
Abstract
Reactive oxygen species (ROS) play both positive and negative roles in the proliferation and survival of a cell. This dual nature has been exploited by leukemia cells to promote growth, survival, and genomic instability—some of the hallmarks of the cancer phenotype. In addition to altered ROS levels, many antioxidants are dysregulated in leukemia cells. Together, the production of ROS and the expression and activity of antioxidant enzymes make up the primary redox control of leukemia cells. By manipulating this system, leukemia cells gain proliferative and survival advantages, even in the face of therapeutic insults. Standard treatment options have improved leukemia patient survival rates in recent years, although relapse and the development of resistance are persistent challenges. Therapies targeting the redox environment show promise for these cases. This review highlights the molecular mechanisms that control the redox milieu of leukemia cells. In particular, ROS production by the mitochondrial electron transport chain, NADPH oxidase, xanthine oxidoreductase, and cytochrome P450 will be addressed. Expression and activation of antioxidant enzymes such as superoxide dismutase, catalase, heme oxygenase, glutathione, thioredoxin, and peroxiredoxin are perturbed in leukemia cells, and the functional consequences of these molecular alterations will be described. Lastly, we delve into how these pathways can be potentially exploited therapeutically to improve treatment regimens and promote better outcomes for leukemia patients. Antioxid. Redox Signal. 18, 1349–1383.
I. Introduction
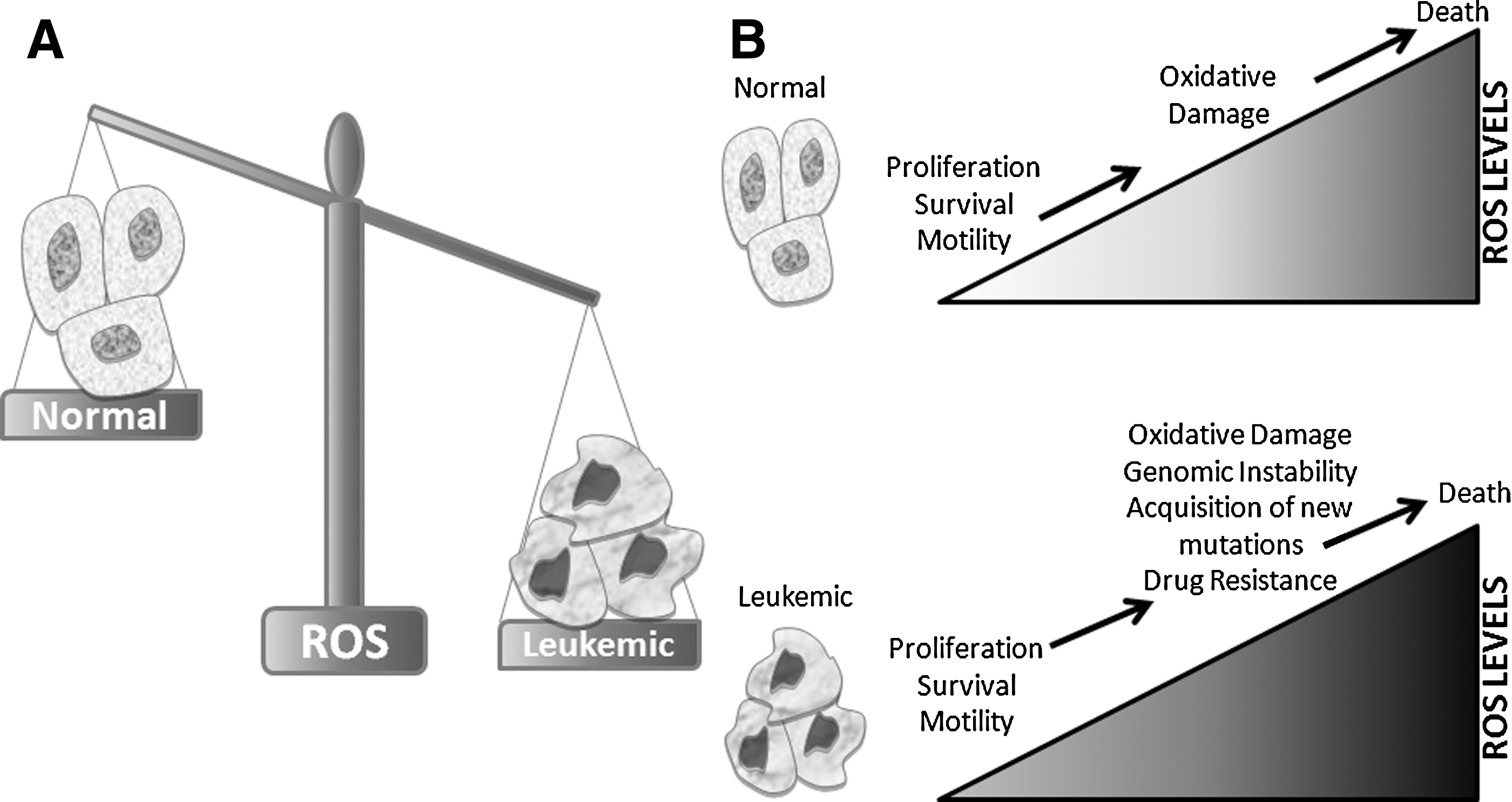
High levels of ROS promote oxidative damage, such as lipid peroxidation, amino acid oxidation, and DNA damage (245). When sustained by leukemia cells, these sequelae can actually promote leukemogenesis. For example, DNA damage produced by ROS can promote genomic instability, leading to advantageous DNA mutations for cancer growth and survival (Fig. 1B, bottom). In addition, leukemia cells frequently alter the expression and activity of a variety of antioxidant pathways (summarized in Table 1), which neutralize free radicals to less-reactive molecular components, preventing a potentially catastrophic redox imbalance. The same amount of oxidative stress is thought to have less of an effect on normal blood cells, because their basal ROS levels are lower.
Parameter indicates how the antioxidant is dysregulated (and the functional outcome).
SOD, superoxide dismutase; HO-1, heme oxygenase 1; GSH, glutathione; Trx, thioredoxin; Prx, peroxiredoxin; CML, chronic myeloid leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; ALL, acute lymphocytic leukemia; APL, acute promyelocytic leukemia; SOD1, Cu/Zn–superoxide dismutase; SOD2, Mn–superoxide dismutase; ATO, arsenic trioxide; EFS, event-free survival; Prx2, peroxiredoxin 2.
In the interest of focusing on leukemia biology and therapy, this review will not cover the effects of ROS on normal hematopoiesis. This topic is covered in a timely and comprehensive review by Hole et al. (103). The impetus to study the redox environment in leukemia is to understand and potentially halt leukemogenesis and to devise selective therapies. These strategies are predicated upon redox alterations unique to leukemia cells and thereby capable of sparing normal blood cells. The first half of this review addresses these alterations and is relevant to leukemogenesis and the discovery of targetable ROS-related molecules.
Altered redox biology in leukemia also has implications for therapeutics. Currently, there are ROS-producing therapeutics in practice and in clinical trials that employ oxidative stress to tip the balance from growth and survival to cell death. Standard and highly utilized leukemia therapeutics approved by the U.S. Food and Drug Administration (FDA) include anthracyclines, cytarabine, vincristine, and arsenic trioxide (ATO); all of these agents have been shown to produce ROS in some capacity (34, 110, 120). Similar reports have documented increased ROS levels by newer agents, such as histone deacetylase inhibitors (HDACi) and proteasome inhibitors (146, 168, 198, 250). Given that these drugs all produce ROS, it is not surprising that upregulation of various antioxidant enzyme systems can alter their effectiveness. Examples of these systems include superoxide dismutase (SOD), heme oxygenase 1 (HO-1), catalase, thioredoxin (Trx), peroxiredoxin (Prx), and glutathione (GSH). The second half of this review explores the use of redox-modulatory drugs as a tool in treating leukemia.
Five-year survival rates for patients with leukemia have improved over recent years, thanks to more effective therapeutic combinations. However, prognosis for specific leukemia types varies greatly. For example, 5-year survival is 24.2% for patients with acute myeloid leukemia (AML), but 78.4% for patients with chronic lymphocytic leukemia (CLL) (107). Resistance and relapse are major issues in the clinical treatment of leukemia and require more effective treatment strategies. Pro- and antioxidant pathways may contribute to the lack of response or resistance to therapeutic agents, and may promote proliferation and survival of leukemia cells, depending upon the context and cell type. Thus, improved understanding of the redox environment in leukemia will lead to benefit for leukemia patients.
II. How Does ROS Affect Leukemia?
A. Background on leukemia
According to the National Cancer Institute's Surveillance, Epidemiology, and End Results (NCI SEER) database, 1 in 80 Americans will develop leukemia in their lifetime (107). Generally defined as cancer of the blood and bone marrow cells, leukemia is categorized based on the primary type of cell affected and the disease course. Myeloid leukemia develops from the common myeloid progenitor lineage, which would otherwise become granulocytes and erythrocytes (Fig. 2A). Lymphocytic leukemia occurs in the common lymphoid progenitor lineage, where cells normally progress to become lymphocytes. Categorization by disease course distinguishes between acute and chronic leukemia. Acute leukemia is characterized by overgrowth and rapid accumulation of immature malignant blood cells. Chronic leukemia is characterized by a slower overgrowth of mature blood cells, and may take months to years to progress. Chronic myeloid leukemia (CML) has an unique and inevitable progression to an accelerated and then a blast phase of disease, which closely resembles acute leukemia. Thus, there are four primary subtypes of leukemia: CML, CLL, AML, and acute lymphocytic leukemia (ALL).

Leukemogenesis, whether acute or chronic, is thought to be a multistep process. Evidence from exposure-based leukemia (due to environmental leukogens such as benzene or treatment related to alkylating agents) has implied that development of a preleukemic state followed by acquisition of further genetic abnormalities leads to leukemia (84, 165). There are a variety of related theories regarding the mechanisms that promote leukemogenesis, suggesting that mutations occur in discrete classes of genes: oncogenes, DNA repair genes, and tumor suppressor genes. One such hypothesis from Kelly and Gilliland proposes that two broad categories of mutations that work together to promote tumorigenesis: Class I mutations, which confer proliferative advantage to the leukemia cell, and Class II mutations, which subvert differentiation (123). However, these theoretical systems exclude many of the complex mutations and epigenetic phenomenon that arise in leukemia.
A more inclusive model of leukemia progression includes driver and passenger alterations. Although there are many genetic abnormalities and epigenetic events in leukemia, few are true drivers of the disease. Driver alterations are defined as abnormalities that confer growth advantage early in hematopoiesis, whereas passenger events occur with, and support, these drivers [Fig. 2A and (91)]. There are many examples of passenger alterations in leukemia, and perhaps more important, many known leukemic oncogenes can be described as driver mutations. Some examples of these oncogenes include breakpoint cluster region–Abelson leukemia virus (BCR/ABL) tyrosine kinase, internal tandem duplication of the juxtamembrane domain of the fms-like receptor tyrosine kinase 3 (Flt3-ITD), Ras, and c-Kit, all of which confer proliferative and survival advantage to leukemia cells (22, 127, 137, 154). As discussed below, some of these driver alterations leverage the redox environment to promote leukemia progression.
It is important to note that ROS have been implicated in normal hematopoiesis (Fig. 2B). Hematopoietic stem cells (HSCs) have self-replicating potential that allows them to constantly replenish blood cells. Fluctuations of relatively low ROS levels control their self-renewal and proliferative capacity (264). HSCs are primarily found in a quiescent state within the stem cell niche in bone marrow (263). However, when HSCs are mobilized to the more oxygen-rich blood stream and begin to undergo proliferation and differentiation, ROS levels increase (Fig. 2B). This elevation of ROS levels can change the phenotype of HSCs in the blood stream (264).
Elevations in ROS can limit the lifespan of HSCs (113). A recent report analyzed lineage-negative CD34+CD38− populations (HSC populations) in a serial transplantation model, and found that the proliferative and repopulating capacity of HSCs was diminished with elevated ROS levels (264). Although the cell surface phenotype remained the same, elevated ROS levels in the HSCs correlated with increased DNA damage and decreased self-renewal capabilities. Further, when separated by ROS level, ROSlow CD34+CD38− cells retained their replicative potential, which was lost in ROShigh cells (264). Elevated ROS levels are also implicated in the maturation of HSCs into common committed lineage cells [Fig. 2B and reviewed in (103)]. Interestingly, a similar phenomenon may occur in leukemic stem cells (LSCs). Specifically, ROSlow LSCs retain the ability to engraft and develop into leukemia with both ROSlow and ROShigh populations. On the other hand, ROShigh LSCs are more actively cycling, have lower in vivo self-renewal potential, and more DNA damage than their ROSlow counterparts (139). Thus, ROS play similar roles in normal hematopoiesis and in leukemogenesis: promoting proliferation, but limiting pluripotency by promoting differentiation.
B. Leukemic oncogenes
Alterations in leukemic driver oncogenes are evident in nearly all forms of leukemia. These modifications can be categorized into three distinct groups: chromosomal translocations, mutations, and amplification or overexpression. Select examples of these groups are cited in Table 2.
BCR/ABL, breakpoint cluster region–Abelson leukemia virus; BCL-2, B-cell lymphoma 2; Flt3, fms-like receptor tyrosine kinase 3; ITD, internal tandem duplication; ZAP70, zeta-chain-associated protein kinase 70.
BCR/ABL is a prime example of a driver mutation caused by chromosomal translocation. The BCR/ABL oncogene is prevalent in CML and a subtype of ALL. This oncogene is the product of a chromosomal translocation between chromosomes 9 and 22 called the Philadelphia chromosome (137). As a driver oncogene for leukemia, BCR/ABL has the well-established functional effect of regulating proliferative and survival signaling in leukemia cells (137). In addition, due to its direct link with CML, BCR/ABL has been targeted therapeutically by a number of kinase inhibitors with excellent initial efficacy. However, relapse and resistance to these inhibitors remain major clinical issues (137).
There are also numerous driver leukemic oncogenes that are activated in leukemia cells by mutation, including Flt3-ITD, Ras, and c-Kit. Expressed in ∼30% of AML cases, Flt3-ITD is the most prevalent mutation associated with acute leukemia (127). Constitutive activity conferred by the ITD mutation increases proliferative and survival signaling in AML cells (127). Ras mutations are also fairly prevalent in AML (∼20%) and have a high incidence in chronic myelomonocytic and juvenile myelomonocytic leukemia (22, 25). As a GTPase, Ras is intricately involved in growth and survival signaling (59). Much like Ras mutations, c-Kit aberrations provide constitutive activity in ∼17% of AML (154). This activity affects proliferative, differentiation, survival, and migratory signaling in leukemia (154). Although these AML mutations are not as frequent as BCR/ABL in CML, they remain driver mutations that, like BCR/ABL (178), promote the hallmarks of cancer (47, 127, 154, 259).
C. How do leukemic oncogenes control the redox environment?
Recent evidence has suggested that oncogenes such as BCR/ABL, Flt3-ITD, Ras, and c-Kit not only directly manipulate leukemia signaling but also alter the redox homeostasis of cancer cells (209). As previously mentioned, redox control of leukemia cells can contribute to proliferative and antiapoptotic effects. It can also contribute to drug resistance. Thus, by regulating both ROS production and antioxidant expression/activity, oncogenes have another mechanism to directly influence leukemia progression, even during anticancer therapy. While the definitive source of ROS production and sustained elevation in leukemia has yet to be deciphered, growing evidence implicates the driver oncogenes described in section II.B.
BCR/ABL, the most studied oncogenic tyrosine kinase, is capable of inducing oxidative stress (222). Initial studies demonstrated that ROS levels are induced by BCR/ABL expression in both human and murine cell lines (222); this observation has been replicated by other researchers (125, 247). Exogenous application of hydrogen peroxide regulated the phosphorylation of BCR/ABL substrates Cbl, Shc, SHP2, and Abl itself, suggesting a role for ROS production in the oncogenic activity of BCR/ABL (222). Moreover, antioxidants such as N-acetylcysteine (NAC) and pyrrolidine dithiocarbamate decreased BCR/Abl kinase activity and phosphorylation of these substrates (222). Using samples from patients with blast-crisis CML as well as murine cell lines transduced with BCR/ABL, ROS formation was found elevated, which contributed to genomic instability and progression to blast crisis (185). BCR/ABL-induced ROS also caused mutations within BCR/ABL itself in human primary CML samples and murine leukemia cell lines (131). These mutations include M244I, E255K, and T315I, which promote resistance to BCR/ABL tyrosine kinase inhibitors (TKIs) (131, 226). The mutation rates were reduced by the antioxidant pyrrolidine dithiocarbamate, suggesting that both ex vivo patient samples and in vivo mouse models of BCR/ABL-positive CML produce mutation-inducing ROS (131).
BCR/ABL-induced ROS can also result in signaling alterations. For example, the nonreceptor tyrosine kinase Fyn is upregulated in a BCR/ABL-dependent, redox-responsive manner in CML cells (12, 77). Specifically, BCR/ABL-induced ROS production has been implicated in the transcriptional upregulation of Fyn by the transcription factor early growth response 1 (77). As evidenced by models of Fyn deficiency in the presence of BCR/ABL expression, Fyn is a mediator of leukemia growth, clonogenicity, and resistance to the BCR/ABL inhibitor imatinib (12, 111, 152). Furthermore, BCR/ABL is capable of influencing ROS production through manipulation of the NADPH oxidase (NOX) complex, in particular through upregulation of p47phox (described in section III.B). Such control of NOX alters downstream signaling to promote antioxidant expression (234), leading to imatinib resistance in CML (163, 164). Together, these results substantiate the idea that BCR/ABL induces ROS, which can lead to proliferation, cellular signaling, and drug resistance in leukemia cells.
The other oncogenic kinases mentioned (Flt3-ITD, Ras, and c-Kit) also have been shown to induce elevated levels of ROS in various model systems. Flt3-ITD causes heightened levels of ROS within murine Ba/F3 or 32D cells expressing Flt3-ITD as well as in human AML cell lines (MOLM-14 and MV-4-11) (221). Oncogenic Ras transgenic mouse models alter regulation of the redox environment, leading to myelodysplastic syndrome (MDS) (207). In addition, Ras mutations can lead to elevated ROS in human hematopoietic progenitor cells, resulting in growth factor independence (104). c-Kit has been noted to regulate NOX activity resulting in production of ROS (155). As to the source of leukemic oncogene-induced ROS, initial indications suggest that Rac1 is activated, leading to increased ROS production in AML cells (221). Rac1 is best known as a cellular GTPase that participates in a large number of physiological functions, including ROS generation (98). In the context of ROS production, Rac1 participates as a critical component in NOX1, NOX2, and NOX4 complexes (15), all of which are associated with leukemic ROS production (15). However, other sources of oncogene-induced ROS in leukemia remain to be explored.
In addition to these well-defined driver events, other alterations have been associated with redox control of leukemia. For example, B-cell lymphoma 2 (BCL-2) is another important factor in hematological malignancies. BCL-2 is an antiapoptotic protein that functions as an oncogene in CLL, AML, and ALL (2, 94, 132). Many compounds and strategies to inhibit BCL-2 have been explored in an effort to abolish BCL-2-dependent leukemia survival. Numerous reports have cited BCL-2 as a potent inhibitor of ROS in cancer cells via induction of antioxidant proteins [reviewed in (240)]. Inhibition of BCL-2 with the agent ABT-737/263 in ALL cells results in GSH depletion and increased oxidative stress (106), which supports the relationship between BCL-2 and GSH established by other researchers (240). More recent evidence has suggested that BCL-2, specifically in human leukemia cell lines, can also promote ROS production (136). In particular, BCL-2 was noted to amplify mitochondrial respiration and cytochrome c oxidase (COX) activity, leading to a prooxidant environment and apoptosis resistance in CEM human ALL cells (136). That study, however, did not analyze the effects of BCL-2 on the antioxidant environment of these cells.
There is no single defining oncogene in CLL that is comparable to BCR/ABL in CML. However, overexpression of the zeta-chain-associated protein kinase 70 (ZAP70), correlates with poor clinical prognosis in CLL. Much like the oncogenes present in other forms of leukemia, ZAP70 has been linked to ROS regulation in lymphocytes (119). The role of ZAP70 in ROS production in Jurkat human T lymphocyte cells has been evaluated. Cells deficient in ZAP70 expression contained less ROS than their ZAP70-expressing counterparts. The model system used was P116 Jurkat cells, which were initially mutagenized with the frameshift mutation agent ICR-191 and then discovered to be ZAP70 negative through a series of control experiments (257). Thus, ZAP70 may appear to be the primary defect of the cells, but it cannot be excluded that other proteins may also be altered in these cells. Therefore, more studies are necessary to directly implicate ZAP70 in the induction of ROS in CLL.
Altering mechanisms of ROS production is not the sole force behind elevations of ROS levels in leukemia. Leukemic oncogenes may also affect the transcription, stability, or activity of antioxidant proteins within leukemia cells. For example, BCR/ABL can increase the transcription of antioxidants such as HO-1, GSH, Trx, and Prx through upregulation of the transcription factor nuclear factor erythroid 2-related factor 2 [Nrf2; reviewed in (95)]. As we will discuss in section IV.A, activation of Nrf2 requires both ROS induction and phosphorylation, which releases the transcription factor from its negative regulators, allowing transcription of the antioxidants just mentioned (95). Transcriptional repressors of antioxidants, including BTB and CNC homology 2 (Bach2), are negatively regulated by BCR/ABL through activation of phosphatidylinositol 3-kinase (PI3K)/S6 kinase signaling (266). As mentioned above, BCL-2 can directly affect production of GSH (106, 240), altering the redox balance of ALL cells. Initial indications also associate c-Kit expression with elevations in GSH concentration in AML samples, although this association is purely correlative (81). Nonetheless, such examples indicate additional pathways by which leukemic oncogenes can influence the redox environment of leukemia cells, and are potential targets for therapeutic intervention.
III. Mechanisms of ROS Production in Leukemia
As with many other cancer cell types, ROS levels are upregulated in leukemia (119, 221, 222, 281). However, the definitive source of increased ROS has yet to be delineated. Elevated production of ROS can occur both as a result of leukemic oncogene activation, as described above (155, 178, 209, 221, 234), and potentially as a result of mutations that are independent of oncogenic activity such as those within the mitochondria (33) and cytochrome p450 (129, 134, 135) systems. Furthermore, downregulation of antioxidants can occur, leading to a net increase in total ROS levels through decreased clearance of ROS. This possibility will be assessed in section IV. While many cellular pathways and subcellular compartments can contribute to the production of ROS in cells, few specific pathways have been noted to directly affect leukemia. In this section, we examine the pathways of heightened endogenous ROS production as a molecular explanation for increased ROS levels in leukemia.
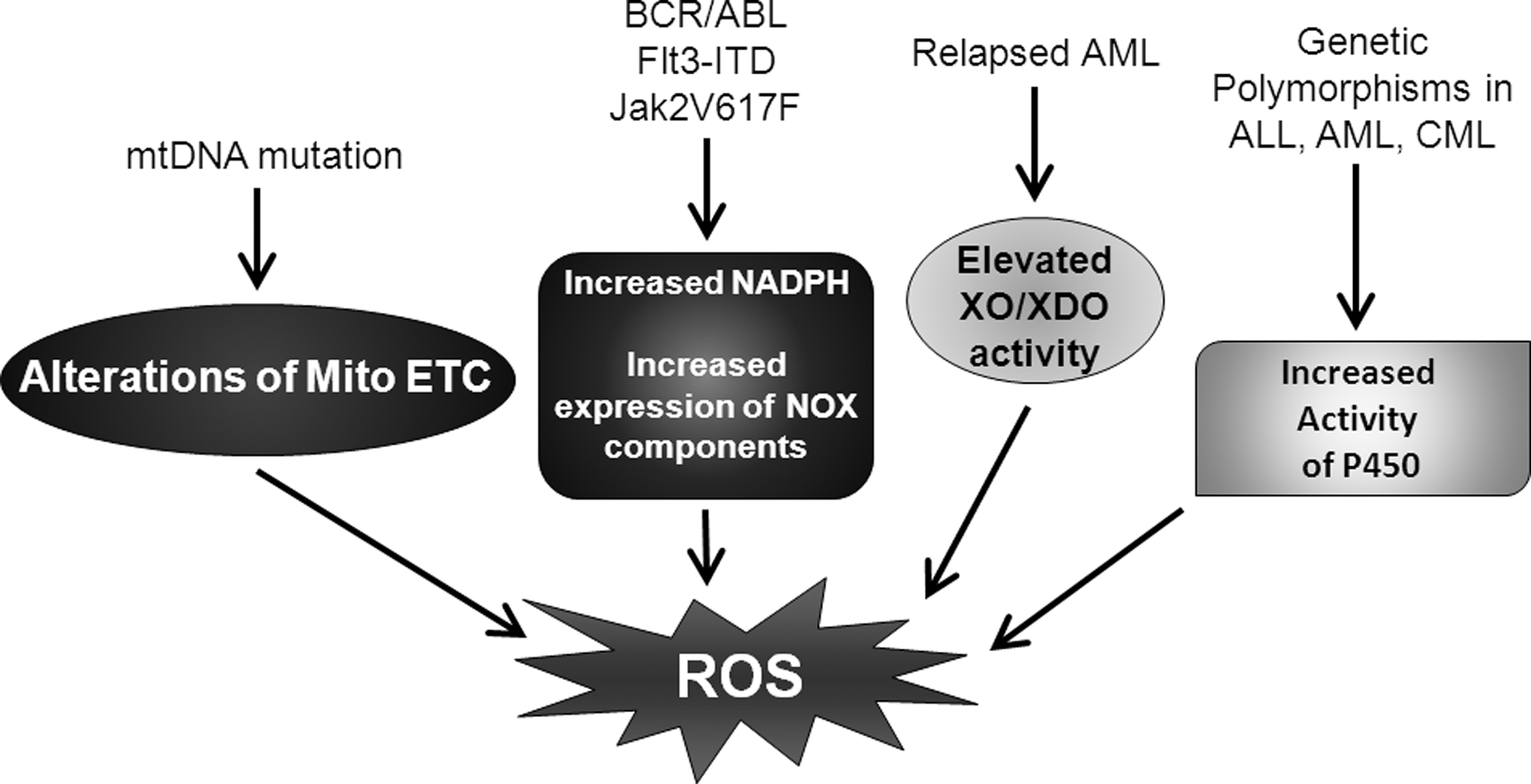
Two of the most studied sources of leukemia cell ROS are the mitochondrial electron transport chain (ETC) and the NOX complex [(155, 209, 232) and Fig. 3]. These are the major endogenous sources of ROS in any cell type. Mutations in mitochondrial DNA (mtDNA) appear in leukemia, resulting in alterations of the mitochondrial ETC and potentially elevated ROS production (30, 33). Leukemic oncogene activity can result in increased expression of a variety of NOX components as well as the production of the rate-limiting substrate NADPH (178, 209, 234). Additionally, metabolic and detoxification pathways, such as xanthine oxidoreductase and cytochrome P450, may contribute to leukemic cell ROS production (262, 278, 279). Elevations in xanthine oxidase (XO) activity are found in relapsed AML (278, 279), which contribute to increased ROS levels (278). Further, as with mutations of mtDNA, genetic polymorphisms of cytochrome p450 associated with leukemia (129, 134, 135, 262) may also result in elevated ROS, though more studies are necessary to validate this suggestion. All of these ROS-producing systems have been suggested to promote genomic instability, survival, growth signaling, and drug resistance in leukemia. Thus, a further understanding of the molecular regulation of ROS production by these pathways is critical to the treatment of leukemia patients.
A. Mitochondrial respiration: a source of life and death for the leukemia cell
The mitochondrial ETC generates an estimated 90% of ROS in a given cell [reviewed in (10)]. In leukemia cells, the mitochondrial ETC is also a major source and target of ROS (204, 232). The ETC is the principal contributor to the production of adenosine triphosphate (ATP) within cells, and is therefore responsible for the viability of both normal and cancer cells. The ETC is located along the inner mitochondrial membrane and contains four membrane protein complexes (I–IV; Fig. 4A) and the ATP synthase. These complexes cooperate to transfer electrons to molecular oxygen, creating a hydrogen gradient in the process. Almost all (98%–99%) of the oxygen consumed during respiration is converted to water; 1%–2% is only partially reduced, leading to generation of superoxide anion (24). Many reactions can produce superoxide during this process, but the main sources are thought to be complexes I and III (249).

Evidence from ALL patient samples has suggested a role for mitochondrial-induced ROS in leukemogenesis (232). ROS production by complex I of the mitochondrial ETC was found to be important for the survival of interleukin 7-dependent T-ALL cells (232). Specifically, treatment with rotenone, which inhibits the transfer of electrons from NADH dehydrogenase (ND) I of complex I to ubiquinone (130), resulted in decreased ROS in T-ALL cells (232). In addition, rotenone treatment abrogated interleukin 7-mediated viability of the cells as measured by annexin V/propidium iodide staining. On the other hand, mitochondrial ROS production is not always stimulatory and protective for leukemia cells. Mitochondrial ROS also play a role in apoptosis induction, rather than survival, in human AML cell lines (204). Specifically, drug-induced ROS, produced by complex III, induced alterations in calcium homeostasis, mitochondrial membrane permeability, and ultimately cell death sensitization in AML cell lines (204). These findings emphasize that leukemia cells rely on higher levels of ROS for viability and highlight the possibility of using this elevated level of ROS and endogenous ROS production as a therapeutic advantage.
Additional data suggest that heightened ROS in leukemia cells may cause mutations in mtDNA (33), and that these mutations can further increase ROS production (8). mtDNA lacks mechanisms of protection that are present in nuclear DNA, such as extensive chromatin organization and multiple repair processes. In addition, due to its sheer proximity to the ROS-producing ETC, mtDNA is highly susceptible to mutations (10, 253). mtDNA encodes for 13 of the 87 subunits required for mitochondrial ETC function: seven subunits of complex I (ND), one subunit of complex III (cytochrome b), three subunits of complex IV (COXs I–III), and two subunits of the ATP synthase (ATPase 6 and 8) [(197) and gray subunits in Fig. 4A]. Figure 4B summarizes the known mtDNA mutations that affect a variety of ETC components in leukemia (33, 79, 96, 200, 210). Such mutational alterations in mtDNA can lead to deregulated expression and activity of these components, resulting in elevated production of ROS (197).
As mentioned, mtDNA is susceptible to mutations that can emanate from endogenous or exogenous sources (253). Chemotherapeutic agents such as fludarabine, which is used in the treatment of CLL, can induce mtDNA mutations (33). Specifically, CLL cells from patients pretreated with chemotherapeutic agents contained mutations in ND1 and ND4, cytochrome b, COX, and ATPase 6 [(33) and Fig. 4B]. Such mutations correlated with increased ROS generation and lack of clinical response to chemotherapy (33). However, these results do not account for the possibility that ROS may also be the cause, rather than the consequence, of mtDNA mutation. Similar findings have been observed with another chemotherapeutic drug used in acute leukemia, doxorubicin. In cardiac cells, doxorubicin decreases levels of mtDNA-encoded complex IV subunits, again potentially affecting ROS production (193). These results have yet to be recapitulated in leukemia cells, but in light of the effects of fludarabine in CLL, there is great potential for a similar effect in acute leukemia. Thus, it seems that mitochondrial ETC may be a dually edged sword for leukemia cells, promoting growth, survival, and drug resistance, but also sensitizing leukemia cells to death by some agents.
B. NOX as a source of oncogene-induced ROS
In addition to mitochondrial respiration, the NOX family of proteins contributes to elevations of ROS in leukemia, leading to leukemia cell proliferation and survival (178, 209, 232), and alterations of therapeutic response (254). The NOX family consists of seven members: NOX 1–5, DUOX1, and DUOX2. NOX enzymes have the common functional activity of producing superoxide anion by transferring an electron from NADPH through the NOX complex to molecular oxygen (15). A number of subunits make up fully functional NOX complexes (Fig. 5A), including organizer subunits (p47phox or NOXO1), activator subunits (p67phox or NOXA1), and nonessential modulatory subunits (e.g., p40phox) [reviewed in (15)]. DUOX1 and DUOX2 also contain specific maturation factors, DUOXA1 and DUOXA2 (15). p22phox is required for stability of NOX1-4. Further, either of the two isoforms of Rac (1 and 2) associates with the assembled NOX1 or NOX2 to create an active complex (association with NOX3 is debatable, and the remaining complexes do not require Rac) (15). The NOX isoforms vary widely by tissue expression. Of particular importance to this discussion, NOX1, NOX2, and NOX4 isoforms are expressed in HSCs and leukemia (15, 194).

NOX isoforms have been described to be altered in leukemia, leading to elevations in ROS levels. Such changes have been specifically linked to sustained leukemia cell viability, proliferation, and migration (178, 209, 232), as well as altered drug susceptibility (254). Leukemic oncogenes are capable of controlling the expression, assembly, and activity of a variety of NOX components. Such control can be found on multiple levels. First, transcriptional regulation can occur (Fig. 5A). NOX4, important in the production of ROS in CML (178), can be induced by the redox-responsive Nrf2 transcription factor. Nrf2 (described in section IV.A) binds to antioxidant-response elements (AREs) upstream of the transcriptional start site within the promoter of the NOX subunit genes. Similarly, mRNA expression of a myriad of components specific to NOX2, NOX4, and NOX5 (including p22phox, p40phox, p47phox, and p67phox) is elevated in human leukemia cell lines that have been transformed by oncogenic tyrosine kinases (209). Elevated expression of NOX components leads to increased NOX activity, as treatment with diphenylene iodonium (DPI, a flavoprotein inhibitor capable of reducing NOX activity) or genetic inhibition of NOX components using small interfering RNA resulted in reduced ROS levels in the CML, AML, and MDS model cell lines (209). Further evidence regarding the regulation of the redox status of leukemia cells by NOX is present in CML, in which inhibition of NOX activity with DPI significantly reduced ROS levels (234). NOX activity also elevated expression of the antioxidant HO-1 (234), suggesting that not only does oncogene-induced NOX activity result in elevated ROS production, but NOX is also capable of inducing antioxidant expression, thereby altering the balance of oxidative stress in the cells.
While leukemic oncogenes can control expression of the individual NOX components, it seems that activation and assembly, rather than expression, are the primary point of regulation of NOX by oncogenes. For example, expression of BCR/ABL, Flt3-ITD, or Jak2V617F oncogenes results in increased availability of NADPH, the rate-limiting substrate for NOX activity (Fig. 5B). Such an elevation in NADPH results in amplified NOX-induced ROS production (209). As a multisubunit enzyme complex, NOX also requires assembly of its constituent components on the membrane. It is thought that phosphorylation of the organizer subunits of NOX by protein kinase C or by Akt and mitogen-activated protein kinase (MAPK) is a critical step in translocation of the components of the complex to the membrane where they can interact to form active NOX (15). While these results remain to be shown specifically in leukemia, enhanced signaling through these phosphorylation pathways is apparent in various leukemia subtypes. Specifically, the PI3K/Akt and MAPK pathways have both been noted to be activated by leukemic oncogenes [for reviews see (55, 82, 159)]. Thus, these pathways may lead to aberrantly active NOX and therefore elevated ROS production in leukemia.
Chemical and genetic inhibition of NOX2, NOX4, and p22phox has revealed a role for NOX-induced ROS in oncogene-induced growth and migration in the CML, AML, and MDS models (209). DPI treatment of the KU812 (BCR/ABL), MOLM-13 (Flt3-ITD), and HEL (JAK2V617F) cell lines resulted in decreased production of ROS, and lentiviral short hairpin RNA constructs against NOX2, NOX4, and p22phox decreased superoxide production in these model cell lines (209). Furthermore, cell growth and migration were also impaired with knockdown of p22phox.
Cytokine-dependent growth of T-ALL cells also requires NOX activity. Interleukin-7-dependent growth induction of T-ALL cells relies on increased levels of intracellular ROS produced by NOX and the mitochondrial respiratory chain (232). The viability of these cells was sufficiently abrogated by inhibition of NOX with apocynin (232), which is not a specific NOX inhibitor, but has been reported to cause disassembly of the NOX complex (15). Thus, cytokine-dependent proliferation of T-ALL cells may require a fully assembled NOX complex. NOX may also play a role in the progression of leukemia and in the development of refractory leukemia. Specifically, NOX-induced ROS was involved in the acquisition of genomic instability in Flt3-ITD-expressing AML (221). Such genomic instability may contribute to drug resistance and the acquisition of new mutations, leading to further progression of the disease.
Elevated NOX activity may also allow a unique therapeutic opportunity for leukemia patients, as it may be indicative of sensitivity to ROS-producing therapeutic agents, at least in acute promyelocytic leukemia (APL). Human APL cells, which contain elevated NOX activity, are highly susceptible to ATO treatment (254). In addition, inhibition of NOX in these cells with DPI decreased their sensitivity to ATO. As mentioned, DPI is a broad flavoprotein inhibitor that is also capable of inhibiting mitochondrial ROS. However, other inhibitors of mitochondrial ROS generation (e.g., rotenone, antimycin A, and potassium cyanide) were unable to inhibit ROS production in these cells, suggesting that NOX is the major contributor to this phenomenon (254). Together, the data from these many studies suggest that NOX expression, activation, and complex formation are critical to leukemogenesis, progression of leukemia, and treatment of the disease.
C. Metabolic/detoxification pathways and ROS production in leukemia
In addition to the mitochondrial ETC and NOX complex, there are other metabolic/detoxification pathways that produce ROS within leukemia cells. Xanthine oxidoreductase, for example, is elevated in expression in some leukemia (278, 279) and is capable of elevating ROS in leukemia cell lines (278). The xanthine oxidoreductase system is composed of two independent species, XO and xanthine dehydrogenase (XDO). XDO prefers NAD+ as a cofactor, while XO cannot reduce NAD+ and thus uses molecular oxygen as a cofactor, resulting in production of either superoxide anion or hydrogen peroxide. XO activity is not alone in its ROS-producing abilities. XDO will also reduce molecular oxygen and produce ROS, although less effectively than XO.
Besides evidence showing that the enzyme xanthine oxidoreductase exists and can produce ROS under hypoxic conditions in lymphocytic leukemia cells (278), little has been discovered regarding the definitive role of the xanthine oxidoreductase system in leukemia, and further studies to elucidate a distinct role for this ROS-producing enzyme are required. However, XO activity is higher in plasma samples from AML patients than from normal control samples (279), and upon disease relapse, there is an additional elevation in XO activity in the plasma, suggesting a role in the recurrence of AML (279). The xanthine oxidoreductase system is also involved in the metabolism of many drugs, including 6-mercaptopurine (6-MP) (11), which is used to treat childhood ALL. Therefore, this enzyme system is potentially important to the treatment of leukemia and presents an interesting target for further evaluation.
The cytochrome P450 (CYP) family of proteins could also be linked to increased ROS production in leukemia. These proteins are encoded by a variety of polymorphic genes important in the activation of polycyclic hydrocarbons, metabolism of carcinogens, and reduction of molecular oxygen to free-radical species (129, 179). Mutations in CYP proteins have the potential to lead to elevated activation and an increased cancer risk as a result of DNA adduct formation (121, 235). Activating polymorphisms in this protein family are associated with leukemia, although the direct role of CYP polymorphisms in leukemia cell ROS production remains to be defined.
Specific activating polymorphisms of CYP associated with an elevated risk for ALL include CYP1A*2A, CYP1A*2B, CYP1A*2, CYP1A*4, and CYP2E1*5 (134, 135, 235). Another CYP family member, CYP2J2, is selectively expressed in K562 (CML), HL-60 (AML), MOLT-4 (ALL), and Jurkat cell lines, as well as in primary cells from patients with either acute or chronic leukemia (40). Such expression was associated with increased leukemic burden in xenograft models (40). Further, inhibition of CYP2J2 with C26 resulted in apoptosis of leukemia cells (40), suggesting that this CYP protein is important for leukemia proliferation and survival. In light of their described functional activity to reduce molecular oxygen, the cytochrome P450 family may be important in the ROS production of leukemia cells. However, further studies are required to reveal the specific role of these activating mutations in the redox control of leukemia.
In summary, there are a variety of mechanisms by which ROS can be produced in leukemia cells. These sources include intricate pathways such as the mitochondrial ETC, complex systems such as the NOX family, and single-metabolic or detoxification enzymes like xanthine oxidoreductase and cytochrome P450. Alterations in these pathways help to maintain and elevate the level of ROS within leukemia cells, leading to a proliferative advantage and alterations in drug susceptibility. These functional roles for endogenous ROS-producing pathways make them interesting targets to explore for therapeutic manipulations.
IV. Cellular Antioxidants in Leukemia
Another source of oxidative stress and potentially of ROS induction in leukemia is the decrease of antioxidant levels. Altered expression of antioxidant proteins or their dysregulation is seen in all four types of leukemia, which suggests the importance of these enzymes to the disease. Altered expression of antioxidants can result in a variety of outcomes. For example, elevated antioxidant expression or activity can promote resistance to therapeutic agents. On the other hand, decreased antioxidants can lead to elevations in ROS levels, resulting in proliferation and genomic instability. The resulting oxidative stress can promote a feedback loop in which antioxidants are upregulated as a response.
Consistent with these scenarios, antioxidants are reported to be both up- and downregulated in leukemia models and primary human leukemia cells (Table 1). Specifically, SOD protein expression and activity are often elevated in serum and plasma samples, respectively, from patients with acute leukemia (184, 282). The human CML cell line K562 also has increased SOD protein expression, conferring resistance to some therapeutic agents (265). Increases in the protein expression of the antioxidants GSH, Trx, and Prx have been illustrated for acute leukemia (Table 1). Catalase is also highly expressed in AML patient samples (149) and in plasma samples from B-cell CLL patients (269). Further, expression of the antioxidant HO-1 protein can also be upregulated (172, 217). Elevations in antioxidants such as those described here may contribute to an imbalance in the redox status of leukemia cells, leading to oxidative stress. Furthermore, elevated antioxidant levels allow leukemia cells to survive otherwise lethal alterations in ROS.
Downregulation of antioxidant activity or expression is also seen in leukemia specimens (Table 1). This downregulation may be transient (i.e., in response to therapy or ROS production) or permanent (i.e., in response to genetic inhibition). Specifically, both SOD activity and catalase activity are reduced in lymphocytes from ALL (223) and CLL patients (187). Further, Prx 4 expression is epigenetically reduced in APL patient samples (190).
Thus, depending on the regulatory event, the expression of some antioxidants is increased, allowing the cells to more readily cope with an increased production of ROS, while the expression of other antioxidants is decreased, allowing ROS to sustain second-messenger signaling. Regardless of the direction of dysregulation, these molecular alterations in redox homeostasis result in progression of the leukemia phenotype (109, 163, 269) and alter drug sensitivity (138, 162, 164, 265). Due to these findings, studies have begun to determine the role antioxidants play in leukemogenesis, progression, and drug resistance. In this section, we review six antioxidant pathways associated with redox imbalance in leukemia cells: SOD, catalase, HO-1, GSH, Trx, and Prx. We also discuss how alterations in these pathways can be both beneficial and detrimental to leukemia cells and treatment of the disease.
A. Nrf2: a master regulator of antioxidant transcription
Before we discuss how each individual antioxidant family contributes to leukemogenesis and treatment options, it is important to understand how they are primarily regulated. In addition to potential post-translational modifications, antioxidants can be regulated at the level of gene expression. Controlling the expression of antioxidant genes are a number of transcription factors, such as activating protein 1 (AP-1), nuclear factor κB (NFκB), and Nrf2. As mentioned earlier, Nrf2 can transcriptionally control activation of constituent subunits of the NOX complex. However, a primary role of Nrf2 is the transcriptional activation of antioxidants such as HO-1, Trx reductase-1 (TrxR1), Trx, Prx, glutathione-S-transferase (GST), GSH reductase, and enzymes involved in the biosynthesis of GSH in response to oxidative stress (95). Indeed, Nrf2 activity itself is elevated in some leukemia types where it contributes to leukemogenesis (219). Elevated nuclear localization of Nrf2 and the subsequent genetic changes result in decreased sensitivity to proteasome inhibitors in AML cell lines (216), suggesting that Nrf2 may also coordinately regulate sensitivity to ROS-producing therapeutic agents.
Molecular analyses have revealed that treatment with stress inducers (e.g., tumor necrosis factor) results in increased Nrf2 activity in THP-1, HL-60, and AML 193 AML cell lines, which in turn increases the transcription of antioxidants (218). A stress stimulus, such as oxidative stress, plus a phosphorylation event, induces release of Nrf2 from Kelch-like ECH-associated protein 1 (Keap1), which then allows Nrf2 to translocate to the nucleus (Fig. 6A). Once inside the nucleus, evidence suggests that Nrf2 binds to AREs as a heterodimer with small Maf proteins (MafF, MafG, and MafK) (95). Maf proteins are required for transcriptional activity of Nrf2, as knockout of all three Mafs abolishes antioxidant induction [reviewed in (95)]. Thus, release of Nrf2 from Keap1 and the subsequent translocation of Nrf2 result in binding of Nrf2 to the antioxidant promoters, enhancing transcription.
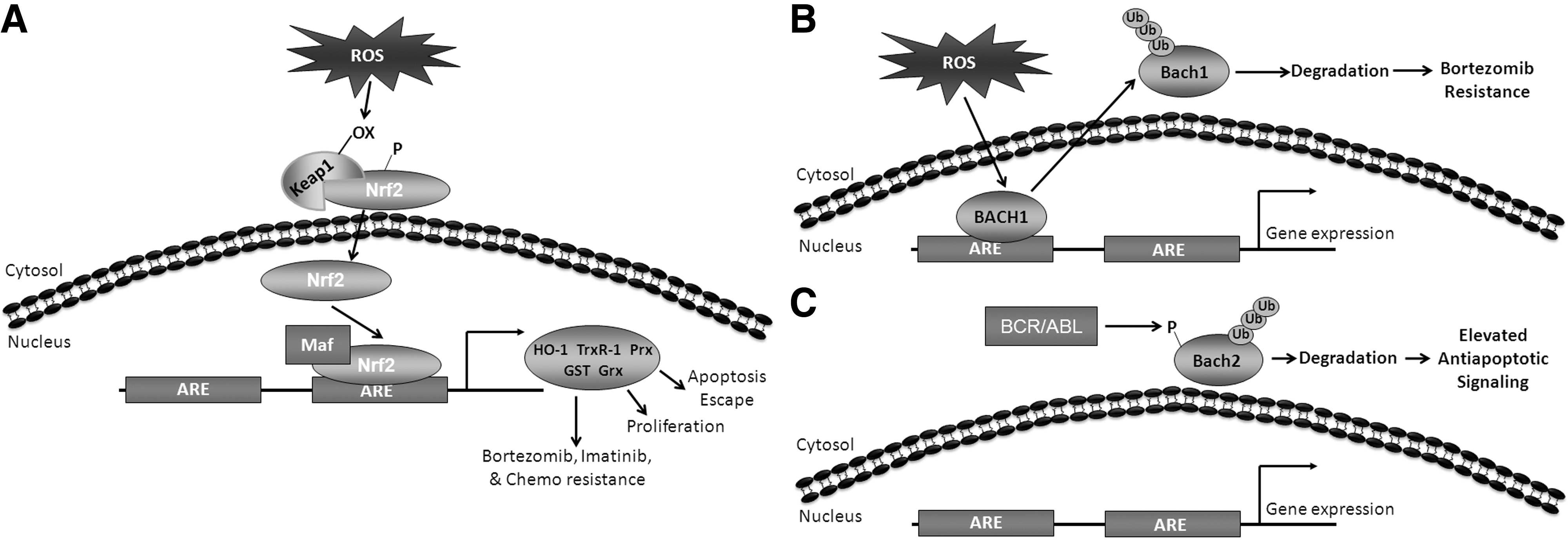
An additional level of regulation can be found within the nucleus, where the Bach1 and Bach2 transcriptional repressors are thought to compete with Nrf2 for binding to AREs to regulate the transcription of antioxidants [reviewed in (95)]. Interplay between Bach1 and Nrf2 is apparent in AML cell lines (216). ROS cause oxidation of cysteine residues within the Bach1 transcriptional repressor, allowing dissociation of Bach1 from ARE, its translocation to the cytosol, and finally its degradation [Fig. 6B and (112)]. Knockdown of Bach1 in THP-1 and bortezomib-resistant AML-307 cell lines allows for induction of Nrf2-specific genes, enhancing drug resistance (216). Bach2 is negatively regulated by BCR/ABL in CML (251, 266). Phosphorylation of Bach2 by BCR/ABL results in its retention in the cytoplasm, where it is degraded (Fig. 6C). However, when active and present in the nucleus, Bach2 represses Nrf2-dependent transcription (266). All of these findings together make it evident that Nrf2 is a particularly powerful regulator of the redox environment of leukemia.
B. Enzymatic elimination of ROS
1. Superoxide dismutase
The primary role of SOD is to catalyze the dismutation of superoxide into molecular oxygen and hydrogen peroxide, which is then further processed by catalase [discussed in section IV.B.2 and reviewed in (73)]. Thus, through the reduction of superoxide, SOD is an antioxidant. Four primary classes of SODs exist and are named based on their metal ion cofactor: Cu/Zn, Mn/Ni, Fe, and Mn (73). The two types of SOD prevalent in mammalian cells are Cu/Zn-SOD (SOD1) and Mn-SOD (SOD2). A third SOD molecule, extracellular Cu/Zn-SOD (SOD3) has been identified, but has not been studied in relation to leukemia (73). SOD1 is present in the cytoplasm, nucleus, and plasma, whereas SOD2 is found at the mitochondria and is the primary isoform associated with oxidative stress (73).
The association between the expression and activity of SOD and leukemia is contentious, primarily because it is only beginning to be examined. As such, it has yet to be clearly defined if SOD is up- or downregulated in various types of leukemia. Differences in the expression of SOD in leukemia blasts versus the serum levels of this enzyme have added further complexity. Lymphocytes from ALL (14, 223) and B-cell CLL (187) patients contain decreased levels of total SOD activities; however, SOD (specifically SOD2) protein expression is increased in serum from acute leukemia patients compared with that from healthy control subjects (184). In addition, disease regression has been associated with a decrease in SOD2 protein level in the serum of acute leukemia patients (184). The discrepancy between whether SODs are elevated or diminished in leukemia could be explained by differences between analyses of SOD in leukemia blasts and in serum from patients. Indeed, serum protein levels of SOD do not correlate well with intracellular SOD protein expression (184). Another issue is the absence of descriptions of SOD isoform specificities. Although such delineations are not usually made in the SOD literature, both SOD1 (282) and SOD2 (184) activities have been independently shown as elevated in samples from acute leukemia patients. These results indicate that inquiries should be made into SOD isoform-specific expression and activity in leukemia samples.
Regardless of these inconsistencies, SOD expression is functionally required in leukemia. In a study assessing the mechanism of cytotoxicity produced by 2-methoxyestradiol (2-ME), inhibition of total SOD function resulted in apoptosis induction in patient samples from all four subtypes of leukemia; this induction was due to elevated superoxide and free radical-mediated mitochondrial damage (109). In addition, inhibition of SOD activity was noted with this cytotoxic agent, and SOD depletion with antisense oligonucleotides resulted in enhanced 2-ME activity, suggesting that SOD and 2-ME inhibit the same pathway. Further investigations of how SOD functional activity can control ROS levels in leukemia are required to fully substantiate these data.
Additional studies have highlighted the importance of SOD in regard to therapeutic resistance. In particular, elevated activity of SOD1 in K562 cells correlated with resistance to radiation, whereas HL-60 cells, which have lower SOD1 activity, were sensitive to radiotherapy (265). Furthermore, when SOD1 activity was elevated in CML cell lines, the cells were more resistant to ROS-inducing drugs. While this study also monitored activity levels of SOD2, there was no correlation between SOD2 activity and response to radiotherapy or ROS induction (265), though further studies are required to confirm these findings. Together, these data suggest that the SOD family of antioxidants may be important therapeutic targets.
With that in mind, researchers have developed therapeutic approaches for treating leukemia that may directly modulate SOD as a part of their mechanisms of action. An example is the use of tannic acid, which reduced SOD activity to increase ROS levels. Tannic acid also induced apoptosis and increased the sensitivity of AML cells to ATO (41). However, the direct connection between reduced SOD activity and sensitivity to ATO was not addressed. Therefore, the association between sensitization to ATO and SOD activity remains correlative, and further studies to directly implicate SOD activity in this effect are required. Nevertheless, the fact remains that some studies suggest that SOD activity and expression may be suppressed in leukemia, while others describe a key role for SOD in potentiating apoptosis escape and drug resistance.
2. Catalase
As we have mentioned, SOD reduces superoxide levels while creating hydrogen peroxide in leukemia. Hydrogen peroxide can be decomposed to a hydroxyl radical and thereby do immense damage to the cell (268). Thus, a mechanism to clear hydrogen peroxide is required. Catalase enzymes coordinate the breakdown of two molecules of hydrogen peroxide to water and molecular oxygen (268). Two primary classes of catalase enzymes exist in humans: monofunctional and catalase peroxidases (268). Catalase enzymes localize primarily within peroxisomes and the cytoplasm; minor levels of these enzymes may also be found within the mitochondrial matrix (39). Overexpression of catalase is protective to cells in that it increases life span and decreases injuries that arise from ROS generation. Like many of its antioxidant counterparts, catalase is altered in leukemia, leading to functional consequences such as increased proliferation, elevated genomic instability, and resistance to therapeutic agents.
Increased expression or activity of catalase has been noted in a number of leukemia subtypes. For example, a fivefold induction of catalase activity was seen in AML cell lines compared with normal granulocytes (133). A proteomic approach revealed a similar elevation in catalase protein in AML patient samples (149). A threefold increase in catalase activity in CML samples compared with normal granulocytes was also shown, which was lost upon induction of differentiation with tunicamycin (133). Catalase activity was increased in the plasma of B-cell CLL patients compared with the plasma of control subjects and correlated with disease progression (269). Functionally, catalase has been suggested to regulate proliferation and resistance to some ROS-producing therapeutic agents (e.g., ATO) in AML cells (49, 92, 116). In addition, chemical inhibition of catalase with 3-amino-1,2,4-triazole, an irreversible inhibitor that binds the active center of catalase (156, 157), increased apoptosis in K562 cells when combined with imatinib, suggesting a role for catalase in enhancing imatinib sensitivity (138).
Unlike myeloid cells, ALL cells contain lower levels of catalase activity compared with normal cells, potentiating genomic instability due to elevations in hydrogen peroxide (223). Also, catalase activity has been reported to be decreased within primary CLL cells compared with normal cells (187). This finding is in contrast to another study that found catalase levels unaltered in blood lymphocytes from B-CLL patients compared to normal counterparts (67). The patient populations did differ between these two studies: Oltra having twice the patient numbers than Farber et al., and the treatment status of the population may have varied; the Farber study patients were untreated, and the Oltra study did not describe the treatment status of their population. Lastly, while both groups used a colorimetric assay to determine activity, Farber et al. measured at a single timepoint, while Oltra et al. determined a rate of activity over time. Together, these differences may explain these contradictory results. Overall, catalase seems to play conflicting roles depending on the type of leukemia in which it is expressed and treatment status. For example, elevated catalase activity plays a tumor-promoting role in myeloid leukemia, contributing to both disease progression and resistance to therapeutics (Fig. 7A). However, in lymphocytic leukemia, decreased catalase activity contributes to the acquisition of genomic instability, leading to mutations and increased disease burden (Fig. 7B), likely because decreased defense against peroxides is providing an environment permissive of secondary mutations.

3. Heme oxygenase
Another enzymatic defense against ROS in leukemia is the HO enzyme family, which has been functionally implicated in both CML and AML and neutralizes the highly cytotoxic, free radical-producing heme (167). Multiple isoforms of HO exist, but the predominant isoform involved in redox biology is HO-1. Functionally, the end products of the enzymatic reaction catalyzed by HO-1 are elevated levels of carbon monoxide and bilirubin, which may have further biological activities. For example, carbon monoxide has signaling properties that can result in decreased apoptotic signaling through activation of Akt (273) and biliverdin, which when converted to bilirubin acts as a physiological antioxidant (13, 237, 238).
HO-1 is regulated primarily at a transcriptional level in response to oxidative stress. Redox-dependent transcription of HO-1 relies heavily on two upstream enhancer regions, E1 and E2, which contain AREs (5, 6). HO-1 can be regulated by a number of transcription factors, including Nrf2, Bach1, NFκB, and AP-1. In leukemia, the primary enhancer of HO-1 gene expression appears to be Nrf2. However, at least in AML, constitutive expression may be limited to distinct molecular subtypes: upregulation of HO-1 was found in samples from the French-American-British (FAB) M4 subcategory of AML patients as compared with samples from other FAB categories (172). In AML cell lines (THP-1 and HL-60), HO-1 expression was shown to be relatively low, although it could be readily induced by cellular stress (218).
In addition to Nrf2 (described more thoroughly in section IV.A), other transcriptional activators play a role in HO-1 induction. For example, NFκB may promote stress-induced HO-1 expression in mice and rats, although the role of NFκB in HO-1 transcription in humans has yet to be elucidated (144, 174). In AML, NFκB plays a primarily inhibitory role in HO-1 expression. Chemical inhibition of NFκB resulted in elevation of HO-1 protein levels in THP-1 and HL-60 cells, demonstrating that the HO-1 promoter can be repressed by NFκB (218). In CML, HO-1 mRNA and protein levels are upregulated due to BCR/ABL in both cell lines and patient samples (163). In this cell type, transcriptional upregulation of HO-1 is a result of PI3K/S6 kinase-negative regulation of the transcriptional repressor Bach2 (Fig. 8A and (266); see also Fig. 6C).

Elevated HO-1 expression also correlates with survival and drug resistance in AML cells [Fig. 8B and (97, 172, 217)]. Specifically, inhibition of HO-1 combined with NFκB inhibition resulted in apoptosis of THP-1 cells and patient samples (217). Perhaps most important, therapeutically, induction of HO-1 was seen in AML cells after treatment with the standard-of-care chemotherapeutic agents cytarabine and daunorubicin (97). In that study, genetic knockdown of HO-1 increased the sensitivity of chemoresistant AML cell lines to these drugs (97), suggesting a role for induced HO-1 in the chemoresistant phenotype.
Similar results have been observed for CML, where HO-1 expression is required for cell survival, proliferation, and TKI resistance. Chemical inhibition of HO-1 with zinc-(II)-deuteroporphyrin-IX-2,4-bisethyleneglycol (a metalloporphryin) resulted in decreased viability of K562 cells (163). The mechanism by which HO-1 is induced in CML cells has only recently been explored. Data suggest that the NOX complex plays a primary role in the upregulation of HO-1 in CML (Fig. 8A). Specifically, ROS-dependent upregulation of HO-1 was noted in murine p210-expressing and K562 CML models (234). Inhibition of the NOX complex via introduction of dominant-negative Rac1, chemical inhibition of Rac1, or small interfering RNA knockdown of p47phox resulted in decreased HO-1 expression. These results indicate that NOX may directly regulate HO-1 expression in CML.
HO-1 may also have a role in promoting resistance to the BCR/ABL-directed small-molecule kinase inhibitor imatinib (163, 164). Chemical or genetic induction of HO-1 expression was found to be sufficient to protect against imatinib-induced cell death (163). In another study, the chemical inhibition of HO-1 was synergistic with either imatinib or nilotinib (a next-generation BCR/ABL TKI) in a mutational-independent imatinib-resistant K562 cell line model, imatinib-resistant BCR/ABL mutant murine models, and primary samples from patients with imatinib-resistant CML; those results suggested that HO-1 may also play an important role in resistance to these inhibitors (164). Resistance to imatinib is a major hurdle clinically and thus makes HO-1 an attractive target for CML. Furthermore, HO-1 is an underlying factor in resistance to tumor necrosis factor α-induced cell death in AML cell lines (218), suggesting that the Nrf2/HO-1 signaling axis may play a role in resistance to apoptosis and therapeutic agents in AML as well.
While little is known about the relationship of HO-1 to lymphocytic leukemia, it is evident that HO-1 is important in both CML and AML. Not only does this enzyme promote proliferation and survival, but it also regulates resistance to standard-of-care treatments for both AML and CML. Thus, it is important to gain a better understanding of the regulation of HO-1 in leukemia and of how induction or constitutive expression of HO-1 contributes to mechanisms of therapeutic resistance in leukemia.
4. Glutathione
Much like the previous antioxidants mentioned, the GSH system is capable of removing ROS from leukemia cells. GSH is, in fact, the most ubiquitous antioxidant present in cells at millimolar concentrations (166). However, the GSH system is primarily utilized to restore other antioxidants and remove oxidative damage within cells [reviewed in (231)]. GSH is a tripeptide consisting of glutamate, cysteine, and glycine (231). These peptides are assembled into active GSH through the enzymatic action of γ-glutamylcysteine synthetase and GSH synthetase. A reactive sulfhydryl group drives the antioxidant function of GSH.
The majority of GSH within the cell is found in a reduced form, but as an electron donor, GSH can reduce disulfide bonds. In this process, glutathione peroxidase (GPX) catalyzes a reaction to convert GSH to the oxidized glutathione disulfide (GSSG). The ratio between GSSG and GSH is often used as an indicator of oxidative stress within cells [reviewed in (205)]. In an effort to alleviate ROS-induced damage, GSTs can conjugate reactive substrates (such as peroxidized lipids) to GSH. To restore reduced GSH levels, GSSG is converted by GSH reductase in a reaction that requires NADPH (166). Through these types of reactions, GSH plays vital roles in a myriad of cellular functions, including DNA and protein synthesis, enzyme activation, and the immune response (231).
Elevated GSH is seen in a variety of cancer types, including hematological malignancies. Two independent studies have found higher GSH levels in CLL patients (69) and in AML and ALL patients (233) compared with healthy individuals. Furthermore, there is a correlation between elevated GSH and an increased risk of disease relapse in children with ALL (122). Several studies have also associated elevated levels of GSH with resistance to daunorubicin, melphalan, and the glucocorticoid receptor antagonist prednisolone in ALL (28, 162). In particular, elevated GSH correlated with resistance to the chemotherapeutics
Other components of the GSH system are also potentially important targets in leukemia. For example, the P1 polymorphism of GST prevents the transfer of reactive substrates to GSH and correlates with susceptibility to AML (38). Additionally, glutaredoxin (GRX) is downregulated in leukemia and is associated with differentiation of the cells (242). Selenite, which has cytotoxic effects on AML cells ex vivo, causes induction of GRX transcription (186) and can also regulate GPX post-translationally (35). On the other hand, GPX activity is elevated, rather than decreased, in acute leukemia (61, 118, 282) and in CLL (187). GPX may be important in CML as well, as Abl can activate GPX activity, potentially protecting CML cells against oxidative DNA damage (29). Regardless, the lack of breadth regarding the role of the GSH system in leukemia is apparent. Thus, it is imperative that more research be performed regarding the biological implications of GSH in this disease.
5. Thioredoxin
Another antioxidant pathway important for maintaining the redox homeostasis of leukemia cells is the Trx pathway. The Trx family of redox proteins was previously named the adult T-cell leukemia-derived factor family, which stressed their apparent roles in leukemia cells (175). Trx aids in the removal of hydrogen peroxide, although catalase is far superior for this function. The primary function of Trx is as an electron donor for GPX (175), and as a reductase to control redox status and to protect proteins from oxidative damage or inactivation [reviewed in (50)]. Oxidized Trx is more stable than reduced Trx, so this reaction is thermodynamically favorable (115). Trx is a protein disulfide reductase, which takes electrons from NADPH and transfers them to the active site of Trx (with help from thioredoxin reductase [TrxR]), and then uses these electrons to reduce protein disulfides (50).
There are two primary isoforms of Trx: Trx1, the cytosolic prototypical member of this family, and Trx2, a mitochondrial-specific isoform that has the primary functional activity of apoptosis inhibition (52, 244). The two other major members of this family include TrxR1, which is located in the cytosol, and TrxR2, which is mitochondrial (175). In addition, there are a variety of specialized subsets of Trx proteins that are found in other subcellular localizations, including the cytosol (p32TrxL), nucleus (nucleoredoxin), mitochondria, plasma membrane, and 10-kDa Trx, which is secreted into the extracellular milleu (202). To maintain homeostasis, Trx-binding protein 2 (TBP-2; also known as Trx-interacting protein [TXNIP]) is a negative regulator of Trx that binds directly to the protein, resulting in decreased antioxidant activity (177).
Extensive studies with this family of proteins have demonstrated that Trx proteins have pleiotropic functions within the cell and its external environment. Trx proteins have been known to act as growth factors, cofactors for DNA synthesis, activators of transcription factors, catalyzers of protein folding, chemotactic agents, and apoptosis controllers (175, 202). Trx expression and activity also confer a survival and growth advantage to cells (62, 151, 181). Due to these favorable functional attributes, it is not surprising that Trx is upregulated in leukemia (228). Specific effects of the Trx pathway in leukemia cells include sustained growth and decreased apoptosis and drug resistance. Studies assessing AML patient samples have determined that elevated Trx mRNA expression correlates with aggressive disease and shorter relapse interval (Fig. 9A), suggesting Trx as a negative prognostic indicator (279). Studies regarding the activation of the Trx pathway in acute leukemia are somewhat contradictory. For example, TBP-2, which inhibits Trx activity, is overexpressed in a small subset of AML patients (∼11%) and is theorized to contribute to the elevation of ROS levels and the acquisition of genomic instability (62). In contrast, TBP-2 is lost in ALL, and re-expression of this molecule results in growth inhibition [Fig. 9B and (183)].
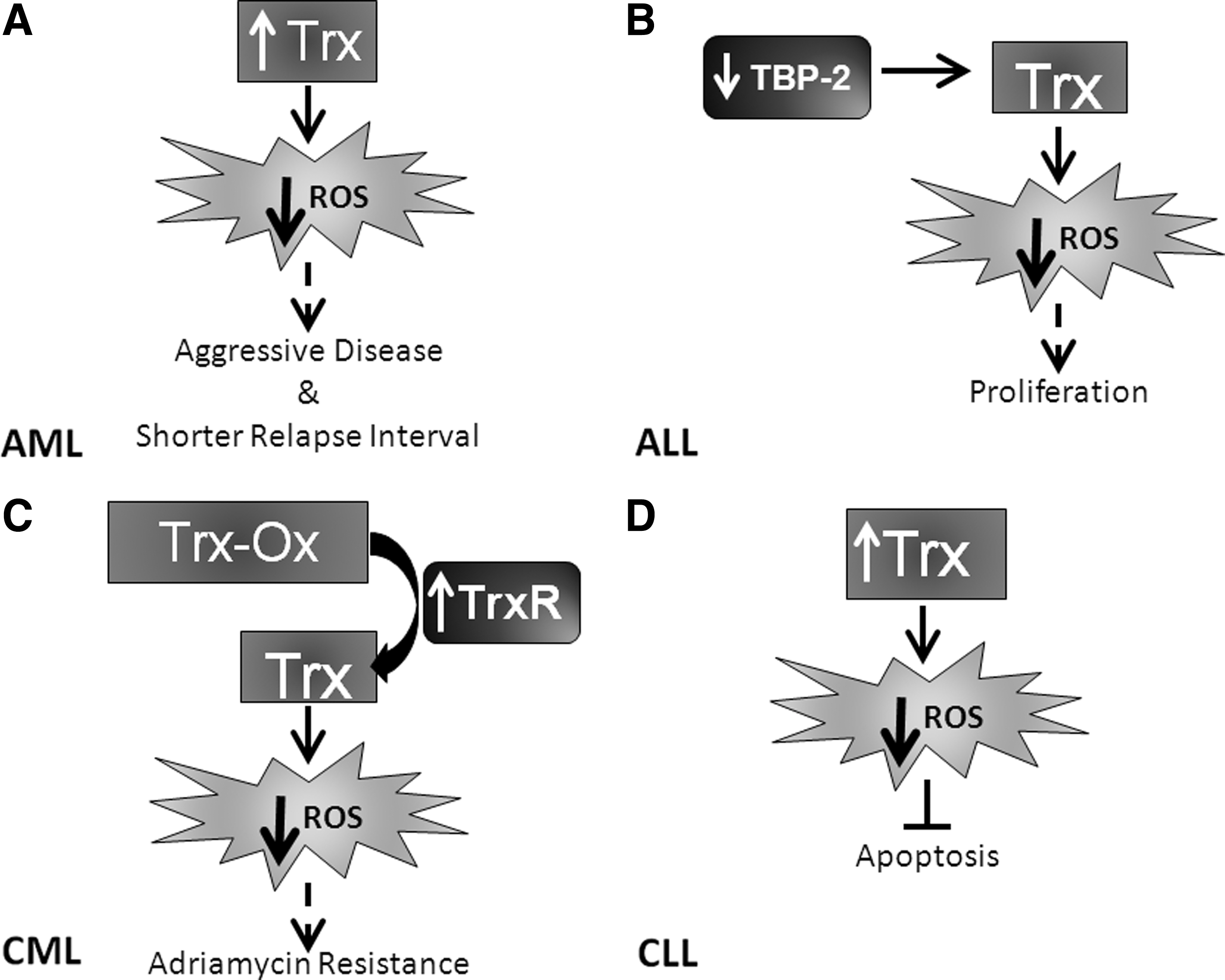
In chronic leukemia, Trx is involved in resistance to apoptotic stimuli. Transfection of mouse lymphocytes with human Trx revealed inhibition of apoptosis induced by different agents (e.g., etoposide and staurosporine) (9). TrxR activity is elevated in adriamycin-resistant CML cell lines (Fig. 9C) compared to sensitive controls, and inhibition of TrxR results in apoptosis, suggesting that it is important for the viability of drug-resistant CML cells (147). Extracellular administration of Trx inhibited apoptosis and supported cellular proliferation in B-type CLL (Fig. 9D) through the release of growth factors (182). Combined, these data suggest an important role of the Trx system in the modulation of leukemia growth and response to therapeutic agents.
6. Peroxiredoxin
Prx proteins belong to the family of Trx-dependent peroxidases, and their expression has been linked to oncogene-induced leukemia. Their major functions include protection against oxidative stress, and induction of cell signaling and proliferation (27). Overexpression of Prx isoforms has been linked to inhibition of apoptosis as well (27). As antioxidants, Prx proteins work mainly by eliminating hydrogen peroxide, using Trx as an electron donor. Prx proteins pick up electrons from Trx and transfer them to degrade hydrogen peroxide to water (27). The involvement of Prx in cell signaling is through events for which hydrogen peroxide molecules serve as second messengers. This family of proteins is divided into two distinct groups based on chemical structure: typical 2-cysteine Prx (Prx I–V) and 1-cysteine Prx (Prx VI). To date, solely typical 2-cysteine Prx isoforms have been associated with leukemia (Table 3).
Prx proteins have variable expression in leukemia, suggesting disparity in functional significance depending on the cellular context. Expression of Prx isoforms can be elevated in leukemia samples, leading to growth and apoptosis resistance (252, 256, 272). Leukemic oncogenes can induce Prx expression. In CML, where Abl is the driving oncogene, Prx I expression is elevated (256). In this context, Prx I decreases the cytostatic and cytotoxic effects of nuclear ABL expression (256). Similarly, when oncogenic c-myc is present, such as in APL, Prx III is highly expressed (252). Downregulation of Prx III in this context resulted in increased sensitivity to ATO (252), suggesting a role for Prx III in drug resistance in APL. Further, proteomic analyses have suggested that Prx II is upregulated in AML cells compared with normal mononuclear cells (149); however, the discrete functional effect of this upregulation has not been discovered. Prx upregulation could be cytoprotective: Prx II induction in MOLT-4 cells was protective against apoptosis induced by serum deprivation, ceramide treatment, and etoposide treatment (272).
However, Prx isoforms are not solely cytoprotective and leukemogenic, and they may also act as tumor suppressors. When samples from 461 myeloid leukemia patients were evaluated by gene expression array, Prx IV was epigenetically downregulated in samples from APL patients relative to patients with other types of AML (190). Prx IV was also shown to be a potential translocation partner for AML1 in a single AML patient (275); how widespread this phenomenon is and the functional consequences of this translocation event are unknown.
More recent evidence has suggested that Prx I, II, and V are downregulated at the mRNA level in AML patient samples (3). Similar to Prx IV in APL, this downregulation was determined to be a result of hypoacetylation and hypermethylation of the gene promoter (3). The functional consequences of this downregulation varied. For example, patient survival rates were increased in patients whose AML cells expressed higher levels of Prx II, but there was no correlation between Prx I expression and disease outcome (Prx V was not analyzed) (3). Prx II was defined as a tumor suppressor gene because forced Prx II expression resulted in decreased leukemogenesis and increased overall survival rates in mouse transplantation models of AML (3). Prx I can interact with BCR/ABL (specifically, Prx I binds to ABL, partially inhibiting its kinase activity in vivo), suggesting a role for Prx in BCR/ABL-positive leukemia (211, 256). This effect may not be specific for Prx I, because Prx III expression has been correlated with response to imatinib (86). These proteins represent a relatively understudied antioxidant family in leukemia, but they may be the key to oncogene-induced leukemia growth and to therapeutic responses. Thus, further studies are needed to elucidate a definitive role for this family in leukemia.
V. ROS-Dependent Therapeutic Agents
As mentioned previously, alterations in redox homeostasis within cells, both normal and oncogenic, can lead to growth and survival as well as death. Because ROS levels are elevated in leukemia cells, drugs promoting further production of ROS to tip the scales toward cellular death have been explored. These drugs include chemotherapeutic agents, HDACi, proteasome inhibitors, and direct ROS-producing agents such as ATO, β-phenylethyl isothiocyanate (PEITC), and adaphostin (20, 120, 145, 153, 168, 227, 246, 281). In sections III and IV, we described how alternative regulation of both pro- and antioxidant mechanisms (both increases and decreases) occurs in leukemia and can lead to resistance to chemotherpeutic agents, relapse of leukemia, and poorer clinical outcomes. These functional effects due to the modulation of the redox environment can and should be exploited therapeutically for the treatment of leukemia.
It is important to note that there are varying opinions on whether ROS production by some of these agents is a cause or a consequence of cell death. Regardless, production of ROS can lead to a feed-forward induction of death in leukemia cells that can be exploited therapeutically. Studies have been initiated to develop inhibitors of or delivery systems for these redox-altering proteins to potentially curb resistance to therapy and to promote death. Here, we have broken these redox control-manipulating agents into three broad categories for further review: standard-of-care chemotherapeutic agents; recently approved ROS-producing agents; and preclinical redox-modifying compounds.
A. Standard-of-care chemotherapeutic agents and modulation of oxidative stress
A number of cytotoxic chemotherapeutic drugs have been approved and are in use as standard-of-care frontline therapeutics for leukemia. Unfortunately, relapse and resistance to such therapies remain critical issues in the treatment of this disease. Thus, a better understanding regarding the mechanisms of action of chemotherapeutic agents is required. Many chemotherapeutic agents have been suggested to induce oxidative stress as a part of their mechanism of action. The primary chemotherapeutic agents used to treat patients with leukemia include purine analogs such as cytarabine, mitotic inhibitors such as vincristine, and anthracyclines such as daunorubicin and idarubicin. All of these agents have been suggested to rely on redox alterations as a part of their cytotoxic mechanism (80, 87, 110, 120). Thus, in this section, we review the redox regulatory mechanisms behind chemotherapeutic drugs as well as the potential mechanisms of resistance to these agents.
1. Purine analogs: cytarabine and fludarabine
Cytarabine is the single most effective agent for remission induction in AML (212), although the response is generally short-lived with 5-year event-free survival and overall survival rates of just 35% and 40%, respectively (150). Cytarabine is an antimetabolite drug that when phosphorylated incorporates into the DNA, thus poisoning the DNA of replicating cells. In addition to the potentially short duration of clinical response, toxicities also hinder its usefulness (19, 150).
Cytarabine causes a substantial increase in ROS in both nonproliferating and leukemia cells (110, 120). For example, in nonproliferating myeloid cells, cytarabine is very potent at inducing apoptosis, which is readily reversible with the addition of the antioxidant NAC or the flavoenzyme inhibitor DPI (110). ROS appear early after treatment with cytarabine, suggesting that induction of ROS is an early step in the stimulation of the apoptotic cascade. The generation of ROS and subsequent apoptosis were inhibited by rotenone, a mitochondrial ETC inhibitor (130), suggesting a role for the mitochondria as a source of cytarabine-induced ROS production. However, the oxidative burst associated with cytarabine treatment was also inhibited by DPI, suggesting that either NOX or xanthine oxidoreductase could also be involved (110). Induction of ROS after cytarabine treatment not only occurs within nonproliferating myeloid cells but also in a variety of leukemia cell lines, including Nalm-6, MOLT-4, Jurkat, and HL-60 (120). Cytarabine can also alter the levels of antioxidant enzymes in leukemia. For example, treatment with cytarabine causes an initial increase in the cellular content of GSH that was followed by a large decrease in GSH (101). Thus, early induction of ROS and possibly alterations in antioxidant expression may contribute to elevations of oxidative stress in leukemia cells after cytarabine treatment, thereby leading to apoptotic death.
Another purine analog, fludarabine, is used to treat patients with CLL. Unlike cytarabine, no research has identified ROS as a mechanism of action for fludarabine. However, in peripheral blood samples from CLL patients treated with fludarabine, elevations in reactive nitrogen species were found to increase mitochondrial biogenesis, leading to resistance to fludarabine treatment and further oxidative stress (32). Those results suggest that the oxidative stress burden within CLL cells may provide a means to resist fludarabine treatment. Thus, it is important to note that the impact of ROS differs between drugs even of the same class and therefore that combination treatments should be adjusted accordingly.
2. Mitotic inhibitor: vincristine
Vincristine, a microtubule-interfering chemotherapeutic, is used to treat patients with ALL (203). The therapeutic regimen of vincristine in combination with steroids, asparaginase, and occasionally anthracyclines has been successful in ALL, with reported outcomes of a 50%–90% remission rate, although relapse within 15 months generally occurs (271). Regardless, the apparent short- and long-term toxicities of this regimen, which include cardiotoxic and neurotoxic effects, hamper the quality of life for patients after treatment (271).
The role that ROS play in the effectiveness of vincristine has not been studied in depth. However, similar to cytarabine, an early release of ROS is seen after treatment with vincristine. When ROS generation was inhibited by the ROS scavenger ascorbic acid in Jurkat cells, apoptosis induced by this agent was prevented (87). ROS also play an important regulatory role in early phases of mitochondrial-controlled, vincristine-induced apoptosis in ALL cell lines (87). ROS induction has been linked to vincristine-induced cytotoxicity in other cancer types as well (42, 248), further suggesting that such a ROS-dependent mechanism exists.
3. Anthracyclines
Anthracyclines, or anthracycline antibiotics, are chemotherapeutic agents derived from bacteria that are in clinical use for a variety of solid tumors and leukemia (170). Specific anthracyclines approved for use in leukemia include daunorubicin, idarubicin, and mitoxantrone (21, 170). Numerous mechanisms of action for anthracyclines are reported and include interference with macromolecular biosynthesis, DNA adduct formation, interference with topoisomerase II, and free radical mechanisms (80).
Anthracyclines are substrates for oxoreductive enzymes such as XO and TrxR. Bioactivation by either of these enzymes can cause anthracyclines such as adriamycin, daunorubicin, and idarubicin to become a free radical (34, 85, 191, 208). These semiquinone free radical forms of anthracyclines induce direct DNA damage themselves, or they may interact with molecular oxygen to promote ROS formation (80). Anthracyclines may also complex with free iron in cells, leading to a chemical reaction called a Fenton reaction, which generates further ROS through the combination of iron and hydrogen peroxide (80). The accumulation of ROS and free-radical damage within leukemia cells can lead to cytotoxic oxidative stress.
Due to the apparent role of redox status and ROS production in the mechanism of anthracycline action, it is no surprise that upregulation of antioxidant enzymes has been suggested as a mechanism of resistance to anthracycline therapy. For example, administration of exogenous catalase, SOD, or GSH has been shown to be protective against anthracycline-induced death (57, 58, 201). Specifically, elevated SOD2 protein expression leads to resistance to anthracyclines (192, 225). Further, levels of GSH are elevated in doxorubicin-resistant leukemia cells compared with their doxorubicin-sensitive counterparts, suggesting a role for GSH in resistance (74). Thus, anthracycline drugs are capable of directly altering the redox environment within a cell, leading to both apoptosis and a feedback antioxidant resistance response.
B. Recently approved ROS-producing agents
1. Arsenic trioxide
Although generally thought of as a poison, arsenic has been used clinically for over two millennia to treat a variety of disorders and diseases (43). As early as the late 1800s, arsenic-containing compounds were reported to have a beneficial role in the treatment of leukemia (43). However, due to concerns about the potential toxicity of the compounds, arsenic-containing drugs were set aside. Years later, ATO has been accepted as a therapeutic agent against cancer (169). Indeed, ATO has shown antitumor benefit in some solid tumors as well as in acute and chronic leukemia (173). The best clinical success with ATO has been for the treatment of relapsed APL, where complete response rates of 50%–85% have been seen in clinical trials (229, 236).
There are a number of suggested mechanisms through which ATO exerts its cytotoxic effects. However, it seems that the primary mechanism of action is the manipulation of intracellular redox control. Microarray analyses performed on myeloid cells treated with ATO show induction of NOX components (p47phox by 400-fold, and p67phox, gp91phox, p40phox, or Rac2 by 2-fold) (45). Small interfering RNA directed toward the p47phox subunit of NOX and a model of deletion of gp91phox by homologous recombination revealed this complex to be the primary source of ROS in APL cells after ATO treatment [Fig. 10A and (45)].
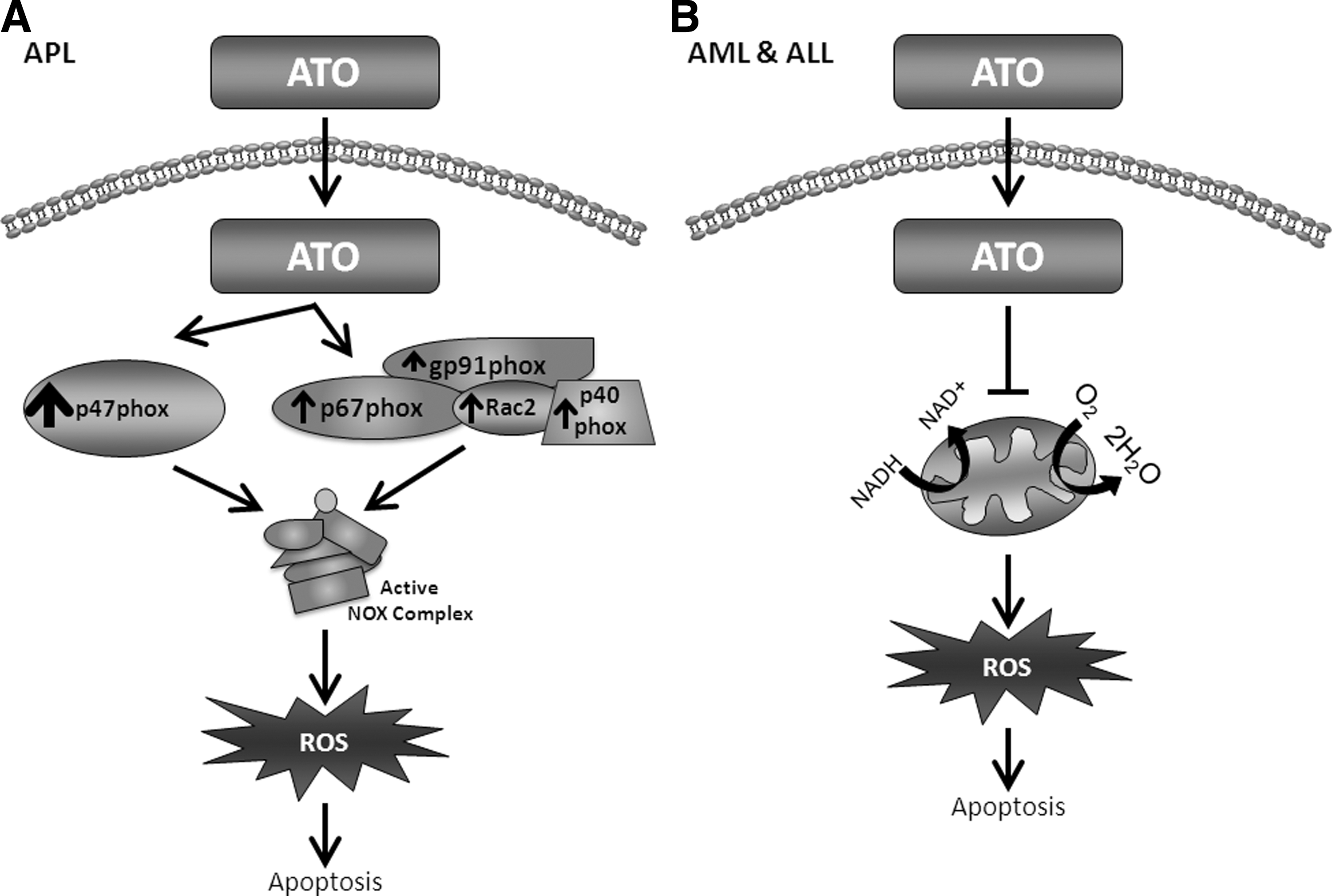
ATO also directly targets the mitochondrial ETC, causing increased ROS production in AML and ALL cells [Fig. 10B and (196)]. Further, a subclone of HL-60 AML cells that are deficient in mitochondrial respiration is resistant to ATO, suggesting again that mitochondrial ROS production is important to the mechanism of ATO-induced death (196). In addition, ATO may affect the expression of antioxidants within leukemia cells. For example, HO-1 is readily induced by arsenic-containing compounds, including ATO (143, 148). Thus, HO-1 may also promote both ATO activity and resistance to ATO effects in leukemia cells.
The GSH/Trx system is also a direct target of ATO treatment. Specifically, ATO was found to irreversibly inhibit Trx function, leading to death of cancer cells (151). These results were discovered in a breast cancer system, but given the importance of the Trx system to leukemia cells (see section IV.B.5), it is plausible that such alterations may also occur in leukemia. Finally, in addition to these antioxidant pathway alterations noted in other cell types, ATO can cause direct depletion of Prx III in APL cells, resulting in elevated ROS levels and cell death (252).
Due to its apparent ability to induce ROS-dependent apoptosis in leukemia cells, ATO is being tested in combination with other leukemia chemotherapeutics. Phase I/II trials have been completed (NCT00081133) to determine the efficacy and safety of the combination of ATO and imatinib in blast-crisis CML and BCR/ABL-positive ALL. ATO was also combined with imatinib in a completed phase I/II trial in chronic-phase CML (NCT00053248). In a newer phase I trial, still actively recruiting, ATO is being combined with imatinib, nilotinib, or dasatinib for the treatment of CML (NCT01397734). ATO is also currently being tested in a phase II trial (NCT00250042) as a potential sensitizing agent for imatinib after its initial failure in CML treatment. Moreover, ATO is being combined with some of the chemotherapeutic drugs discussed earlier, such as cytarabine and idarubicin, in patients with AML (NCT00513305, NCT00195104, and NCT00093483) or APL (NCT00528450) as a part of phase I and phase III trials. The results of all of these trials have not been published, but it stands to reason that increasing levels of ROS may overcome resistance mechanisms to these drugs, thereby allowing cell death to occur.
2. Histone deacetylase inhibitors
Another class of ROS-producing therapeutic agents is HDACi. Histone deacetylases (HDACs) themselves are so named because they function to remove acetyl groups on histones. HDACs form molecular complexes with transcriptional coactivators and corepressors to modulate gene transcription through altering the pattern of histone modifications. Specifically, HDACs and histone acetyl transferases control the availability of chromatin for transcription and gene expression through the removal and addition of acetyl groups, respectively.
HDACs can be divided into four distinct subgroups that are targeted differentially by HDACi. Class I HDACs (HDACs 1, 2, 3, and 8) are ubiquitously expressed, while Class II HDACs, including class IIa HDACs (4, 5, 7, and 9) and class IIb HDACS (6 and 10), are more restricted in their tissue expression. Class III HDACs (sirtuins) differ in that they do not contain zinc in their catalytic domains and have an absolute dependence on NAD+ (56). Lastly, there is a fourth class of HDACs that currently only describes HDAC 11. The name lysine deacetylases is a more accurate name for these enzymes, because they have the ability to remove acetyl groups from many other proteins in addition to histones (158). In fact, HDACs target ∼10% of the 3600 acetylation sites on 1750 proteins within leukemia cells (46).
The biological functions of HDACs suggest that they may be important to the cancer phenotype. As such, four classes of HDACi have been developed based on the selectivity for these HDAC substrates: hydroxamates (e.g., vorinostat and PCI-24781), cyclic peptides (romidepsin), aliphatic acids (valproic acid and butyrate), and benzamides (entinostat). Vorinostat, romidepsin, butyrate, and PCI-24781 are pan-HDACi compounds that block the activity of both class I and II HDACs. Entinostat and valproic acid, in contrast, are specific for class I HDACs. Despite differences in the innate specificity of these agents, a product of treatment with a variety of HDACi is ROS. Vorinostat, for example, was the first HDACi shown to affect ROS as a part of its cytotoxic mechanism. In particular, vorinostat affects Bid, a BCL-2 family member, leading to a mitochondrial leak of ROS in lymphoid leukemia cells [Fig. 11 and (215)]. Since then, vorinostat, butyrate, and entinostat all have been described to produce ROS accumulation in cancer cells (215, 250, 261). Thus, the production of ROS as a result of HDACi treatment is not specific to any particular structural class of HDACi.

There remains some controversy as to whether ROS are a cause or consequence of apoptosis induced by HDACi. Some researchers have suggested that ROS accumulation occurs as an early step in the pathway toward HDACi-induced apoptosis (under 2 h), and that inhibition of ROS with NAC can effectively inhibit HDACi-induced apoptosis (213, 215). Other researchers have observed that inhibition of caspases with z-VAD-fmk blocks entinostat-induced cell death and ROS accumulation: thus, ROS may also be a consequence of HDACi-induced apoptosis in CLL cells (153). Although these studies have shown an effect of HDACi on ROS levels, the source of ROS produced by HDACi also remains debated.
Direct control of ROS production is not the only way that HDACi contribute to alterations in the redox environment. HDACi can also alter the chromatin structure, thereby controlling the transcription of antioxidant genes. Such changes produced by vorinostat and entinostat, for example, can promote upregulation of antioxidant regulators such as TBP-2 (Fig. 11), which binds to Trx and inactivates it. In addition to quenching the antioxidant effect of Trx, TBP-2 may blunt the inhibition of the proapoptotic molecule ASK1 by Trx (Fig. 11), leading to cell death (26, 220, 250).
HDACs are upregulated in many types of cancers, including leukemia (23, 171). In a phase I clinical trial for patients with advanced AML, the hematological response rate to vorinostat treatment was 17% (78). Because phase I trials typically do not measure overall response rates, these data are quite striking, but also suggest the presence of a large cohort of patients that may not respond to HDACi treatment alone. Microarray-based gene profiling performed with patient samples from this study revealed that the expression of 17 antioxidant genes correlated with resistance to vorinostat (78). A separate study showed that AML cells resistant to HDACi contain increased levels of Nrf2-dependent antioxidants and therefore have decreased levels of ROS (108). In those cells, vorinostat was capable of inducing ROS by activating NOX, as inhibition of NOX with DPI blocked vorinostat-induced ROS generation (108). In addition, the natural ROS-producing compound PEITC, which disables the GSH antioxidant system by inhibiting GPX activity (247), was capable of modulating the cellular redox state in AML cell lines and primary samples, resulting in a synergistic interaction with vorinostat (108). In light of these findings, it may be more beneficial to combine HDACi with other therapeutic agents that induce ROS or inhibit oncogenes than to use HDACi alone.
One outstanding example of using HDACi in combination therapeutics is with standard-of-care chemotherapeutics. In a Jurkat cell model system, synergism was found between the HDACi LAQ-824 and fludarabine (214). This combinatorial effect required ROS produced at a low level by the HDACi, which caused a mild amount of DNA damage within leukemia cells, but was not cytotoxic on its own. When combined with fludarabine, however, treatment with LAQ-824 resulted in cytotoxicity (214). Oxidative stress is also a marker for synergy between the HDACi entinostat and the chemotherapeutic agent 5-azacytidine in AML and ALL, suggesting that redox regulation by these two drugs may be important in the treatment of acute leukemia (76). Such preclinical evidence suggests that the combination of traditional chemotherapeutics with ROS-producing HDACi will improve treatment outcomes for patients. Combinations of various HDACi (e.g., entinostat, panobinostat, valproic acid, and vorinostat) with standard chemotherapeutic agents are being tested in numerous phase I and II clinical trials for all four subtypes of leukemia.
HDACi may potentiate the effects of not only standard-of-care therapies but also newer TKIs developed for the treatment of imatinib-resistant, BCR/ABL-positive leukemia. For example, when combined with the novel small-molecule multiprotein kinase inhibitor KW-2449 (which is capable of inhibiting Flt3, Abl, T315I-Abl, and aurora kinase), HDACi caused a significant rise in ROS levels, leading to enhanced DNA damage and cell death, specifically within the ALL and CML cell lines and patient samples (180). These effects were specific to BCR/ABL-expressing cells, because combination treatment did not harm normal CD34+ cells (180).
Further studies have also suggested combining HDACi with inhibitors of autophagy or MAPK/PI3K for the treatment of imatinib-resistant leukemia. In the case of autophagy, this cellular process was synergistically inhibited by the combination of chloroquine and HDACi, promoting apoptosis within CML cell lines (31). NAC was capable of inhibiting the apoptosis that occurred, suggesting that this drug combination required the elevated ROS level to produce cell death. When the mechanism behind this effect was examined, it was found that the treatment with HDACi and chloroquine had resulted in upregulation of cathepsin D protein and activity, leading to cleavage of Trx and thus elevation of ROS (31). Low concentrations of HDACi have been combined with MAPK and PI3K/Akt inhibitors to promote leukemia cell death. Specifically, this combination resulted in elevated levels of ROS-induced oxidative stress in CML cells, regardless of their BCR/ABL TKI sensitivity, and resulted in cell death that was blocked by NAC (189). Due in part to these types of findings, clinical trials combining entinostat or panobinostat with imatinib are ongoing for patients with BCR/ABL-positive ALL (NCT01383447) and CML (NCT00686218). It is clear that combining ROS-inducing HDACi with other agents could be vital to the assessment and clinical use of HDACi in treating all types of leukemia.
3. Proteasome inhibitors
Much like HDACi, inhibitors of the proteasome have been recognized to generate ROS as a portion of their cytotoxic mechanism of action. The proteasome itself is a molecular complex within the cell that degrades a vast majority of cellular proteins. E1, E2, and E3 enzymes prepare proteins for degradation by the proteasome through activation and conjugation of ubiquitin to substrates. Polyubiquitination results in direct targeting of proteins for proteasomal degradation (102).
Leukemia cells contain elevated levels of ubiquitinated proteins compared with normal cells (160), suggesting that leukemia cells may have increased misfolded, unfolded, or damaged protein levels and are more reliant on proteasomal activity for cellular survival. Also, inhibitors of the proteasome exhibit selectivity toward cancer cells, including those of hematopoietic origin, implying that the use of proteasome inhibitors may benefit cancer patients (4). Thus, a number of proteasome inhibitors have been developed to target leukemia, including synthetic aldehydes (e.g., lactacystin, MG132, marizomib, carfilzomib, and PSI) and boronic acid derivatives (e.g., bortezomib), which inhibit proteasomal activity by binding, irreversibly or reversibly, to active sites within the proteasomal catalytic core (224).
Inhibition of the proteasomal complex can result in cell cycle arrest, apoptosis induction, and ROS generation (53, 54, 72). Initial indications for a role of ROS in proteasomal inhibition-mediated cell death came from studies of solid tumors treated with bortezomib, the first FDA-approved proteasome inhibitor. In solid tumors, ROS induction by proteasome inhibitors remains controversial, but for hematopoietic malignancies, the association between ROS induction and proteasome inhibition is better established (168, 195, 198).
In addition to mantle cell lymphoma studies (198), ROS have been indicated as a product of proteasome inhibition in ALL. A role for ROS was noted in the effects of bortezomib and marizomib (NPI-0052) treatment in ALL cell lines (168). Both agents were found capable of inducing superoxide production in Jurkat cells. Further, the apoptotic effects of marizomib on leukemia cell lines and patient samples were inhibited by the antioxidant NAC, suggesting that ROS are innately involved in the leukemia cell death-promoting effects of this drug (168). The mitochondrial ETC has been suggested as a source of proteasome inhibitor-induced ROS (142, 146); however, further analyses of the contributions of the mitochondrial ETC and NOX to ROS production as a result of proteasome inhibition are required.
Because bortezomib has shown clinical success in the treatment of some hematological malignancies, including myeloma and mantle cell lymphoma, scientists have been eager to apply these therapies to leukemia. For chronic leukemia, there are preclinical data suggesting the efficacy of proteasome inhibitors. Bortezomib has shown promise as a single agent in a number of studies on CLL and CML (1, 60). However, phase II studies have shown only modest effects of single-agent bortezomib in patients with refractory CLL, although the patients' highly pretreated state may have hindered firm conclusions (63). Indeed, resistance to single-agent treatment seems to be an issue, at least in acute leukemia (105, 241). Resistance to proteasome inhibitors in AML may be due to the downstream actions of the inhibitors themselves, because in addition to directly influencing the generation of ROS, proteasome inhibitors can increase the antioxidant response by stimulating Nrf2 induction [Fig. 12 and (216)]. Proteasome inhibitor resistance was correlated with stabilization of Nrf2 and upregulation of genes downstream, including those encoding GST, glutamyl cysteine ligase modulator, the glutamyl cysteine ligase catalytic subunit, NADPH:quinone oxidoreductase, and HO-1 (216). Thus, by altering the balance of redox homeostasis in AML cells, bortezomib promotes resistance to itself. It may be more beneficial to combine proteasome inhibitors with other ROS-producing agents to overcome this increase in antioxidant signaling.
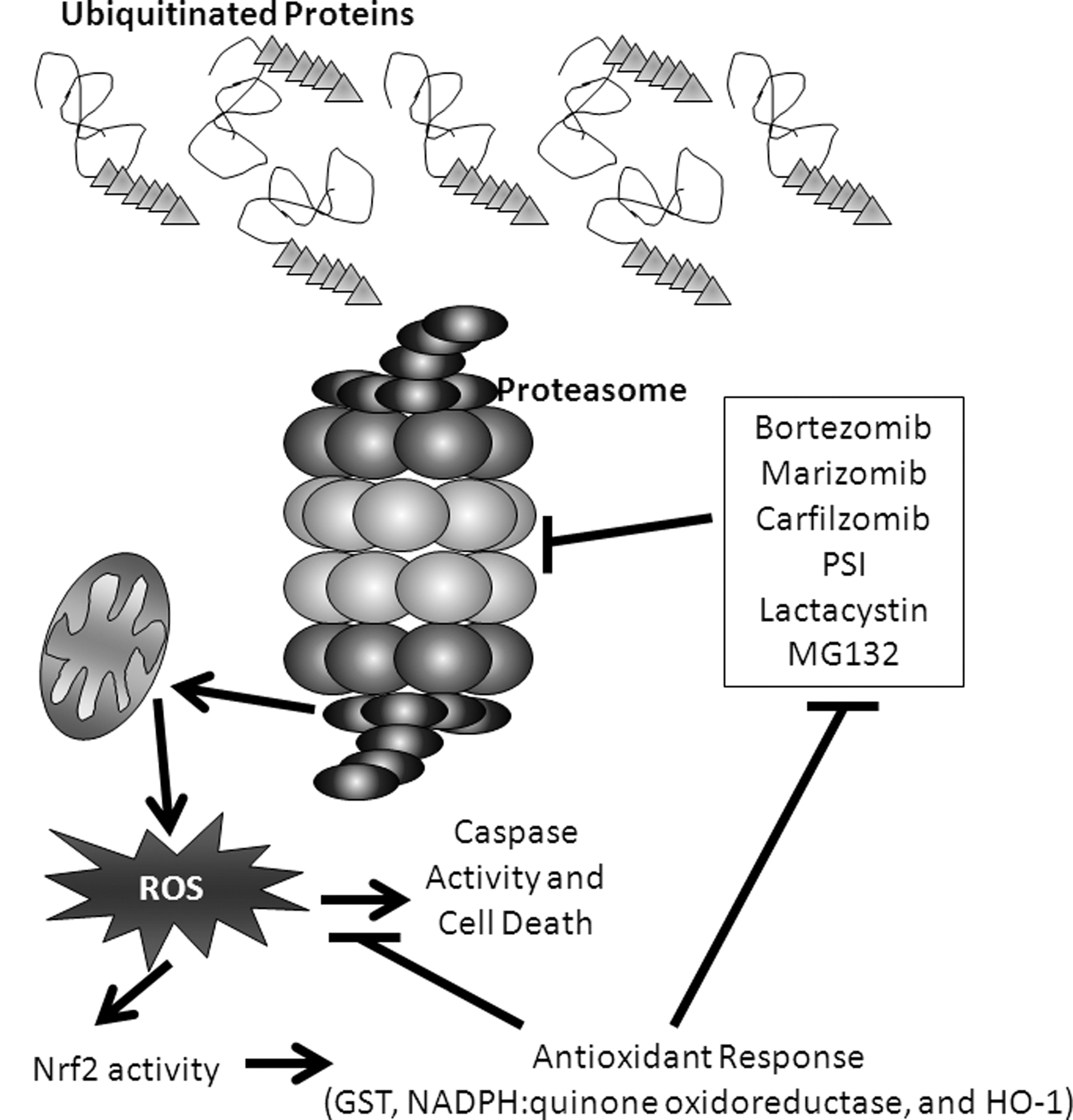
Although little clinical response has been observed with proteasome inhibitors alone in leukemia, using them with other ROS-producing agents seems to increase therapeutic benefit. Multiple research groups have suggested that the combination of proteasomal inhibition and HDAC inhibition may be a viable treatment option for leukemia. Initial indications from studies in myeloma suggested that this combination can lead to a synergistic induction of oxidative cell injury, suggesting that these ROS-producing therapeutic agents may enhance the efficacy of each other (195). Indeed, several laboratories, including our own, have shown that the combination of these two classes of drugs is advantageous for the treatment of leukemia. As early as 2003, it was suggested that the combination of the proteasome inhibitor bortezomib with vorinostat or sodium butyrate may increase the effects on ROS production, mitochondrial injury, caspase activation, and ultimately apoptosis in a variety of human leukemia cell lines (267). Further, the combinatorial effects of proteasomal and HDAC inhibition were blocked by NAC, suggesting that ROS are a major player in the synergy between these therapeutic agents (267). A similar ROS-dependent mechanism of action occurs between marizomib, an irreversible proteasome inhibitor, and HDACi. In particular, the combination of marizomib with entinostat or valproic acid results in even greater levels of cell death than does the combination of these HDACi with bortezomib (168). Thus, it seems that combination treatment of HDACi with various proteasome inhibitors leads to further induction of oxidative stress and results in the death of leukemia cells. This body of findings has led to ongoing phase II clinical studies on the combination of the HDACi vorinostat and the proteasome inhibitor bortezomib for the treatment of AML (NCT00818649) and ALL (NCT01312818).
C. Preclinical redox modifying compounds for the treatment of leukemia
The therapeutic agents we have mentioned thus far are all in use or are undergoing clinical trials for use in leukemia. However, many of the pathways we have described earlier may also be modulated for disease treatment. On the one hand, potentiating ROS production (e.g., with isothiocyanates, HO-1 inhibitors, adaphostin, piperlongumine, or parthenolide [PTL]) can push the oxidative balance, resulting in death, while on the other hand, inhibition of ROS production (e.g., with NOX and XO inhibitors, SOD, and catalase delivery systems) may lead to less genomic instability and increased drug sensitivity. Although not all of these potential therapeutic avenues are currently undergoing clinical evaluation, they may provide insight into pathways that could be used as targets for direct therapeutic intervention in leukemia.
1. Preclinical ROS inhibition
a. NOX inhibitors
The field of potential therapeutic agents directed toward the NOX complex has primarily focused on the decrease of ROS levels and its harmful effects within diseased states. As such, a number of variable inhibitor types have been used in regards to NOX activity. PR-39, which is a peptide-based inhibitor, has proven useful in preventing the association of the p47phox subunit of NOX with gp91phox, an important step in activation of the complex (230). However, this peptide compound works by binding the SH3 domain of p47phox, so it is likely that it may bind other SH3 domains in a promiscuous manner, making it a rather unspecific inhibitor (36, 243). Another peptide inhibitor of NOX, gp91ds-Tat, contains nine amino acids from the human immune virus coat protein and nine amino acids from gp91phox. This peptide acts as a competitive inhibitor of gp91phox by competing for complex formation with p47phox, much like PR-39. This peptide is more specific than PR-39 and readily enters cells, but because it cannot be administered orally, the usefulness of gp91ds-Tat as a clinical inhibitor is also limited [reviewed in (258)].
Other chemical inhibitors of NOX, such as apocynin and DPI, have been used preclinically. Apocynin blocks the assembly of NOX and is orally bioavailable (7), but it can have off-target effects such as induction of AP-1 (140), which may limit its affectivity. DPI is a pan-NOX inhibitor capable of reducing genomic instability caused by ionizing radiation, a known carcinogen. Unfortunately, much like apocynin, DPI can be fairly nonspecific, because it is capable of inhibiting all flavoenzymes and therefore of altering a number of pathways therapeutically (7).
Triphenylmethane dyes, which share structural homology with DPI, have shown effectiveness with inhibiting NOX2 and NOX4. Treatment with brilliant green or gentian violet resulted in reduced NOX2 and NOX4 activities in vitro and decreased hemangioma formation in vivo (199). However, these dyes are poorly tolerated in vivo. Thus, from these dyes, a novel class of potential NOX inhibitors was recently synthesized. Specifically, fulvene-5 is a water-soluble fulvene that, much like brilliant green and gentian violet, inhibits NOX2 and NOX4 activity and reduces hemangioma formation in vivo (17). While this new class of drug may be useful in treating hematological malignancies, there remains the question of specificity, which has not been addressed to date. Because NOX may be an important factor in many disease states, still other NOX inhibitors are undergoing preclinical development (124).
b. Xanthine oxidoreductase inhibitors
There are already inhibitors of the xanthine oxidoreductase pathway in clinical development, primarily for the treatment of gout. Allopurinol, a pyrazolopyrimidine derivative, is a structural analog of alloxanthine and is currently in clinical use. However, there are issues with the use of this therapeutic agent, because its effect does not seem to be dose dependent: at high levels, it becomes a substrate for XO, leading to elevated, rather than reduced, levels of ROS. Another pyrimidine derivative, 4-amino-6-hyroxypyrazolo[3,4-d] pyrimidine (AHPP), has a lower inhibitory concentration than allopurinol and does not become a substrate for XO at high levels; however, it has poor water solubility. To make up for this lack of solubility, researchers have derived a styrene maleic acid copolymer version of AHPP (SMA-AHPP) (66). Conjugation of the SMA micelle component to this drug not only improves its water solubility but limits its intracellular release (176).
For leukemia treatment, inhibition of XO may be beneficial on multiple levels. First, it would inhibit the production of ROS and other free radicals from XO activity, potentially reducing signaling and genomic instability downstream of ROS production. Second, XO is involved in the metabolism of many agents, including 6-MP, which is used in the treatment of pediatric ALL (11). Therefore, inhibitors of this ROS-producing compound may, in addition to decreasing ROS levels, increase the efficacy and half-life of currently available therapeutic agents.
c. SOD and catalase delivery
Another method of reducing the oxidative burden within leukemia cells in an effort to decrease its beneficial effects is by elevating the levels of antioxidants. Studies regarding the therapeutic potential of SOD and catalase have focused on the delivery of these molecules to the oxidatively stressed cancer cell in the hopes of combating the elevated levels of ROS within. Administration of SOD directly to combat ROS is challenging because SOD has a short half-life, is quite unstable, and is immunogenic. Furthermore, SOD can create hydrogen peroxide and metal ions, which participate in reactions that increase ROS. Therefore, coadministration with catalase has been suggested. To combat the difficulties of SOD administration, conjugates, including pyram copolymer–, polyethylene glycol– (PEG-), Ficoll–, lecithinized, SMA–, gelatin–, or albumin–SOD, have been generated and tested [reviewed in (66)]. These conjugate versions of SOD have improved its circulation half-life, increased stability, and decreased immunogenicity (66). In clinical practice, lecithinized SOD1 has shown efficacy against ischemic injury, neuronal injury, inflammation, and HIV infection (66). In addition to direct administration of SOD through these methods, 2-ME, as mentioned in section IV.B.1, was shown to work primarily through induction of SOD in all four subtypes of leukemia (109). Thus, administration or induction of SOD may be a worthwhile therapeutic avenue to investigate for the treatment of leukemia.
2. Preclinical ROS induction
a. HO-1 inhibitors (protoporphyrins)
While the previous three pathways have been modulated in an effort to decrease ROS, many other therapeutic agents aim to provide a net increase in ROS to induce cellular death. HO-1 is elevated in leukemia, resulting in resistance to therapeutics and apoptosis (see section IV.B.3). Thus, inhibitors of HO-1 have been developed for preclinical use and testing. Zinc protoporphyrin (ZnPP) is able to inhibit HO-1 and accumulates within tumor tissues, possibly due to an effect called enhanced permeability and retention (66). Chemical conjugation of ZnPP with PEG or SMA is required for solubility of the molecule in aqueous solutions. The PEG-ylated form of ZnPP (PEG-ZnPP) has shown anticancer effects in colon cancer cells in vitro and in vivo as well as in other solid tumor models, and has shown synergism with conventional therapeutics (66). The SMA conjugate of ZnPP (SMA-ZnPP) shows antitumor efficacy in some solid tumor models with little toxicity (66), and acts synergistically with cytarabine and fludarabine in AML cells in vitro (83). In addition, SMA-ZnPP is capable of inhibiting tumor growth without affecting normal cells (83). PEG-ZnPP or SMA-ZnPP may also have efficacy in imatinib-resistant CML and ALL by inducing ROS-related cytotoxicity without being toxic to normal cells (93, 128, 164). However, there are concerns regarding the use of protoporphyrins in vivo due to potential off-target effects. Thus, further drug testing and discovery will be necessary to effectively target HO-1 clinically for leukemia.
b. Trx pathway inhibitors
The involvement of Trx in redox regulation of leukemia makes this protein a good potential therapeutic target. Trx inhibitors, such as PX-12, have been developed and preclinically proven to be effective in solid tumors. A phase I/II clinical trial of PX-12 in advanced/metastatic cancers and lymphomas was recently completed (NCT00736372), but the results of this study have yet to be published.
Inhibition of the Trx system with the compound 13-hydroxy-15-oxo-zoaptlin results in proapoptotic signaling and cell cycle arrest in leukemia cells, suggesting again that this pathway might be a desirable target for leukemia treatment (181). Other newer agents work to genetically reactivate TBP-2 expression, which inhibits Trx antioxidant activity (see section IV.B.5), in leukemia cell lines. In particular, the histone methyltransferase inhibitor 3-deazaneplanocin A (DZNep) reactivates TBP-2 expression in AML cells, leading to inhibition of Trx, elevation of ROS, and apoptosis induction in AML cell lines, xenograft models, and primary cells (280).
In addition to specific Trx inhibitors, several conventional anticancer therapies, such as cisplatin (18), doxorubicin (161), and cyclophosphamide (255), inhibit Trx in vitro and in vivo. More recently, studies have demonstrated Trx to be a part of the mechanism of action of HDACi as well (250). Thus, through direct or indirect inhibition of Trx, these agents may increase ROS levels and lead to leukemia death.
c. Isothiocyanates
Belonging to a family of proteins called isothiocyanates, PEITC is a chemically active, naturally derived compound with ROS-producing anticancer effects. PEITC has shown preclinical evidence of anticancer activity in Jurkat (ALL), HL-60 (AML), ML-1 (AML), KBM5-T315I (CML) cell lines, and primary leukemia samples (44, 75, 117, 260, 270, 277). Isothiocyanates in general are derived exclusively from plants and fungi (64). Humans regularly ingest these compounds through various dietary sources, primarily cruciferous vegetables such as dikon, broccoli, and watercress (48, 64, 65). Multiple reviews have been written that highlight the potential benefit of isothiocyanate compounds (51, 99, 100, 276), so we restrict our discussion here to their role as ROS-producing therapeutic agents in leukemia.
It is generally thought that isothiocyanates work to decrease survival and proliferation by modulating the oxidative environment within the cancer cells (274). In a study of CLL cells treated with PEITC, there was severe GSH depletion, ROS accumulation, and oxidative injury leading to degradation of prosurvival factors and induction of cell death (246). This alteration of redox pathways and increase in death were specific to CLL cells; normal lymphocytes were unaffected by the treatment. PEITC is also biologically active in imatinib-resistant CML cells. In particular, treatment of CML cells with the T315I mutation of BCR/ABL with PEITC resulted in elevated ROS levels and redox-mediated degradation of BCR/ABL (270). Unfortunately, these studies have not determined effects of PEITC on specific ROS-producing pathways nor antioxidant responses.
PEITC has been used clinically in a completed phase I trial in lung cancer patients. In addition, this compound will soon be undergoing phase I trial with patients with hematological malignancies that have been treated with fludarabine (NCT00968461) in the hopes of improving leukemia treatment regimens through its redox-modulatory effects.
d. Adaphostin
Adaphostin has been identified as a potential agent to treat acute leukemia. This molecule is part of the tyrphostin family of TKIs, which affect BCR/ABL kinase, but not through the ATP-binding site. Interestingly, both cells lacking BCR/ABL (e.g., ML-1, OCI/AML2, and OCI/AML1) and cells in which BCR/ABL is present (e.g., KBM5, KBM7, and K562) are sensitive to adaphostin treatment (37, 188). Thus, the antileukemic activity of adaphostin is not selective for the presence of BCR/ABL. Instead, the mechanism of action is primarily due to depletion of GSH, which would increase the levels of ROS and thereby result in apoptosis (37).
Further studies have been devoted to elucidating the specific mechanism by which adaphostin induces oxidative stress. Using K562 cells, adaphostin was noted to bind to complex III within the mitochondrial ETC, resulting in accumulation of ROS (141). In addition, adaphostin has been linked to the upregulation of genes encoding proteins involved in oxidative stress (e.g., GST, SOD, and heat shock proteins) in both AML and CML cell lines (239). Interestingly, the Nrf2/HO-1 pathway has also been implicated in the mechanisms of action of adaphostin (68). Adaphostin is selective for cancer cells, which may be explained by the observed high levels of ROS in leukemia cells compared with normal cells. Together, these observations indicate that adaphostin is a redox-modulatory agent in leukemia.
e. Piperlongumine
Originally isolated from the plant species Piper longum L., piperlongumine (or piplartine) effectively alters the redox environment within cancer cells and is cytotoxic to a number of a cancer cell types (206). Piperlongumine is capable of inducing both apoptosis and necrosis pathways in the leukemia cell lines HL-60 (AML), K562 (CML), Jurkat (ALL), and MOLT-4 (ALL) (16). When these leukemia lines were treated with increasing doses of this compound over a 24-h period, DNA synthesis was inhibited, and apoptosis and necrosis were actively induced.
It was not until more recently that a defined mechanism was described for piperlongumine. Specifically, this drug selectively kills a variety of cancer cells by targeting oxidative stress. The viability of human cancer cell lines of endometrioid adenocarcinoma (EJ), colon (HCT116, SW620, and DLD-1), breast (BR-474, MDA-MB-231, and MDA-MB435), pancreatic (Panc-1, Mia PaCa-2), osteosarcoma (Saos-2), and lung (A549 and H1975) origin is greatly inhibited by piperlongumine (206). In addition, studies combining affinity enrichment with stable isotope labeling and quantitative proteomics found that piperlongumine binds to several members of the GSH antioxidant pathway (GST pi, GST Z1, GST M3, GST O1, and glyoxalase 1 [GLO1]) and the Prx family (Prx 1). Additional studies confirmed that the compound could bind to and inhibit these various redox control enzymes (206). This inhibition resulted in elevated ROS levels (as determined by staining with the redox-sensitive fluorescent probe 2′-,7′-dichlorodihydrofluorescein diacetate) in the solid tumor cell lines and mouse models tested. Piperlongumine had no effect on the proliferation or ROS levels of normal blood cells or of cell lines representing normal tissues, suggesting that this drug selectively alters the redox status of transformed cells (206). Piperlongumine has been tested in a mouse model of breast cancer (MMTV-PyMT), for which it was found to have in vivo activity with very little toxicity. In light of its apparent cytotoxic effects in leukemia and other cancer types as well as its high selectivity for transformed cells, piperlongumine may be an important ROS-producing therapeutic agent for the treatment of leukemia.
f. Parthenolide
PTL is another natural product with potential redox-altering effects that may be beneficial for leukemia treatment. PTL is derived from the feverfew plant and has been used for the treatment of inflammatory conditions such as rheumatoid arthritis and migraine (88). In addition to its anti-inflammatory effects, PTL induces apoptosis specifically in AML stem and progenitor cells with no effect on normal hematopoiesis (89). The primary mechanism of action in leukemia cells is thought to be inhibition of NFκB and upregulation of p53, leading to the induction of different proapoptotic genes and generation of ROS (88). In a study of AML cells, NAC was capable of inhibiting PTL-induced apoptosis, demonstrating that the increase in ROS is a major player in the cytotoxic mechanism of this drug. NAC also blocked the NFκB inhibitory effect of PTL, suggesting a possible link between these two events (88). In addition, PTL causes ROS levels to increase, GSH depletion, and NFκB downregulation in AML cells with elevated levels of myeloperoxidase (126). These results suggest that myeloperoxidase expression in leukemia cells may be a biomarker of susceptibility to PTL.
Despite the efficacy of PTL in leukemia cells, its pharmacological properties make clinical use a challenge. The dimethylaminoparthenolide derivative LC-1, which was developed to overcome this obstacle, demonstrates better pharmacokinetic properties (90). Similar to PTL, this analog inhibits NFκB, activates p53, and increases ROS levels, resulting in cell death of myeloid and lymphoid leukemia cells (90, 114). With better oral bioavailability, LC-1 represents a promising agent for the treatment of leukemia.
VI. Summary/Remarks
The evidence provided herein suggests a strong role for the redox environment in leukemia survival, growth, progression, relapse, and drug resistance. Redox homeostasis in leukemia cells is already tipped toward ROS: enzyme complexes such as the mitochondrial ETC, NOX, and xanthine oxidoreductase are expressed and highly active in leukemia cells. In some cases, the oncogenes responsible for transformation of normal blasts to leukemia blasts control the assembly of these complexes, either through phosphorylation events or transcriptional alteration. Some clinical relevance can be found in these ROS-producing complexes, either by further induction to poison the cell with ROS, or through inhibition to limit the growth and survival potential of the cell. For the most part, antioxidants are downregulated in leukemia cells and thereby promote ROS signaling and genomic instability. However, upon treatment with ROS-producing agents, many antioxidant enzymes have the potential to be upregulated and thus to promote resistance to therapy.
Traditional chemotherapeutic agents also alter the redox status of leukemia cells, but have the added negative effects of cardiotoxicity and neurotoxicities that can severely degrade the quality of life for patients. Further, after current treatment regimens, the risk of secondary malignancies is greatly elevated due to the DNA-damaging effects of standard-of-care chemotherapeutic agents. The initial response rates to these agents may be as high at 90%, but toxicities, relapse, and secondary leukemia limit their true effectiveness. Therefore, further study regarding the role of redox modulation in the resistance to and toxicities of chemotherapeutic drugs is necessary.
Direct inhibitors of antioxidants or prooxidants are still fairly young in development. Targeting of the dysregulated redox environment within leukemia cells could provide further selectivity to treatment regimens, allowing for a decrease in overall toxicities. Unfortunately, current techniques to quantitatively measure levels of oxidative stress in vitro and in vivo are limited, which decreases the ability of researchers to fully exploit redox-modulation therapeutically. Thus, further study of how the redox system is controlled and balanced toward a proliferative advantage in leukemia cells and the creation of new techniques to more accurately measure ROS and oxidative stress are strongly warranted, as these players may hold the key to unlocking therapeutic resistance in leukemia.
Footnotes
Acknowledgments
This work was supported, in part, by the National Institutes of Health core grant CA16672, F32 CA171905 to M.E.I., and R01 CA115811 to J.C.