
Abstract
Introduction
Mitochondrial ferritin (MtFt) was identified as a ferritin heavy-chain-like (H-ferritin) protein in 2001 (15). Both human and mouse MtFt precursors are imported into the mitochondria and processed into an ∼22-kDa mature protein in the mitochondrial matrix (22). In the matrix, these proteins assemble into homopolymeric ferritin shells with ferroxidase activity (14). Both H-ferritin and L-ferritin can sequester and store iron in a safe yet accessible form (1). Excess free iron can generate reactive oxygen species (ROS) during mitochondrial electron transport. Under these conditions, ferritins exert cellular protective roles against iron-mediated free radical damage (8). MtFt has been shown to dramatically modulate cellular iron metabolism (15), and our previous studies have shown that MtFt exerts a neuroprotective effect against 6-hydroxydopamine (6-OHDA)–induced dopaminergic cell damage (27).
In this study, we show that the endogenous expression of MtFt in the hippocampus of rats is age related. Other reports have shown that excessive activation of the mitogen-activated protein kinase (MAPK) pathway is one of the most important signals for neuronal cell death (5, 7). Inhibition of the MAPK pathway could be beneficial in the treatment of neurodegenerative diseases, including AD (23). A recent study confirmed that MtFt expression was significantly increased in the cerebral cortex of AD patients (34). However, the neuroprotective roles and the underlying mechanisms of MtFt action in AD and AD-like syndromes have not been studied. To explore these mechanisms, the Aβ25–35 peptide was injected into the hippocampus of rats to produce an AD-like animal model. As in many studies (40), Aβ-treated SH-SY5Y cells were used as an AD cellular model. For in vitro studies, the mouse MtFt gene was stably transfected into SH-SY5Y cells to investigate the role of MtFt in a cell model of AD induced by Aβ25–35. We demonstrated that MtFt could dramatically affect neuronal cell iron metabolism and prevent the neuronal cell damage induced by Aβ25–35. We further demonstrate that the MAPK pathway plays an important role in this process.
Results
The expression of ferritins in the hippocampus at different ages in the rat
Ferritin is the major iron storage protein in almost all mammalian cells. We detected three forms of ferritin—H-ferritin, L-ferritin, and MtFt—in the rat hippocampus. As shown in Figure 1A and B, MtFt expression in the hippocampus was low at week 1 and increased with age (at 12, 44, and 88 weeks). L-ferritin expression peaked at week 44. H-ferritin expression increased from week 1 to week 12, reached its maximal value at week 44, and remained at this high level through week 88 (Fig. 1A, B).

The effect of MtFt short interfering RNA on the expression of MtFt and the release of cytochrome C in rats
To determine whether MtFt has protective effects against Aβ25–35 neurotoxicity, rats were treated with MtFt short interfering RNA (siRNA). This siRNA specifically knocked down MtFt expression (Fig. 2Ai, Aii) without affecting the ferritin H or L chains (Supplementary Fig. S1; Supplementary Data are available online at
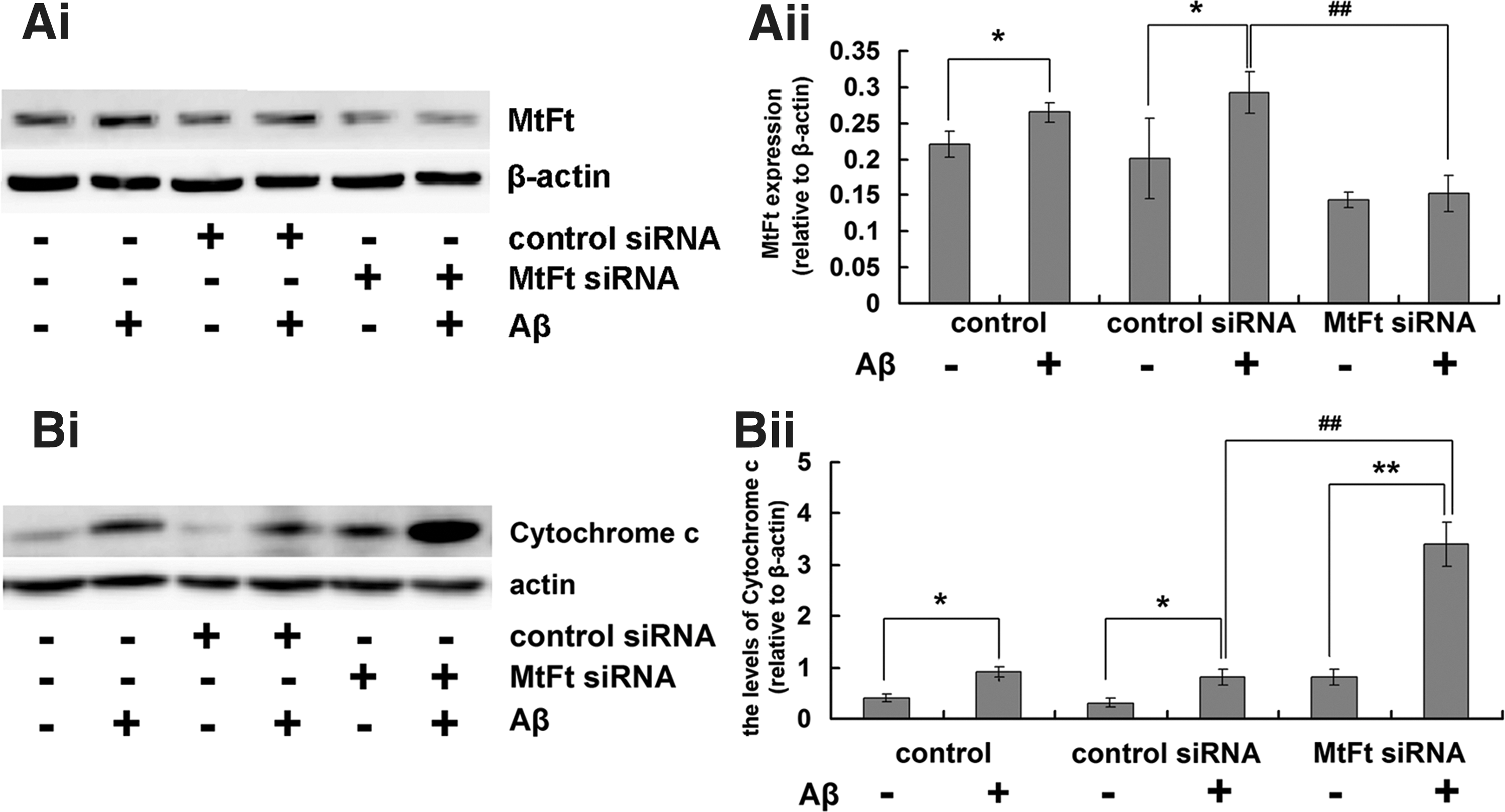
Aβ accumulation in mitochondria is associated with mitochondrial dysfunctions (12) that include changes in membrane potential, which leads to the release of cytochrome C from the mitochondrial intermembrane space into the cytoplasm. This release of cytochrome C is a sign of mitochondrial-dependent apoptosis (33). Figure 2Bi and Bii shows that Aβ25–35 increases the release of cytochrome C. Knockdown of MtFt induced more cytochrome C release into the cytoplasm, which exacerbated the toxicity of Aβ25–35. These results indicate that Aβ25–35 can induce mitochondrial dysfunction and that MtFt can play a protective role in the process.
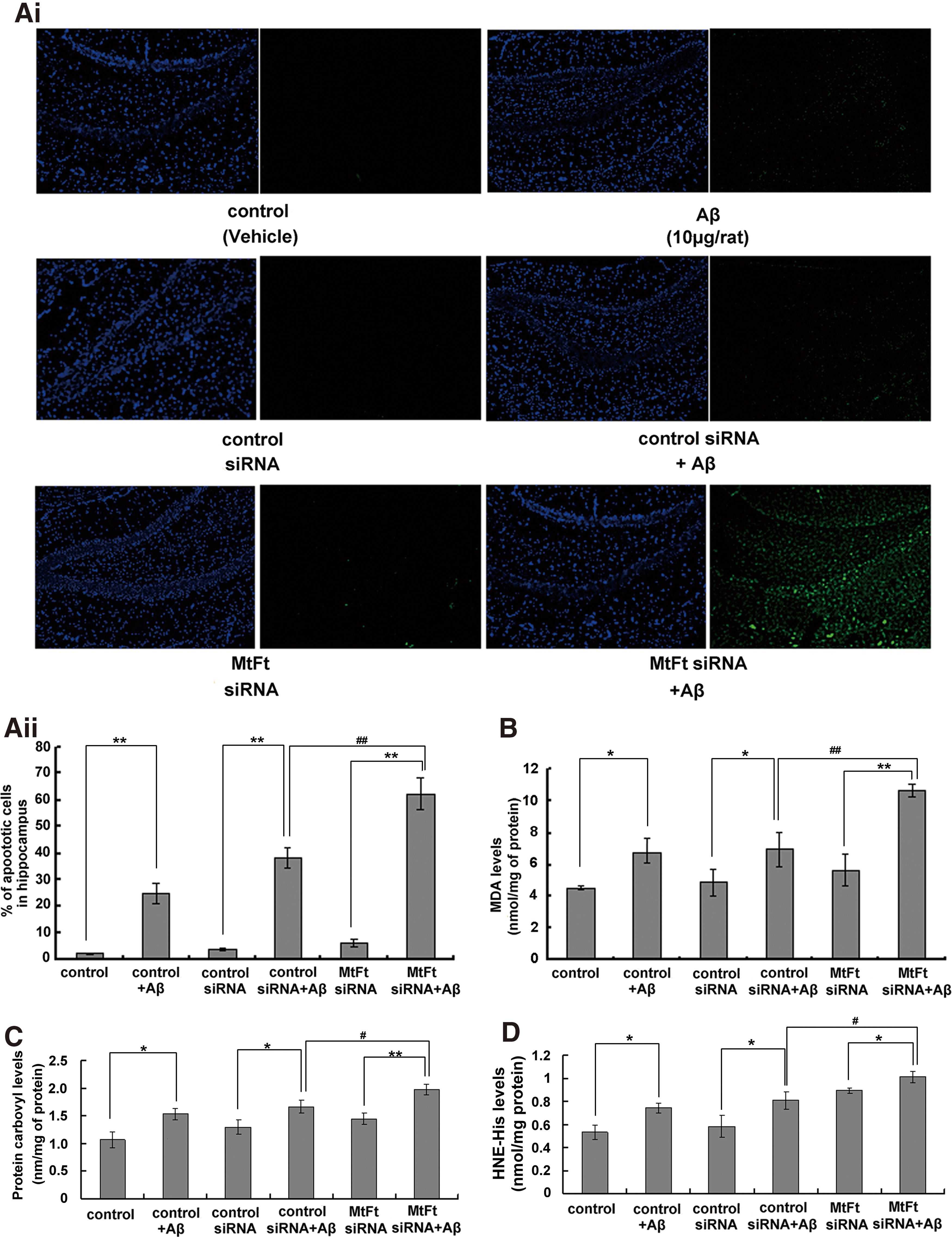
The effect of MtFt on Aβ25–35-induced neuronal cell apoptosis, malonyl dialdehyde levels, protein carbonyls, and hydroxynonenal–histidine in the hippocampus of rats
Aβ25–35 induced prominent cell apoptosis (Fig. 3Ai, Aii). Knockdown of MtFt exacerbated the neuronal cell apoptosis induced by Aβ25–35 in the dentate gyrus of the hippocampus (∼62%). The predominant cells affected were neurons (Supplementary Fig. S2). Our results also show that the concentrations of malonyl dialdehyde (MDA), protein carbonyls, and hydroxynonenal–histidine (HNE–His) increased in the hippocampus of the control group and control siRNA group after injection of Aβ25–35, but this increase was significantly greater when MtFt was downregulated (Fig. 3B–D). This finding is consistent with a previous report that lipid peroxidation increases after treatment with Aβ (13).
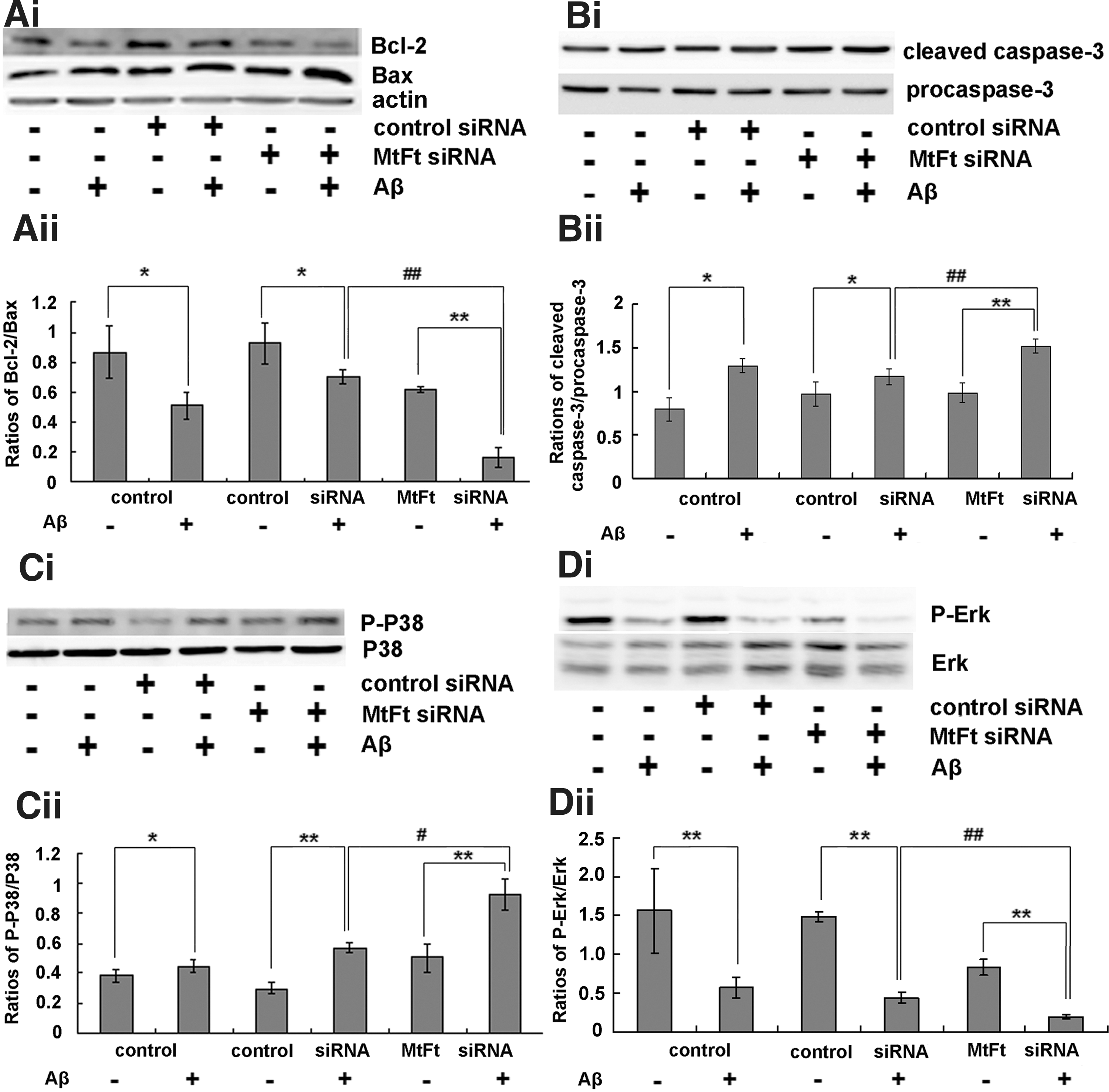
The effect of MtFt on the Bcl-2/Bax ratio, caspase-3 activation, and extracellular signal-regulated kinase/P38 MAPK cell signaling in rats
The ratio of Bcl-2/Bax has been widely used to monitor the degree of apoptosis (27). Injection of MtFt siRNA significantly decreased the ratio of Bcl-2 to Bax (Fig. 4Ai, Aii) and increased the levels of cleaved caspase-3 (Fig. 4Bi, Bii) after Aβ25–35 treatment in rats. These results indicate that decreased expression of MtFt can prominently influence the Bcl-2/Bax ratio and lead to caspase-3 activation and a resulting increase in apoptosis after Aβ25–35 treatment in rats. The activation of MAPK is implicated in oxidative-stress-induced cell death. As shown in Figure 4Ci and Cii, Aβ25–35 significantly induced the activation of P-P38 in the hippocampus. After treatment with MtFt siRNA, P-P38 levels were remarkably enhanced, and the level of P-extracellular signal-regulated kinase (Erk) was less than that of the control and control siRNA groups after treatment with Aβ25–35 in hippocampus (Fig. 4Di, Dii).
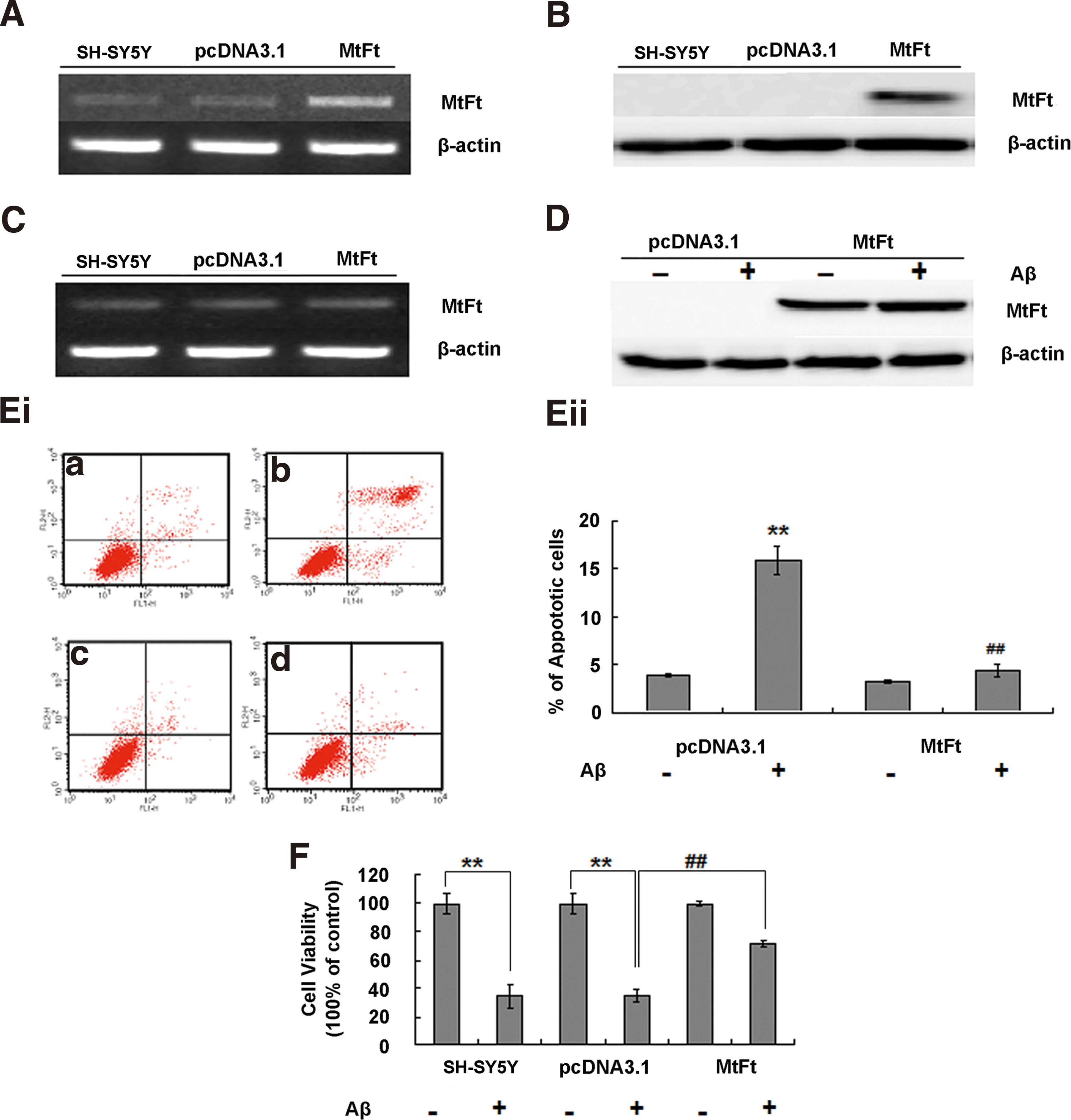
The effect of MtFt on Aβ25–35-induced neuronal cell toxicity in vitro
Cells stably transfected with MtFt showed increased expression of MtFt mRNA and protein (Fig. 5A, B). The endogenous expression of human MtFt was not changed by MtFt transfection (Fig. 5C). MtFt was slightly increased by Aβ25–35 treatment in MtFt-overexpressing cells (Fig. 5D). The ratios of apoptotic control (pcDNA3.1 transfected) cells increased (to ∼15%), but MtFt-overexpressing cells showed no significant difference (Fig. 5Ei, Eii) following Aβ25–35 treatment. These findings suggest that MtFt has a neuroprotective role against Aβ25–35-induced cell damage. We further examined the effect of MtFt on cell viability using the MTT method. Treatment with Aβ25–35 significantly decreased the viability of SH-SY5Y and pcDNA3.1-transfected SH-SY5Y cells (Fig. 5F) by ∼65% in each case; however, in the MtFt-transfected cells, viability decreased only slightly, and this change in viability was significantly different than that in the pcDNA3.1-transfected group. A similar protective role of MtFt on Aβ1–42-induced neurotoxicity was also detected (Supplementary Fig. S3).
The effect of MtFt on Aβ25–35-induced ROS generation in SH-SY5Y cells
6-OHDA can significantly increase ROS production in SH-SY5Y and pcDNA3.1 cells, resulting in cell apoptosis (27). ROS production increased more than twofold in untransfected and pcDNA3.1-transfected SH-SY5Y cells after Aβ25–35 treatment; however, ROS production did not change in MtFt-overexpressing cells (Fig. 6A). Thus, overexpression of MtFt can largely prevent Aβ25–35-induced ROS generation in cells.

MtFt expression attenuates the Aβ25–35-induced reduction in mitochondrial membrane potential and prevents cytochrome C release
Measuring the mitochondrial membrane potential (MMP) in living cells is commonly used to assess the metabolic activity and functional integrity of mitochondria (41). To examine the underlying protective mechanism of MtFt in mitochondria, MMPs were measured. Aβ25–35 significantly decreased the MMP in SH-SY5Y and control cells but not in MtFt-overexpressing cells (Fig. 6B). These results indicate that MtFt expression is able to prevent the loss of membrane integrity and protects against the mitochondrial damage induced by Aβ25–35.
Release of cytochrome C into the cytoplasm is characteristic of apoptosis, and it can activate a series of apoptosis cascades leading to cell death. Figure 6Ci and Cii shows that Aβ25–35 significantly increased the release of cytochrome C in SH-SY5Y and control cells but not in MtFt-overexpressing cells. These results further emphasize that MtFt expression strongly prevents the apoptosis of SH-SY5Y cells associated with Aβ25–35 treatment.
The effect of MtFt on the Bcl-2/Bax ratio and caspase-3 activation in cells
In vivo, we found that the ratio of Bcl-2 to Bax decreased and that cleaved caspase-3 levels increased significantly after knocking down MtFt in the Aβ25–35-treated rat hippocampus. In contrast, the ratio of Bcl-2 to Bax did not change noticeably in MtFt-overexpressing cells when cells were treated with Aβ25–35. However, in SH-SY5Y and pcDNA3.1 cells, the Bcl-2/Bax ratio decreased significantly compared with the control group (Fig. 7Ai, Aii). Aβ25–35 treatment dramatically activated caspase-3 in SH-SY5Y and pcDNA3.1 cells but not in MtFt cells (Fig. 7Bi, Bii). These observations indicate that MtFt can maintain the normal ratio of Bcl-2 to Bax and prevent caspase-3 activation to protect the cells against Aβ25–35 toxicity.

MtFt represses Aβ25–35-induced activation of Erk/P38/MAPK in cultured cells
The activation of MAPK is implicated in oxidative-stress-induced cell death. MtFt overexpression blocked the activation of P-P38 and P-Erk (Fig. 7C, D). These results indicate that Erk/P38/MAPK signaling may be involved in MtFt-mediated protection against Aβ25–35 toxicity in cultured cells.
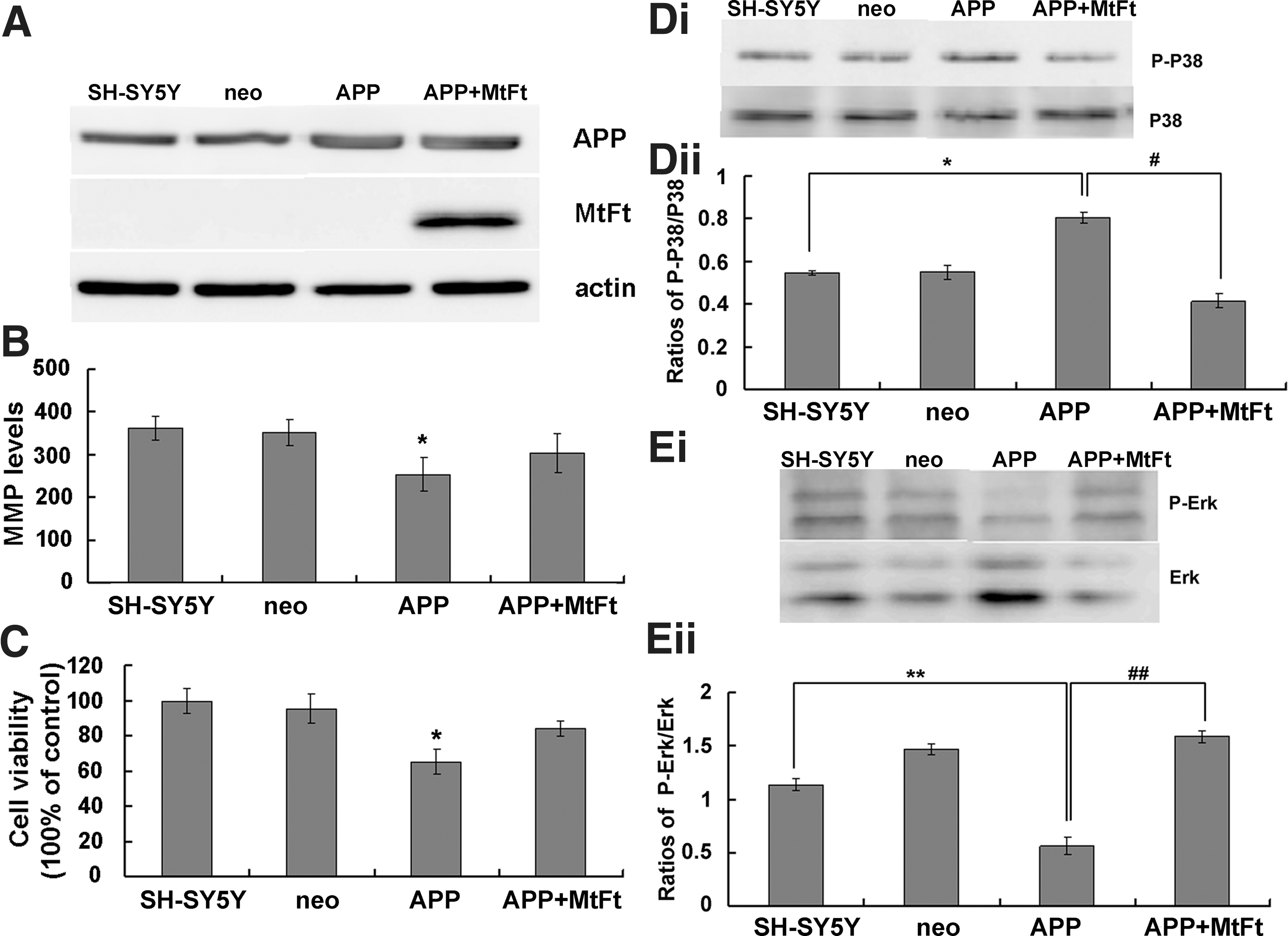
MtFt contributes to the oxidative damage and signaling associated with endogenous amyloid precursor protein
To determine whether MtFt can block endogenously derived amyloid precursor protein (APP)–induced oxidative damage, we transiently transfected plasmids that expressed APP into MtFt cells (Fig. 8A). MMP and cell viability analyses indicated that MtFt expression reduced the damage induced by APP (Fig. 8B, C). The P-P38/P38 and P-Erk/Erk ratios also changed significantly after APP transfection into MtFt cells (Fig. 8D, E). These results indicate that MtFt has a role not only in ameliorating Aβ25–35 damage but also in reducing the toxicity associated with APP.
MtFt decreases labile iron pool levels by regulating iron redistribution in Aβ25–35-treated cells
Iron is an essential cofactor for many proteins, particularly those involved in oxidative metabolism; however, excess free iron contributes to the enhanced generation of ROS and oxidative stress (39). To clarify the mechanisms by which MtFt depresses ROS levels in Aβ25–35-treated cells, we measured the levels of the labile iron pool (LIP). MtFt overexpression slightly decreased LIP levels but dramatically inhibited the elevation of LIP levels in cells treated with Aβ25–35 (Fig. 9A).

To clarify the change of LIP in MtFt protection, iron-related proteins were also measured. The iron uptake proteins TfR1 and DMT1 (+IRE) decreased and the iron release protein FPN1 increased in pcDNA3.1 cells after Aβ25–35 treatment (Fig. 9B, C). These changes in protein expression are consistent with increased levels and regulation of cytoplasmic iron via the IRE–IRP system. Consistent with this idea, H-ferritin levels also increased (Fig. 9B). Interestingly, DMT1 (−IRE) expression also significantly increased (Fig. 9C), which may explain why iron levels increased in Aβ25–35-treated cells. Overexpression of MtFt increased TfR1 and decreased H-ferritin levels (Fig. 9Bi, Bii), suggesting a reduction in cytoplasmic iron levels, which is in accord with our previous studies (27). After treatment with Aβ25–35, TfR1, DMT1 (+IRE) or DMT1 (−IRE), FPN1, and ferritin did not change significantly in the MtFt cells (Fig. 9B, C). These results may explain why LIP increases in the SH-SY5Y and pcDNA3.1 cells but not in the MtFt cells; MtFt protein acts to redistribute iron from the cytoplasm to the mitochondria, thereby decreasing LIP after treatment with Aβ25–35. The increase in DMT1 (−IRE) levels was restrained by MtFt overexpression, which decreased the iron uptake induced by Aβ25–35 treatment.
Discussion
Iron is absolutely required by virtually every cell, and cells of the central nervous system, which have high levels of metabolic activity and oxygen consumption, have particularly high iron requirements. However, excess iron can catalyze the generation of ROS (27); therefore, iron is considered an important factor contributing to neurotoxicity in several neurodegenerative disorders, including AD (17). Oxidative stress has been reported in the brains of AD patients. Aβ-associated free radicals can initiate lipid peroxidation, protein oxidation, ROS formation, and mitochondrial Ca2+ accumulation, eventually leading to the death of neurons. Other potential sources of oxidative stress in the pathogenesis of AD include the concentration of iron, the activation of microglial cells, and a decrease of complex IV activity. There is strong evidence to suggest that the MAPK pathway is strongly activated in AD (16).
MtFt, a recently discovered H-ferritin-like protein that is expressed only in mitochondria, is found in tissues with high levels of metabolic activity and oxygen consumption, including the testes, brain, heart, and kidneys. This tissue distribution pattern suggests that the major role of MtFt may be to protect mitochondria from iron-dependent oxidative damage. Recent research suggests that MtFt is involved in the pathology of AD and may play a neuroprotective role by reducing oxidative stress; however, the mechanism of this action has not been reported (35).
In the current study, we found that the expression of MtFt increased with age over the period from weeks 12 to 88 in the hippocampi of rats (Fig. 1). Cytochrome C and oxidative stress play a central role in apoptosis by signaling the cell to begin the process of programmed cell death (9). To confirm the protective role of MtFt in Aβ-induced neurotoxicity, siRNA was injected into the hippocampus of rats. We demonstrated that decreased expression of MtFt significantly increased the release of cytochrome C into the cytoplasm (Fig. 2B), thereby increasing the number of apoptotic cells (Fig. 3Aii) and decreasing the Bcl-2/Bax ratio (Fig. 4Aii). Thus, MtFt plays a clear neuroprotective role in vivo. After treatment with Aβ25–35, MtFt knockdown aggravated apoptosis in the hippocampus and oxidative damage to the tissue, as evidenced by increased levels of MDA, protein carbonyls, and HNE–His (Fig. 3A–D). The activities of the mitochondrial complex enzymes I–III (A), II (B), and IV (C) were also significantly decreased (Supplementary Fig. S1). To verify that the increased apoptosis was related to the modulation of MtFt levels, we carried out further studies using SH-SY5Y cells that stably overexpressed MtFt. MtFt overexpression reduced cellular apoptosis in response to Aβ25–35 and reduced the production of ROS, as predicted by the animal studies (Figs. 5 and 6). These observations demonstrate that MtFt has a protective effect in the brain, and the regulation of MtFt expression is a potential approach to diminish Aβ25–35-induced oxidation and cell apoptosis.
It is well documented that developmental neuronal cell body degeneration requires the apoptotic effectors Bax and caspase-3 (36) and that Bcl-2 protects neurons against oxidative stress and apoptosis (31). In our studies, we observed an increase in caspase-3 protein, a reduction in the Bcl-2/Bax protein ratio, and a loss of MMP following Aβ25–35 treatment (6). MtFt gene knockdown decreased the Bcl-2/Bax ratio and enhanced caspase-3 activation (Fig. 4A, B). Opposite effects were observed when MtFt was overexpressed in SH-SY5Y cells that were treated with Aβ25–35 (Fig. 7A, B).
There is a large amount of evidence that demonstrates that Aβ-induced neuronal injury triggers transcriptional and post-transcriptional processes that regulate neuronal fate, including the activation of MAPK pathways by neurotrophins and neurotransmitters and the induction or activation of pro- and antiapoptotic proteins (e.g., those of the Bcl-2 family) (37). Among the MAPK pathways, P38 is often implicated in cell death (4), and Erk has been widely associated with cell survival (19). Recent reports have shown that Erk may act as a physiological Bcl-2 kinase to prevent cell death in response to cytotoxic stimuli (10) and that activation of the P38 MAPK pathway leads to P53 phosphorylation (30), which, in turn, stimulates the expression of Bax in Aβ-induced cells (11). This suggests that Erk plays a neuroprotective role in cortical neurons and that P38 exerts toxic effects (37). Our study showed that MtFt siRNA treatment decreased P-Erk expression and increased P-P38 levels after the injection of Aβ25–35. In SH-SY5Y cells, after the transfection of MtFt gene, the opposite results were obtained. In summary, MtFt may have direct neuroprotective effects against Aβ25–35 toxicity via activation of the MAPK pathway in neurons.
An increasing number of recent studies have suggested that oxidative stress may be associated with AD neurodegeneration. Metals, such as iron, copper, and zinc, have been shown to be key factors in biochemical reactions that produce free radicals, which lead to peroxidation of cellular lipids and neuronal damage or death (3). Increased iron concentrations are found in specific brain areas of AD patients (20). In this study, we found that overexpression of MtFt decreased LIP levels and significantly reduced Aβ25–35-induced ROS levels. The increase in LIP levels that occurred in response to Aβ25–35 treatment decreased the expression of the iron import proteins TfR1 and DMT1 (+IRE) and increased the expression of the iron export protein FPN1. A schematic representation of the mechanisms suggested by our study is provided in Figure 10. We propose that Aβ25–35 increases DMT1 (−IRE) expression, which in turn enhances cellular iron uptake, leading to an increase in LIP levels and enhanced ferritin levels. The excess LIP donates electrons for the generation of ROS and lipid peroxidation. These changes may cause the cell to begin the process of programmed cell death through the MAPK pathway (by increasing P38 phosphorylation and decreasing that of Erk). MtFt could lead to cellular iron redistribution, wherein large amounts of iron are translocated to the mitochondria and LIP levels are decreased (21). Our data suggest that MtFt can attenuate Aβ25–35-induced neurotoxicity and reduce oxidative damage through Erk/P38 kinase signaling and that these effects are coordinately regulated by intracellular LIP levels.

The current work is the first to demonstrate that the expression of MtFt in the rat hippocampus is age related and that its neuroprotective effect on the toxicity induced by Aβ25–35 may depend on the MAPK pathway. Appropriate regulation of MtFt expression may prevent the damage to neuronal cells induced by neurotoxins and pathologic conditions such as those observed in patients with AD.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium, fetal calf serum, and MTT were purchased from Invitrogen. Aβ25–35, annexin V/PI, rhodamine 123, calcein-AM, and 2,7-dichlorofluorescein diacetate (DCFH-DA) were purchased from Sigma. TRIzol reagent, Lipofectamine 2000, and rat MtFt siRNA were purchased from Invitrogen. Anti-HA, Bcl-2, Bax, and cytochrome C antibodies were purchased from Santa Cruz Biotechnology. Antibodies to Erk, phosphorylated Erk (P-Erk), P38, phosphorylated P38 (P-P38), and caspase-3 were purchased from Cell Signaling Technology. H-ferritin, L-ferritin, DMT1 (+IRE), DMT1 (−IRE), and β-actin antibodies were purchased from Alpha Diagnostic International. The anti-MtFt antibody was a gift from Professor Sonia Levi.
Animals
Adult male Sprague Dawley rats were obtained from the Experimental Animal Center of Hebei Medical University. Animals were housed in a temperature- and humidity-controlled environment on a 12-h light–dark cycle with free access to food and water. All experiments were approved by the Animal Care and Use Committee of the Hebei Science and Technical Bureau in the RPC.
siRNA generation and treatment
siRNA targeting rat MtFt was obtained commercially from Invitrogen. The sequences are UAAGCUUCUCUGCAUGCUCCCUCUC (siRNA1) and GAGAGGGAGCAUGCAGAGAAGCUUA (siRNA2). Transfection of siRNAs into the hippocampus of rats was carried out using Lipofectamine 2000, and the reagent was delivered as described in the following section.
Preparation of aggregated Aβ25–35 and rat surgery
Aggregated Aβ25–35 was solubilized in sterile isotonic saline solution [pH 7.4 (28)] to a final concentration of 2 μg/μl and then incubated at 37°C for 36 h. Five microliters of this solution was injected over 5 min into the rat hippocampus (18). The rats were divided randomly into six groups with six rats in each group. The control group was injected with 0.9% saline. The second group was injected with Aβ25–35. The third group was injected with control siRNA. The fourth group was injected with Aβ25–35 24 h after the injection of control siRNA. The fifth group was injected with MtFt siRNA. The last group was injected with Aβ25–35 24 h after the injection of MtFt siRNA. The rats were then kept for 10 days under normal conditions. Three rats from each group were dissected for biochemical and molecular analysis of the hippocampus, and the brains of the other three rats were fixed in 50 ml of phosphate buffered solution containing 4% paraformaldehyde. Serial coronal sections of the brains (20 μm) were cut with a freezing microtome (13).
Cell culture and treatment
Plasmids expressing mouse MtFt (MtFt-pcDNA3.1) or the corresponding empty vector were transfected into SH-SY5Y cells as previously described (27). After SH-SY5Y cells (SH-SY5Y), MtFt-transfected cells (MtFt), and vector-transfected cells (pcDNA3.1) had grown to ∼70% confluency, 25 μM Aβ25–35 was added. The cells were then incubated at 37°C for 24 h prior to analysis.
Western blot and reverse transcription-PCR analysis
Protein expression was assessed by western blotting as previously described (27). mRNA expression was assessed by reverse transcription-PCR using total RNA extracted with TRIzol reagent according to the literature (27). The primer sequences used are listed in Supplementary Table S1. All of the experiments were performed at least three times. The relative band intensities of the proteins are expressed as the ratio of each to β-actin.
Detection of apoptosis
The presence of apoptosis in the dentate gyrus of rat hippocampi was assessed by the terminal deoxynucleotidyl transferase-mediated FITC-dUDP nick-end labeling method (13), and apoptosis in SH-SY5Y cells was measured by flow cytometry using annexin V/PI staining as previously described (27).
Measurement of lipid peroxidation and protein carbonyl levels
MDA, a reliable marker of lipid peroxidation, was determined with thiobarbituric acid according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute). The levels of protein carbonyls and HNE-His were determined as previously described (13).
Measurement of intracellular ROS and MMP
The level of intracellular ROS was quantified by measuring the fluorescence of DCFH-DA according to the methods of Bass et al. (2). Changes in MMP were assayed by measuring the retention of rhodamine 123 as previously described (27).
Measurement of the intracellular LIP
Intracellular LIP levels were measured according to the literature (27). For these studies, cells were treated with 25 μM Aβ25–35.
Statistical analysis
All experiments were performed at least in triplicate. One-way analysis of variance was used to estimate overall significance and was followed by Tukey's post hoc test corrected for multiple comparisons. Data are presented as the mean±standard deviation. A probability level of 95% (p<0.05) was considered significant.
Footnotes
Acknowledgments
This work was supported by the National Natural Sciences Foundation of China (30871260, 10979025, 30930036), the Excellent Youth Foundation of Hebei Province (C2010002032), and the Natural Science Foundation of Hebei Province (10966120D). Anti-MtFt antibody was a generous gift from Professor Sonia Levi and Dr. Paolo Santambrogio, Milan.
Author Disclosure Statement
The authors declare no competing financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.