was also directed to the luminal compartment; the presence of tyrosine in the 358 position is crucial for the activity (32). The expression of ER-catalase and its mutant form was controlled in HEK293 cells (Fig. 5A) and the ER localization was verified with immunofluorescence microscopy (Fig. 5B). Both versions of catalase remained co-localized with ER-markers calreticulin, and did not co-localize with ERGIC-53; thus it was retained in the ER lumen and did not appear in the early secretory compartments.
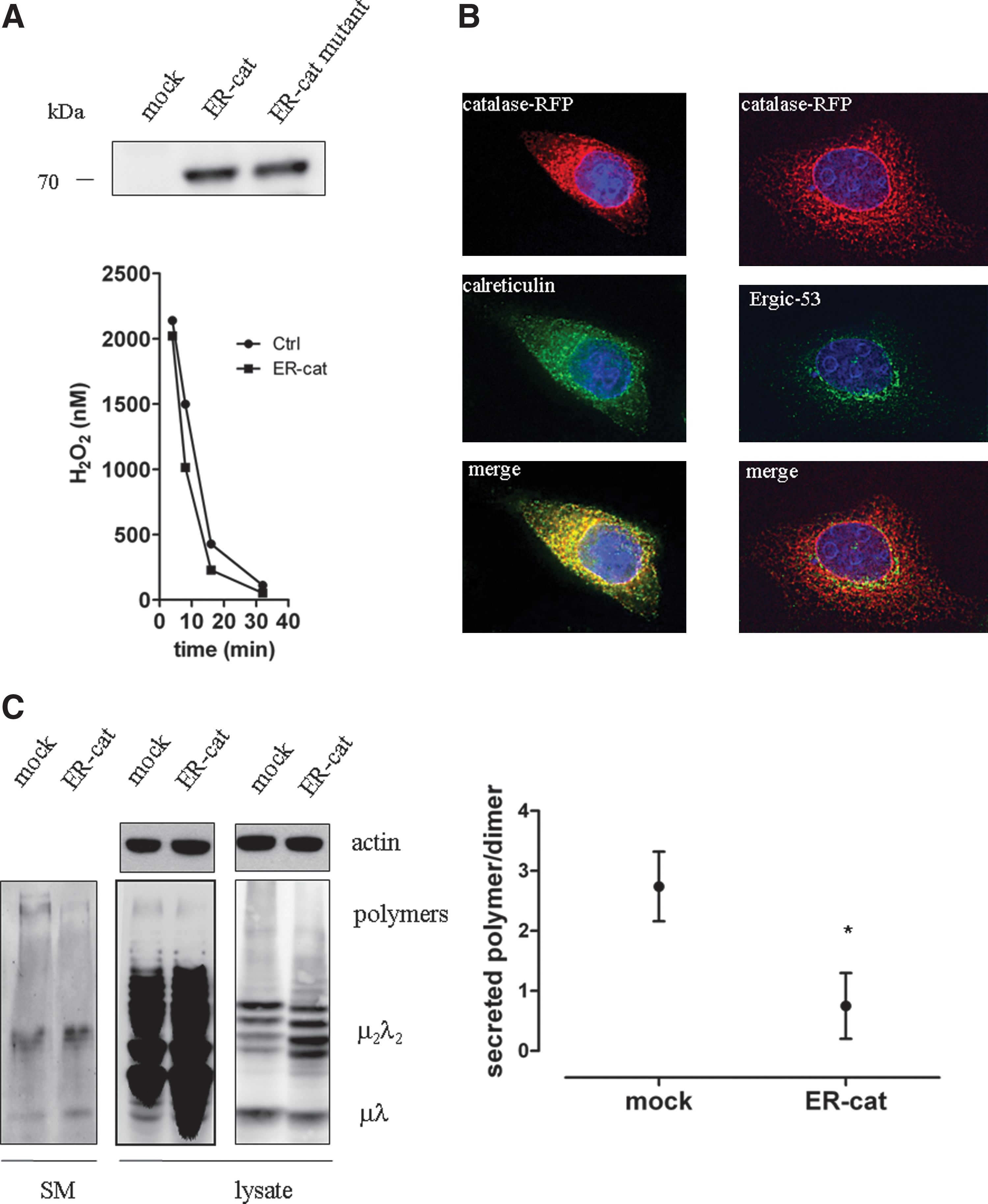
Multimeric glycoproteins (e.g., IgM) undergo a structural maturation in the ER until the fully assembled polymer —containing hundreds of disulfides —is released (7). When expressed in HEK293 cells, μ- and λ-chains of IgM experience polymerization and form disulfide-linked polymers, dimers, and monomers, until the mature immunoglobulin and —due to imperfect quality control mechanisms with respect to designated antibody secreting cells —some dimers are secreted into the medium (9). Antibody secretion of HEK293 cells were analyzed and compared with immunoblot after co-transfecting IgM components with wild- type or mutant ER-targeted catalase. We found that cells expressing ER-targeted catalase secreted less IgM polymers than mock-transfected cells; densitometric analysis showed a significantly decreased polymer/dimer ratio in the medium of ER-catalase-containing cells. The ER-targeted catalase expressing cells showed a decreased intracellular level of assembled polymers and a slightly increased amount of nonassembled intermediates (Figs. 5C and 5D). Cells expressing mutant catalase in the ER did not exhibit significant alteration either in polymer secretion or in intracellular assembly intermediates (data not shown).
The effect of H2O2 elimination on IgM assembly was also investigated in I29μ+ lymphocytic cells. To investigate consequences of capturing immediately the ER-derived H2O2, I29μ+ cells expressing ER-targeted catalase and its mutant version were created by stable transfection with lentiviral vectors. Plasma cells, being designated secretors, released only IgM polymers and not any assembly intermediates. Polymer secretion significantly decreased in those lines where H2O2 was captured within the lumen (Fig. 6A). Cell lysates derived from ER-catalase transfected I29μ+ cells contained less IgM polymers as well, alike to the data obtained on the secreted material. Polymer assembly was also checked in I29μ+ cells transfected with the mutant catalase, and was found to be not significantly different from controls (Fig. 6B). Decreased polymer secretion was not due to an increased cell death (Fig. 6C), as assessed by propidium iodide staining.
Discussion
Hydrogen peroxide, the side-product of oxidative protein folding, is an effective electron acceptor. On the basis of its pro-oxidant behavior, it has been supposed that enhanced oxidative folding represents an oxidative burden in designated secretory cells. However, hydrogen peroxide as an alternative electron acceptor can also promote disulfide bond formation. Indeed, H2O2 in vitro was able to promote oxidative folding, resulting in the efficient formation of a natively folded protein containing proper disulfide bonds. The recent identification of ER peroxidases (PRDX4, GPx7, and GPx8) as PDI oxidants further strengthened the view that hydrogen peroxide can be productively used in oxidative folding.
The results obtained in vivo and reported here demonstrate that hydrogen peroxide contributes to the process of disulfide bond formation; its removal rather than its generation can hinder the oxidative folding. ER-targeted overproduction of hydrogen peroxide, by the exogenous substrate supply of the ER resident gulonolactone oxidase in vivo in mice resulted in decreased glutathione and protein thiol content in the ER of the liver (Figs. 3A and 4A) and in a redox shift of ER oxidoreductases towards the oxidized direction (Figs. 4B and 4C). On the contrary, the stimulated decomposition of ER luminal hydrogen peroxide by the ER-targeted overexpression of catalase resulted in an impaired secretion of IgM in HEK293 (Fig. 5C) and I29μ+ cells (Fig. 6A).
Manipulation of luminal hydrogen peroxide levels however was not accompanied with altered viability either in vivo or in the cellular models, indicating the robustness of ER redox control. Gulonolactone administration, although resulting in an acute, dramatic liver swelling and dilation of ER cisternae (Fig. 1) due to a temporary change of the permeability of the ER membrane (25), provoked only minor signs of ER stress; a transient increase in eIF2α phosphorylation was observed that was not accompanied with XBP1 activation or chaperon induction (Figs. 4D–4F). Thus, it can be supposed that altered proteostasis rather than redox imbalance is the main factor of ER stress upon protein overproduction.
ER-targeted hydrogen peroxide overproduction compromised not only the thiol/disulfide, but also the pyridine nucleotide redox system. Consumption of luminal NADPH was detected on the basis of microsomal cortisone-to-cortisol conversion and was verified directly in liver microsomes by hydrogen peroxide addition. The biochemical basis of this phenomenon needs further studies. GSSG-mediated NADPH oxidation is not probable, due to the absence of glutathione reductase in the compartment (30). A peroxiredoxin-mediated pathway nevertheless can be taken into account (17). Moreover, the difference in the protective effect of DTT supplies a further proof for the uncoupling of the thiol–disulfide and the pyridine nucleotide redox systems. DTT co-administered with gulonolactone prevented liver swelling, morphological alterations, protein thiol oxidation, ERp72 redox shift, and eIF2α phosphorylation (i.e., the effects presumably mediated by the oxidation of protein thiols), but did not counterbalance the altered redox state of pyridine nucleotides. Although the antagonistic effect of gulonolactone and DTT can be explained by their opposite redox effects, the different profile in the activation of the signaling pathways of ER stress needs further studies. However, several findings confirm the selective activation and divergent role of ER stress signaling pathways (21, 26, 33).
While hydrogen peroxide generation in the ER stimulated the oxidation of microsomal protein thiols in our in vivo model, the stimulated decomposition of ER luminal hydrogen peroxide by the ER-targeted overexpression of catalase resulted in an impaired secretion of IgM in cellular models (Figs. 5 and 6.). However, the vast majority of disulfide bonds necessary to fold μ2λ2 intermediates are formed also in cells expressing active catalase. Only those mediating the polymerization or the retention of immature intermediates seem to be impaired; the subtle effect of lower levels of hydrogen peroxide on IgM polymerization suggests that further compartmentation or biochemical circuits are at work to assist the biogenesis of the complex molecule. The decreased secretion of IgM polymers and increased retention of assembly intermediates also suggest the significant contribution of quality control mechanisms, which are presumably activated by the impaired disulfide bond formation. The decreased IgM secretion and the activation of the quality control however cannot be merely attributed to the luminal overexpression of a protein, since the catalase mutant did not produce the same effects.
The results confirm the assumption that local oxidases other than Ero1 can contribute to the functioning of oxidative folding by providing hydrogen peroxide. Gulonolactone oxidase, however, cannot be regarded as a general hydrogen peroxide supplier; the enzyme is expressed usually only in the liver and in some species— including primates and guinea pig— is even absent. Apart from this aspect, hydrogen peroxide produced in the ER can be efficiently channeled into the electron transfer chain of oxidative protein folding, by using multiple routes involving ascorbate, PDI peroxidases, peroxiredoxins, or other electron carriers to be identified.
Materials and Methods
Materials
L-Gulono-γ-lactone, 5,5’-dithio-bis(2-nitrobenzoic acid), α,α’-dipyridyl, ascorbate, dithiothreitol, diamide, cortisone, cortisol, NADPH, MOPS, hexadimethrine bromide, lipopolysaccharides, and hydrogen peroxide were obtained from Sigma Chemical Co (St. Louis, MO).
Animal experiments
Male CD-1 mice (20–25 g body weight) and male Hartley guinea pigs (250–300 g body weight) were purchased from Charles River Hungary (Isaszeg). Animal treatments were approved by the Committee on Animal Experiments of Semmelweis University, Budapest. Animals were kept with ad libitum access to food and water. Gulonolactone (3.5 g/kg body weight) and dithiothreitol (150 mg/kg body weight) were dissolved in physiological saline and was injected intraperitoneally in a volume of 1 ml (in mice) or 10 ml (in guinea pigs). Control animals received an equivalent volume of the vehicle. Treatments with different agents were tolerated well by the animals. At various time points after treatment (0.5 or 12 h) animals were sacrificed. Liver samples were taken and processed for electron microscopy and for RNA isolation. The remnant of the liver was homogenized in sucrose–HEPES buffer (0.3 M sucrose, 0.02 M HEPES, pH 7.2) with a glass-Teflon homogenizer. The microsomal fraction was then isolated using fractional centrifugation as detailed elsewhere (23). Microsomes were resuspended in a KCl- MOPS buffer (100 mM KCl, 20 mM NaCl, 1 mM MgCl2, 20 mM MOPS, pH 7.2), were immediately frozen in liquid nitrogen, and kept in liquid nitrogen until use. The integrity of the microsomal membranes was assessed by using the mannose-6-phosphatase assay (6) and by measuring p-nitrophenol glucuronidation (15), which showed latency greater than 95%.
Electron microscopy
For transmission electron microscopy investigation, upon dissection after an in vivo treatment, the liver samples were immediately fixed in 0.5% glutaraldehyde+2% paraformaldehyde (PolySciences Europe GmbH., Eppelheim, Germany) in 0.1 M cacodylate buffer (pH 7.2) containing 0.25 M sucrose and 2 mM CaCl2 for 2 h at room temperature, then washed in 0.1 M cacodylate buffer. After postfixation in 1% osmium tetroxide (Serva Feinbiochemica GmbH., Heidelberg, Germany) in 0.1 M cacodylate buffer, liver samples were dehydrated in ascending series of ethanol, infiltrated in propylene oxide, and embedded in Durcupan ACM resin (Fluka AG, Buchs, Switzerland). Ultrathin sections were contrasted with uranyl acetate and lead citrate and electron micrographs were taken by a Jeol JEM1011 electron microscope operating at 60 kV.
Immunoblotting, antibodies
The protein concentration of samples was measured using the Bradford protein assay (Fermentas, Hanover, MD). Equal amounts of microsomal proteins (25–100 μg) were separated in SDS–PAGE and transferred to nitrocellulose filter membranes (Bio-Rad, Hercules, CA) by electroblotting. The filter membranes were incubated overnight with the primary antibodies (anti-eIF2α and anti-phospho-eIF2α are from Cell Signaling Technology, anti-ERp72 is from Calbiochem; sec61α, PDI, GRp78, GRp94, and H6PDH are from Santa Cruz Biotechnology; 11βHSD1 is from Cayman Chemical), and for 1 h with the species-specific peroxidase-conjugated secondary antibodies (anti-goat, anti-mouse, and anti-rabbit IgGs from Santa Cruz Biotechnology). The anti-myc murine monoclonal antibody 9E10 (8) was used to detect tagged proteins. The antibodies were detected using a chemiluminescence reagent (ECL) kit (Pierce, Rockford, IL) and blue-sensitive X-ray film. Equal protein loading was also verified by Ponceau-staining of the membrane and by β-actin detection. Band intensities were quantified by densitometry using Image Quant 5.2 software (Molecular Dynamic, Sunnyvale, CA). The thiol redox state of ER foldases was investigated by alkylation with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS; Molecular Probes–Invitrogen, Eugene, OR) as reported in (14). 200 μg microsomal proteins were precipitated with 5% trichloroacetic acid, washed two times with 70% acetone and two times with 0.1 M Tris buffer (pH 7.2). The resuspended proteins were reacted with 20 mM AMS for 15 min on ice, then for 15 min at 37°C. For the reduced and oxidized controls, microsomal samples from the liver of control mice corresponding to 200 μg protein were incubated at 37°C for 15 min with 50 mM dithiothreitol or with 25 mM diamide, respectively.
Analysis of XBP1 splicing
The splicing of XBP1 mRNA was analyzed as described by Yoshida et al. (38) with the following primers: sense 5′ CCT TGTGGTTGAGAACCAGG 3′ and antisense 5′ AGGCTTGGT GTATACATGG 3′ obtained from Sigma-Aldrich Co.
Metabolite measurements
Ascorbate contents were measured in trichloroacetic acid-soluble supernatants of the total liver homogenates by the method of Omaye et al. (29), based on the reduction of Fe3+ with the oxidation of ascorbate and the subsequent determination of the Fe2+-α,α’-dipyridyl complex. The results were controlled by measuring the ascorbate contents with HPLC (40); these measurements confirmed the data gained by the dipyridyl method. Protein thiols were measured in the washed and resuspended pellets by the Ellman method (11) (Supplementary Fig. S4). Determination of glutathione content of liver or microsomes were performed by HPLC analysis as described earlier (25). Glutathione measurement from freeze–thawed microsomes gives a good approximation of the original ER glutathione levels because permeation of both GSH and GSSG through microsomal membranes is relatively slow. Hydrogen peroxide content of cellular supernatants was measured by Amplex® Red Assay Kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction.
Measurement of cortisone–cortisol conversion
Microsomal cortisone–cortisol conversion (in both directions) was measured by incubating intact mouse liver microsomes (5 mg protein/ml) at 37°C in KCl/MOPS buffer in the presence of 10 μM cortisone or cortisol, respectively. Samples were incubated for 0, 5, 10, and 20 min, the reactions were terminated by adding ice-cold methanol. Samples were stored at −20°C until analysis. After sedimentation of the precipitates by centrifugation (20 000 g for 10 min at 4°C), the cortisol and cortisone content of the supernatants was measured by HPLC (Alliance 2690; Waters Corp., Milford, MA) using a Nucleosil 100 C18 column (5 μm 25×0.46) (Teknokroma). The eluent was 58% methanol, samples were eluted for 20 min and the absorbance was detected at 245 nm wavelength (Dual l Absorbance Detector 2487). The retention times of cortisol (approx. 13.5 min) and cortisone (approx. 15.5 min) were determined by injecting standards.
Fluorimetric detection of reduced pyridine nucleotides
Reduced pyridine nucleotides were detected based on their characteristic fluorescent spectrum in microsomes (30). Murine liver microsomal vesicles (1 mg/ml protein) from control mice were incubated in in KCl/MOPS buffer (pH 7.2) at 22°C. Fluorescence was monitored at 350 nm excitation and 460 nm emission wavelengths by using a Cary Eclipse fluorescence spectrophotometer (Varian). The activity of 11βHSD1 and H6PDH was measured on the basis of NADPH formation, as described earlier (2, 30).
Cell lines and culturing
HEK293 were grown in α-minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS) at 37°C, 5% CO2. The mouse B lymphoma cell line I29μ+ cells were cultured in RPMI media (Invitrogen) supplemented with 10% defined FBS (HyClone Lab.), glutamax (1 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml), and activated with LPS as described earlier (37).
Cell lysis and secretion assay
Cells were lysed in 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 1% NP40, plus freshly added protease inhibitors (Roche, Basel, CH), and 10 mM N-ethylmaleimide (19) (Sigma Chemical Co), then centrifuged 15 min at 4°C 20,000 g to pellet nuclei. For secretion assay, HEK293 cells or differentiating I29μ+ cells were washed twice in phosphate-buffered saline and cultured in OptiMem Medium (Invitrogen) in the absence of FCS for 4 h at a cell density of 1×106/ml. After centrifugation, spent media were supplemented with protease inhibitors and 10 mM NEM and analyzed. To detect IgM assembly intermediates, total cell lysates and cell supernatants were separated on NuPAGE 4-12% Bis-Tris gel (Invitrogen) and IgM intermediates were detected with polyclonal rabbit anti-mouse IgM from ZYMED (San Francisco, CA).
Vectors, cell transfections
Vectors encoding μ -chain and λ –chain were described earlier. The pCMV-myc-ER expression vectors (Invitrogen V823-20) encoding catalase, catalase-RFP, catalase-Y358F, and catalase-Y358F-RFP were kind gifts of M. Geiszt (Semmelweis University, Dept. of Physiology, Budapest, Hungary). The inserts were amplified with the primers sense 5’ CGTGTACGGTGGGAGGTCTA 3’ and antisense 5’ AGGCACAGTCGAGGCTGAT 3’ and were introduced with Sal I./Sma I. sites into a bidirectional lentiviral vector #945 (which was a kind gift of L. Naldini, Vita Salute San Raffaele University, Milan, Italy) expressing GFP in one direction and the introduced sequence in the other. Cloning was performed with StrataClone PCR cloning kit (Stratagene, La Jolla, CA). Virus packaging and cell infection was performed according to Follenzi et al. (13). Transfected I29 μ+ cells were selected on the basis of GFP expression. HEK-293 cells were transiently transfected with a solution of polyethyleneimine “MAX” linear MW 25000, (Polysciences, Inc, Warrington, PA), plasmid DNA, and PBS. The mix solution was incubated for 20 min at RT before addition to cultured cells for 6 hours.
Immunofluorescence
HEK293 cells were grown directly (and, when indicated, transiently transfected) on 10 mm2 cover-slips, were fixed in 3% paraformaldehyde and permeabilized with 0.1% Triton×100. After decoration with the indicated antibodies—rabbit anti-calreticulin was from Stressgen (Victoria, BC Canada), Alexa Fluor 488 goat anti-rabbit IgG (H+L) was from Molecular Probes—cells were stained with Hoechst (Molecular Probes), and then samples wereprepared in Mowiol.
Flow cytometry
About 1×106 differentiating B cells were washed in phosphate-buffered saline/0.5% bovine serum albumin, and cell death rate was assessed by propidium iodide (PI) staining following the manufacturer's instructions (BD Bioscience, San Jose, CA). After extensive washing, samples were analyzed by FACSCalibur and data were analyzed by the CellQuestPRO (BD Bioscience) or FCS Express (De Novo Software, Los Angeles, CA) software.
Statistical analysis
The quantified results are shown as mean values±S.D. Statistical analysis for enzyme activities was performed with one-way ANOVA followed by the Tukey–Kramer Multiple Comparison Test (GraphPad InStat).
Footnotes
Acknowledgments
We thank Zsófia Kováts for skillful technical assistance in the electron microscopy work, Valéria Mile for expert technical assistance, M. Geiszt for the catalase plasmids and L. Naldini for the lentiviral vectors. This work was supported by the Hungarian Scientific Research Fund (OTKA NN 78300), by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (to ÉM), by the New Széchenyi Plan (TÁMOP-4.2.1./B-09/1/KMR-2010-0001) and by FEBS short term fellowhip (to ÉM). We thank AIRC (IG and 5x1000 program), Regione Lombardia (ASTIL program), and Telethon Italy (GGP06155). The acquisition of a JEOL JEM-1011 transmission electron microscope was supported by a grant of Széchenyi Plan (TÁMOP-4.2.1./B-09/1/KMR-2010-0003).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.